
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceByproduct formation in heterogeneous catalytic ozonation processes
Tingting
Wu
 *
*
Civil and Environmental Engineering Department, The University of Alabama in Huntsville, Huntsville, AL 35899, USA. E-mail: Tingting.Wu@uah.edu; Fax: +1-256-824-6724; Tel: +1-256-824-6423
First published on 14th February 2023
Abstract
Heterogeneous catalytic ozonation (HCO) is a promising advanced oxidation process (AOP) that can effectively degrade recalcitrant organic pollutants. While research efforts have been mainly devoted to the development of different catalysts to enhance the process efficiency, more studies are needed to investigate and address the other challenge faced by AOPs, i.e. generation of harmful byproducts. Bromate is the major inorganic byproduct of concern when ozone is involved. While most studies have reported less bromate formation in HCO than ozonation alone, the effects of catalysts depend on their interaction with O3 and the dominant bromate formation pathway (direct O3 oxidation vs. indirect ˙OH oxidation) in the system. Production of H2O2 and cyclic redox reactions on the catalyst surface can also reduce different Br species leading to a lower bromate yield. Generation of organic byproducts (OBPs; e.g. aldehydes, keto-acids, carboxylic acids) in HCO depends on the reactivity of precursors (e.g. dissolved organic matter/DOM) and OBPs with O3/˙OH, interactions between DOM/OBPs and catalysts, characteristics of DOM, and O3 dose. HCO generally increased the removal of dissolved organic carbon (DOC) and the biodegradability of the bulk organics. HCO treatment may also decrease the formation potential of some disinfection byproducts (DBPs) such as trihalomethanes (THMs) and haloacetic acids (HAAs) but may increase the brominated species of the DBPs and also the formation potential of haloacetonitrile (HAN) under certain conditions. This review discusses the current status of studies on both organic and inorganic byproduct formation in HCO as well as transformation of bulk organics and the effects on DBP formation in the downstream disinfection process, and further provides recommendations for future research and development. A standardized experimental protocol and rigorous experimental design is important to deepen our understanding and gain insights on the byproduct formation in HCO from different studies collectively.
Environmental significanceHeterogeneous catalytic ozonation (HCO), which does not require addition of other chemical agents and avoids the following separation step, can be an effective advanced oxidation process (AOP) for the abatement of organic pollutants. Since AOPs are generally considered energy intensive, research on HCO has been focusing on developing different catalysts to enhance the degradation efficiency of target pollutants. However, generation of harmful byproducts is another challenge of AOPs and more attention in this regard is needed to facilitate practical applications of newly developed processes. This paper aims to present a review of the up-to-date knowledge of both inorganic and organic byproduct formation in HCO and recommendations on future research. |
1. Introduction
Ozone-based advanced oxidation processes (AOPs) usually involve agents that can promote ozone decomposition to initiate and propagate the formation of hydroxyl radical (˙OH) that is extremely reactive with organic compounds in water. One of the most commonly used chemical agents is hydrogen peroxide (H2O2),1–3 which, along with its storage and transportation, can represent substantial cost. Hence electro-peroxone with in situ generation of H2O2 through O2 reduction at carbon cathodes was also developed.4–7 On the other hand, transition metal ions such as Fe2+, Cu2+ can be used as homogeneous catalysts for such a purpose but need to be separated from treated water.8 Reactive species can also be generated during O3 decomposition in the presence of solid catalysts, i.e. heterogeneous catalytic ozonation (HCO), which does not require addition of other chemical agents and avoids the following separation step. Since conventional AOPs are considered energy intensive, investigation of novel AOPs including HCO often concentrates on enhancing process efficiencies for the removal of target pollutants, thus reducing the energy consumption. However, in addition to the intensive use of energy and chemicals, the risk of harmful by-products formation is another concern for AOPs and calls for more attention. It is worth emphasizing that, analogous to disinfection byproducts (DBPs) that are formed during reactions of disinfectants with naturally occurring organic matter (NOM) or inorganic species (e.g. halide anions) present in the water, here the byproducts refer to the unintended products formed due to the reaction of the co-existing species in the environmental matrices with oxidants in AOPs, not the transformation products of the target compound such as emerging contaminants.Both inorganic and organic byproducts may be formed in AOPs. When ozone is involved, bromate (BrO3−), a probable human carcinogen, can be generated through the reaction of O3/˙OH with naturally occurring Br−, with HOBr/OBr− being the critical intermediates.9–12 HCO has been reported to form less bromate than that of single ozonation in most studies but different mechanisms have been proposed.13–18 Such apparent discrepancies may arise from several causes: (1) the different catalysts used; (2) the different experimental conditions and experimental design employed; (3) the multiple possible pathways for bromate formation. While it is possible that the dominant mechanism is different in studies with different catalysts, it is worth mentioning that since HCO is a highly heterogeneous system, to understand the underlying mechanism and delineate the reaction pathways is a challenging task. The multiple species and pathways responsible for bromate formation, along with the possible interactions among them, certainly add more difficulties in reaching any convincing conclusions especially when a rigorous experimental design and comprehensive investigation is lacking. For example, very often the presence of catalysts accelerates O3 decomposition and consequently ˙OH generation. However, this has been used to explain completely opposite experimental observations, i.e., inhibition and promotion of bromate formation, because both species (i.e. O3 and ˙OH) can contribute to the oxidation of bromide to bromate.10,11,19
O3 and ˙OH can also react with NOM present in water, thus decreasing the efficiency of target contaminants' removal. When organic byproducts are concerned, attention should be paid to the “direct byproducts” as well as the transformation and changes of bulk organics because very likely this will affect the formation of DBPs in the downstream disinfection process. Ozonation may change the structure and biodegradability of NOM.20,21 Usually the transformation products are oxygenated compounds such as aldehydes, alcohols, ketones and carboxylic acids.22,23 These compounds react slowly with ozone and may accumulate as oxidation byproducts (OBPs) in the finishing water. When catalysts are used to promote the generation of ˙OH, their removal can be increased,24 but ˙OH can also effectively oxidize the precursors leading to higher accumulation of some OBPs.25 Furthermore, as complete mineralization usually cannot be achieved cost-effectively, transformation products of the organic matters may form disinfection byproducts in the following disinfection process. Studies on the DBPs formation by chlorination and ozonation/postchlorination treatment indicated ozonated/postchlorinated water had lower concentrations of total organic halogen, trihalomethanes (THMs), haloacetic acids (HAAs), and higher concentrations of bromate, and aldehydes.26 However, higher DBP concentrations were reported in water pretreated with electro-peroxone than that pretreated with ozonation because the major precursors, NOM with high UV254 absorbance, were more efficiently removed in ozonation.7 Since both peroxone and HCO involve agents (i.e. H2O2 and catalysts respectively) to promote ozone decomposition, these results indicate that evaluation and optimization of HCO should concentrate on not only abatement of the target pollutants, but also transformation of background organic content. Moreover, co-existence of organic and inorganic precursors can further affect the formation of byproducts (e.g. bromate, halogenated organics) as well as the species distribution of DBPs.27,28 Although there have been some reports on NOM transformation and subsequent DBP formation in the literature (which are reviewed in Section 3), HCO studies have been more focusing on developing different catalysts that can effectively improve the process efficiency for the destruction of recalcitrant organic pollutants.29
Hence, this tutorial review aims to discuss the current status of studies on byproduct formation and transformation of bulk organics in HCO processes and further provide recommendations for future research and development.
2. Formation of inorganic byproducts in HCO
Toxic oxyanions (e.g. bromate, chlorate, and perchlorate) may be produced during oxidation processes in the presence of some inorganic ions especially halides. For ozone-based AOPs, bromate is a characteristic inorganic byproduct which is regulated at 10 μg L−1 in drinking water in several countries.30 Since the bromide level in natural waters varies from ∼10 μg L−1 to >1000 μg L−1 in fresh waters,31 formation of bromate during water treatment is practically relevant and thus has been studied in various oxidation processes including heterogeneous catalytic ozonation. On the other hand, chloride is ubiquitous in the environment and commonly exists at mg L−1 level in natural waters, with chlorate and perchlorate being two possible oxidation byproducts of concern. The yield and rate of formation of these harmful oxyanions in ozonation processes depends on the chemical conditions (e.g. pH, DOC), doses, and most importantly, the aqueous Cl species (e.g. Cl−, HOCl/OCl−, ClO2−etc.).32 While reactions between chlorine (HOCl/OCl−) and ozone via free radical pathways can lead to the formation of chlorate in low-DOC water, the very slow rate of perchlorate production from Cl− even at high concentrations of O3 and Cl− implies negligible potential of perchlorate formation in water/wastewater treatment processes involving ozone.33 This is because O3 interacts with Cl− very slowly (k = 2.2 × 10−3 M−1 S−1 at 20 °C, pH > 3) to produce OCl− for chlorate and perchlorate generation.34 Although ˙OH may react with Cl− and lead to the Cl2˙− intermediate for chlorate formation, the product (ClOH−) of the starting reaction has a high dissociation rate constant (Cl− + OH ↔ ClOH−, kf = 4.3 × 109 M−1 S−1, kr = 6.1 × 109 M−1 S−1),35 resulting in limited chlorate production under water/wastewater relevant conditions (e.g. many species competing for ˙OH and low Cl− level). Hence, when O3-based AOPs are used for the removal of recalcitrant organic pollutants in the absence of active Cl species such as HOCl/OCl− (e.g. before disinfection), formation of chlorate and perchlorate is unlikely a big concern. Because of this and also because of the less regulation pressure, chlorate/perchlorate has drawn much less attention compared to bromate as an ozonation byproduct. To the author's knowledge, there is no report on chlorate/perchlorate formation in HCO yet. Moreover, while it can be formed during ozonation of iodide-containing water, iodate is considered non-problematic because it is reduced back to iodide endogenically. Therefore, this review focuses on bromate.2.1 Overview of bromate formation in ozone-based processes
Bromate is a long-known byproduct of oxidative treatment of bromide containing water and its formation has been intensively studied since early 1990s. Fig. 1 shows the formation pathways of bromate in the reaction of bromide with ozone and hydroxyl radicals. It was originally proposed by von Gunten et al. and later revised by Fischbacher et al., where the last reaction between ozone and bromite was recommended to be via electron transfer instead of the previously assumed O transfer.10,11,19 The pathways/reactions which have been suggested to influence the bromate formation in HCO are discussed in details in the following sections and illustrated in Fig. 2. The related studies are summarized in Table 1. | ||
| Fig. 1 Reaction pathways for bromate formation during ozone-based AOPs (adapted from Fischbacher et al., 2015;19 Von Gunten and Hoigne, 1994;10 von Gunten and Oliveras, 1998.11 | ||
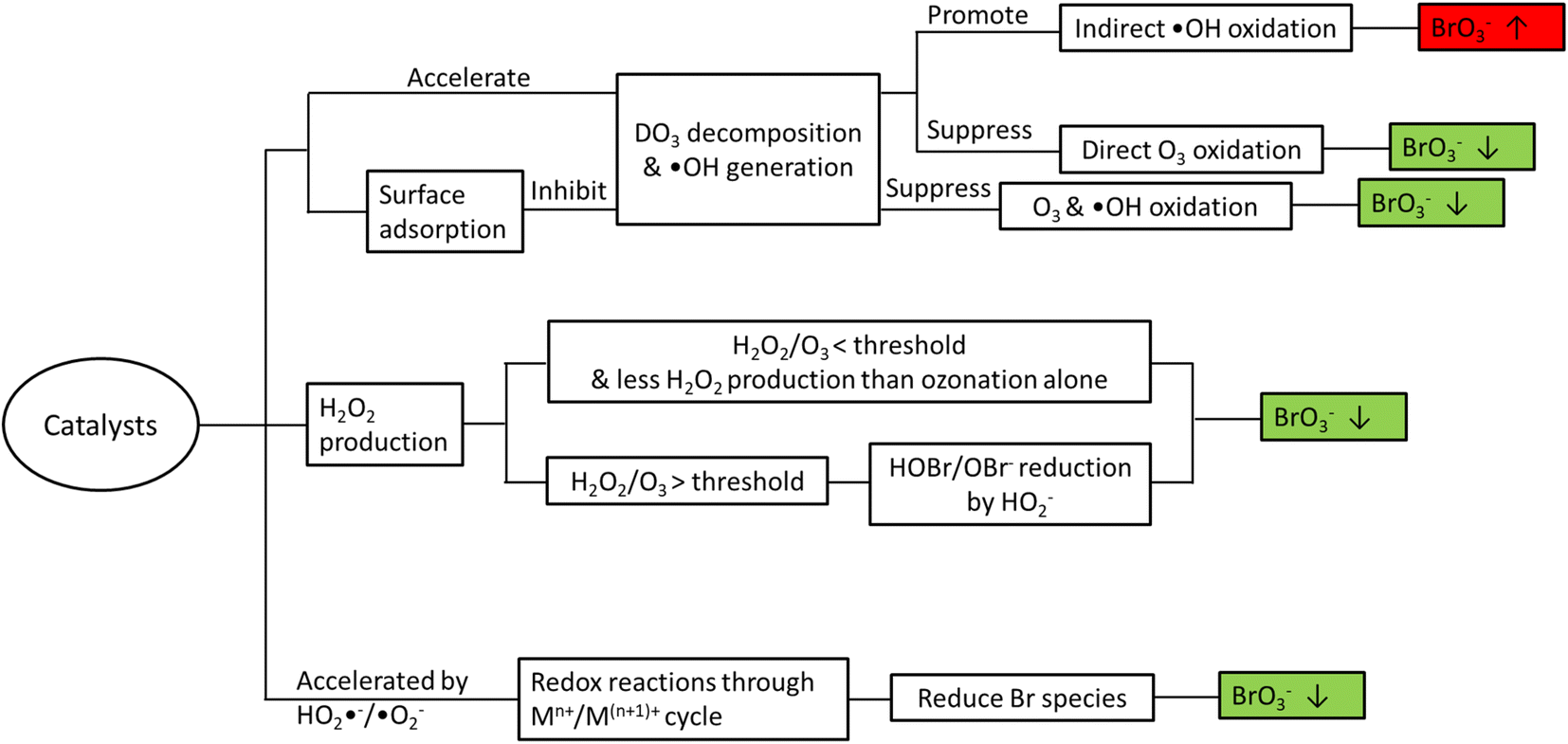 | ||
| Fig. 2 Formation and inhibition of bromate in HCO processes. | ||
| Catalyst | Experimental conditions | Results/mechanism | Ref. |
|---|---|---|---|
| a 2,4-Dichlorophenoxyacetic acid (2,4-D), phenazone (PZ), diphenhydramine (DP), amitrole (AMT). b Benzotriazole (BZA), 4-chlorobenzoic acid (p-CBA). | |||
| Fe-Cu-MCM-41/Fe-MCM-41/Cu-MCM-41 | Semi-batch reactor; catalyst: 1 g L; [Br−]0: 1000 μg L−1, initial pH: 5.0 | HCO inhibited bromate formation: (1) quick O3 decomposition led to less O3 exposure; (2) high ˙OH generation caused more bromate formation using Fe-Cu-MCM-41 than Fe-MCM-41 or Cu-MCM-41 | 39 |
| Fe-Cu-MCM-41 | Semi-batch reactor; catalyst: 1 g L; [Br−]0: 1000 μg L−1, initial pH: 6.8 | HCO inhibited bromate formation: (1) quick O3 decomposition led to less O3 exposure; (2) H2O2 was generated and reduced HOBr/OBr− | 40 |
| Fe–Al LDH/Al2O3 | Batch reactor; [O3]0: 2 mg L; [Br−]0: 1.25 μmol L; [2,4-dichlorophenoxyacetic acid]0 ([2,4-D]0): 45.2 μmol L−1; catalyst: 0.25 g L; initial pH: 6 | HCO with Fe–Al LDH/Al2O3 was more efficient in bromate inhibition and pollutant removal: reduction of bromate by surface Fe(II), which was generated from the reaction between Fe(III)-intermediates complex and HO2˙−/˙O2− | 13 |
| β-FeOOH/Al2O3 | Batch reactor; [O3]0: 2 mg L; [Br−]0: 1.25 μmol L; [pollutant]0a: 45.2 μmol L−1; catalyst: 0.25 g L; pH: 6 | No significant bromate formation and higher removal of TOC were observed during HCO of the model pollutants except AMT: (1) adsorbed bromate was reduced to Br− by surface Fe(II), which was generated from reactions between Fe(III) and HO2˙−/˙O2−; (2) complexation of surface Fe(III) with O-containing functional groups of pollutants and their transformation products enhanced surface Fe(II) generation, resulting in higher bromate reduction | 50 |
| Faceted CeO2 [(1 0 0), (1 1 0), and (1 1 1)] | Batch reactor; [O3]0: 4 mg L; [Br−]0: 2 mg L; sulfamethoxazole: 10 mg L; catalyst: 0.25–0.5 g L; initial pH: 6.3 | Faceted CeO2 inhibited bromate formation and enhanced the pollutant removal: (1) promoted O3 decomposition; (2) less H2O2 generated in the presence of catalysts; (3) more ˙O2− generated in the presence of CeO2 (1 0 0), which reduced Ce4+ to Ce3+and Ce3+ reduced different Br species | 37 |
| CeO2 | Batch reactor; [O3]0: 5.21 mg L; [Br−]0: 1.8 mg L; catalyst: 0.1 g L; initial pH: 6.3 | The low lattice O and adsorbed O of CeO2 promoted the adsorption of O-containing intermediates (from O3 decomposition) on the active sites and inhibited ˙OH generation in solution and consequent bromate formation; inhibition of ˙OH generation also led to less organic degradation | 17 |
| CeO2 | Batch reactor; [O3]0: 4.51 mg L; [Br−]0: 2.0 mg L; catalyst: 0.1 g L; initial pH: 6.2 | CeO2 minimized bromate formation possibly due to lower H2O2 formation and reduction of BrO˙ to HOBr/OBr− during H2O2 decomposition | 18 |
| Ce-MCM-48 | Batch reactor; [O3]0: 2 mg L; [Br−]0: 1 mg L; catalyst: 0.5 g L; initial pH: 7.5 | Ce66-MCM-48 inhibited bromate formation: (1) promoted O3 decomposition to generate ˙OH thus suppressing initial oxidation of Br− to HOBr/OBr−; (2) ˙OH may form H2O2 which reacted with ˙OH and O3; (3) circulating reactions between Ce4+ to Ce3+ inhibited the oxidation of Br− and HOBr/OBr− | 30 |
| CeO2/rGO | Semi-batch reactor; [O3]: 10 mg (L−1 min); [Br−]0: 12.5 μM; catalyst: 0.02 g L; pH 7 with 10 mM PBS | Ce3+ on CeO2/rGO quenched Br˙ and BrO˙, and inhibited bromate formation | 36 |
| Lattice O-rich MnOOH | Semi-batch reactor; catalyst: 0.1 g L; Br−: 1.1 mg L; 4-nitrophenol (4-NP): 20 mg L−1 | HCO with MnOOH inhibited bromate formation and boosted 4-NP degradation: (1) accelerated O3 decomposition and decreased DO3; (2) O3 decomposition led to generation of ˙O2− instead of ˙OH; (3) direct reduction of formed bromate by the catalyst | 38 |
| MnOx/Al2O3 | Batch reactor; [O3]0: 2 mg L; [Br−]0: 100 μg L; [2,4-D]0: 10 mg L−1; catalyst: 0.25 g L; initial pH: 6.5 | MnOx/Al2O3 exhibited inhibition of bromate formation and efficient catalytic ozonation of 2,4-D: reduction of HOBr and bromate was facilitated by redox reactions between Mn2+/Mn3+/Mn4+ | 49 |
| Nano-TiO2 | Batch reactor; [O3]0: 3.38 mg L; [Br−]0: 0.4 mg L; catalyst: 0.1 g L; initial pH: 6.0 | The catalysts accelerated O3 decomposition into ˙OH and generated H2O2 which reduced HOBr/OBr− to Br− | 14 |
| H+-form high silica zeolite (HZSM-5) | Batch reactor; [O3]0: 1.3 mg L; [Br−]0: 300 μg L; catalyst: 1 g L; filtered surface water with pH 8.0 & [DOC]0 1.4 mg L−1 | HZSM-5 reduced bromate formation was possibly ascribed to the selective OBr− adsorption and the inhibited formation of H2O2 | 43 |
| LaCoO3 & LaFeO3 | Semi-batch reactor; [Br−]0: 100 μg L; [BZA]0: 10 m L; catalyst: 0.5 g L; initial pH: 6.4 | LaCoO3 exhibited catalytic activity for BZA degradation and inhibited bromate formation: (1) produced ˙OH and accelerated BZA degradation; (2) production of HO2˙−/˙O2− led to more H2O2 generation, contributing to the reduction of bromate to HOBr/OBr−; (3) cyclic reaction of Co3+/Co4+ facilitated O3 decomposition and accelerated the above reactionsLaFeO3 exhibited no catalytic activity for BZA degradation but inhibited bromate formation: (1) didn't accelerate ROS production; (2) surface –OH combined with generated H2O2 to form Fe–H2O2 which reduced bromate to HOBr/OBr− easily | 42 |
| g-C3N4 | Semi-batch reactor; [Br−]0: 100 μg L; [pollutant]0b: 0.084 mM; catalyst: 0.25 g L; initial pH: 4.75 for p-CBA solution and 6.01 for BZA solution | g-C3N4 inhibited bromate formation due to the formation of H2O2 | 44 |
| Heteroatom-doped GO | Semi-batch reactor; [Br−]0: 100 μg L; [pollutant]0b: 0.084 mM; catalyst: 0.25 g L; initial pH: 4.75 for p-CBA solution and 6.01 for BZA solution | Bromate elimination followed the order N-rGO > P-rGO > B-rGO > rGO > S-rGO: formation of H2O2 favored bromate elimination | 45 |
2.2 Bromate formation in HCO
While most relevant studies have reported less bromate formation in HCO compared with ozonation alone under similar conditions, there are a few exceptions.17,36 In general, addition of heterogeneous catalysts may affect bromate formation due to one or several of the following reasons (Fig. 2).3. Formation of organic byproducts and transformation of bulk organics in HCO
While AOPs including HCO often target at recalcitrant organic pollutants which usually exist at very low concentrations in environmental waters, oxidation of bulk organics (e.g. DOM and EfOM) is also unavoidable in these processes especially when non-selective oxidants (e.g. ˙OH) are generated. Hence the transformation of bulk organics and formation of organic byproducts are also of great importance as they may affect the treated water quality as well as the formation of DBPs in the following disinfection process. Hence, in this review direct formation of organic oxidation byproducts (OBPs), removal and transformation of bulk organics, and removal of DBP precursors will be discussed with the main findings summarized in Table 2.| a Limited studies available. | |
|---|---|
| OBPs formation | • Aldehydes production more depends on O3 dose |
| • Keto-acids production usually is lower in HCO | |
| • Carboxylic acids production depends on O3 dose and interactions between DOM and catalysts | |
| • Similar TOX production in the presence of Cl− as in ozonation; less TOX production in the presence of Cl− and Br−a | |
| DOM removal & transformation | • Enhance TOC removal |
| • Increase biodegradability of bulk organics | |
| • UV254 removal depends on catalysts & DOM characteristics | |
| Removal of DBP precursors | • Decrease THMFP and HAAFP |
| • May increase BIF of THMs & HAAs | |
| • Effects on HANEP depend on matrices & O3 dose | |
3.1 OBPs formation
Carbonyl compounds, carboxylic acids, and keto-acids have been reported to be the main OBPs formed due to the reaction between ozone and DOM.21,51–53 The addition of solid catalysts may alter the exposure of bulk organics to O3, increase the production of ˙OH, and introduce surface adsorption and complexation, thus changing the OBPs formation.In the study of catalytic ozonation of BZA and reduction of toxic by-products in a primary/secondary effluent matrix with LaCoO3, at a lower O3 dose (e.g. 1 mg L−1) the production of total aldehydes increased with time, but was always lower in catalytic ozonation than in ozonation, whereas at higher O3 doses (e.g. 2 mg L−1 and 5 mg L−1), total aldehydes increased then decreased and the production was similar with or without catalysts.27 Formation of oxalic acid, on the other hand, was higher in catalytic ozonation with LaCoO3 than ozonation at the O3 dose of 1 mg L−1 but lower at the O3 dose of 2 mg L−1 and 5 mg L−1.27 In catalytic ozonation of filtered river water, the yields of aldehydes and acetone were higher in the process of O3/α-FeOOH than that of ozonation alone because of the enhanced production of ˙OH which can effectively oxidize DOM into low molecular weight (MW) molecules such as aldehydes. In contrast, such yields in the O3/CeO2 were close to that of ozonation.54 In the same study, the production of keto-acids was lower with O3/CeO2 than with O3/α-FeOOH and both catalytic processes yielded less total keto-acids than ozonation alone when O3 was in excess.54 Moreover, the two catalysts exhibited different influence on the yield of carboxyl acids, which was in the order of O3/α-FeOOH > O3 > O3/CeO2.54 Such different behavior was ascribed to the fact that unlike α-FeOOH promoting ˙OH generation, CeO2 enhanced the O3 degradation of low MW compounds via adsorption and surface complexes.54,55
Similarly, in a comparative study of ozonation and FeOOH catalyzed ozonation of different DOM fractions isolated from filtered river water, lower yields of keto-acids were reported after catalytic ozonation of hydrophobic acid and neutral (HOA and HON) and hydrophilic acid (HIA) fractions, whereas the yields of carboxylic acids from these three fractions were higher in the process of O3/FeOOH as compared to ozonation alone.56 In the meantime, no significant difference was observed for the production of keto-acids and carboxylic acids from the hydrophilic base (HIB) fraction in the O3 and O3/FeOOH processes.56 As for aldehydes and acetone, addition of FeOOH led to higher yields from HIA and HIB fractions, lower yields from HON fractions, and no effect on HOA fraction.56 Further, in a parallel study of fluorescence spectroscopic characterization of DOM fractions after ozonation and catalytic ozonation (FeOOH and CeO2), it was found catalytic ozonation substantially enhanced the formation of low aromaticity and low MW by-products from HIA fraction and improved the destruction of highly polycyclic aromatic structures for all DOM fractions.57
Lastly, halogenated organic byproducts may be formed during oxidation treatment in the presence of Cl− and Br−. Interestingly, HCO produced similar amount of total organic halogen (TOX) as ozonation of EfOM matrix (Cl−: 121 mg L−1); but with the addition of 0.1 mg L−1 Br−, HCO generated much less TOX than sole ozonation.27 It was suspected that ˙OH may change the EfOM structure, making it less reactive to Br˙ and HOBr/OBr− and can also degrade the formed halogenated organic byproducts.27
Production of OBPs depends on the reactivity of precursors and OBPs with O3/˙OH, interactions between DOM/OBPs and catalysts, characteristics of DOM, and O3 dose. ˙OH generally has higher oxidizing capability and is more effective in degrading DOM into low MW compounds. For the low MW compounds that have similar reactivity with O3 and ˙OH such as aldehydes, yields of these OBPs may more depend on O3 dose than the addition of catalysts that promotes ˙OH generation. For OBPs with higher reactivity towards ˙OH such as keto-acids, the production usually is lower in the presence of catalyst especially at a sufficient O3 dose. Short-chain carboxylic acids such as oxalic acid are considered as the “bottleneck” and main OBPs of AOPs. Very often accumulation of carboxylic acids is observed in HCO, which is sometimes higher than ozonation alone due to more effective degradation of precursors such as keto-acids by ˙OH. While these OBPs are described as refectory to both O3 and ˙OH in some studies, it should be noted although much lower than those of many other organic compounds, their reaction rate constants with ˙OH are still several orders of magnitude higher than that with O3 (for example, oxalic acid was frequently used as the model compounds in HCO studies).58,59 Hence, eventually it might be the high energy consumption that makes the elimination of OBPs economically inhibitive in HCO, and integration with other treatment processes such as biofiltration may be a more cost-effective approach to further polish the treated water quality.28,60,61
3.2 Removal and transformation of bulk organics
Humic substances represent a major fraction of NOM in virtually all drinking water sources as well as effluent organic matter (EfOM) from biological wastewater treatment. Hence humic acid (HA) has been chosen as a target compound in several HCO studies and a higher mineralization rate was reported for the HCO processes in comparison to ozonation alone under otherwise same conditions, owing to higher ˙OH generation.62–64 In particular, Tang et al. examined the acute toxicity of the treated water by ozonation and catalytic ozonation with Cu/Ce–Al2O3 and found that the toxicity of HCO treated water was 56% lower than that of ozonation ascribed to the effective removal of toxic intermediates of HA degradation (e.g. phenolic compounds or long-chain hydrocarbon compounds). It was also suggested that minimizing metallic ion leaching from the catalyst was important for further reduction of the treated water toxicity.64Furthermore, several studies were conducted to investigate the degradation and transformation of DOM from natural water or wastewater in HCO processes. Compared to ozonation alone, HCO generally led to a higher TOC/DOC removal20,56,65–67 but the ratio of assimilable organic carbon (AOC) was reported to increase in HCO treated water especially in the hydrophilic acid and base fractions of DOM.56,65,68 Dabuth et al. investigated molecular changes of DOM after catalytic ozonation with TiO2 coated activated carbon (TiO2-AC) employing unknown screening analysis with Orbitrap mass spectrometry, and reported that catalytic ozonation decreased or removed more CHO features than that of ozonation alone owing to the generation of more reactive species (e.g. ˙OH and ˙O2−).68 Furthermore, a few hundred CHO features were increased or newly formed after oxidation treatment, indicating the formation of OBPs, whereas the number of OBPs was also lower for O3/TiO2-AC processes than ozonation.68 Reduction of UV254 (mainly denoting organic compounds with double bonds or aromatic structures) seemed to depend on the characteristics of DOM as well as catalysts. An enhanced UV254 removal was observed in catalytic ozonaton of filtered water NOM with TiO2/Zeolite and CeO2,55,65 but a lower removal than ozonation was reported for MgO and goethite.55 It was also found that HCO improved the UV254 reduction for fractions of MW >1000 Da but not much for lower-MW components of organic matter from a filtered river water.56 A study on catalytic ozonation (copper supported alumina) of different DOM fractions from secondary effluent of a petrochemical wastewater plant reported lower SUV254 reduction than ozonation alone for both hydrophilic and hydrophobic substances but a higher DOC removal for all fractions.67 Often ozonation was only effective for hydrophobic fractions and may transform them to hydrophilic substances.20,66,67 Furthermore, interactions between DOM and catalysts can also affect the oxidation processes. Although the enhanced DOC reduction in both HCO processes was ascribed to ˙OH formation, the higher efficiency of the O3/FeOOH system than the O3/MgO was explained by higher DOM adsorption on MgO, which may adversely affect O3 mass transfer and access to the active sites of the catalyst.55 However, Sun et al. suggested that O3 and generated ˙OH could react with DOM adsorbed on the catalyst surface and allow for further adsorption from the bulk solution, and a dynamic balance between oxidation and re-adsorption was important for DOM removal in HCO processes.67 On the other hand, degradation of DOM in CeO2 catalytic ozonation was attributed to surface complexation, not ˙OH formation, resulting in a low efficiency.55
3.3 Removal of DBP precursors
Hydrophobic fractions of NOM have been reported to be the major precursors of trihalomethanes (THMs).69,70 Since both ozone and HCO can remove hydrophobic substances, THM formation potential (THMFP) can be reduced in these processes as well. For example, TiO2-catalyzed ozonation and ozonation alone removed 80% and 45% THM precursors respectively, during the treatment of a groundwater sample dominated by fulvic acid fraction.20 Similar trend was also observed for TiO2-catalyzed ozonation of river water and the higher the ozone dose, the greater the THMFP reduction.71 Catalytic ozonation with LaCoO3 showed better efficiency than sole ozonation on the reduction of formation potential of two THMs, trichloromethane (TCM) and bromodichloromethane (BDCM), during the treatment of a mixture of primary and secondary effluent.27 Moreover, Fe–Mn oxide, TiO2/Al2O3, and synthetic goethite (α-FeOOH) catalyzed ozonation were also found to remove more THMs and nine haloacetic acids (HAA9) precursors than ozonation alone in the treatment of filtered river water.28,61 During the treatment of 11 filtered natural water samples sampled from upstream to downstream along Jingmi Channel (China), Ru/AC-O3 process was reported to reduce HAAFP much more efficiently than ozonation alone, but such enhancement was less pronounced for THMs, probably because the reduction of THMPF was already high in ozonation.72 In addition to reduction of DBPFP, HCO can also affect the species distribution of DBPs. While the THMFP and HAAFP decreased, the bromine incorporation factor (BIF) of THMs and HAAs increased with catalyst dosage during chlorination after HCO treatment.28 On the other hand, in a study investigating DBP formation during chlorination of a HCO treated Diclofenac (DCF) solution with co-existing Br−, Chen et al. found that Fe-Cu-MCM-41/O3 pretreatment led to less bromate, THMs, and HAAs as well as Br-DBP formation than O3 pretreatment, owing to the more effective removal of hydrophobic and transphilic intermediates generated from DCF degradation.40Hydrophilic fractions of NOM have been found to be rich in nitrogenous matters as the potential precursors of N-DBPs,73 hence oxidation processes may increase the N-DBP formation due to the oxidation of hydrophobic fractions and consequent increase of hydrophilic fractions. An increased haloacetonitrile formation potential (HANFP) compared with raw water was reported after ozonation and TiO2-catalyzed ozonation treatment of groundwater.20 Further, while TiO2/O3 produced less HANs than O3 direct oxidation by O3 was found to be more effective for specific HANFP reduction compared to the radical pathway.20 In contrast, ozonation and catalytic ozonation with LaCoO3 decreased the HANFP during the treatment of a primary/secondary effluent mixture, where the reduction by catalytic ozonation was higher than ozonation at lower O3 doses (1 mg L−1 and 2 mg L−1) but lower at a higher O3 dose (5 mg L−1).27 In addition to the different matrices (groundwater20vs. primary/secondary effluent27), the O3 dose may be one important reason for the different observations. Unfortunately, it is difficult to make direct comparisons as one was reported as 3 mg O3/mg DOC20 and the other 1–5 mg L−1 O3.27 Moreover, O3 and LaCoO3/O3 can also reduce the precursors of emerging DBPs, haloacetamides (HAcAms), with the LaCoO3/O3 process being more efficient.27
Lastly, it should be pointed out that the observed enhancement of DBPFP removal in HCO may not all be ascribed to the high ˙OH in the presence of catalysts because it has been showed that adsorption by the catalysts (e.g. Fe–Mn oxide) can also remove a significant amount of precursors.61 However, most studies did not look into the adsorption effects of catalysts with only a few exceptions.61,72
4. Summary and perspective
While heterogeneous catalytic ozonation processes have shown a great potential in enhancing the removal of recalcitrant organic pollutants, formation of byproducts as well as transformation of bulk organics and the consequent impacts on DBPs formation calls for more attention. In this regard, the main findings of the relevant literature can be summarized as follows:• As the major inorganic byproduct of concern in ozonation processes, bromate can be formed via both direct O3 oxidation and indirect ˙OH oxidation pathways. Very often the presence of catalysts accelerates the O3 decomposition and increases ˙OH generation, and thus bromate formation may be inhibited or promoted depending on the dominant reaction pathway (direct vs. indirect) in the system. The addition of catalysts can also affect the generation of H2O2 which may reduce HOBr/OBr−. Decomposition of O3 can also generate reactive species (e.g. HO2˙−/˙O2−) that accelerate the redox cycle of the metal species on the catalyst surface, during which Br species (e.g. Br˙/BrO˙, HOBr/OBr−, and BrO3−) are reduced resulting in less bromate formation. Lastly, some catalysts (e.g. CeO2) may adsorb O3/O2-containing species and inhibit O3 decomposition into ˙OH and subsequent bromate formation.
• For organic byproducts, aldehydes yield may more depend on O3 dose than the addition of catalysts. Keto-acids production is usually lower in HCO than ozonation alone. Carboxylic acids can accumulate in both ozonation and HCO processes and the yields are influenced by O3 dose and interactions between NOM/OBPs and catalysts.
• HOC can increases DOC removal and biodegradability of the bulk organics. HCO may also decrease THMFP and HAAFP, but may increase bromine incorporation factor of THMs and HAAs. Its effect on HANFP depends on the matrices and O3 dose.
Despite some progress has been made in understanding byproducts formation during HCO, there are still some important issues that need to be further investigated and addressed:
• It is encouraging that most of the studies reported an inhibition of bromate formation in HCO. However, elucidation of the underlying mechanism can be challenging due to the high heterogeneity of the system and the complex interactions between different species as well as the solid catalysts. Also because of the multiple reaction pathways possibly involved, it is relatively easy to formulate a hypothesis to explain the experimental observations but difficult to confirm and validate it. Therefore, a rigorous and comprehensive experimental design is required to probe the mechanism (which is likely to be system-specific due to the diversity of catalysts) and facilitate further process enhancement and optimization. Moreover, most of the studies reported in the literature were carried out in synthetic solutions not environmental matrices. Although starting with a “clean” matrix is a reasonable approach for initial evaluation and mechanism investigation, further examination of the matrix effects is needed, especially as it has been shown that co-existence of organic and inorganic precursors can affect the final byproducts formation.
• Due to the ubiquity and diversity of NOM in the environmental waters, investigation on the organic byproducts' formation and transformation of bulk organics in HCO can be more challenging. In order to enhance our understanding and gain insights from different studies collectively, it is important for the research community to standardize the experimental protocols and provide enough information so to enable comparisons and systematic evaluation. For example, regardless of the addition of catalysts, O3 dose is one most influencing factor that determines process performance and byproducts formation. Due to the limitation of mass transfer in most laboratory setup, it is recommended O3 dose to be reported as total transferred dose during the treatment not just the concentrations in the feeding gas or aqueous phase.
• Some catalysts that promote surface complexation with DOM instead of ˙OH production (e.g. CeO2) seem to perform better in OBPs control. However, the efficiency for the removal of recalcitrant pollutants as well as possible catalyst fouling in long-term performance in real environmental waters may need further evaluation. In the meantime, direct interactions with catalysts (e.g. adsorption) can play an important role and should be evaluated in all HCO studies to truly understand the underlying mechanism.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The author appreciates the support from U.S. National Science Foundation (CBET-1606117).References
- I. A. Katsoyiannis, S. Canonica and U. von Gunten, Efficiency and energy requirements for the transformation of organic micropollutants by ozone, O3/H2O2 and UV/H2O2, Water Res., 2011, 45(13), 3811–3822, DOI:10.1016/j.watres.2011.04.038.
- E. J. Rosenfeldt, K. G. Linden, S. Canonica and U. von Gunten, Comparison of the efficiency of {radical dot}OH radical formation during ozonation and the advanced oxidation processes O3/H2O2 and UV/H2O2, Water Res., 2006, 40(20), 3695–3704, DOI:10.1016/j.watres.2006.09.008.
- T. Wu and J. D. Englehardt, Mineralizing urban net-zero water treatment: Field experience for energy-positive water management, Water Res., 2016, 106, 352–363, DOI:10.1016/j.watres.2016.10.015.
- S. Yuan, Z. Li and Y. Wang, Effective degradation of methylene blue by a novel electrochemically driven process, Electrochem. Commun., 2013, 29, 48–51, DOI:10.1016/j.elecom.2013.01.012.
- H. Wang, J. Zhan, W. Yao, B. Wang, S. Deng, J. Huang, G. Yu and Y. Wang, Comparison of pharmaceutical abatement in various water matrices by conventional ozonation, peroxone (O3/H2O2), and an electro-peroxone process, Water Res., 2018, 130, 127–138, DOI:10.1016/j.watres.2017.11.054.
- W. Yao, S. W. Ur Rehman, H. Wang, H. Yang, G. Yu and Y. Wang, Pilot-scale evaluation of micropollutant abatements by conventional ozonation, UV/O3, and an electro-peroxone process, Water Res., 2018, 138, 106–117, DOI:10.1016/j.watres.2018.03.044.
- Y. Mao, D. Guo, W. Yao, X. Wang, H. Yang, Y. F. Xie, S. Komarneni, G. Yu and Y. Wang, Effects of conventional ozonation and electro-peroxone pretreatment of surface water on disinfection by-product formation during subsequent chlorination, Water Res., 2018, 130, 322–332, DOI:10.1016/j.watres.2017.12.019.
- C. Canton, S. Esplugas and J. Casado, Mineralization of phenol in aqueous solution by ozonation using iron or copper salts and light, Appl. Catal., B, 2003, 43(2), 139–149, DOI:10.1016/S0926-3373(02)00276-X.
- U. Pinkernell and U. Von Gunten, Bromate minimization during ozonation: mechanistic considerations, Environ. Sci. Technol., 2001, 35(12), 2525–2531, DOI:10.1021/es001502f.
- U. Von Gunten and J. Hoigne, Bromate formation during ozonation of bromide-containing waters: Interaction of ozone and hydroxyl radical reactions, Environ. Sci. Technol., 1994, 28(7), 1234–1242, DOI:10.1021/es00056a009.
- U. von Gunten and Y. Oliveras, Advanced Oxidation of Bromide-Containing Waters : Bromate Formation Mechanisms, Environ. Sci. Technol., 1998, 32(1), 63–70, DOI:10.1021/es970477j.
- P. Westerhoff, R. Song, G. Amy and R. Minear, NOM's role in bromine and bromate formation during ozonation, J. - Am. Water Works Assoc., 1998, 90(2), 82–94 CrossRef CAS.
- Y. Nie, N. Li and C. Hu, Enhanced inhibition of bromate formation in catalytic ozonation of organic pollutants over Fe–Al LDH/Al2O3, Sep. Purif. Technol., 2015, 151, 256–261, DOI:10.1016/j.seppur.2015.07.057.
- Y. Wu, C. Wu, Y. Wang and C. Hu, Inhibition of Nano-Metal Oxides on Bromate Formation during Ozonation Process, Ozone: Sci. Eng., 2014, 36(6), 549–559, DOI:10.1080/01919512.2014.904735.
- H. W. Yang, S. X. Yang, L. Wu and W. J. Liu, CexZr1−xO2 mixed oxides applied to minimize the bromate formation in the catalytic ozonation of a filtered water, Catal. Commun., 2011, 15(1), 99–102, DOI:10.1016/j.catcom.2011.08.032.
- Y. Nie, C. Hu, L. Yang and J. Hu, Inhibition mechanism of BrO? 3 formation over MnOx/Al2O3 during the catalytic ozonation of 2,4-dichlorophenoxyacetic acid in water, Sep. Purif. Technol., 2013, 117, 41–45, DOI:10.1016/j.seppur.2013.03.045.
- Q. Wang, Z. Yang, J. Ma, J. Wang and L. Wang, Study on the mechanism of cerium oxide catalytic ozonation for controlling the formation of bromate in drinking water, Desalin. Water Treat., 2015, 1–15, DOI:10.1080/19443994.2015.1079261.
- T. Zhang, W. Chen, J. Ma and Z. Qiang, Minimizing bromate formation with cerium dioxide during ozonation of bromide-containing water, Water Res., 2008, 42(14), 3651–3658, DOI:10.1016/j.watres.2008.05.021.
- A. Fischbacher, K. Löppenberg, C. von Sonntag and T. C. Schmidt, A New Reaction Pathway for Bromite to Bromate in the Ozonation of Bromide, Environ. Sci. Technol., 2015, 49(19), 11714–11720, DOI:10.1021/acs.est.5b02634.
- J. J. Molnar, J. R. Agbaba, B. D. Dalmacija, M. T. Klašnja, M. B. Dalmacija and M. M. Kragulj, A comparative study of the effects of ozonation and TiO2-catalyzed ozonation on the selected chlorine disinfection by-product precursor content and structure, Sci. Total Environ., 2012, 425, 169–175, DOI:10.1016/j.scitotenv.2012.03.020.
- P. Westerhoff, J. Debroux, G. Aiken and G. Amy, Ozone-induced changes in natural organic matter (NOM) structure, Ozone: Sci. Eng., 1999, 21(6), 551–570, DOI:10.1080/01919512.1999.10382893.
- W. H. Glaze, Reaction products of ozone: a review, Environ. Health Perspect., 1986, 69, 151–157, DOI:10.1289/ehp.8669151.
- U. von Gunten, Ozonation of drinking water: part I. Oxidation kinetics and product formation, Water Res., 2003, 37(7), 1443–1467, DOI:10.1016/S0043-1354(02)00457-8.
- B. Kasprzyk-Hordern, U. Raczyk-Stanisławiak, J. Świetlik and J. Nawrocki, Catalytic ozonation of natural organic matter on alumina, Appl. Catal., B, 2006, 62(3–4), 345–358, DOI:10.1016/j.apcatb.2005.09.002.
- L. Wei, K. Wen, J. Lu and J. Ma, Quantification of low molecular weight oxidation byproducts produced from real filtered water after catalytic ozonation with different pathways, J. Hazard. Mater., 2021, 405, 124674, DOI:10.1016/j.jhazmat.2020.124674.
- R. J. Miltner, T. F. Speth, S. D. Richardson, S. W. Krasner, H. S. Weinberg and J. E. Simmons, Integrated disinfection by-products mixtures research: disinfection of drinking waters by chlorination and ozonation/postchlorination treatment scenarios, J. Toxicol. Environ. Health, Part A, 2008, 71(17), 1133–1148, DOI:10.1080/15287390802182060.
- Y. Zhang, Y. An, C. Liu, Y. Wang, Z. Song, Y. Li, W. Meng, F. Qi, B. Xu, J. P. Croue, D. Yuan and A. Ikhlaq, Catalytic ozonation of emerging pollutant and reduction of toxic by-products in secondary effluent matrix and effluent organic matter reaction activity, Water Res., 2019, 166, 115026, DOI:10.1016/j.watres.2019.115026.
- Y. H. Wang and K. C. Chen, Removal of disinfection by-products from contaminated water using a synthetic goethite catalyst via catalytic ozonation and a biofiltration system, Int. J. Environ. Res. Public Health, 2014, 11(9), 9325–9344, DOI:10.3390/ijerph110909325.
- G. Yu, Y. Wang, H. Cao, H. Zhao and Y. Xie, Reactive Oxygen Species and Catalytic Active Sites in Heterogeneous Catalytic Ozonation for Water Purification, Environ. Sci. Technol., 2020, 54(10), 5931–5946, DOI:10.1021/acs.est.0c00575.
- W. Li, X. Lu, K. Xu, J. Qu and Z. Qiang, Cerium incorporated MCM-48 (Ce-MCM-48) as a catalyst to inhibit bromate formation during ozonation of bromide-containing water: Efficacy and mechanism, Water Res., 2015, 48, 1–7, DOI:10.1016/j.watres.2015.05.052.
- M. B. Heeb, J. Criquet, S. G. Zimmermann-Steffens and U. Von Gunten, Oxidative treatment of bromide-containing waters: Formation of bromine and its reactions with inorganic and organic compounds - A critical review, Water Res., 2014, 48(1), 15–42, DOI:10.1016/j.watres.2013.08.030.
- M. S. Siddiqui, Chlorine-ozone interactions : chlorate formation of, Water Res., 1996, 30(9), 2160–2170 CrossRef CAS.
- B. Rao, T. A. Anderson, A. Redder and W. A. Jackson, Perchlorate formation by ozone oxidation of aqueous chlorine/oxy-chlorine species: Role of ClxOy radicals, Environ. Sci. Technol., 2010, 44(8), 2961–2967, DOI:10.1021/es903065f.
- A. V. Levanov and O. Y. Isaikina, Mechanism and Kinetic Model of Chlorate and Perchlorate Formation during Ozonation of Aqueous Chloride Solutions, Ind. Eng. Chem. Res., 2020, 59(32), 14278–14287, DOI:10.1021/acs.iecr.0c02770.
- G. G. Jayson, B. J. Parsons and A. J. Swallow, Some simple, highly reactive, inorganic chlorne derivatives in aqueous solution, J. Chem. Soc., Faraday Trans. 1., 1973, 69, 1597–1607 RSC.
- B. Ye, Z. Chen, X. Li, J. Liu, Q. Wu, C. Yang, H. Hu and R. Wang, Inhibition of bromate formation by reduced graphene oxide supported cerium dioxide during ozonation of bromide-containing water, Front. Environ. Sci. Eng., 2019, 13(6), 86, DOI:10.1007/s11783-019-1170-z.
- X. Chen, H. Yang, C. Au, S. Tian, Y. Xiong and Y. Chang, Efficiency and mechanism of pollutant degradation and bromate inhibition by faceted CeO2 catalyzed ozonation: Experimental and theoretical study, Chem. Eng. J., 2020, 390, 124480, DOI:10.1016/j.cej.2020.124480.
- Y. Huang, M. Luo, S. Li, D. Xia, Z. Tang, S. Hu, S. Ye, M. Sun, C. He and D. Shu, Efficient catalytic activity and bromate minimization over lattice oxygen-rich MnOOH nanorods in catalytic ozonation of bromide-containing organic pollutants: Lattice oxygen-directed redox cycle and bromate reduction, J. Hazard. Mater., 2021, 410, 124545, DOI:10.1016/j.jhazmat.2020.124545.
- W. Chen, X. Li, Y. Tang, J. Zhou, D. Wu, Y. Wu and L. Li, Mechanism insight of pollutant degradation and bromate inhibition by Fe-Cu-MCM-41 catalyzed ozonation, J. Hazard. Mater., 2018, 346, 226–233, DOI:10.1016/j.jhazmat.2017.12.036.
- W. Chen, Z. Zhang, X. Li, D. Wu, Y. Xue and L. Li, Reducing DBPs formation in chlorination of Br-containing Diclofenac via Fe-Cu-MCM-41/O3 peroxidation: Efficiency, characterization DBPs precursors and mechanism, J. Taiwan Inst. Chem. Eng., 2018, 84, 212–221, DOI:10.1016/j.jtice.2018.01.019.
- H. Yang, S. Yang, L. Wu and W. Liu, CexZr1−xO2 mixed oxides applied to minimize the bromate formation in the catalytic ozonation of a filtered water, Catal. Commun., 2011, 15(1), 99–102, DOI:10.1016/j.catcom.2011.08.032.
- Y. Zhang, Y. Xia, Q. Li, F. Qi, B. Xu and Z. Chen, Synchronously degradation benzotriazole and elimination bromate by perovskite oxides catalytic ozonation: Performance and reaction mechanism, Sep. Purif. Technol., 2018, 197, 261–270, DOI:10.1016/j.seppur.2018.01.019.
- T. Zhang, P. Hou, Z. Qiang, X. Lu and Q. Wang, Reducing bromate formation with H+-form high silica zeolites during ozonation of bromide-containing water: Effectiveness and mechanisms, Chemosphere, 2011, 82(4), 608–612, DOI:10.1016/j.chemosphere.2010.10.078.
- Z. Song, Y. Zhang, C. Liu, B. Xu, F. Qi, D. Yuan and S. Pu, Insight into[rad]OH and O2[rad]−formation in heterogeneous catalytic ozonation by delocalized electrons and surface oxygen-containing functional groups in layered-structure nanocarbons, Chem. Eng. J., 2019, 357, 655–666, DOI:10.1016/j.cej.2018.09.182.
- Z. Song, M. Wang, Z. Wang, Y. Wang, R. Li, Y. Zhang, C. Liu, Y. Liu, B. Xu and F. Qi, Insights into Heteroatom-Doped Graphene for Catalytic Ozonation: Active Centers, Reactive Oxygen Species Evolution, and Catalytic Mechanism, Environ. Sci. Technol., 2019, 53(9), 5337–5348, DOI:10.1021/acs.est.9b01361.
- Y. Pi, J. Schumacher and M. Jekel, Decomposition of aqueous ozone in the presence of aromatic organic solutes, Water Res., 2005, 39(1), 83–88, DOI:10.1016/j.watres.2004.09.004.
- K. Pelle, M. Wittmann, K. Lovrics, Z. Noszticzius, M. L. Turco Liveri and R. Lombardo, Mechanistic Investigations of the BZ Reaction with Oxalic Acid Substrate. I. The Oscillatory Parameter Region and Rate Constants Measured for the Reactions of HOBr, HBrO2, and Acidic BrO3− with Oxalic Acid, J. Phys. Chem. A, 2004, 108(25), 5377–5385, DOI:10.1021/jp048817s.
- E. G. Heckert, S. Seal and W. T. Self, Fenton-Like Reaction Catalyzed by the Rare Earth Inner Transition Metal Cerium, Environ. Sci. Technol., 2008, 42(13), 5014–5019, DOI:10.1021/es8001508.
- Y. Nie, C. Hu, L. Yang and J. Hu, Inhibition mechanism of BrO-3 formation over MnOx/Al2O3 during the catalytic ozonation of 2,4-dichlorophenoxyacetic acid in water, Sep. Purif. Technol., 2013, 117, 41–45, DOI:10.1016/j.seppur.2013.03.045.
- Y. Nie, C. Hu, N. Li, L. Yang and J. Qu, Inhibition of bromate formation by surface reduction in catalytic ozonation of organic pollutants over β-FeOOH/Al2O3, Appl. Catal., B, 2014, 147, 287–292, DOI:10.1016/j.apcatb.2013.09.005.
- B. Jurado-Sánchez, E. Ballesteros and M. Gallego, Occurrence of carboxylic acids in different steps of two drinking-water treatment plants using different disinfectants, Water Res., 2014, 51, 186–197, DOI:10.1016/j.watres.2013.10.059.
- A. Papageorgiou, D. Voutsa and N. Papadakis, Occurrence and fate of ozonation by-products at a full-scale drinking water treatment plant, Sci. Total Environ., 2014, 481(1), 392–400, DOI:10.1016/j.scitotenv.2014.02.069.
- W. H. Glaze, Drinking-water treatment with ozone, Environ. Sci. Technol., 1987, 21(3), 224–230, DOI:10.1021/es00157a001.
- L. Wei, K. Wen, J. Lu and J. Ma, Quantification of low molecular weight oxidation byproducts produced from real filtered water after catalytic ozonation with different pathways, J. Hazard. Mater., 2021, 405, DOI:10.1016/j.jhazmat.2020.124674.
- Q. Wang, Z. Yang, B. Chai, S. Cheng, X. Lu and X. Bai, Heterogeneous catalytic ozonation of natural organic matter with goethite, cerium oxide and magnesium oxide, RSC Adv., 2016, 6(18), 14730–14740, 10.1039/c5ra21674e.
- T. Zhang, J. Lu, J. Ma and Z. Qiang, Comparative study of ozonation and synthetic goethite-catalyzed ozonation of individual NOM fractions isolated and fractionated from a filtered river water, Water Res., 2008, 42(6–7), 1563–1570, DOI:10.1016/j.watres.2007.11.005.
- T. Zhang, J. Lu, J. Ma and Z. Qiang, Fluorescence spectroscopic characterization of DOM fractions isolated from a filtered river water after ozonation and catalytic ozonation, Chemosphere, 2008, 71(5), 911–921, DOI:10.1016/j.chemosphere.2007.11.030.
- J. Hoigne and H. Bader, Rate constants of reactions of ozone with organic and inorganic compounds in water—II: Dissociating organic compounds, Water Res., 1983, 17, 185–194 CrossRef CAS.
- B. G. Ershov, E. Janata, M. S. Alam and A. V. Gordeev, Studies of the reaction of the hydroxyl radical with the oxalate ion in an acidic aqueous solution by pulse radiolysis, Russ. Chem. Bull., 2008, 57(6), 6–8 Search PubMed.
- K. C. Chen, Y. H. Wang and Y. H. Chang, Using catalytic ozonation and biofiltration to decrease the formation of disinfection by-products, Desalination, 2009, 249(3), 929–935, DOI:10.1016/j.desal.2009.06.066.
- K. Chen and Y. Wang, The effects of Fe–Mn oxide and TiO2/a-Al2O3 on the formation of disinfection by-products in catalytic ozonation, Chem. Eng. J., 2014, 253, 84–92, DOI:10.1016/j.cej.2014.04.111.
- A. Alver and A. Kılıç, Catalytic ozonation by iron coated pumice for the degradation of natural organic matters, Catalysts, 2018, 8(5), 219, DOI:10.3390/catal8050219.
- S. B. Mortazavi, G. Asgari, S. J. Hashemian and G. Moussavi, Degradation of humic acids through heterogeneous catalytic ozonation with bone charcoal, React. Kinet. Mech. Catal., 2010, 100(2), 471–485, DOI:10.1007/s11144-010-0192-0.
- X. Tang, Y. Zhang, W. Li, J. Geng, H. Ren and K. Xu, Mechanism and toxicity evaluation of catalytic ozonation over Cu/Ce–Al2O3 system aiming at degradation of humic acid in real wastewater, Sci. Rep., 2021, 11(1), 1–13, DOI:10.1038/s41598-021-83804-x.
- S. J. Wang, J. Ma, Y. X. Yang, J. Zhang and T. Liang, Degradation and transformation of organic compounds in songhua river water by catalytic ozonation in the presence of TiO2/zeolite, Ozone: Sci. Eng., 2011, 33(3), 236–242, DOI:10.1080/01919512.2011.560561.
- J. S. Park, H. Choi, K. H. Ahn and J. W. Kang, Removal mechanism of natural organic matter and organic acid by ozone in the presence of goethite, Ozone: Sci. Eng., 2004, 26(2), 141–151, DOI:10.1080/01919510490439285.
- X. Sun, C. Wu, Y. Zhou and W. Han, Using DOM fraction method to investigate the mechanism of catalytic ozonation for real wastewater, Chem. Eng. J., 2019, 369, 100–108, DOI:10.1016/j.cej.2019.03.074.
- N. Dabuth, S. Thuangchon, T. Prasert, V. Yuthawong and P. Phungsai, Effects of catalytic ozonation catalyzed by TiO2 activated carbon and biochar on dissolved organic matter removal and disinfection by-product formations investigated by Orbitrap mass spectrometry, J. Environ. Chem. Eng., 2022, 10(2), 107215, DOI:10.1016/j.jece.2022.107215.
- M. Y. Z. Abouleish and M. J. M. Wells, Trihalomethane formation potential of aquatic and terrestrial fulvic and humic acids: Sorption on activated carbon, Sci. Total Environ., 2015, 521–522, 293–304, DOI:10.1016/j.scitotenv.2015.03.090.
- M. Kitis, J. E. Kilduff and T. Karanfil, Isolation of dissolved organic matter (dom) from surface waters using reverse osmosis and its impact on the reactivity of dom to formation and speciation of disinfection by-products, Water Res., 2001, 35(9), 2225–2234, DOI:10.1016/S0043-1354(00)00509-1.
- R. Gracia, S. Cortes, J. Sarasa, P. Ormad and J. L. Ovelleiro, TiO2-catalysed ozonation of raw Ebro river water, Water Res., 2000, 34(5), 1525–1532, DOI:10.1016/S0043-1354(99)00297-3.
- J. Wang, Y. Zhou, W. Zhu and X. He, Catalytic ozonation of dimethyl phthalate and chlorination disinfection by-product precursors over Ru/AC, J. Hazard. Mater., 2009, 166(1), 502–507, DOI:10.1016/j.jhazmat.2008.11.046.
- T. Bond, J. Huang, M. R. Templeton and N. Graham, Occurrence and control of nitrogenous disinfection by-products in drinking water – A review, Water Res., 2011, 45(15), 4341–4354, DOI:10.1016/j.watres.2011.05.034.
| This journal is © The Royal Society of Chemistry 2023 |