
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparative analysis of supercritical fluid-based and chemical-based decellularization techniques for nerve tissue regeneration†
Beom-Seok
Kim
a,
Jeong-Uk
Kim
b,
Jae Woo
Lee
b,
Kyung Min
Ryu
b,
Rachel H.
Koh
b,
Kyoung-Ha
So
*bc and
Nathaniel S.
Hwang
 *abcd
*abcd
aInterdisciplinary Program in Bioengineering, Seoul National University, Seoul, 08826, Republic of Korea
bSchool of Chemical and Biological Engineering, Institute of Chemical Processes, Seoul National University, Seoul, 08826, Republic of Korea
cBio-MAX Institute, Institute of Bio-Engineering, Seoul National University, Seoul, 08826, Republic of Korea
dInstitute of Engineering Research, Seoul National University, Seoul, 08826, Republic of Korea
First published on 12th February 2024
Abstract
Axon regeneration and Schwann cell proliferation are critical processes in the repair and functional recovery of damaged neural tissues. Biomaterials can play a crucial role in facilitating cell proliferative processes that can significantly impact the target tissue repair. Chemical decellularization and supercritical fluid-based decellularization methods are similar approaches that eliminate DNA from native tissues for tissue-mimetic biomaterial production by using different solvents and procedures to achieve the final products. In this study, we conducted a comparative analysis of these two methods in the context of nerve regeneration and neuron cell differentiation efficiency. We evaluated the efficacy of each method in terms of biomaterial quality, preservation of extracellular matrix components, promotion of neuronal cell differentiation and nerve tissue repair ability in vivo. Our results indicate that while both methods produce high-quality biomaterials, supercritical fluid-based methods have several advantages over conventional chemical decellularization, including better preservation of extracellular matrix components and mechanical properties and superior promotion of cellular responses. We conclude that supercritical fluid-based methods show great promise for biomaterial production for nerve regeneration and neuron cell differentiation applications.
1 Introduction
A peripheral nerve injury (PNI) represents a traumatic pathological condition that can manifest in various regions of nervous tissues in the human body, excluding the cerebral and spinal components (CNS: central nervous tissue). Incidences of traumatic PNI account for more than 80% of reported nerve injuries within the upper extremities category.1 Due to their abundant lateral location in the extremities and vacancies of the skeletal protection framework, PNIs occur more frequently than CNS injuries. The cost of PNI treatments has been estimated to be about 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 USD, and over 0.5 million casualties were reported for traumatic PNI cases in the USA.2 To treat PNI, stem cell therapies have been actively investigated for nerve tissue regeneration to accelerate the intrinsic growth function of peripheral neurons in response to supply of stem cells.3 Stem cell therapy provides a stem cell derived Schwann cell population to PNI and supports neuronal axon growth and neuronal cell differentiation in vivo.4,5 However, in these cell-based therapies, it is hard to maintain their self-renewal and multipotent ability under shear stress conditions and cells can easily dissipate in vulnerable liquid-like tissue environments.6 Implanted stem cells are expected to contribute partial neuro-regeneration by promoting inherent cellular reactivity, but it is difficult to recapitulate the cylindrical tissue shape of the original nerve structure that bridges the distal and proximal ends of the peripheral nerve gap injury.7
000 USD, and over 0.5 million casualties were reported for traumatic PNI cases in the USA.2 To treat PNI, stem cell therapies have been actively investigated for nerve tissue regeneration to accelerate the intrinsic growth function of peripheral neurons in response to supply of stem cells.3 Stem cell therapy provides a stem cell derived Schwann cell population to PNI and supports neuronal axon growth and neuronal cell differentiation in vivo.4,5 However, in these cell-based therapies, it is hard to maintain their self-renewal and multipotent ability under shear stress conditions and cells can easily dissipate in vulnerable liquid-like tissue environments.6 Implanted stem cells are expected to contribute partial neuro-regeneration by promoting inherent cellular reactivity, but it is difficult to recapitulate the cylindrical tissue shape of the original nerve structure that bridges the distal and proximal ends of the peripheral nerve gap injury.7
For this reason, to achieve ideal neuronal tissue reformation, polymeric cylindrical shape scaffolds were investigated with biocompatible materials with high mechanical properties to supply tissue like objects. The polyester family (PVA, PGA, PLGA, PCL and their copolymers) has been utilized for nerve scaffold fabrication because of their easy handling property and hydrolyzing property, which degraded naturally in specific wet circumstances.8,9 However, these synthetic materials lack cell binding moieties and tissue comprising polysaccharide and proteins which are essential to attract cell migration, one of the crucial steps of tissue formation. To overcome these limitations, simple blending of native tissue-derived proteins, the cell binding arginyl-glycyl-aspartic acid (RGD) peptide, and extracellular matrix (ECM) components has been implemented in nerve tissue regeneration studies.10,11 However, the polymeric material mixture and cell responsive material conjugation strategies are still not enough to replace complex living tissues that comprised functional proteins and the extracellular matrix (ECM) in terms of various material compositions and structures. To overcome the limitations of artificial nerve scaffolds, in clinical assessments, autologous nerve transplantation has been applied as the most preferable method for nerve defect treatment; however, the gold standard trials also faced challenges of insufficient supply of autologous tissues due to donor tissue-dependent neuroma and variations in size among autologous nerves.12 To address the quality of autologous nerve tissue in PNI treatments, allografts and xenografts have been employed. These approaches involve the use of immune suppressive agents to mitigate tissue rejection in recipients, albeit at the cost of reduced overall immune function.13 The host's immune system is considered as one of the major obstacles for the tissue regeneration studies using xenografts and allografts. The main cause of immune reaction derived regeneration impairments is known to originate from the cellular components like lipids and nucleic acids within the transplanted tissues. For this reason, to fabricate non-immunogenic native tissue derived scaffolds, the chemical decellularization technology was incorporated to fabricate an implantable nerve scaffold using Triton X-100, 200, sodium dodecyl sulfate (SDS), or sulfo betaine (SB) which could eliminate the cellular materials from tissue compartments.14,15 The chemical decellularization technology extracts cell responsive polysaccharides and proteins in tissues with detergents while eliminating immunogenic materials from them.16,17 For this selective elimination process, the decellularized tissues were applied to biomedical applications and clinical research works offering an ideal tissue microenvironment with a minimal risk of generic material derived immune rejection or inflammation.18 However, the chemical decellularization technique also caused original tissue material loss and damage to the ECM structural integrity as a result of harsh detergent based chemical processing and solvent changes. Furthermore, the anionic detergents used in the decellularization process have been reported to degrade the ECM components in tissues, which consequently decreases the biological responses and regenerative potential of the decellularized ECM.19
To decrease ECM and functional protein loss during the decellularization process, stable and non-ionic solvents, such as supercritical CO2, have been utilized in the tissue decellularization process.20 Supercritical phase CO2 has liquid and gas-like characteristics that make it diffuse instantaneously into solid structures and solubilize chemicals efficiently with a short reaction time.21 Furthermore, supercritical CO2 sustains mild operating conditions (10.3 to 27.6 MPa, 10 to 37 °C) during the process and also shows low reactivity towards polar proteins and ECM materials in tissues.22 Thus, the supercritical fluid-based decellularization method is expected to eliminate DNA materials more selectively and better preserve the ECM components in the native tissue than chemical-based methods with its own supercritical fluid dynamics characteristics and more shorter reaction time than chemical processes.
In this study, we have conducted chemical (SDS)-based and supercritical state fluid-based decellularization methods for the fabrication of sciatic nerve scaffolds. The two different decellularization methods were compared in terms of decellularization efficiency, scaffold–cell responses and therapeutic potential in vivo. We hypothesized that the supercritical fluid-based method can preserve more native tissue like the ECM, protein components and structural and mechanical characteristics after decellularization than chemical-based methods. The ECM and protein components in native and decellularized tissue were characterized with biochemical assays and proteomic analysis. Mechanical and structural changes were quantified via measuring the modulus of the ECM. The regenerative ability of the ECM was quantified with the neuronal cell reactivity via in vitro neuronal outgrowth tests and in vivo sciatic nerve injury recovery and muscle function rehabilitation. The supercritical fluid-based decellularization method preserved more quantitatively the remaining biomaterial components and their mechanical integrity and promoted advanced neuronal cell migration and axonal outgrowth and practical nerve regeneration.
2 Materials & methods
2.1 Reagents
Sodium dodecyl sulfate (Biorad, USA, 1610302), papain (Worthington, USA, 9001-73-4), sGAG (Biosynth Carbosynth, England, YC31458), 4% paraformaldehyde (PFA; Biosesang, Republic of Korea, BP031a), chloramine T (MP, USA, 7080-50-4), p-dimethylaminobenzal aldehyde (MP, USA, 100-10-7), bovine serum albumin (Gen Depot, USA, A0100-005), and Triton X-100 (Sigma Aldrich, USA, 9036-19-5).Phosphate buffered saline (PBS, Thermo Fisher Scientific, USA, 10010023), trypsin-EDTA (Thermo Fisher Scientific, USA, 27250018), normal goat serum (Thermo Fisher, USA, 31872), and RIPA lysis buffer (Thermo Fisher, USA, 89901) were purchased from Thermo Fisher Scientific.
Sucrose (Sigma Aldrich, USA, S0389), DMMB (Sigma Aldrich, USA, 341088), cysteine (Sigma Aldrich, USA, C7352), hydroxy-proline (Sigma Aldrich, USA, 51-35-4), Canada balsam (Sigma Aldrich, USA, 8007-47-4), and proteinase K (Sigma Aldrich, USA, 3115887001) were purchased from Sigma Aldrich.
2.2 Isolation and decellularization of sciatic nerve tissues
Sciatic nerves were isolated from porcine (Yorkshire, 6 month) biceps femoris after elimination of fat and skin. Isolated sciatic nerves were cleaned with PBS and treated with different concentrations of detergent solution (0.1, 0.5, 1% w/v of sodium dodecyl sulphate, SDS) at 37 °C for 30 h with gentle mixing. The nerve tissues were collected and stored at −80 °C until further use. The native porcine nerve tissues were prepared as described above. Tissues were then transferred into a medical container in supercritical fluid devices. 50 ml of pure ethanol was added to the container as a co-solvent. CO2 fluid was injected into the medical container at 200–400 bar. After 3 h of treatment, decellularized tissue was washed with PBS and stored in −80 °C freezer. DOF Inc, Korea conducted the supercritical fluid-based decellularization process and provided the processed samples.2.3 Histological assessment of sciatic nerve tissues
2.4 ECM characterization in nerve tissues
:1 volume ratio). Absorbance was measured using a microplate reader (Tecan, Switzerland, Infinite 200) at 525 nm wavelength. Standard lines for the calculation of related sGAG contents in nerve tissue were made with chondroitin sulfate sodium salt.
:1 volume ratio). Fluorescence of the dye was measured with a microplate reader (Tecan) at 480 nm excitation and 520 nm emission wavelength. A standard line for the calculation of relative DNA contents in nerve tissue was made with the Quant-iT Pico Green dsDNA stock.
000g for 60 min). And 100 μl of 0.05 M iodoacetamide in 8 M urea were added to purified protein solution over 25 min at room temperature. And 50 mM ammonium bicarbonate (pH 8.0) was added to the protein samples and centrifuged (14000g for 20 min) three times. After trypsin digestion (20 ng ml−1) at 37 °C overnight, the samples were vacuum dried. The dried samples were dissolved in trichloroacetic acid and transferred to LC/MS (Orbitrap Exploris 240, Thermo Fisher Scientific). The proteomic data were processed by the National Instrumentation Center for Environment Management mass spectrometry research department. Total protein contents, coverage, unique peptides, and proteomic scoring raw data were extracted and used for proteomic analysis.
2.5 Mechanical property characterization of nerve tissues
2.6 Neuronal cell culture
000 cells per slice and differentiated with nerve growth factor-containing (NGF; 100 ng ml−1) differentiation medium as described above.
:1, Abcam ab75810). Primary antibody solution was washed with buffer and incubated with secondary antibodies (500:1 Alexa fluoro 488). The stained tissue slices were imaged with a Zeiss 700 (Zeiss, Germany, LSM780).
2.7 Surface imaging of decellularized ECM with SEM
The decellularized nerve tissues were fixed with 2.5% (w/v) glutaraldehyde and 2% (v/v) aldehyde for 16 h. The fixed samples were transferred to 1% (v/v) osmium tetroxide solution and incubated for 1 h at 4 °C. After washing with deionized water 3 times for 5 min, each sample was dehydrated with different concentrations of ethanol (50, 70, 80, 90, 100, 100% w/v) and specimens were dried in a critical-point dryer (Thermo) and stored in the vacuum state until imaging by SEM. Dried tissue samples were fixed with carbon tape on the SEM mount and coated with Pt sputter for 1 min. The coated samples were imaged with Mini SEM (JCM-6000).2.8 In vivo experiment and characterization of regeneration of tissues
:1, abcam ab283654) and imaged by microscopy (EVOS 3000).
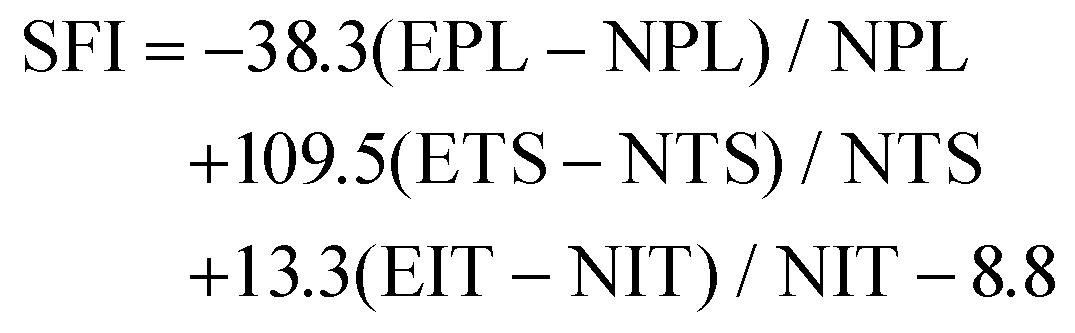 | (1) |
The variables were measured with ImageJ analysis tools by recording videos of the stride patterns of each rat for 8 weeks.
| Muscle weight ratio = (weight of (E)/weight of (N)) | (2) |
And muscular tissues (gastrocnemius) were stained with hematoxylin and eosin to measure the muscle fiber diameter and muscle fiber areas (whole 2D area of fibers). The quantification was performed with ImageJ software.
:1, Abcam ab75810) and S 100 (500:1, Abcam ab52642) for 16 h at 4 °C. The tissue samples were washed with buffer and then incubated with secondary antibodies (500:1 Alexa Fluoro 488, 594 ab150077, ab150080). The stained tissue slices were imaged with a Ti2 Eclipse microscope (Nikon, Japan, Eclipse Ti2)
2.9 Statistical analysis
All data were processed with Graphpad prism 5.0 and a two-tailed t-test and one-way ANOVA. Statistical calculations were performed. P-value = *p < 0.05, **p < 0.01, ***p < 0.005, #p < 0.05, ##p < 0.01, ###p < 0.005. The *symbol compared the control group versus SDS 0.5, SC groups. The #symbol compared the SDS0.5 group versus SC groups. Biorender SW was utilized to draw the schemes.3 Results
3.1. Decellularized ECM scaffold fabrication and characterization of ECM
The decellularized ECM scaffolds were prepared by using two different decellularization methods to remove genetic elements from animal tissues that may trigger immune reactions upon transplantation. A conventional decellularization technique using a chemical (SDS) and a novel technique using a supercritical fluid (SC) were compared, in terms of effective DNA removal while preserving protein and ECM contents of native tissues. To determine the optimal SDS concentration, porcine nerve tissues were treated with different concentrations of SDS solutions. Tissue deformation occurred from the outer surface of nerve tissues (Fig. 1A) and the extent of deformation increased with the SDS concentration (Fig. S1A†). The swelling ratio, which represents higher water contents in the polymeric network with lower crosslinking density, was determined to describe the tissue inter-network deformation. The swelling ratio of tissues increased with SDS treatment as compared with native tissues and SC samples and increased with higher concentration of SDS in the decellularization process (Fig. 1B and Fig. S1B†). The DNA remnants were not observed in H&E staining for groups that were treated with over 0.5% SDS (Fig. 1C). In SDS-based decellularization process, we confirmed that the concentration of immunogenic DNA in the tissues decreased to less than 50 ng mg−1 when the SDS concentration was maintained at or above 0.5% (w/v) (Fig. 1D). The tissue degradation and deformation during the process were further characterized using MTC, safranin-O staining and ECM-quantifying assays (hydroxyproline: SDS 0.5: 52.21 ± 3.32, SC: 67.39 ± 4.23), (sGAG: SDS 0.5: 68.33 ± 6.03, SC: 82.09 ± 8.11%), (Fig. 1E–H). It seems that the ECM content generally decreased with the increase in SDS concentration, while the ECM content of supercritical fluid-based decellularized tissue (SC group) remained quantitatively higher than that of chemical-based decellularized tissue (SDS group). Therefore, we optimized the SDS concentration for chemical-based decellularization to 0.5% (w/v), fulfilling minimum thresholds of <50 ng ml−1 DNA contents and ECM degradation. So, in further experiments, the 0.5% (w/v) SDS group was directly compared with the SC group.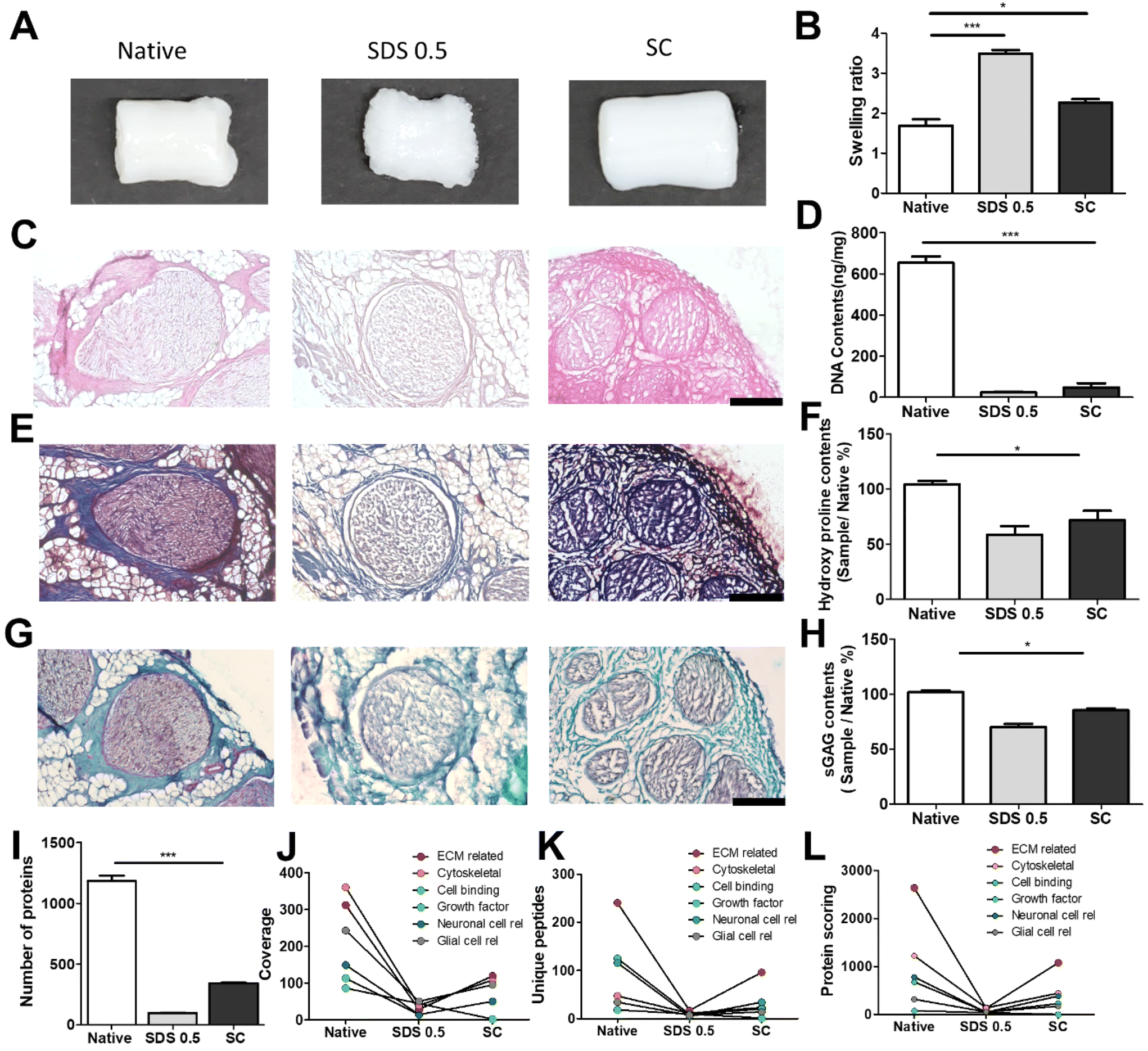 | ||
| Fig. 1 ECM and proteomic component analysis of native tissue and decellularized tissues by two different decellularization methods. (A) representative images of native and decellularized nerve tissues. (B) The swelling ratio of native and decellularized tissues, (C) representative images of H&E staining of native and decellularized tissues, (D) measurements of DNA contents of native and decellularized tissues, (E) representative images of MTC staining of native and decellularized tissues, (F) measurements of hydroxyproline contents of native and decellularized tissues, (G) representative images of safranin-O staining of native and decellularized tissues, (H) measurements of sGAG content of native and decellularized tissues, and (I) quantification of how many kinds of proteins were found in native tissue and decellularized nerve tissues. (J–L) Proteomic analysis data of proteins in native and decellularized tissues (scale bar = 200 μm). Error bars indicate SD. n = 3, *p < 0.05, **p < 0.01, ***p < 0.005. | ||
To compare more specific protein contents in processed tissue, total protein content quantification and proteomic analysis were conducted on native and decellularized tissues. While total protein contents decreased in both decellularization methods compared to native tissues (1098 ± 125.12), the SC group (345 ± 48.23) showed higher preservation of protein contents after decellularization than the SDS 0.5 group (103 ± 22.38) (Fig. 1I). The ECM-related proteins were the most abundant protein type in decellularized nerve tissues, followed by cytoskeletal proteins. Other cell-responsive and neuronal, glial cell related proteins were also observed. In a similar manner of the total protein content results of tissues, the overall types of tissue derived proteins were reduced after the decellularization process. Additionally, the SC groups exhibited higher proteomic scoring compared to the SDS 0.5 group (Fig. 1J–L).
3.2 Mechanical characterization of decellularized tissue samples
It has been previously confirmed that the surface and microstructure of tissues were damaged by SDS treatment, which affects its mechanical properties. To measure the mechanical properties of decellularized and native tissues, the extension test was performed using a bidirectional extension method (Fig. 2A). The mechanical properties of the decellularized tissues were characterized with Young's modulus and critical tension strength parameters (Fig. 2B and C). The SC group had a relatively higher modulus compared to the SDS 0.5 group (SDS 0.5: 42 ± 3.5, SC: 57 ± 5.3 MPa). The critical strength of the SC group was higher than that of the SDS 0.5 group (SDS 0.5: 3.64 ± 0.89, SC: 4.13 ± 0.46 N). To investigate repeated external force-derived tissue deformation, repeated compressive indentation tests were conducted. The strain–stress curve data showed that the SDS group has significantly lower properties than the native or SC groups (Fig. 2D–F). As the number of cycles increased, the sequentially measured modulus gradually decreased in all the samples (Fig. 2G–I). There were larger modulus decreases in native tissue than in the SC group as the indentation cycle increased. The sequential modulus data of SC group show that it can maintain the modulus of initial stages even with repeated indentation procedures.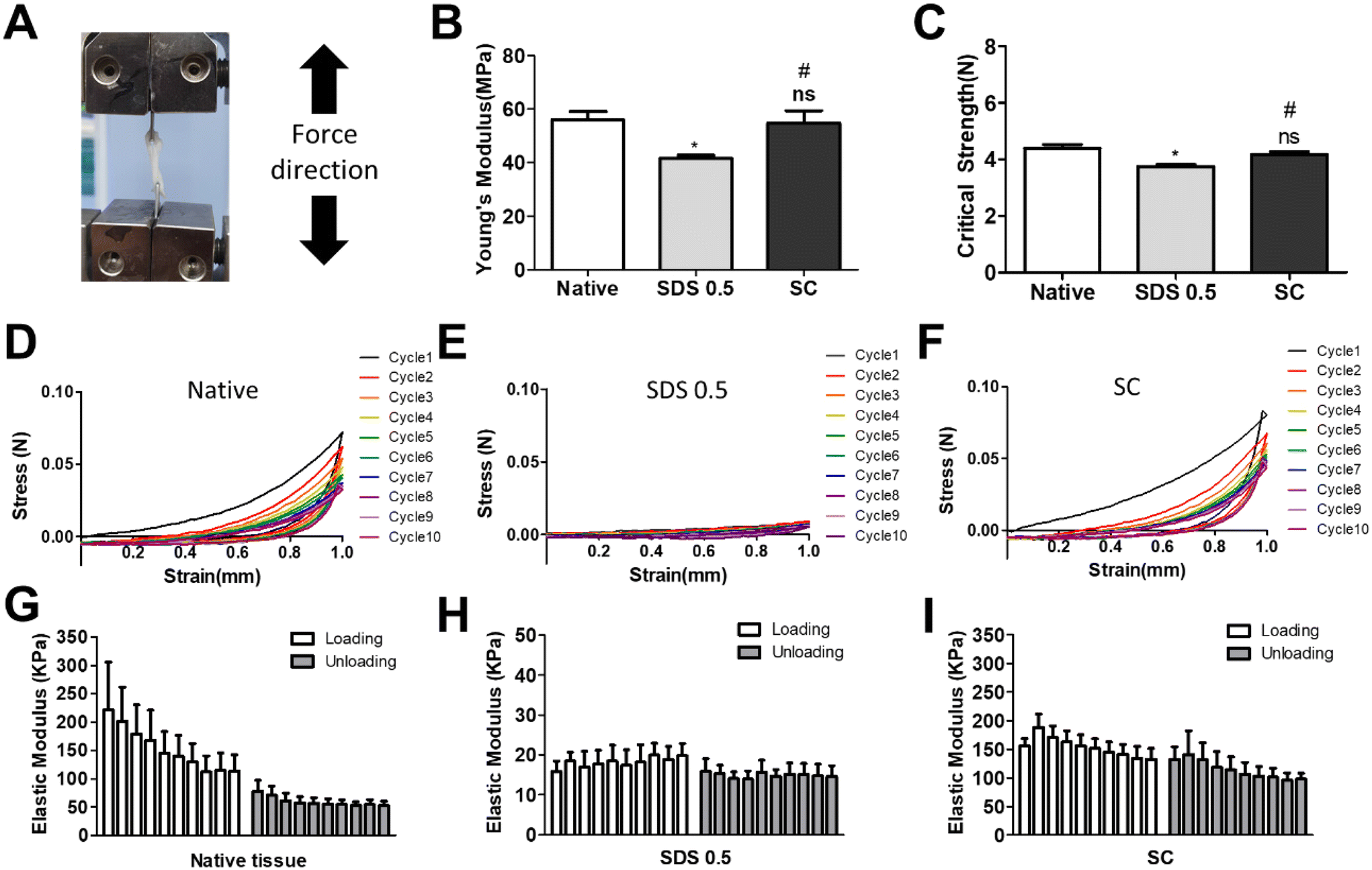 | ||
| Fig. 2 Mechanical characterization of native and decellularized tissues. (A) Representative image of teh extension test method of wet tissues. (B) Measurements of Young's modulus and (C) critical strength of native and decellularized tissues under extended conditions. (D–I) Compression test cycles and elastic modulus of calculation of native tissues (D and G), decellularized tissues with 0.5% SDS treatments (E and H), and decellularized tissues with supercritical fluid process (F and I). Error bars indicate SD. n = 3, P-value = *p < 0.05, **p < 0.01, ***p < 0.005. #p < 0.05, ##p < 0.01, ###p < 0.005. | ||
3.3 PC12 differentiation on tissue slice samples
In order to investigate the cellular reactivity of decellularized tissues, PC12 cells were cultured on the tissue slices. After PC12 differentiation, the amount of microfilament attachment to the tissue slices and the degree of differentiation were assessed through class III β-tubulin and actin staining (Fig. 3A and B). The value of the class III β-tubulin signal, which stands for neuronal microtubule formation, was significantly increased in the supercritical-fluid-treated decellularized tissue compared to the SDS-treated tissue (SDS 0.5: 28 ± 1.56, SC: 37 ± 2.33%). But the microtubule signal of the SC group was lower than that of the native group (native: 48 ± 6.92, SC: 37 ± 2.33%) (Fig. 3C). The actin signals were also measured after PC12 differentiation on nerve tissue slices. Higher actin signal was observed in the SC group than in SDS 0.5 and native groups (native: 42 ± 4.32, SDS 0.5: 37 ± 9.88, SC: 72 ± 20.65%) (Fig. 3D). To measure the neurite extension and outgrowth of differentiated PC12 cells, PC12 cells were cultured for 21 days under the same conditions as the short term culture (3, 7 days) to characterize the fully differentiated aspect of neuronal cells (Fig. 4A). As a result, the signal of class III β-tubulin was highest in the SC group (native: 175 ± 21.71, SDS 0.5: 183 ± 7.36, SC: 224 ± 37.65%) (Fig. 4B). Neurite length of PC12 cells was measured to confirm the most efficient microenvironment conditions for neuronal cell culture (Fig. 4C). As anticipated, the native group showed the longest neurites after neural differentiation of PC12 cells. The SC group exhibited neurites slightly shorter than those of the native group, but longer than those of the SDS 0.5 group (native: 84 ± 11.28, SDS 0.5: 43 ± 3.36, SC: 74 ± 18.35 μm).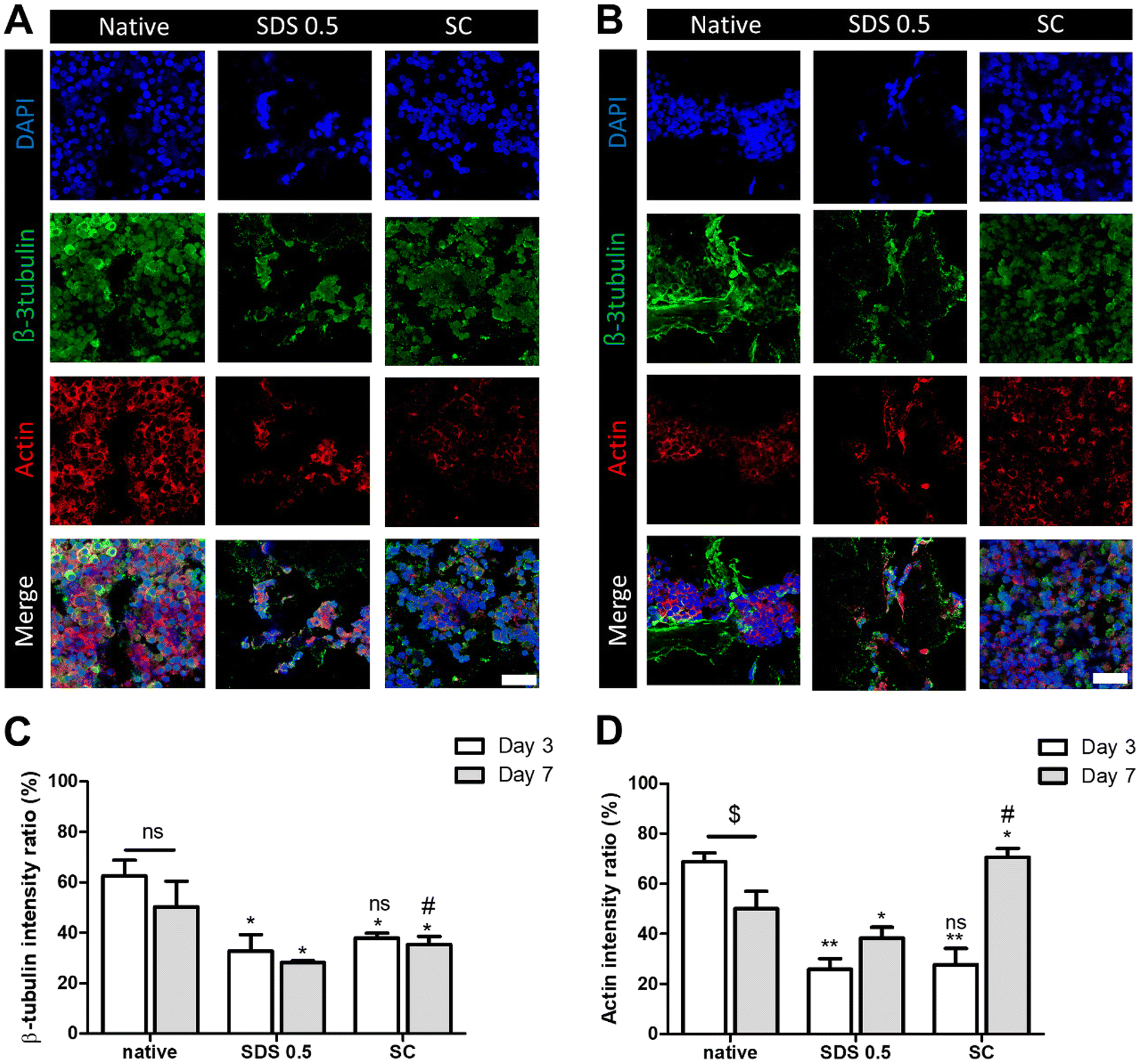 | ||
| Fig. 3 The PC12 cell culture and differentiation on nerve tissue slices. (A and B) The class III β-tubulin and actin staining of PC12 cells cultured on native and decellularized tissues at (A) day 3 and (B) day 7. (C and D) Relative fluorescence data of (C) class III β-tubulin and (D) actin staining (scale bar = 50 μm). Error bars indicate SD. n = 3, P-value = *p < 0.05, **p < 0.01, ***p < 0.005. #p < 0.05, ##p < 0.01, ###p < 0.005. | ||
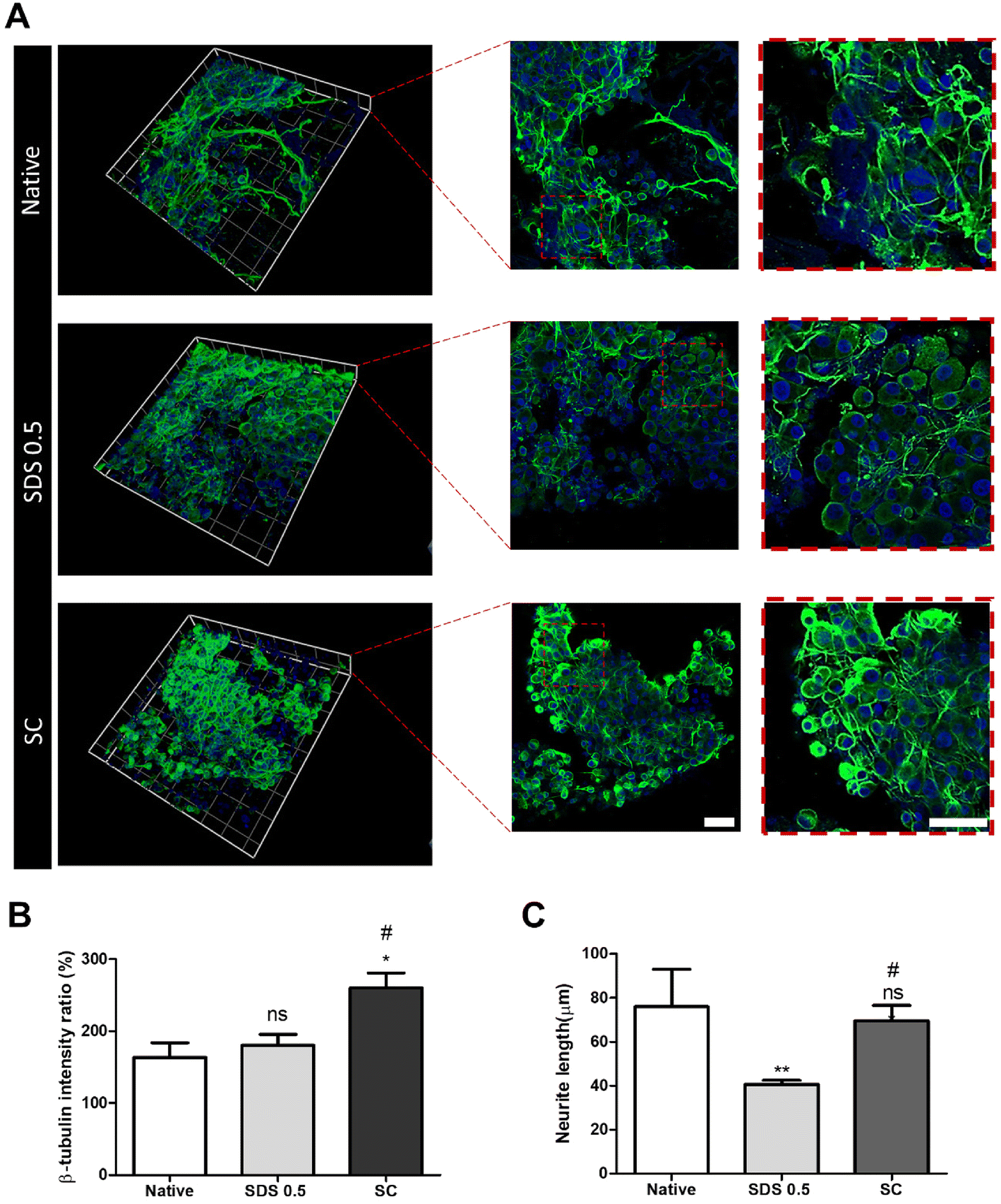 | ||
| Fig. 4 The PC12 cell culture and differentiation on nerve tissue slices for long-term period (21 days). (A) Representative images of the PC12 cell differentiation on nerve tissue slices for 21 days (B) relative fluorescence intensity measurements of the differentiated PC12 cells (Class III β-tubulin). (C) The neurite length measurements of differentiated PC12 cells (scale bar = 50 μm). Error bars indicate SD. n = 3, P-value = *p < 0.05, **p < 0.01, ***p < 0.005. #p < 0.05, ##p < 0.01, ###p < 0.005. | ||
3.5 Immune cell infiltration of implanted tissue samples
The decellularized and native tissue samples were subcutaneously implanted to verify the level of host immune response activation towards implanted decellularized tissue compared to native tissues (Fig. 5). In the first week post implantation, the ratios of immune cells were much higher in the native tissue group than any in other decellularized sample implantation groups (native: 103.36 ± 5.28, SDS 0.5: 48.29 ± 3.36, SC: 31.65 ± 8.12%) (Fig. 5B). At week 2 of implantation, the immune reactions were decreased in all experimental groups than one-week time point. The native tissue implanted group still showed higher population of immune cells in the implanted areas than the decellularized tissue implanted groups. Additionally, the SC group showed significantly decreased immune cells compared to both native and SDS groups (native: 34.65. 11 ± 2.15, SDS 0.5: 23.75 ± 8.26, SC: 8.65 ± 3.95%) (Fig. 5C). After 4 weeks of implantation of tissues, the immune reaction was maintained at a similarly high level to week 2 for the native group (native: 32.13 ± 4.32%), while the immune responses were increased in SDS 0.5 and SC groups at the 4 week time point compared to 2 weeks (SDS 0.5: 33.65 ± 11.36, SC: 16.29 ± 4.60%). The SC group showed lower host immune response than native and SDS 0.5 groups at week 4 which confirms the relatively low immunogenic characteristics of supercritical fluid-based decellularized tissues (SC group).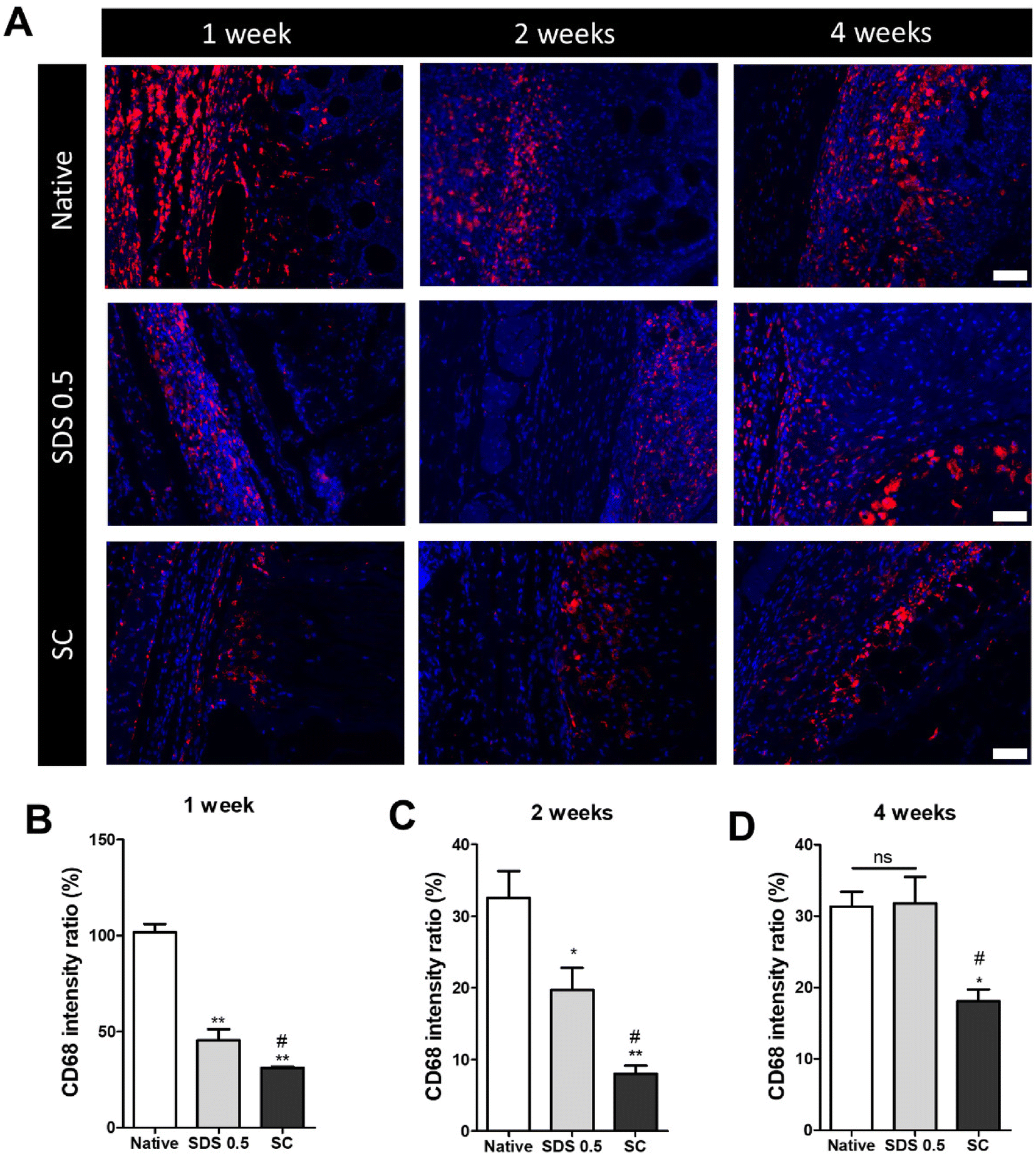 | ||
| Fig. 5 Immune cell infiltration ratio characterization of post implantation of native tissues and decellularized tissue to mouse skin tissues. (A) Representative images of CD68 marker staining of nerve tissue implanted area (B) relative fluorescence data of CD68 at 1 week (C) relative fluorescence data of CD68 at 2 weeks. (D) Relative fluorescence data of CD68 at 4 weeks (scale bar = 50 μm). Error bars indicate SD. n = 3, P-value = *p < 0.05, **p < 0.01, ***p < 0.005. #p < 0.05, ##p < 0.01, ###p < 0.005. | ||
3.6 Motor function and muscular tissue regeneration after implantation in vivo
To assess the regenerative potential of nerve tissue in animals, each decellularized tissue sample (SDS 0.5, SC) was transplanted into a rat nerve defect model. The decellularized tissues were transplanted and directly sutured to host nerve tissues after a nerve gap of 1 cm was made by medial sciatic nerve elimination. No graft group (control) was made with the same defect as the above experimental groups without any treatments. Motor function recovery was characterized by the sciatic function index test (Fig. 6B). The index was increased in all groups until 4 weeks. But the function recovery ratio was gradually decreased in the No graft group at 6 and 8 weeks. A higher motor function recovery index was noted in the SC group than in the SDS 0.5 group at the final regenerative phase (at 8 weeks) (SDS 0.5: −70.69 ± 3.36, SC: −66.79 ± 5.33). The muscle size of the surgery leg (left), from which the sciatic nerve had been dissected, was decreased compared to the opposite muscle (right), in which surgery had not been performed (Fig. 6C). The muscle volumes of the SDS 0.5 group and SC group were restored more than in the No graft group. Furthermore, the muscular tissue recovery was characterized by weight ratio calculation and muscular tissue histological assessments after surgery. The muscle weight ratio of the surgery legs was recorded higher in the SC groups than in the SDS 0.5 and control samples at 8 weeks post implantation (No graft: 16.36 ± 4.88, SDS 0.5: 25.33 ± 1.32, SC: 31.11 ± 4.33%) (Fig. 6D). Muscle fiber diameters and areas were quantified using histological images (Fig. 6E–G). The SC group showed the highest muscle fiber diameters at 4 weeks (No graft: 10.12 ± 1.33, SDS 0.5: 11.39 ± 1.32, SC: 14.11 ± 4.33 μm) (Fig. 6F). The muscle diameters were gradually increased as the time elapsed in the decellularized tissue implanted groups at 8 weeks (SDS 0.5 and SC group). However, the muscle fiber diameter of the No graft group was decreased in 8 weeks compared to 4 weeks. (No graft: 6.80 ± 2.36, SDS 0.5: 16.11 ± 5.31, SC: 22.09 ± 3.98 μm) (Fig. 6F). In a similar pattern with that of muscle fiber diameter data, the muscle fiber areas were recorded as the highest in the SC group at 4 weeks after implantation (Fig. 6G). There was no significant difference between the SDS 0.5 group and control at 4 weeks (No graft: 388.59 ± 73.67, SDS 0.5: 423 ± 62.20, SC: 796 ± 106.66 μm2) (Fig. 6F). The highest muscle fiber area was found in the SC group at 8 weeks after implantation also (No graft: 313 ± 68.36, SDS 0.5: 815 ± 121.22, SC: 1496 ± 134.52 μm2) (Fig. 6G).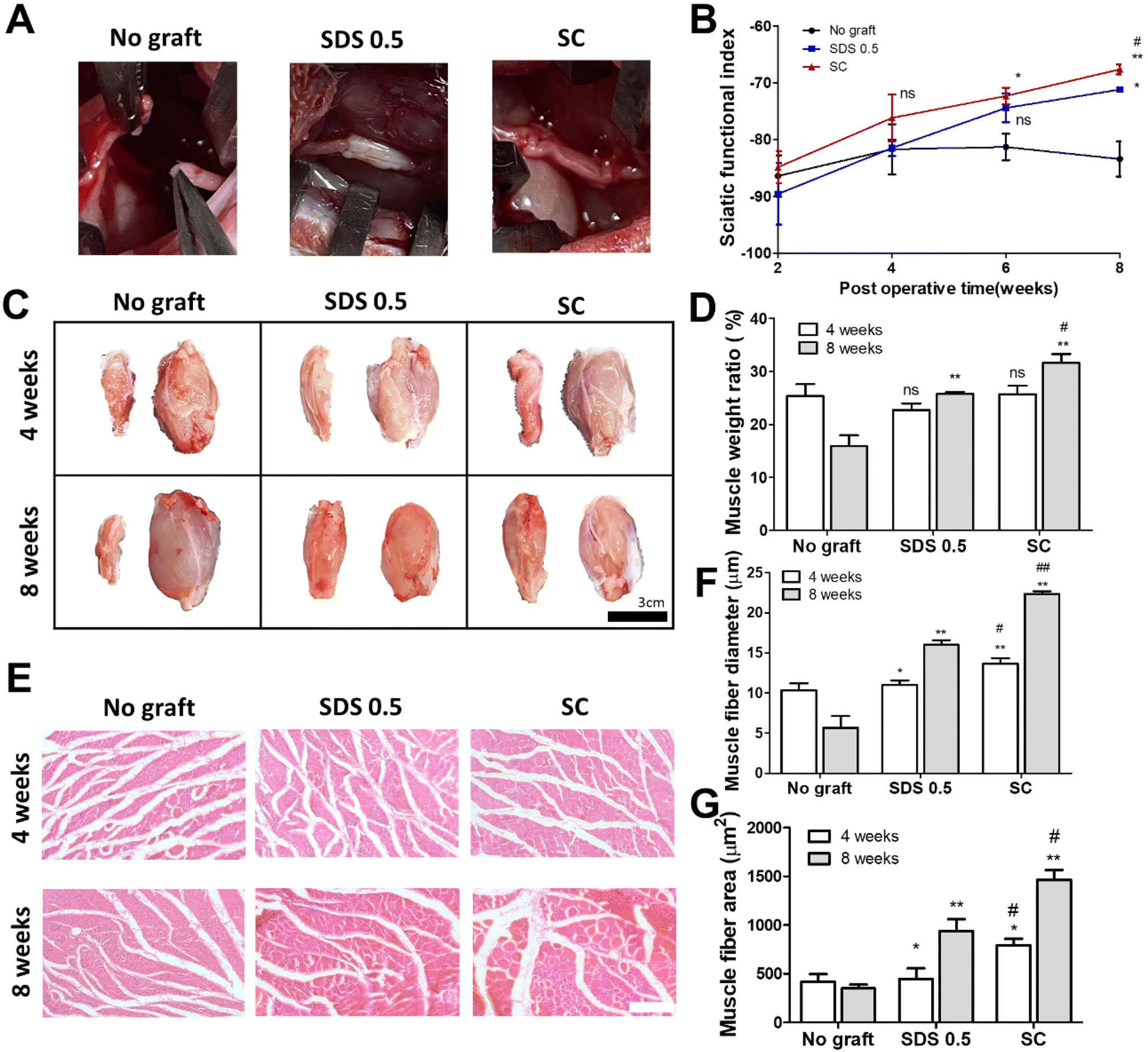 | ||
| Fig. 6 Muscular regeneration and motor function recovery assessments in vivo. (A) Development of nerve gap defects and direct nerve tissue implantation. (B) Sciatic functional index characterization 8 weeks after nerve tissue implantation. (C) Representative images of rat gastrocnemius muscle 4 and 8 weeks after implantation (scale bar = 50 μm). (D) Weight ratio of experimental muscle normalized by weight of normal muscle (opposite limb). (E) Representative images of H&E staining of gastrocnemius muscle (scale bar = 100 μm). (F and G) Measurements of muscle fiber diameter (F) and areas (G) of gastrocnemius muscle after regeneration. Error bars indicate SD. n = 3, P-value = *p < 0.05, **p < 0.01, ***p < 0.005. #p < 0.05, ##p < 0.01, ###p < 0.005. | ||
3.7 Nerve tissue regeneration assessment after tissue implantation in vivo
To evaluate the regeneration of sciatic nerve tissues, fluorescence staining was performed to confirm the presence of Schwan cell marker (S100) and neuronal synaptic marker (GAP-43). Staining and analysis were conducted by dividing the transplant site into distal and proximal sections. This division aimed to ascertain whether both sides of the neural junction were connected with the implanted tissue after creating the defect. There was no significant difference of the S-100 intensity at 4 weeks post implantation in all groups (No graft: 27.58 ± 3.88, SDS 0.5: 22.62 ± 5.32, SC: 28.11 ± 4.03%) (Fig. 7C). At 8 weeks, the overall cell response was decreased in the No graft control group; however the host cells were infiltrated into the dECM scaffold and proliferated in the SDS 0.5 group. The S-100 expression was decreased in both the control and SDS 0.5 groups compared to that at the 4 week timepoint. However, the SC group showed relatively increased S-100 intensities compared to that at 4 weeks (No graft: 5.21 ± 1.38, SDS 0.5: 8.69 ± 3.09, SC: 28.55 ± 6.83%) (Fig. 7B). The decellularized tissue implanted groups showed increased synaptic signaling in the tissue defect area than the No graft group at 4 weeks. In the SC group, Gap 43 intensities were recorded higher than in SDS 0.5 (No graft: 36.79 ± 9.31, SDS 0.5: 41.64 ± 2.71, SC: 53.85 ± 13.23%) (Fig. 7D). The Gap 43 signals were decreased at 8 weeks in all groups compared to that at the 4-week time point; however, the intensity trend of the most abundant signals showing in the SC group did not change. Although, the highest intensity was observed in the SC group, there was still a notable increase of Gap 43 intensities in the SDS 0.5 group (No graft: 11.01 ± 2.11, SDS 0.5: 16.26 ± 4.44, SC: 24.92 ± 4.13%) (Fig. 7D).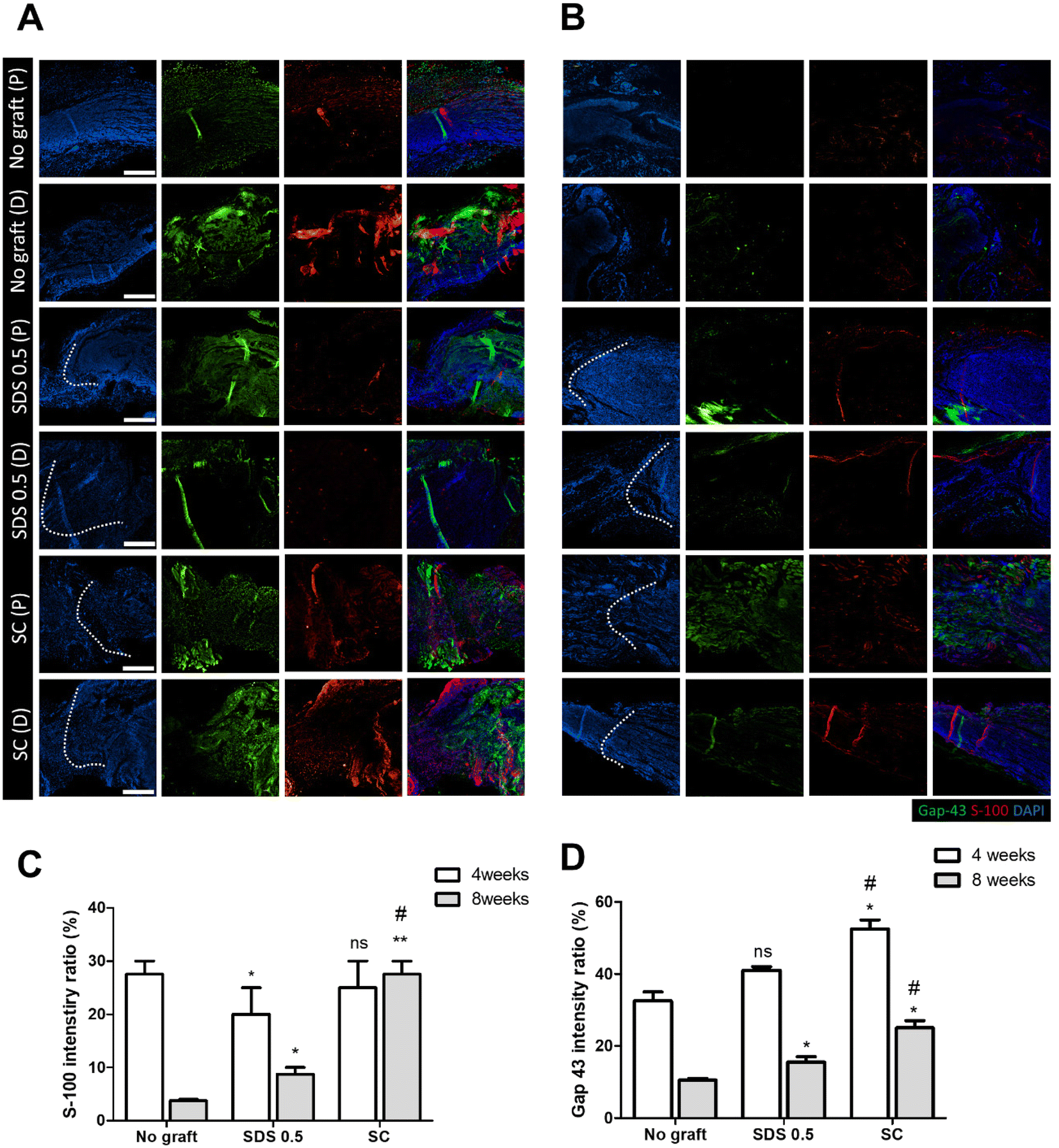 | ||
| Fig. 7 Gap-43 and S-100 fluorescence staining of sciatic nerves post-implantation at 4 and 8 weeks (P: proximal, D: distal). (A) Gap-43 and S-100 fluorescence staining of sciatic nerves 4 weeks after regeneration. (B) Gap-43 and S-100 fluorescence staining of sciatic nerves 8 weeks regeneration. (C) Measurements of S-100 intensity characterization of regenerated sciatic nerves. (D) Measurements of Gap-43 intensity characterization of regenerated sciatic nerves (scale bar = 400 μm). Error bars indicate SD. n = 3, P-value = *p < 0.05, **p < 0.01, ***p < 0.005. #p < 0.05, ##p < 0.01, ###p < 0.005. | ||
To confirm whether the transplanted allogenic decellularized tissues were connected to the host nerves by forming myelinated nerve fibers, S-100 staining was performed in cross sectioned elongated nerve tissues (Fig. 8). In the control group (No graft), weak myelin signals and oval circle shaped myelin sheaths were observed in the overall histologic area (proximal, medial, distal) (Fig. 8A). In the SDS group, nerve bundles were well observed up to the medial part, but partially myelinated nerves were found in the distal part of the transplanted scaffold. In the SC group, well-formed neural tissue and myelin sheath were observed in all three histological areas of the nerve tissues (Fig. 8A). The G-ratio of the No graft and SDS 0.5 groups was distributed sporadically and showed smaller diameters of axons together with low G-ratio scores. The SC groups showed an undispersed status along with various axon diameters and relatively large diameters of axons were found than in other experimental groups (Fig. 8B–D) (axon diameter: No graft: 2.86 ± 1.16, SDS 0.5: 1.47 ± 0.03, SC: 7.49 ± 1.03).
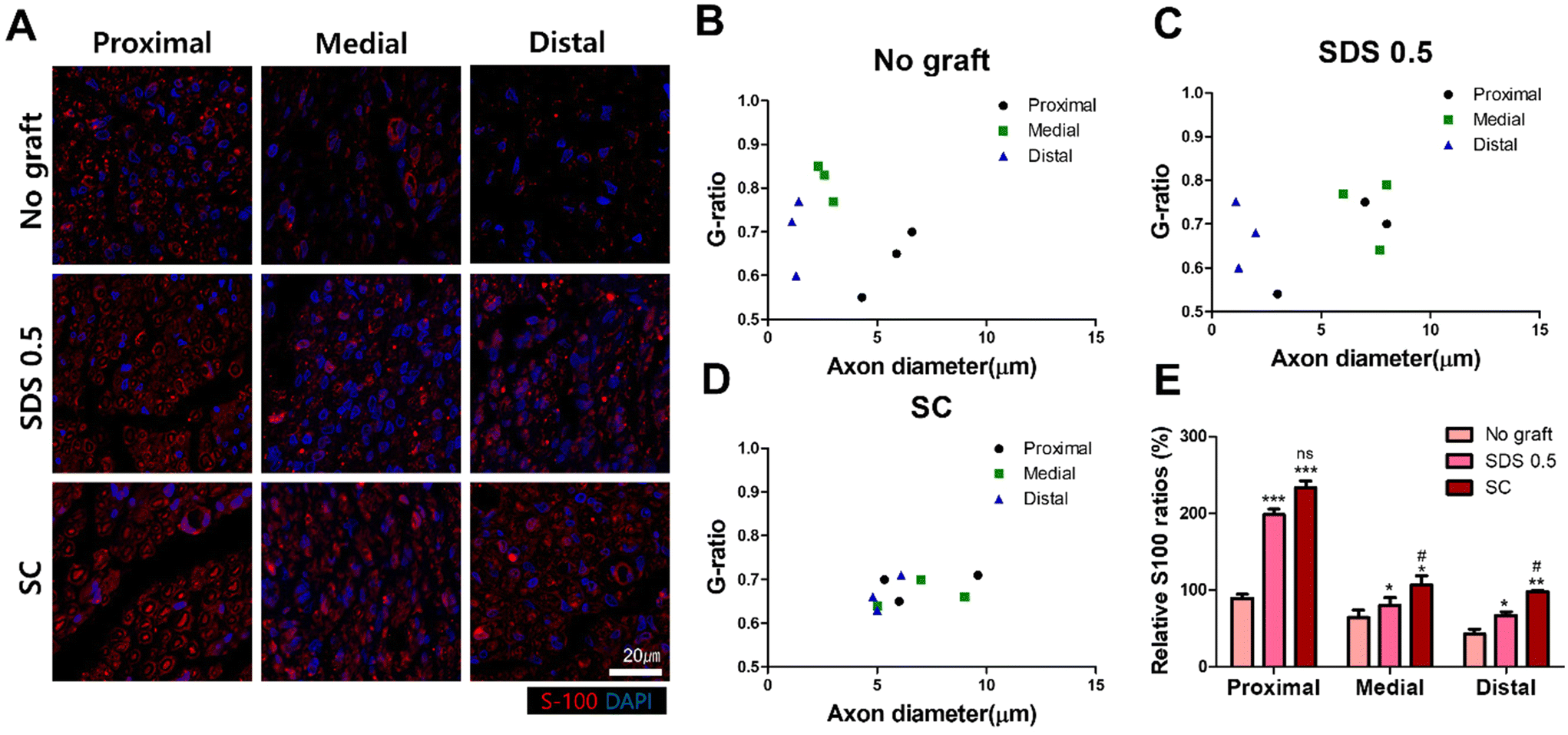 | ||
| Fig. 8 S-100 staining of cross sectioned regenerated nerve tissues 8 weeks after decellularized tissue implantation. (A) Representative images of S-100 staining of regenerated nerve tissues with and without treatments (proximal, medial, distal areas). (B) Myelin sheath G-ratio calculation of the non-treated group (C). Myelin sheath G-ratio calculation of the SDS 0.5 group (D). Myelin sheath G-ratio calculation of the SC group. (E) The S-100 signal ratio of myelin sheath in the regenerated nerve tissue with or without treatments (scale bar = 50 μm). Error bars indicate SD. n = 3, *p < 0.05, **p < 0.01, ***p < 0.005. *Indicates statistical analysis about comparing No graft group versus SDS 0.5 and SC. | ||
4 Discussion
The ECM and DNA concentrations have been verified after decellularization due to the preservation of major ECM and removal of genetic components are important criteria for dECM fabrication. To apply dECM to surgical wounds in regenerative medicine, the decellularization protocol should be verified with the aim of finding the appropriate decellularization conditions for not only the preservation of ECM components (regeneration inducer) but also removal of DNA (immunogenic) from tissues at the same time. The DNA concentration for decellularization is a standard parameter of optimizing the decellularization processes. Successful decellularization was considered to occur under 10–50 ng mg−1 DNA concentrations in the dECM.23 The sciatic nerve tissues were treated with various concentrations of chemical (SDS) solutions to find out the optimal chemical treatment conditions of decellularization which fit the DNA elimination standard. In this study, SDS was chosen as the conventional detergent for decellularization, considering both regenerative capabilities and commercial viability in terms of performance and short production time that took short process time as compared to supercritical fluid based decellularization. Triton-X is also extensively applied for decellularization studies; however, Triton-X has low DNA removal efficiency for whole tissue decellularization as in a nerve conduit which needs high tissue penetration and lipid extraction ratio. For this reason, sodium deoxycholate or SB detergent and DNA degradable enzymes were incorporated together with Triton for the decellularization of sciatic nerve tissues.24,25The DNA concentration of less than 50 ng mg−1 was recorded in 0.5%, 1% (w/v) SDS treated nerve tissues and customized supercritical fluid-based decellularization (SDS 0.5: 26.26 ± 10.06, SDS 1.0: 18.15 ± 7.81, SC: 38.07 ± 16.88). The various experimental groups showed the successful elimination of DNA; however, there was considerable tissue surface degradation and ECM loss in the SDS treated tissues. The tissue degradation and ECM loss increased with higher SDS concentrations, which is why a specific SDS concentration value (0.5% w/v) was chosen as the optimal condition for chemical decellularization not only to minimize ECM loss but also to record for 50 ng mg−1 DNA concentrations.26 The supercritical fluid-based method recorded more ECM preservative characteristics versus the optimized SDS treated protocol while both methods achieved the DNA elimination standard. This trend was also found in the tissue protein profiling results that recorded the types of proteins and their relative quantities in each dECM and native tissues. Although both decellularization processes (SDS, supercritical fluid-based) could not completely preserve all proteins found in native tissues during decellularization, the supercritical fluid-based decellularization group showed higher preservation ability towards proteins than the chemical-based decellularization group. The total protein preservation tendency in the decreasing order of native, SC, and SDS 0.5 groups also showed in the protein score data that totally proved the advanced decellularization efficiency of the supercritical fluid-based method compared to the traditional (SDS) method.
Both SDS and supercritical fluid-based decellularization processes successfully removed genetic material from the native tissues, accompanied by an inevitable loss in the protein and the ECM contents. However, the extent of decrease was greater in the group treated with SDS compared to the group decellularized using supercritical fluid. The reduction in these components affected the mechanical properties of tissues, as shown by relatively higher compressive and tensile properties of the SC group compared to that of the SDS 0.5 group (Fig. 2B and C). During the repeated indentation experiments, after 10 cycles, the SDS 0.5 group recorded approximately 10 times weaker elastic modulus than the native tissue and SC groups. These results demonstrate that the decrease in tissue mechanical properties is proportional to the amount of tissue loss, suggesting that the formal decellularization method that best preserves the ECM content helps maintain compositional and structural integrity of processed tissues.
Next, the biological activities of the dECM were investigated by culturing PC12 cells in vitro on tissue slices to assess nerve cell migration and elongation capacity. Until day 3, relatively high levels of actin and class III β-tubulin were detected in PC12 cells cultured on the native group (Fig. 3). However, in the SDS 0.5 group, the cells were found to not adhere to tissue surfaces as much as in the other groups (native, SC), possibly due to the loss of cell binding proteins during chemical-based decellularization. The expression level of class III β-tubulin was found to be higher in the native group than in both decellularized groups on day 3 and day 7; however, SC group showed a higher axon signal ratio versus the SDS 0.5 group. Furthermore, the signal ratio of actin found in the native tissue and SC group recorded relatively higher than that of the SDS 0.5 group, in the same manner as class III β-tubulin expression in short-term periods. It was reported that the expression of actin at the neurite tip relatively increased on the growth cones of sprouting neurons,27 and even during differentiation, a high amount of actin proteins moved to the neurite tips from cytoplasmic areas that promote the outgrowth of neurite tips.28 This implies that there is a correlation between the increase in the expression level of class III β-tubulin and a significant amount of actin expression. This correlation was observed in actively differentiating PC12 cells of the native group, as well as in the SC group, which provided a superior differentiating environment compared to the SDS group. Furthermore, there is no significant difference in DAPI signal changes in the decellularized group during culture period when compared to the native group (ESI Fig. 4†). However, the DAPI signal decreased in the native tissue group that eventually affected protein marker expression calculation. This phenomenon has been observed to occur during the early stages of cell culture. The reason is that lipid–proteins or temporal glycol protein cell binding used for initial cell attachment in the native tissue is still present, leading to a temporary and substantial cell attachment until day 3–4.29,30,31 As time progresses, it is speculated that these proteins located in the outer surface of tissues lose their temporal cell binding function easily, by serial washing or dissipation, eventually inducing cell detachment from the tissue surface. In contrast, in the decellularized group, cells have attached in a stable state with a larger amount of exposed ECM proteins and strongly conjugated, after an amount of proteins have already been removed during decellularization.32 Therefore, it is presumed that there is less occurrence of cell detachment over time in the SC group compared to the native tissue.
The differentiation of PC12 cells was observed in the outgrowth of neurites and related protein expression under long term culture conditions (day 21). In the 3D culture system, the fully differentiated PC12 cells were hardly found in the short-term culture period (∼Day 7) which was set as the standard differentiation periods in the 2D culture method (ESI Fig. 3†). Specifically, the proper outgrowth of neurites was not observed in the 3D culture by day 7, potentially due to the distinct culture conditions associated with 3D scaffolds and ECM surface that are different from those used in the 2D TCP culture.33,34 In addition, the appropriate cell density required for cell–cell interaction which is related to formal differentiation and cell cycles is different in the 3D platform due to the formation of a volume, rather than simply along with a single z-axis in 2D systems.35–37 Additionally, the three-dimensional nature of the scaffold may slow cell migration and tubulin formation because of their structural turbulences.38 Analysis of class III β-tubulin signaling over a prolonged culture period (∼Day 21) revealed that the SC group exhibited the strongest signal of β3-tubulin. And there was no significant difference between the native and SDS groups in the signal intensity of β3-tubulin. However, the longest neurites were observed in the native tissues, followed by the SC and then SDS 0.5 groups. This may be attributed to the remaining ECM components and cell responsive proteins in the native and SC groups, providing an environment conducive to neuronal growth.39 The SDS 0.5 group exhibited considerably high class III β-tubulin signals also when compared to the native group; however, the SDS 0.5 group showed relatively lower neurite outgrowth than other experimental groups. The SDS group exhibited tubulin signals primarily concentrated around the cell nucleus, and an increased cytoplasm size compared to the other two groups cultured under the same conditions (Fig. 4A). This means the tubulin proteins were still synthesized in cells by NGF signals but could not progress to the designated directions and were captured in the cytoplasm. The condensed tubulin signals in the nucleus suggested high internal class III β-tubulin levels which support relatively higher tubulin amounts in the cell cytoplasm and not in the neurites. This is hypothesized to be due to the inability of neurites to extend, resulting in clustering around the nucleus and increased signal intensity of tubulin proteins.40
In order to achieve the ultimate goal of neural regeneration, it is necessary to minimize host immune response against the implanted dECM. Immune reactions that occur during material transplantation are mainly mediated by monocytes and macrophages, unlike B and T cells that are mediated by adaptive immunity.41,42 Appropriate immune responses play a role in defense against external substances and in preventing fibrosis, but excessive immune reactions associated with foreign materials can potentially accelerate the disintegration of transplanted tissues and provide a non-homogeneous environment for surrounding normal tissues, which can hinder tissue formation.43,44 The data presented here show that excessive immune reactions were induced at 1 week after transplantation of native tissues, higher than the immune response of SDS and SC group implantation showing whole tissue material derived immune response increase. This indicates that tissue fragments such as DNA and damaged ECM fragments that originated from SDS-treated tissue degradation, induced increased immune reactions compared to the SC group during tissue transplantation.45–47 After 4 weeks of transplantation, the native group maintained a lower level of immune response compared to that at the 1-week time point, but still showed higher than that of the other two groups. The SDS and SC groups showed increased immune cell infiltration compared to that at 2 weeks. This is expected to be due to tissue disintegration and increased immune cell infiltration, with the SC group showing a relatively lower level of immune cell infiltration, as demonstrated in the earlier mechanical evaluation, due to the ability to maintain strong stiffness even after the cell removal process.46,47
When the total immune response of SDS 0.5 and SC group implantation results were compared, SC groups showed lower immune response than the SDS 0.5 group which recorded a lower concentration of DNA in it. It is well-known that residual DNA in the ECM can induce inflammation and immune responses.48 However, from the perspective of tissue transplantation, inflammation and immune reactions arise not only from DNA but also from the molecular recognition of all high-molecular-weight materials, including proteins, lipids, and carbohydrates, by immune cells.49 Consequently, it is inferred that the probability of releasing substances triggering inflammation has increased in the SDS 0.5 group, leading to an increase in inflammatory cell response, as evidenced in the graph, possibly due to the absolute quantity of immune activating material increased by harsh degradation in the SDS 0.5 group (Fig. 2).
Based on the evaluation of protein preservation, mechanical strength, and cell reactivity, as well as immunogenicity, the supercritical fluid-based decellularization method is deemed to be more superior to fabricate native tissue like scaffold for further nerve regeneration. Next, we conducted nerve gap defect transplantation with the fabricated dECM to evaluate the treatment potential of the dECM. The gait analysis results showed that the SC group exhibited better recovery of motor functions compared to the SDS 0.5 group, and the volume and weight ratios of the gastrocnemius (muscular tissue) were also greater compared to the No graft and SDS 0.5 groups (Fig. 6C and D). It was confirmed through muscle images of the gastrocnemius where the nerve gap defect made did not function properly, while the gastrocnemius of the normal leg (opposite limb) was particularly overdeveloped to compensate for reduced motor function of limbs which had undergone surgery, and these differences were reflected in the relative muscle weight ratio data showing dramatic difference (Fig. 6D).
After 2 months post nerve dECM transplantation, the nerve tissues were collected and stained with S-100 and Gap-43 antibodies to confirm nerve tissue regeneration. Gap-43 is known to activate the signaling system that regulates neuron sprouting and connectivity during the development of nerve tissues.50,51 The results obtained 4 weeks after nerve transection showed no significant differences in Gap-43 between the No graft group, SDS 0.5 group, and SC group. It seems that host nerve cells were still active even with the intervention made and this expectation was also deduced from muscle in vivo data collected at the 4 week time point that still sustain the volumetric state of the muscle in Fig. 6. Strong Gap-43 signals in the two groups (SDS 0.5 and SC) at 4 weeks post-implantation was expected to support growth and migration of neuronal, nerve supportive glial cells towards the transplanted decellularized tissues (Fig. 7A). At the 8-week time point, the No graft group showed a decrease of Gap-43 signals, and the degenerated morphology in both proximal and distal areas in the nerve gap. It might be due to immune cell related tissue degradation and fibrotic scar formation after injury at the harmed tissue sites.52 In the SDS 0.5 groups, the Gap-43 signals were found in the surface area of elongated gap junctions which means neuronal cell infiltration may happen but not in the correct direction and depth in the transplanted tissues because of neuronal cell binding protein loss after the process.53,54 However, the SC group showed evenly concentrated Gap-43 signals in the transplanted areas which stand for formal neuronal tissue formation in implanted biomaterials.
S-100 is a marker for Schwann cells in peripheral nerves that is known to be involved in nerve metabolism, bundle formation, and nerve tissue atrophy.55,56 Therefore, the formation of Schwann cells during nerve regeneration is an important factor in evaluating healthy neuron elongation and formal neural tissue formations in peripheral nervous systems.57 The results for S-100 were very similar to the expression pattern of Gap-43 at both time points (4 and 8 weeks). However, at 4 weeks, the SDS 0.5 group showed a lower S-100 signal ratio compared to the No graft group. It was expected because of the remaining Schwann cells that were active in the native tissues to sustain damaged nerve functions in early time points of surgery and a low level of cell binding proteins and ECM contents in the SDS 0.5 group that could induce time consuming migration of Schwann cells, expressing S-100, toward the implanted dECM.58
To confirm whether the neural tissue was properly formed in transplanted decellularized tissues, histological staining was performed on cross-sections of tissues to visualize the myelin sheath formed by Schwann cells, rather than sagittal sections that were previously used to confirm the expression of Gap-43 and S-100.59 In the No graft group (Fig. 8A), myelin sheath was still present in the proximal area of sciatic nerve, but it was not observed in a formal cylindrical shape at the medial and distal areas of dissected nerve tissues. In the SDS 0.5 group, the myelin sheath signal was found in proximal and medial area of nerve tissues, and the smaller axon with myelin sheaths were found in the distal area (Fig. 8A). These results showed that the physical, biochemical cues of decellularized tissues induced formal tissue regeneration with the scaffolds–host tissue interaction. Furthermore, the myelin sheaths had thicker and larger tissue morphology in the proximal area than those in the distal direction of the sciatic nerves in both the No graft and SDS 0.5 groups. It was expected that myelin would be more easily maintained or rejuvenated in the proximal area of peripheral nerve where the fast release of nerve regenerative molecules and strong regeneration signal transition could have occurred for the closely located neuronal cell body in the spine.60 In the SC group, condensed myelin sheath populations were observed in the whole anatomical area in the newly regenerated axon including in the distal area. This result indicates that myelinated axons from the proximal origin had grown into the grafted tissue (SC) and the regenerative microenvironment formation by the grafted tissues had helped to maintain the activity of the intrinsic axons in the distal area also. The G-ratios of myelin sheath formation presented the superior regeneration ability of the SC group as shown by their higher proteomic components, ECM preservation ratio and cell reactivity. The non dispersed G-ratio distribution and increasing tendency of G-ratio along with the larger axon diameter in the SC group showed the formation of more functional neuronal tissues compared to that in the chemically processed scaffolds.61
5 Conclusion
Supercritical fluid-based decellularization and SDS-based decellularization are two different approaches used in the decellularization of the sciatic nerves with the aim of enhancing their regeneration ability. The supercritical fluid-based method showed higher preservation of the ECM in the processed tissue compared to the SDS-based method. By preserving the native ECM architecture, supercritical fluid-based decellularized nerve scaffolds retain their mechanical integrity like native tissue. In contrast, SDS-based decellularized scaffolds may experience a decrease in mechanical strength due to the potential damage to the ECM structure during the decellularization process. This structural stability was crucial for surgical handling and implantation, as well as for withstanding physiological loading, cellular reactivity and facilitating the regeneration process. The higher ECM and protein preservation of the supercritical fluid-based method resulted in providing more appropriate neuronal cell culture scaffolds that supported the outgrowth of axons under culture conditions. In terms of advanced regeneration ability, the supercritical fluid-based decellularization method had shown promising results of neural tissue regeneration. Studies have demonstrated that the supercritical fluid-based decellularized sciatic nerve scaffolds exhibited a favorable microenvironment for neuronal cell growth and Schwann cell proliferation in tissues.Author contributions
Beom Seok kim: conceptualization, investigation, methodology, writing – original draft, visualization, and validation; Jeong-Uk Kim: reviewing, data curation, and investigation; Jae Woo Lee: reviewing; Kyung Min Ryu: reviewing and data curation; Rachel H Koh: reviewing; Kyoung-Ha So: reviewing; Nathaniel S. Hwang: supervising, reviewing, and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Ministry of Trade, Industry and Energy grant funded by the Korean Government (MOTIE-20016553). This work was financially supported by the Ministry of Science and ICT (NRF – 2021R1A2C2008821, NRF – 2023R1A2C2007283).References
- B. J. Pfister, T. Gordon, J. R. Loverde, A. S. Kochar, S. E. Mackinnon and D. K. Cullen, Crit. Rev. Biomedicaleng., 2011, 39, 2 Search PubMed.
- M. Tapp, E. Wenzinger, S. Tarabishy, J. Ricci and F. A. Herrera, Ann. Plast. Surg., 2019, 83, 676–680 CrossRef CAS PubMed.
- R.-C. Zhang, W.-Q. Du, J.-Y. Zhang, S.-X. Yu, F.-Z. Lu, H.-M. Ding, Y.-B. Cheng, C. Ren and D.-Q. Geng, Neural Regener. Res., 2021, 16, 2170 CrossRef CAS PubMed.
- A. D. Sharma, J. Wiederin, M. Uz, P. Ciborowski, S. K. Mallapragada, H. E. Gendelman and D. S. Sakaguchi, J. Proteomics, 2017, 165, 93–101 CrossRef CAS PubMed.
- Å. Fex Svennigsen and L. B. Dahlin, Brain Sci., 2013, 3, 1182–1197 CrossRef PubMed.
- J. Gorecka, V. Kostiuk, A. Fereydooni, L. Gonzalez, J. Luo, B. Dash, T. Isaji, S. Ono, S. Liu and S. R. Lee, Stem Cell Res. Ther., 2019, 10, 1–10 CrossRef PubMed.
- W. Sulaiman and T. Gordon, Ochsner J., 2013, 13, 100–108 Search PubMed.
- S. Wang and L. Cai, Int. J. Polym. Sci., 2010, 2010(speical), 1–20 Search PubMed.
- P. Chrząszcz, K. Derbisz, K. Suszyński, J. Miodoński, R. Trybulski, J. Lewin-Kowalik and W. Marcol, Neurol. Neurochir. Pol., 2018, 52, 427–435 CrossRef PubMed.
- B.-K. Lee, Y. M. Ju, J.-G. Cho, J. D. Jackson, S. J. Lee, A. Atala and J. J. Yoo, Biomaterials, 2012, 33, 9027–9036 CrossRef CAS PubMed.
- Y. Qian, X. Zhao, Q. Han, W. Chen, H. Li and W. Yuan, Nat. Commun., 2018, 9, 323 CrossRef PubMed.
- J. Ijkema-Paassen, K. Jansen, A. Gramsbergen and M. Meek, Biomaterials, 2004, 25, 1583–1592 CrossRef CAS PubMed.
- D. Hebebrand, G. Zohman and N. Jones, J. Hand Surg., 1997, 22, 304–307 CrossRef CAS PubMed.
- M. Sondell, G. Lundborg and M. Kanje, Brain Res., 1998, 795, 44–54 CrossRef CAS PubMed.
- T. W. Hudson, S. Zawko, C. Deister, S. Lundy, C. Y. Hu, K. Lee and C. E. Schmidt, Tissue Eng., 2004, 10, 1641–1651 CrossRef CAS PubMed.
- U. Mendibil, R. Ruiz-Hernandez, S. Retegi-Carrion, N. Garcia-Urquia, B. Olalde-Graells and A. Abarrategi, Int. J. Mol. Sci., 2020, 21, 5447 CrossRef PubMed.
- T. W. Gilbert, T. L. Sellaro and S. F. Badylak, Biomaterials, 2006, 27, 3675–3683 CAS.
- S. R. Meyer, J. Nagendran, L. S. Desai, G. R. Rayat, T. A. Churchill, C. C. Anderson, R. V. Rajotte, J. R. Lakey and D. B. Ross, J. Thorac. Cardiovasc. Surg., 2005, 130, 469–476 CrossRef PubMed.
- J. Chakraborty, S. Roy and S. Ghosh, Biomater. Sci., 2020, 8, 1194–1215 RSC.
- B. S. Kim, J. U. Kim, K. H. So and N. S. Hwang, Macromol. Biosci., 2021, 21, 2100160 CrossRef CAS PubMed.
- T. Ahmad, F. Masoodi, S. A. Rather, S. Wani and A. Gull, J. Biol. Chem. Chron., 2019, 5, 114–122 Search PubMed.
- D. M. Casali, R. M. Handleton, T. Shazly and M. A. Matthews, J. Supercrit. Fluids, 2018, 131, 72–81 CrossRef CAS.
- D. C. Sullivan, S.-H. Mirmalek-Sani, D. B. Deegan, P. M. Baptista, T. Aboushwareb, A. Atala and J. J. Yoo, Biomaterials, 2012, 33, 7756–7764 CrossRef CAS PubMed.
- A. Zaminy, S. Sayad-Fathi, F. M. Kasmaie, Z. Jahromi and A. Zendedel, Neural Regener. Res., 2021, 16, 1086 CrossRef CAS PubMed.
- C. Philips, F. Campos, A. Roosens, M. D. C. Sánchez-Quevedo, H. Declercq and V. Carriel, Ann. Biomed. Eng., 2018, 46, 1921–1937 CrossRef PubMed.
- Y. Snyder and S. Jana, Biomaterials, 2022, 121675 CrossRef CAS PubMed.
- O. Benzina, T. Cloitre, M. Martin, C. Raoul, C. Gergely and F. Scamps, PLoS One, 2014, 9, e110687 CrossRef PubMed.
- J. X. Chia, N. Efimova and T. M. Svitkina, Mol. Biol. Cell, 2016, 27, 3695–3704 CrossRef CAS PubMed.
- M. Abdolahi, E. Karimi, P. Sarraf, A. Tafakhori, G. Siri, F. Salehinia, M. Sedighiyan, B. Asanjarani, M. Badeli and H. Abdollahi, BMC Res. Notes, 2021, 14, 1–7 CrossRef PubMed.
- C. L. Merry, U. Lindahl, J. Couchman and J. D. Esko, Essentials of Glycobiology, 4th edn, 2022 Search PubMed.
- K. Sugitani, D. Egorova, S. Mizumoto, S. Nishio, S. Yamada, H. Kitagawa, K. Oshima, D. Nadano, T. Matsuda and S. Miyata, Biochim. Biophys. Acta, Gen. Subj., 2021, 1865, 129804 CrossRef CAS PubMed.
- A. Ramanathan and N. Karuri, Biochem. Biophys. Res. Commun., 2015, 459, 246–251 CrossRef CAS PubMed.
- P. Chua and W. K. Lim, Cell Biol. Int., 2023, 47, 367–373 CrossRef CAS PubMed.
- B. T. Mehl and R. S. Martin, Anal. Methods, 2019, 11, 1064–1072 RSC.
- M. Kapałczyńska, T. Kolenda, W. Przybyła, M. Zajączkowska, A. Teresiak, V. Filas, M. Ibbs, R. Bliźniak, Ł. Łuczewski and K. Lamperska, Arch. Med. Sci., 2018, 14, 910–919 Search PubMed.
- M. Ravi, V. Paramesh, S. Kaviya, E. Anuradha and F. P. Solomon, J. Cell. Physiol., 2015, 230, 16–26 CrossRef CAS PubMed.
- E. Knight and S. Przyborski, J. Anat., 2015, 227, 746–756 CrossRef PubMed.
- C.-Y. Yang, W.-Y. Huang, L.-H. Chen, N.-W. Liang, H.-C. Wang, J. Lu, X. Wang and T.-W. Wang, J. Mater. Chem. B, 2021, 9, 567–584 RSC.
- C.-F. V. Latchoumane, P. Chopra, L. Sun, A. Ahmed, F. Palmieri, H.-F. Wu, R. Guerreso, K. Thorne, N. Zeltner and G.-J. Boons, ACS Appl. Mater. Interfaces, 2022, 14, 28476–28488 CrossRef CAS PubMed.
- L. Dehmelt, F. M. Smart, R. S. Ozer and S. Halpain, J. Neurosci., 2003, 23, 9479–9490 CrossRef CAS PubMed.
- K. Sadtler, B. W. Allen, K. Estrellas, F. Housseau, D. M. Pardoll and J. H. Elisseeff, Tissue Eng., Part A, 2017, 23, 1044–1053 CrossRef CAS PubMed.
- M. C. Cramer and S. F. Badylak, Ann. Biomed. Eng., 2020, 48, 2132–2153 CrossRef PubMed.
- A. Aloysius, S. Saxena and A. W. Seifert, Curr. Opin. Immunol., 2021, 68, 72–82 CrossRef CAS PubMed.
- B. Kim, S.-H. Kim, K. Kim, Y.-H. An, K.-H. So, B.-G. Kim and N. Hwang, Mater. Today Bio, 2020, 8, 100079 CrossRef CAS PubMed.
- A. M. Padma, A. B. Alshaikh, M. J. Song, R. Akouri, M. Oltean, M. Brännström and M. Hellström, J. Tissue Eng. Regener. Med., 2021, 15, 674–685 CrossRef CAS PubMed.
- D. Tang, R. Kang, C. B. Coyne, H. J. Zeh and M. T. Lotze, Immunol. Rev., 2012, 249, 158–175 CrossRef CAS PubMed.
- A. F. Malik, R. Hoque, X. Ouyang, A. Ghani, E. Hong, K. Khan, L. B. Moore, G. Ng, F. Munro, R. A. Flavell, Y. Shi, T. R. Kyriakides and W. Z. Mehal, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20095–20100 CrossRef CAS PubMed.
- B. Briard, D. E. Place and T.-D. Kanneganti, Physiology, 2020, 35, 112–124 CrossRef CAS PubMed.
- J. E. Woodell-May and S. D. Sommerfeld, J. Orthop. Res., 2020, 38, 253–257 CrossRef PubMed.
- L. Burello, P. De Bartolo, F. Gelfo, F. Foti, F. Angelucci and L. Petrosini, Exp. Neurol., 2012, 233, 273–282 CrossRef CAS PubMed.
- D. Chung, A. Shum and G. Caraveo, Front. Cell Dev. Biol., 2020, 8, 567537 CrossRef PubMed.
- A. Lemke, J. Ferguson, K. Gross, C. Penzenstadler, M. Bradl, R. L. Mayer, C. Gerner, H. Redl and S. Wolbank, Acta Biomater., 2018, 66, 335–349 CrossRef CAS PubMed.
- T. Bongolan, J. Whiteley, J. Castillo-Prado, A. Fantin, B. Larsen, C. J. Wong, L. Mazilescu, M. Kawamura, P. Urbanellis and A. Jonebring, Biomater. Sci., 2022, 10, 2972–2990 RSC.
- X. Zhou, Y. Qian, L. Chen, T. Li, X. Sun, X. Ma, J. Wang and C. He, ACS Nano, 2023, 17, 5140–5156 CrossRef CAS PubMed.
- Q. Min, D. B. Parkinson and X. P. Dun, Glia, 2021, 69, 235–254 CrossRef PubMed.
- K. R. Jessen and R. Mirsky, Front. Mol. Neurosci., 2019, 12, 69 CrossRef CAS PubMed.
- G. Nocera and C. Jacob, Cell. Mol. Life Sci., 2020, 77, 3977–3989 CrossRef CAS PubMed.
- E. Schnell, K. Klinkhammer, S. Balzer, G. Brook, D. Klee, P. Dalton and J. Mey, Biomaterials, 2007, 28, 3012–3025 CrossRef CAS PubMed.
- A. J. Moszczynski, K. Volkening and M. J. Strong, Appl. Immunohistochem. Mol. Morphol., 2020, 28, 562–565 CrossRef PubMed.
- B. Chen, Q. Chen, D. B. Parkinson and X.-P. Dun, Front. Mol. Neurosci., 2019, 12, 308 CrossRef CAS PubMed.
- M. C. Ford, O. Alexandrova, L. Cossell, A. Stange-Marten, J. Sinclair, C. Kopp-Scheinpflug, M. Pecka, D. Attwell and B. Grothe, Nat. Commun., 2015, 6, 8073 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm02072j |
| This journal is © The Royal Society of Chemistry 2024 |