
Antimony doped tin(IV) hybrid metal halides with high-efficiency tunable emission, WLED and information encryption†
Wenchao
Lin
,
Qilin
Wei
,
Tao
Huang
,
Xianfu
Meng
 ,
Ye
Tian
,
Hui
Peng
* and
Bingsuo
Zou
*
,
Ye
Tian
,
Hui
Peng
* and
Bingsuo
Zou
*
State Key Laboratory of Featured Metal Materials and Life-cycle Safety for Composite Structures, School of Resources, Environments and Materials, Guangxi University, Nanning 530004, China. E-mail: zoubs@gxu.edu.cn
First published on 23rd March 2023
Abstract
Although single-component white light emitting perovskites have emerged as a new star material with great potential in solid-state lighting, the synthesis of lead-free halide perovskites with high efficiency and high color rendering index (CRI) remains challenging. Here, we report a series of zero-dimensional antimony-doped tin(IV)-based hybrid metal halides (C13H30N)2SnCl6:x%Sb with wavelength-dependent tunable emission. (C13H30N)2SnCl6:20% Sb produces a strong white emission upon excitation at 325 nm, with nearly 100% photoluminescence quantum yield (PLQY), and produces bright red emission with 80.98% PLQY under 380 nm excitation at room temperature (RT). This profile shows that there are two types of emission centers, resulting from high-efficiency dual emission bands, one out of the self-trapped exciton (STE) of 3P1 to 1S0 from SbCl6 octahedra with green emission at 510 nm, and another due to the radiative recombination of the STE of 3P1 to 1S0 from the SbCl5 pyramid with broad red emission around 666 nm, which also contain some DAP (D-donor, A-acceptor, P: pair) transitions around 740 nm between Sb and Sn sites. The single-component light-emitting diode (LED) device based on (C13H30N)2SnCl6:20% Sb has an excellent color rendering index as high as 96.7, and the corresponding color coordinates are (0.346, 0.380). The excitation wavelength-dependent tunable emission profile makes (C13H30N)2SnCl6:x%Sb a high-performance material for applications in single component white light-emitting diodes (WLEDs), anti-counterfeiting, information encryption, and flexible lighting displays.
Introduction
White light-emitting diode (WLED), as a new generation of solid-state lighting source, has the advantages of low power consumption, long life, and human friendliness.1,2 They have completely changed people's lives in the past ten years. For many years, WLEDs were used that combined blue-emitting chips with yellow-emitting phosphors.3,4 However, the above strategy has disadvantages such as a low color rendering index (CRI), poor color stability, and blue light damage to human eyes. Therefore, a very promising strategy is to search for single-component broadband white-emitting phosphor materials.Recently, low-dimensional organic–inorganic hybrid metal halides (LOHMHs) have been extensively studied in the field of solid-state lighting due to their low-cost preparation process, facile synthesis, and excellent properties.5–8 Among them, lead-based LOHMHs have attracted much attention due to their excellent optical properties. Meanwhile, lead-based LOHMHs with white light emission have also been extensively reported. For example, the white-emitting one-dimensional compound (2,6-dmpz)3Pb2Br10 was reported by Mao et al. with the highest photoluminescence quantum yield (PLQY) of 12%.9 Yuan et al. reported a one-dimensional organic lead bromide perovskite C4N2H14PbBr4 with unique blue-white light emission via strong quantum confinement.10–17 However, the toxicity and instability of lead have not been resolved, and lead-based LOHMHs with white light-emission often exhibit low photoluminescence quantum yield (PLQY),9,14–16,18 which hinders their large-scale commercial applications. To solve the problem, replacing Pb with other environmentally friendly metal ions is a feasible strategy.19–21 As a metal element with similar chemical properties to lead, tin is considered to be an effective substitute for lead.22–24 However, Sn2+ is easily oxidized to Sn4+, which is the same IVA metal element as Pb, and tin(IV)-based metal halides have often been found to exhibit weak PL emission.25,26 Therefore, obtaining high-efficiency luminescent tin-based metal halides has become a challenge. The doping technique is considered an effective strategy to tune the optical bandgap of metal halide perovskites and improve their luminescence properties. Recently, metal ions (such as Sb3+, Te4+, Bi3+, and Sn2+) with the ns2 electron configuration were demonstrated to readily generate broadband and efficient self-trapped excitons (STEs) in LOHMH lattices with strong electron–phonon coupling. Metal Sb3+ ions with a unique singlet and triplet exciton emission show a blue and yellow double emission band, which provides the possibility for the preparation of efficient and stable white light emission.27 Interestingly, pure Sb halides usually exhibit different luminescent behaviors due to their different crystal structures. The green emission of [Bzmim]3SbCl6 (Bzmim = 1-benzyl-3-methylimidazolium) reported by Huang et al.28 comes from SbCl6 clusters in the lattice, and Peng et al. reported a series of Sb halides, whose red emission comes from SbCl5 clusters in the compound.6,29 After doping Sb3+ in other metal halides, it can achieve blue,30 green,31–36 yellow,33,35–40 orange,31,33 red,25,39,41,42 near-infrared,43 and white emission.38,40 Li et al. reported that Sb3+ doped (BTPP)2MnCl4 (BTPP = benzyltriphenylphosphonium) halides with excitation-dependent dual emission achieve tunable emission colors from green to orange, and (BTPP)2MnCl4:2.0%Sb crystals exhibit green and orange emission under 254 and 365 nm UV lamps, respectively.31 In addition, Shi et al. found that methanol can coordinate with the Sb(III) center of the (C6H18N2)InCl5·H2O:Sb halide and undergo structural deformation, red-shifting the emission center from 565 nm to 663 nm.39 Although Sb3+-doped metal halides have been widely studied, there are few reports of high-efficiency emissive Sb3+-doped tin(IV)-based metal halides, especially white-light emitting metal halides.
Here, we report a novel Sb3+-doped zero-dimensional (0D) organic–inorganic hybrid halide perovskite (C13H30N)2SnCl6:Sb, and the 20% Sb3+ doped sample exhibits efficient warm white light (PLQY = 99.32%, λex = 325 nm) and red light (PLQY = 80.98%, λex = 380 nm) emission at room temperature (RT). Its high-efficiency warm white light originates from dual emission bands under high-energy excitation, and its profile exhibits two type emission centers, resulting from highly efficient dual emission bands, one out of the STE of 3P1 to 1S0 from SbCl6 octahedra with green emission at 510 nm, and another due to the radiative recombination of the STE of 3P1 to 1S0 from the SbCl5 pyramid with broad red emission around 666 nm, which also contain some DAP (D-donor, A-acceptor, P: pair) transitions around 740 nm between Sb and Sn sites. Interestingly, (C13H30N)2SnCl6:Sb exhibits a wide range of tunable emission properties by changing the temperature and excitation wavelength. With the change of the excitation wavelength, it can achieve tunable emission from warm white, white, to red, and the color temperature is changed from 1287 K to 5342 K. Its unique dual emission bands can be monitored over a wide temperature range (80–320 K) under high-energy excitation (λex =325 nm). In addition, we also used Sb3+ doped (C13H30N)2SnCl6 to make a single component WLED lamp, which has good optical performance. The exploration of color-changing anti-counterfeiting applications shows that it also has great potential in the field of anti-counterfeiting encryption. Our work provides a direction for designing white luminescent materials to facilitate the application of Sb3+ in solid-state lighting, anti-counterfeiting, information encryption, and other fields.
Experimental method and characterization
Materials
Dichloromethane (DCM), n-hexane and methanol, N,N-dimethylformamide (DMF), and HCl were purchased from Nanning Yuanlai Instrument Co., Ltd. Methyl tributyl ammonium chloride (C13H30ClN, 95%), antimony trichloride (SbCl3 AR,99%) and stannic chloride hydrate (SnCl4·5H2O, 99.995%) were purchased from Macklin. All reagents and solvents were used without further purification and treatment.Synthesis of (C13H30N)2SnCl6:x% Sb with a variety of Sb3+ doping concentrations (Sb/Sn = x = 0, 5, 10, 15, 20, 30, x always represents the feed ratio) and (C13H30N)xSnCly
Sb3+ doped (C13H30N)2SnCl6 crystal powder was synthesized by the following method: 1 mmol SnCl4·5H2O, 2 mmol C13H30ClN, 3 ml DCM, 3 ml DMF and SbCl3 with different molar ratios were added into 50 ml beakers, fully stirred to dissolve, and then maintained at 35 °C volatilized on a heating platform at constant temperature for 48 h. The precipitated crystals were collected, washed with n-hexane, and dried at 60 °C for 24 h. (C13H30N)xSbCly was synthesized by adding 2 mmol C13H30ClN, 10 ml methanol, 1 ml HCl and 1 mmol SbCl3. The precipitated crystals were collected, washed with n-hexane, and dried at 60 °C for 24 h.Measurement and characterization
Details of measurement and characterization are as follows: X-ray diffraction (XRD) patterns were obtained with an X-ray diffractometer manufactured by Rigaku Corporation (model: Rigaku D/MAX 2500V). The scanning range is 5–60°, the speed is 6° min−1, and the step size is 0.02°. The photoluminescence excitation (PLE) and photoluminescence (PL) spectra were measured as a function of temperature using a HORIBA fluorescence spectrometer test system. The fluorescence, phosphorescence lifetime, and PLQY of these samples were obtained by using an Edinburgh FLS 1000 fluorescence spectrometer and a HORIBA spectrometer. Their Raman spectra were acquired on a confocal Raman imaging system (WITec alpha300 R) at an excitation wavelength of 633 nm. Optical absorption spectra were obtained with a LAMBDA 750 UV spectrophotometer. Energy-dispersive X-ray spectroscopy (EDS) characterization was performed using a new high-resolution field emission scanning electron microscope SU8020. X-ray photoelectron spectroscopy (XPS) characterization was performed using an X-ray photoelectron spectrometer (model: ESCALAB 250XI+) manufactured by Thermo Fisher Scientific. Thermogravimetric analysis (TGA) was characterized by using a thermogravimetric analyzer DTG-60(H) manufactured in Shimadzu, Japan.Computational methods
All calculations by density functional theory are carried out using the Vienna Ab initio simulation package (VASP).44 The generalized gradient approximation of the Perdew–Burke–Ernzerhof (PBE)45,46 parameterization with projector-augmented wave47 method is performed for the exchange–correlation functional. The kinetic-energy cutoff of 400 eV and a 2 × 4 × 2 Monkhorst–Pack k-mesh for the wavefunction basis set is employed. For the elements C, H, N, Sn, Sb and Cl, ultra-soft pseudopotentials are used. The energy convergence criterion is set as 1.0 × 10−5 eV for structural relaxations.Results and discussion
All samples were synthesized by volatilizing a mixed solution of N,N-dimethylformamide (DMF) and dichloromethane at room temperature. The single crystal structure of the (C13H30N)2SnCl6 crystal was determined by using a single crystal X-ray diffractometer (SCXRD). The results show that the (C13H30N)2SnCl6 unit cell belongs to the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group of the triclinic crystal system, and the unit cell parameters are a = 9.8400(6) Å, b = 10.8858(7) Å, c = 18.0546(9) Å, and the details can be found in the ESI† (Tables S1–S5). As shown in Fig. 1(a), tin(IV) ions and six chloride ions form the [SnCl6]2− octahedron, and the large-sized [C13H30N]+ cations separate the [SnCl6]2− octahedron in space to form a single cluster [SnCl6]2− octahedral array. The nearest distance between the two adjacent tin(IV) ions is 9.84 Å and the furthest neighboring cluster distance is 14.51 Å, which may avoid the dipole interaction between the [SnCl6]2− octahedron and forms a 0D quantum dot crystal structure in space. Fig. 1(b) shows the bond length of the [SnCl6]2−octahedron and the structure of the organic cation [C13H30N]+, the different bond lengths indicate that the [SnCl6]2− octahedral structure is distorted, and the organic cation [C13H30N]+ has an asymmetric structure in space. Fig. 1(c) shows the structural diagram of the Sb-doped (C13H30N)2SnCl6 crystal, in which Sb3+ replaces part of Sn4+, forming a six-coordination octahedron [SbCl6]3− centered in the lattice. After Sb3+ incorporation the crystal structure of Sb-doped (C13H30N)2SnCl6 did not change compared with Fig. 1(a). We characterized a series of powder X-ray diffractometers (PXRDs) of Sb-doped (C13H30N)2SnCl6 with different ratios (Fig. 1(d)). The results are in good agreement with the calculated results, proving the formation of high-purity crystals. As the doping concentration increases, the peak position at 12.76° gradually shifts to a smaller angle slightly, and the peak shape becomes wider after Sb incorporation. This is because the radius of Sb3+ (0.76 Å) is larger than that of Sn4+ (0.69 Å). With the increase of the doping concentration, more Sb3+ enters the lattice, causing lattice expansion.43 It is found that the XRD pattern has minor variations after Sb doping. Furthermore, we characterized the XRD of as prepared (C13H30N)xSbCly for comparison as shown in the ESI,† Fig. S1, which shows a different crystal structure from that of (C13H30N)2SnCl6, further confirming that the successfully synthesized pure Sn(IV)-based metal halides have a different local structure or symmetry in this hybrid structure. Subsequently, we performed X-ray photoelectron spectroscopy and EDS characterization. Among them, the sample of (C13H30N)2SnCl6:20%Sb was studied. The EDS energy spectrum shows that Sn, Sb, and Cl elements are uniformly distributed in the sample as shown in Fig. S2 and Table S6 (ESI†), proving that Sb3+ was successfully incorporated into the product lattice. The fitted XPS spectrum shown in Fig. S3 (ESI†) shows that the peaks at 495.4 eV and 487 eV correspond to Sn4+ 3d3/2 and 3d5/2, and the peaks at 539.6 eV and 530.25 eV correspond to Sb3+ 3d3/2 and 3d5/2, confirming the 4+ oxidation state of Sn and 3+ oxidation state of Sb in the crystalline product.
space group of the triclinic crystal system, and the unit cell parameters are a = 9.8400(6) Å, b = 10.8858(7) Å, c = 18.0546(9) Å, and the details can be found in the ESI† (Tables S1–S5). As shown in Fig. 1(a), tin(IV) ions and six chloride ions form the [SnCl6]2− octahedron, and the large-sized [C13H30N]+ cations separate the [SnCl6]2− octahedron in space to form a single cluster [SnCl6]2− octahedral array. The nearest distance between the two adjacent tin(IV) ions is 9.84 Å and the furthest neighboring cluster distance is 14.51 Å, which may avoid the dipole interaction between the [SnCl6]2− octahedron and forms a 0D quantum dot crystal structure in space. Fig. 1(b) shows the bond length of the [SnCl6]2−octahedron and the structure of the organic cation [C13H30N]+, the different bond lengths indicate that the [SnCl6]2− octahedral structure is distorted, and the organic cation [C13H30N]+ has an asymmetric structure in space. Fig. 1(c) shows the structural diagram of the Sb-doped (C13H30N)2SnCl6 crystal, in which Sb3+ replaces part of Sn4+, forming a six-coordination octahedron [SbCl6]3− centered in the lattice. After Sb3+ incorporation the crystal structure of Sb-doped (C13H30N)2SnCl6 did not change compared with Fig. 1(a). We characterized a series of powder X-ray diffractometers (PXRDs) of Sb-doped (C13H30N)2SnCl6 with different ratios (Fig. 1(d)). The results are in good agreement with the calculated results, proving the formation of high-purity crystals. As the doping concentration increases, the peak position at 12.76° gradually shifts to a smaller angle slightly, and the peak shape becomes wider after Sb incorporation. This is because the radius of Sb3+ (0.76 Å) is larger than that of Sn4+ (0.69 Å). With the increase of the doping concentration, more Sb3+ enters the lattice, causing lattice expansion.43 It is found that the XRD pattern has minor variations after Sb doping. Furthermore, we characterized the XRD of as prepared (C13H30N)xSbCly for comparison as shown in the ESI,† Fig. S1, which shows a different crystal structure from that of (C13H30N)2SnCl6, further confirming that the successfully synthesized pure Sn(IV)-based metal halides have a different local structure or symmetry in this hybrid structure. Subsequently, we performed X-ray photoelectron spectroscopy and EDS characterization. Among them, the sample of (C13H30N)2SnCl6:20%Sb was studied. The EDS energy spectrum shows that Sn, Sb, and Cl elements are uniformly distributed in the sample as shown in Fig. S2 and Table S6 (ESI†), proving that Sb3+ was successfully incorporated into the product lattice. The fitted XPS spectrum shown in Fig. S3 (ESI†) shows that the peaks at 495.4 eV and 487 eV correspond to Sn4+ 3d3/2 and 3d5/2, and the peaks at 539.6 eV and 530.25 eV correspond to Sb3+ 3d3/2 and 3d5/2, confirming the 4+ oxidation state of Sn and 3+ oxidation state of Sb in the crystalline product.
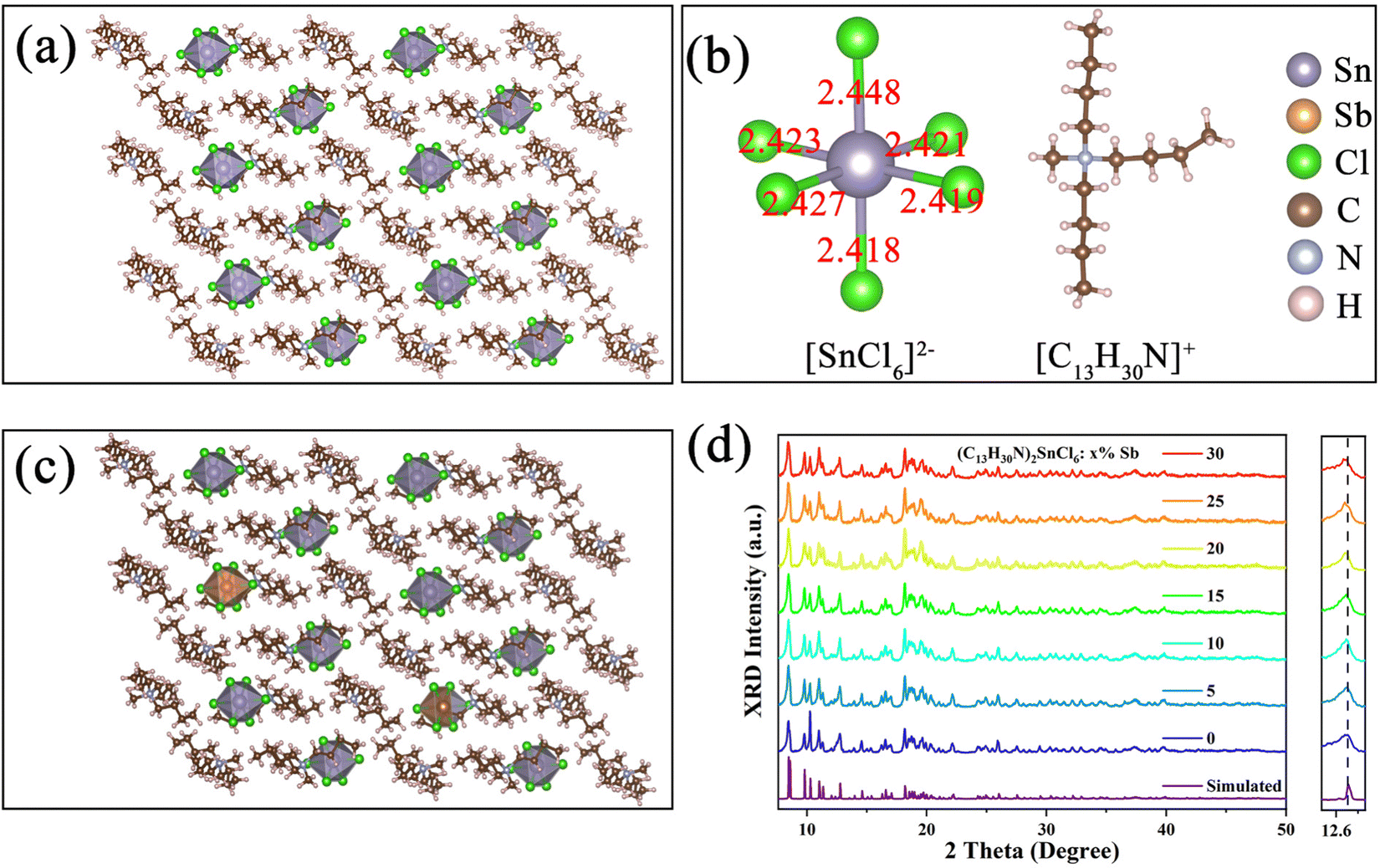 | ||
| Fig. 1 (a) Crystal structure of undoped (C13H30N)2SnCl6. (b) Schematic diagram of the [SnCl6]2− octahedron and the organic cation [C13H30N]+. (c) Crystal structure of Sb3+ doped (C13H30N)2SnCl6. (d) The simulated and experimental powder X-ray diffraction patterns of (C13H30N)2SnCl6 and (C13H30N)2SnCl6:20%Sb. | ||
In order to study the photophysical properties of (C13H30N)2SnCl6 and Sb3+ doped (C13H30N)2SnCl6, we characterized their optical properties using a UV-Vis spectrophotometer and a HORIBA spectrometer. The UV-Vis absorption spectrum (Fig. 2(a)) shows that the band edge of the undoped sample is at about 320 nm, with the increasing Sb3+ doping amount, the absorption band red shifts, indicating that the optical bandgap of Sb doped (C13H30N)2SnCl6 decreases, which proves that the stereochemically active Sb3+ cannot modify the structure, but effectively modify the (C13H30N)2SnCl6 bandgap (320 nm) as shown in Fig. 2(a), and the absorption band after Sb3+ incorporation usually appear at 270–320 nm and 320–390 nm, as shown in previous reports.40,42 Both undoped (C13H30N)2SnCl6 and (C13H30N)2SnCl6:20% Sb are colorless powders under natural light (Fig. 2(b)). Under 302 nm and 365 nm UV lamps, (C13H30N)2SnCl6 cannot be well distinguished with the naked eye because it emits only weak light. (C13H30N)2SnCl6:20% Sb exhibits bright warm white emission under a 302 nm UV lamp and red emission under a 365 nm UV lamp irradiation, which has a relationship with the new absorption peaks at 297 nm and 336 nm in the absorption spectrum after doping Sb3+. The former band possibly due to the 1S0 to 1P1 from Sb3+ is merged into the band tail of (C13H30N)2SnCl6, so the excitation for the doped sample cannot excite only the SbCl6 cluster itself but also excite the SnCl6 cluster in the lattice. The wavelength longer than 320 nm may be assigned to the absorption of 1S0 to 3P1 of Sb3+ in this lattice, which can be used to reflect the usual role of Sb doping in a host perovskite lattice. As shown in Fig. S4 (ESI†), the absorption spectra of (C13H30N)xSbCly, (C13H30N)2SnCl6 and (C13H30N)2SnCl6:20%Sb prove the above possibility. (C13H30N)xSbCly exhibits typical absorption bands of 1S0 to 1P1 (220–270 nm), 1S0 to 3P2 (270–340 nm) and 3P1 (340–470 nm) for Sb halides, in which SbClx–SbClx strong coupling between octahedra red shifts significantly.
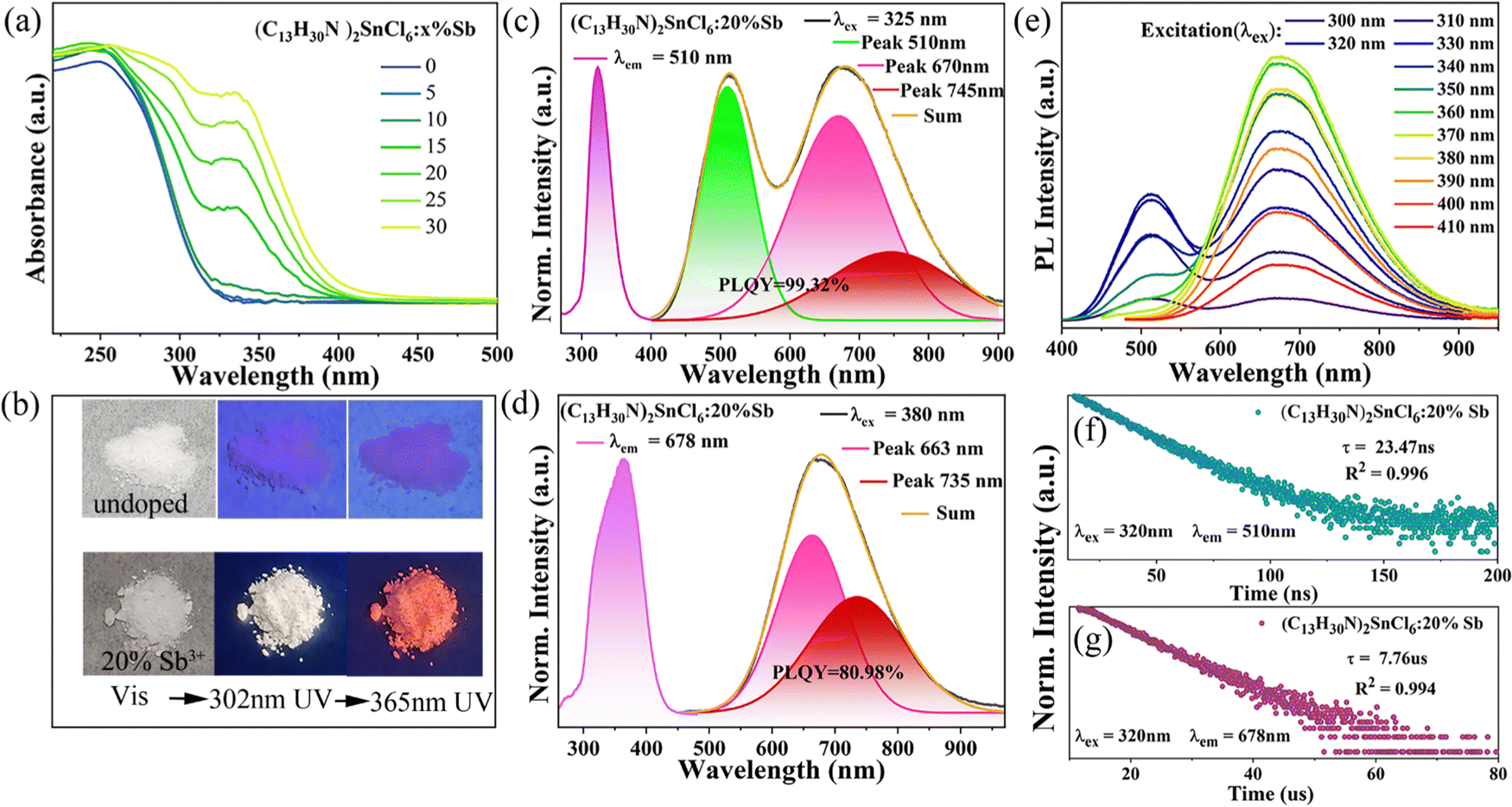 | ||
| Fig. 2 (a) Absorption spectra of (C13H30N)2SnCl6:20%Sb. (b) Optical photographs of undoped (C13H30N)2SnCl6 and (C13H30N)2SnCl6:20%Sb samples under visible light, 302 nm UV lamp, and 365 nm UV lamp, respectively. (c) Normalized PL spectrum of (C13H30N)2SnCl6:20%Sb excited at 325 nm and PLE spectrum excited at 510 nm. (d) Normalized PL spectrum of (C13H30N)2SnCl6:20%Sb excited at 380 nm and PLE spectrum excited at 678 nm. (e) PL spectra of (C13H30N)2SnCl6:20%Sb at different excitation wavelengths (λex = 300–410 nm). The time-resolved PL spectrum of (C13H30N)2SnCl6:20%Sb was monitored at 510 (f) and 678 nm (g), respectively. | ||
We characterized its photoluminescence (PL) spectra at different excitation wavelengths in Fig. S5 (ESI†) in order to verify whether (C13H30N)2SnCl6 emits light. By comparing with the PL spectrum monitored by the xenon lamp at 250 nm, we found that the emission peaks at 320, 397 and 520 nm originate from the luminescence of (C13H30N)2SnCl6. (C13H30N)2SnCl6 exhibits very weak luminescence, which is consistent with its optical photographs under different UV lamps. Among them, the emission peak at 320 nm originates from confined excitons (S1–S0), the 397 nm emission band may be the emission of triplet emission (1P1–S0) and the emission peak at 520 nm may originate from the self-bound states. The associated PLE spectra show an absorption center at 250 nm monitored at 397 nm, and another absorption (excitation) center at 320 nm monitored at 520 nm. (C13H30N)2SnCl6 has a PLQY of 1.56% excited at 260 nm (Fig. S23, ESI†). As a host with metal ion doping, the emission doped product may show a relation to the PL profile of (C13H30N)2SnCl6 under UV excitation. In addition, we also characterized the PL and PLE spectra of the organic compound C13H30NCl shown in Fig. S6 (ESI†), organic compound C13H30NCl has a weak emission peak at 505 nm at 316 nm excitation. The lifetime decay curve (Fig. S7, ESI†) monitored at 505 nm shows that the organic compound C13H30NCl has a nanosecond lifetime of 484.62 ns, which fits a single exponential fit.
Doping has been proven to be an effective strategy to modify the wide-band crystal environment, band structure and electronic states, which may generate new excited states and give new luminescent properties.31,32,42,48,49 Here, we introduce antimony ions into the lattice of (C13H30N)2SnCl6, which can give different emission behaviors at varied excitation with dual emission profiles. The results effectively reflect the improvement of the emission properties of (C13H30N)2SnCl6. Here, we mainly present the properties of the (C13H30N)2SnCl6:20%Sb sample. As shown in Fig. 2(c), it gives a very strong dual emission band at 510 nm and 678 nm at 325 nm excitation. As shown in Fig. 2(c), (d) and Fig. S8, S9 in the ESI,† the excitation center at 325 nm in the PLE spectrum does not change for emission at 510 nm (λem = 510 nm) as the dopant ratio increases, which is in contrast to that of the host. This emission has three absorption ranges (300–360 nm, 270–297 nm and 230–270 nm), For the emission at 678 nm (λem = 678 nm), its PLE absorption band redshifts, with a broader excitation band centered at 332 nm at minor doping, and gradually red shifts to 370 nm with increasing doping ratio, whose edge can extend to 440 nm. This PLE profile variation corresponds to the measured absorption band-edge change with Sb doping. As the doping ratio increases, the absorption band in the longer range of >320 nm has been observed, and the absorption band edge red-shifted to about 440 nm as shown in Fig. 2(a), which matches the PLE in Fig. S9 (ESI†). Under 325 nm excitation (Fig. 2(c)), it exhibits a bright and broad dual-band emission centered at 510 nm and 678 nm. The latter band at 678 nm has even broader width, both bands can span 400–910 nm, manifested as a very strong white emission. Under 380 nm excitation, it exhibits an ultra-broad emission band centered at 678 nm, spanning 490–950 nm (full width at half-maximum (FWHM) = 171 nm, Stokes shift of 298 nm) (Fig. 2(d)). When the doping amount reaches 20% (Sb/Sn = 20%), the sample has the strongest PL emission, the PLQY of red emission under 380 nm excitation can be as high as 80.98%, and the quantum efficiency of white emission under 325 nm excitation is close to 100% (PLQY = 99.32%, CRI = 84), the color coordinates are (0.36,0.42). It is worth mentioning that this is the highest white-light PLQY reported so far for hybrid tin(IV)-based metal halides (as shown in Table S7, ESI†).42
The PLQY values corresponding to samples with different doping ratios are shown in Fig. S10 in the ESI.† The normalized PL spectra in Fig. S9 (ESI†) show that the addition of more Sb3+ does not produce a shift of the emission maximum, however, the PLE band for 510 nm emission band become broader with increasing Sb dopants, and the PLE band for 678 nm emission shows a redshift from 330 nm to 370 nm if the dopant Sb ratio ranges from 5 to 30%. In the low doping case, the dual emission bands have a clear overlap of PLE profiles in the 240–360 nm range; while for all doping cases, PLE at longer wavelengths beyond 360 nm mainly contributes to the 678 nm emission band rather than 510 nm emission.
We monitored the wavelength-dependent PL emission spectra in Fig. 2(e), which has a dual-band emission when using high-energy excitation (λex ≤ 370 nm) and only a broadband emission centered at 678 nm when using longer wavelength excitation (λex ≥ 370 nm). The PL emission for high energy excitation has a double band, in which the second band has a larger FWHM than that of the first band. The second band is always present at all above excitations. The strong emission profile and its corresponding color coordinate diagram in Fig. S11 (ESI†) show that the introduction of Sb3+ into (C13H30N)2SnCl6 can obtain modified tin(IV) halides with excellent luminescence properties and adjustable color temperature (1287–5342 K). In addition, we monitored the PL lifetimes of the (C13H30N)2SnCl6:20%Sb sample at 510 nm and 678 nm (Fig. 2(f) and (g)), which were 23.47 ns and 7.76 μs, respectively, which were all obtained with the single exponential fitting. As the doping amount increases, the lifetimes of both emission centers first increase and then decrease (Fig. S12, ESI†), but the changes are little. The lifetime decrease of samples with high doping ratio may be related to the non-radiative transition caused by concentration quenching.40 Significant differences in the lifetimes of the high-energy and low-energy emission peaks suggest that they originate from different emission state (ES). Moreover, the lifetime values are strongly dependent on the facility and its time scale. However, we did not obtain a lifetime of microseconds for 510 nm emission, but only nanoseconds. This fact is not common in the Sb doped halides with STE emission. The linear dependence of PL emission intensity on the excitation power is observed in Fig. S13 (ESI†), which rules out the effect of permanent defects.
Interestingly, we observed different emission bands at 468 nm and 654 nm under continuous wave (CW) 405 nm laser excitation, which is due to the relatively transient luminescent states produced by high power laser excitation, the STE state filling may blueshift its emission maximum for both emission states. Large Stokes shift, wide FWHM, long lifetime, and similar excitation and emission characteristics to (C13H30N)2SnCl6 indicate that both of the broadband emissions (= 510 nm and 678 nm) of (C13H30N)2SnCl6:20%Sb originate from the STE. Its short lifetime of 23.47 ns at 510 nm emission is significantly different from the lifetime of 484.62 ns of organic C13H30NCl, indicating that this high-energy emission peak is not derived from the lowest level, among the states near the band edge out of Sb, Sn, Cl and organic cation [C13H30N]+, but from a self-trapped exciton (STE), possibly the state in 0-d SbCl6 cluster29 in this system. Interestingly, it was determined that confined single SbCl6 cluster kinetically generates this state and emits green light in the 500–530 nm range.28,50,51 This was also confirmed in previous reports. Sun et al. achieved green emission up to 89.29% and 85.84% PLQY by doping Sb3+ into [DAPEDA]InCl6·Cl·H2O and [DPA]3InCl6 crystals, respectively.52 Huang et al. observed green emission with PLQY up to 90% in Sb3+-doped (Cs0.29Rb0.71)3InCl6, which was attributed to relatively weak electron–phonon coupling and Jahn–Teller distortion.36 Wei et al. achieved high-efficiency green emission by doping Sb3+ in Cd-based halides. The emission peaks of these halides are all between 500–530 nm.32,53,54 In this system, Sb3+ ion has three specific characters compared with Sn4+: (1) its ion radius is slightly larger than Sn ion in the octahedral site; the chemical covalent bonding is much stronger in this matrix Sn–Cl6, hence the octahedral for Sb is favored to be present at this 0-d structure; (2) the valence state is smaller than Sn4+, which leaves excess charge, that is important the long time STE and its DOS; (3) 5s2 electron configuration of Sb in the lattice may introduce a novel Sb pyramid site with distorted lattice due to strong sp–d hybridization and d–d correlation, which may increase the emission bandwidth and larger redshift. This has been reported in our previous research,6,29 upon introducing large-sized organic cations, they tend to form five-coordinated SbCl5 clusters in the crystal. This shows that in our system, there are both SbCl5 and SbCl6 clusters. In addition, we also calculated the formation energy required for the simultaneous existence of SbCl5 and SbCl6 in the crystal lattice by DFT (Fig. S25, ESI†). The presence of both SbCl5 and SbCl6 clusters is more likely than SbCl6 clusters alone, with a difference of 4.99 meV. All these pieces of evidence indicate that the emission peak at 510 nm originates from SbCl6 clusters. Hence this green band may be the high energy emission band at 510 nm in our system.
This band in our system may be a STE state located at the Sb site from 3P1 to 1S0 under these strongly confined conditions in these organic Sn halides, whose lifetime is short because its intrinsic location is not the lowest. If so what is the origin of the next low energy band at 678 nm? Here, we notice that the emission peak of (C13H30N)2SnCl6:20%Sb at 678 nm presents an asymmetric emission peak shape, and the lifetime (Fig. S14, ESI†) monitored at 810 nm is different from that at 678 nm. At the same time, we also detected an additional emission side-band at 800 nm from its LED device, which will be shown in the following section. By fitting the PL peaks by 325 nm excitation (Fig. 2(c) and (d)), the emission peaks at 510 (ES1), 670 (ES2) and 745 nm (ES3) could be obtained when excited at 325 nm, with the half-peak widths of 87, 156, and 212 nm, respectively. When excited by 380 nm, the fitted emission peaks at 663 (ES2) and 735 nm (ES3) were obtained, with the half-peak widths of 129 and 180 nm, respectively. These close fitting results indicate that there are two different emission bands at around 666 (ES2) and 740 nm (ES3) respectively. The 666 nm emission band may be from the Sb in the 5-coordination site, because the Sb3+ ion in organic confined halides can often form the SbCl5 cluster.11,28,29 Its emission is usually with a double band profile, in which the high energy band is very weak in the 400–500 nm range, due to the 1P1–1S0 of Sb3+, though not observed here; while its low energy band is often very strong around the 570–680 nm region due to its large lattice distortion with defection, which is in agreement with the emission profile of state ES2 due to the STE radiative recombination of 3P1 to 1S0 from SbCl5 clusters. For this coordination, these compounds often have a reversible emission band or color under heating and cooling. Sometimes they can be synthesized in different thermodynamic environments.55 Xia et al. also indicated that the SbCl5 cluster can be present in the Sb doped all-inorganic metal halides contributing to its efficient multi-STE emission bands.37 therefore the 666 nm emission band should be the triplet STE of the Sb3+ ion in halides with long lifetime.
Then what is the source of the sideband emission state ES3 at 740 nm? In an early report, we found an independent DAP band in Sb doped NH4SnCl456 because of the clear difference of electronegativity and electronic density of Sb and Sn ions as well as close separation in halides. Therefore the 740 nm band may be the Sn and Sb site charge-transfer radiation transition between the SbCl5 site(D) and Sn site(A).57 We see that the former state has almost 100% quantum yield (QY) and the latter one has about 80% QY because its excitation is near the absorption band edge. The 510 nm emission has a very short lifetime, which is a different order of magnitude lifetime compared to 678 and 810 nm, and all these can also reflect this process in this system. All of the above results indicate the existence of another emission center from the Sn site involved in the emission processes, which is the radiative recombination of the STE from the Sn site through the DAP transition at around 740 nm. The emission band at 800 nm detected in the LED device in following section should be the relaxed and accumulated spectral shift within the active layer in the device.
The 510 nm emission is not from a typical steady STE, because rigid SnCl6 and organic molecules work together to confine its exciton out of 1S0 to 3P1 in the SbCl6 cluster, and its level is higher than that from the SbCl5 cluster and DAP, so it has a short lifetime due to fast relaxation to the lower levels. As shown in PLE in Fig. 2(c), this transition occurs at 325 nm, that is also seen in the absorption band at about 335 nm; the other component in the absorption band-edge for >10% Sb ratio sample should be made up of the other components or interactions in this compound, Sb–Cl, Sn–Cl and organic group-Cl charge transfer, as well as the possible donor–acceptor pair (DAP) transition between SbCl6–SnCl6 clusters. These interactions can be reflected by comparing with their PL and PLE spectra of (C13H30N)xSbCly (Fig. S15, ESI†). Compared with (C13H30N)xSbCly, it is supposed that it has no Sb–Cl–Sb coupling, the high energy PL band of (C13H30N)2SnCl6:20%Sb is seen to red-shift from 493 to 510 nm, and the low energy PL band red shifts from 642 to 678 nm, therefore, some DAP effect should be present to contribute to more such redshifts. So the STE (ES3) state out of the DAP transition may merge with the STE (ES2) state of the SbCl5 cluster to produce a new asymmetric broad PL band in the longer wavelength range as shown in Fig. 2(d).
To further understand the relevant PL mechanism of (C13H30N)2SnCl6:x%Sb, we studied temperature-dependent spectra of (C13H30N)2SnCl6:20%Sb at different excitation wavelengths (Fig. 3(a) and (b)). As the temperature increases, the PL intensities of the high-energy emission band (peak 1, 510 nm) at 325 nm excitation increase first and then decrease, and their position redshifted. This implies that this state needs phonons to assist in this emission, so it has definitely STE or exciton–phonon coupling nature. The PL intensities of the low-energy band emission (peak 2) gradually decrease, and the position redshifts first, then blueshifts, which implies that this state is intrinsically formed with STE character at low temperature and shows a strong temperature dependence by phonon scattering for its low symmetry. The bands’ blueshifts come from the lattice expansion and local STE nature. Its energy depends on the quantum confinement, electron–phonon coupling and phonon scattering. The pseudo color mapping of temperature-dependent PL intensities with different excitation wavelengths as shown in Fig. 3(c) and (d) reveals that under high-energy excitation at λex = 325 nm, the high-energy (ES1) and low-energy emission (ES2) peaks associated with STE can originate from the SbCl6 and SbCl5 cluster, respectively, and the DAP may be easily formed via coulomb potential for some of the lowest SnCl6 level occurrence to interact with SbCl5, that is strongly coupled with phonons. As the temperature increases, the dual emission bands always exist in the temperature range of 80–320 K. Under low-energy (λex = 380 nm) excitation, only low-energy emission bands associated with SbCl5 clusters and DAP were detected. This indicates that these broad bands are related to the STE; whereas the dual emission bands arise because the SbCl5 and SnCl6 clusters simultaneously trap photogenerated excitons,37,41,58 while also facing competition from low-energy DAP. As shown in Fig. 3(e), we studied the temperature dependence of the peak position and luminescence intensity. As the temperature increases, peak 2 at 80–240 K and peak 1 positions (Fig. 3(a)) undergo a red shift by phonon scattering, while peak 2 above 260 K and peak 3 move to higher energy by lattice expansion.41 Their intensity increase indicates the phonon assistance to form STE dominantly, and the intensity reduction reflect the dominant phonon scattering in this system. Such behavior reflected the complication of their electronic structure and microscopic interactions inside.
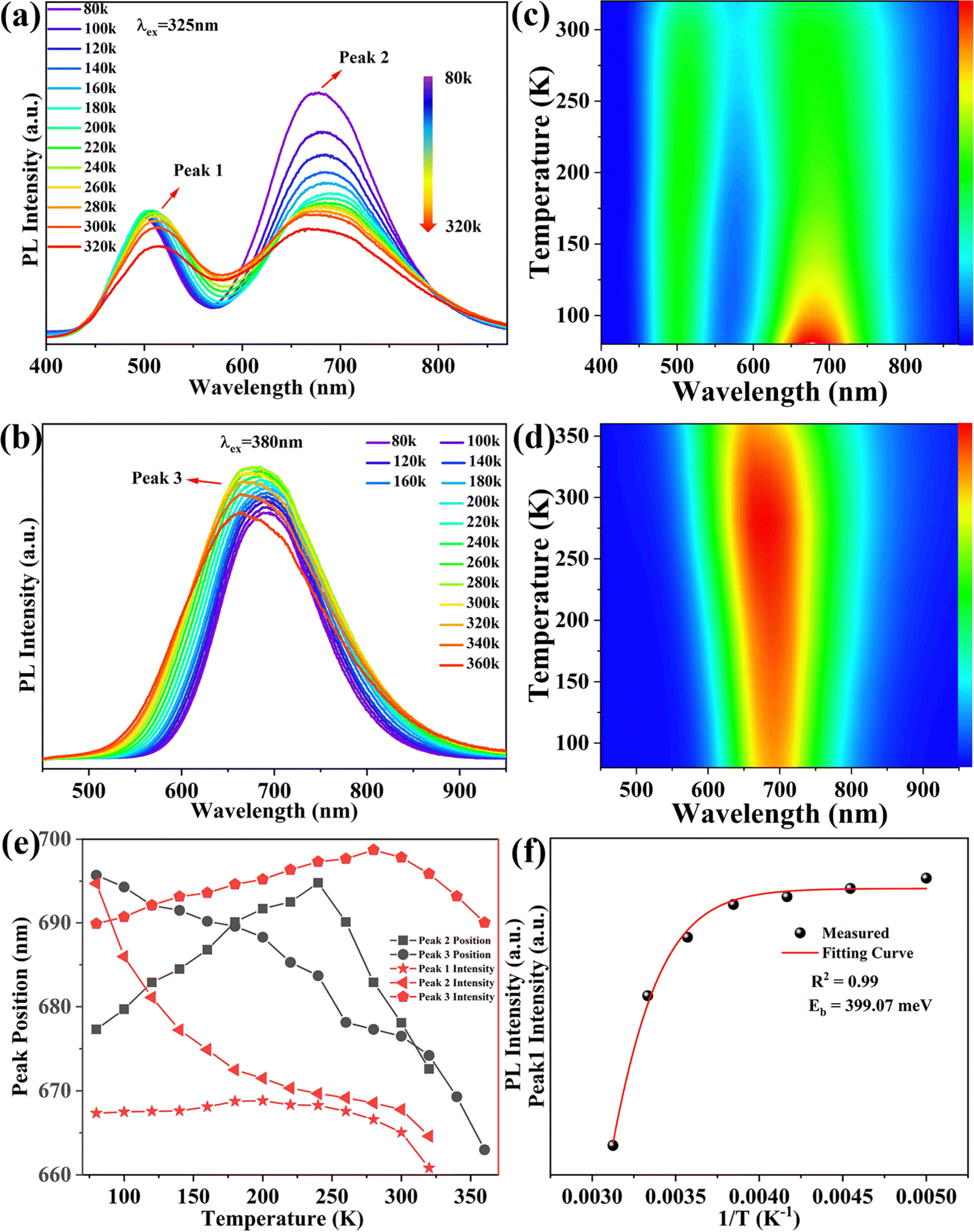 | ||
| Fig. 3 Temperature-dependent steady-state photoluminescence spectra of (C13H30N)2SnCl6:20%Sb excited at (a) 325 nm and (b) 380 nm in the temperature range of 80–320 K and 80–360 K, respectively. Pseudo color mapping of temperature-dependent PL intensity at excitation wavelengths of (c) 325 nm and (d) 380 nm. (e) Schematic diagram of the relationship between the PL intensity, peak position and wavelength of peak 1, peak 2, and peak 3. (f) PL intensity at 510 nm for (C13H30N)2SnCl6:20% Sb fitting results with 1/T and lining. | ||
With increasing temperature, their same temperature dependence indicates that they originate from the same state. The variation of PL emission intensities is related to the STE formation and phonon scattering for peak 1 and peak 3.59 The emission band becomes wider with increasing temperature, which may be caused by more phonon scattering at increasing temperature.25 Previous reports have shown that in antimony doped halide systems, the state energy is more easily transferred from the singlet state to the triplet state by intersystem channeling (ISC) at high temperatures than at low temperatures, resulting in PL emission which usually appears as a triplet STE state dominated at high temperatures.38,40,42 But this diagram is inconsistent with our observation that peak 2 exhibits significant temperature sensitivity under high-energy excitation, dopant concentration dependent excitation redshift and low energy excited temperature dependent emission intensity increase; all these support that the peaks 1 and 3 originate from the STE initialized emission transition from the normal Sb site in this tin(IV) halide, but peak 2 shows some specific profiles.
The Huang–Rhys factor (S) is often used to describe the strength of the electron–phonon coupling and can be fitted using the following formula:60
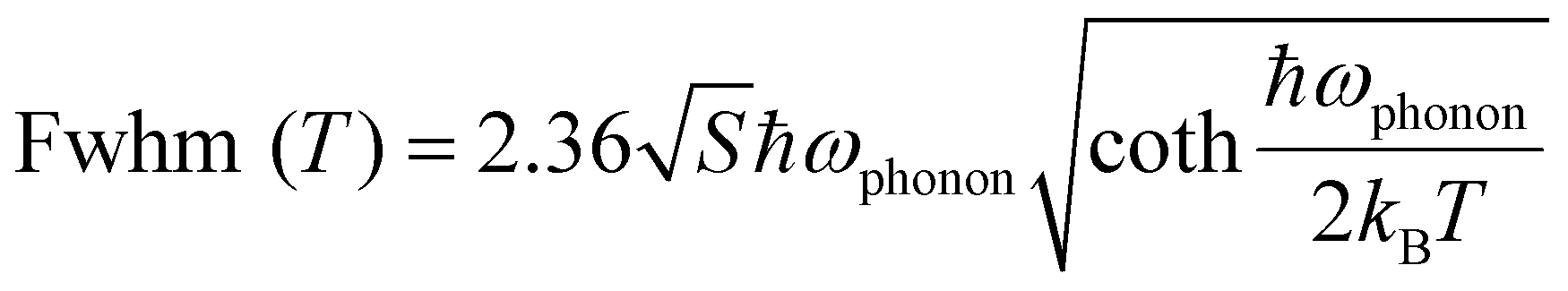 | (1) |
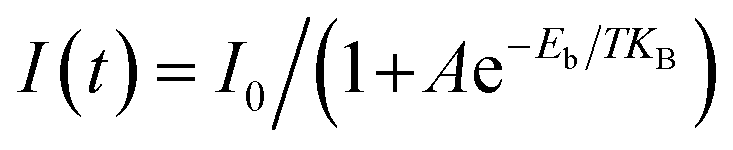 | (2) |
The Raman spectrum (Fig. S17, ESI†) was used to elucidate the vibrational modes of (C13H30N)2SnCl6:x%Sb. We found that the dominant Raman peak at 311.16 cm−1 (38.6 meV) corresponds to the A1g vibrational mode,61 which is about twice the peak at 158.64 cm−1 (19.7 meV) and belongs to the overtone of this Raman peak at 158 cm−1, which is critical to promote the formation of STE and contributes to the efficient luminescence. The Raman peak at 233.78 cm−1(29 meV) belongs to the Eg vibration mode.62 The Raman peaks at 88.87 cm−1 (11.02 meV) and 254.69 cm−1 (31.6 meV) originate from [SnCl6]2− inorganic units. The Raman peak at 254.69 cm−1 gradually disappears with increasing Sb doping ratio, which may be due to the interaction between [SnCl6]2− and [SbCl6]3− octahedra,63,64 and the carrier screening after Sb3+ incorporation. The combination of above Raman modes may constitute the three phonon energy values obtained by eqn (1) for the STE formation in the above section. These all indicate the existence of strong electro–phonon coupling in the lattice for STE formation. The Raman peaks between 850–1550 cm−1 and 2800–3050 cm−1 originate from the organic part in the compound. The organic cations in the lattice help to form a softer lattice, enhance the electro–phonon coupling, promote the formation of local STE, and generate high-efficiency PL emission.
The first-principles calculation (Fig. 4) based on density functional theory (DFT) was used to study the electronic structure of (C13H30N)2SnCl6 and Sb3+ doped (C13H30N)2SnCl6. According to DFT calculation, both undoped and Sb3+ doped (C13H30N)2SnCl6 have direct band gaps, and their band gaps are 3.13 eV and 1.38 eV, respectively, Sb incorporation has reduced the bandgap significantly. The bandgap value decreases by 1.75 eV, which is consistent with the red shift of the absorption spectrum measured experimentally. In addition, the experimental bandgap values of (C13H30N)2SnCl6 and (C13H30N)2SnCl6:20%Sb are 4.11 eV and 3.37 eV, respectively (Fig. S24, ESI†), which further proves that the doping of Sb3+ significantly reduces the bandgap. It is worth noting that the closest distance between Sb3+ and Sn4+ is about 9.8 Å, which limits the exchange interaction between them and allows for a direct radiation energy transfer. The flat electronic energy bands indicate that the excitons are highly localized on the individual Sn/Sb–Cl polyhedra.30 The calculated density of states (DOS) (Fig. 4(c) and (d)) shows that the valence band maximum (VBM) of undoped (C13H30N)2SnCl6 consists of C and H orbitals of organic and Cl-p electronic states, and the conduction band minimum (CBM) consists of Sn-s and Cl-p electronic states, the effect of the organic matter on the CBM is almost negligible. After doping with Sb3+, the product VBM becomes a new electronic state composed mainly of Sb-s and Cl-p orbitals, while its CBM is still composed of Sn-s and Cl-p electronic states and both levels move much closer in energy, indicating that the introduction of Sb3+ can effectively reduce the band gap. After doping with Sb3+, the Sb-5s state at the VBM and the Sn-s state at the CBM are highlighted with the lowest energy separation in the Sb–Cl and Sn–Cl octahedron (Fig. S18, ESI†), respectively, which may work to form their respective D–A pair excited states between separated clusters, further confirming their possible radiation energy transfer between respective highly localized excitons. Therefore, the PL emission could originate from within or between Sb/Sn–Cl octahedra, where the high-energy emission band originates from the STE of Sb3+ sites, and the low-energy even broader emission band may originate from the STE of DAP transition between SnCl6 and SbCl5 clusters in the Sb doped (C13H30N)2SnCl6. From these energy DOS we can clearly see that the transition between Sb states itself is larger than that of the transition between Sb and Sn sites, which is consistent with our results and analysis we obtained from the above spectra.
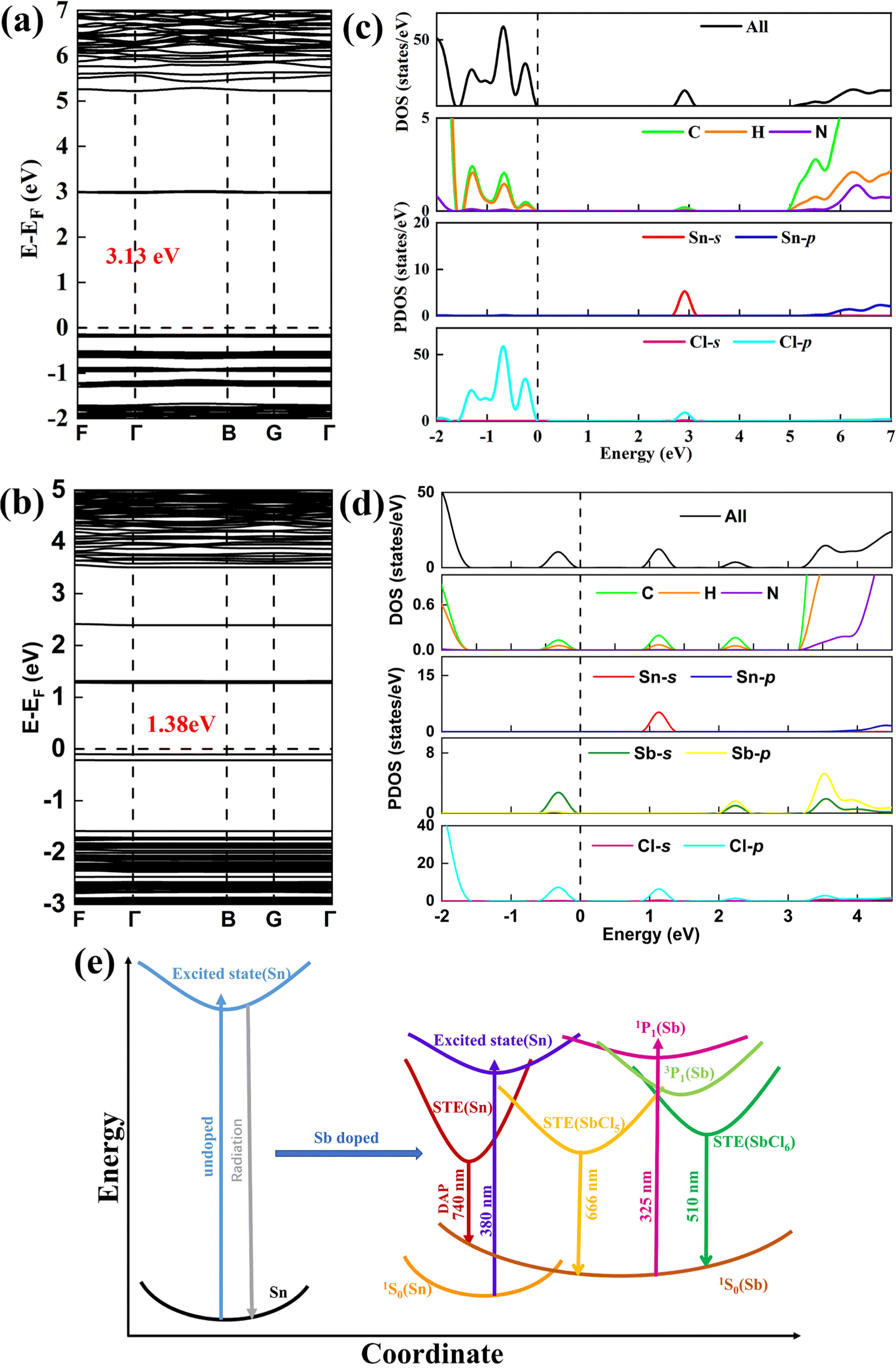 | ||
| Fig. 4 Electronic structure properties calculated based on density functional theory (DFT). (a) Band structures of (C13H30N)2SnCl6. (b) Band structures of (C13H30N)2SnCl6:Sb. (c) Density of states of (C13H30N)2SnCl6. (d) Density of states of (C13H30N)2SnCl6:Sb. (e) Schematic diagram of the energy-transfer process of (C13H30N)2SnCl6:Sb. | ||
Based on the above discussion, theoretical calculation and photophysical properties, we proposed the following photophysical processes in Sb3+ doped (C13H30N)2SnCl6 (Fig. 4(e)). When high-energy light (λex = 310–370 nm) is used to excite Sb3+-doped (C13H30N)2SnCl6 samples, the compound absorbs light energy and exhibits transition to the higher energy excited states mainly at Sb sites. Both SbCl6 and SbCl5 sites have been excited. For the former site, the emission band at 510 nm have been seen, which originates from the confined STE from the 3P1 to 1S0 in the 0-d SbCl6 cluster under the strongly confined condition of organic molecules and Sn halides. Another SbCl5 site gives emission at 666 nm, which is assigned to the radiative recombination of 3P1 to 1S0 from SbCl5 clusters, and accompaying DAP transition of Sn to 1S0 (Sb) can be seen with the combination of the above band for the close energy level of Sn. They merge into an asymmetric ultra-broadband red emission band at 678 nm under the interaction of organic molecules, Sn–Cl, etc. Low-energy (λex = 380–390 nm) excitation does not allow electrons to transition to higher energy levels of Sb3+, and its energy cannot satisfy the formation conditions of confined excitons originating from 0-d SbCl6 clusters, but that at the Sn site. As shown in Fig. 4(d), the Sn-s state at the CBM is located at a lower energy level than the Sb-p state, so only DAP transitions to 1S0 of the SbCl5 clusters could be seen, finally generating ultrabroadband red emission at 678 nm.
In order to explore the environmental stability of Sb3+doped (C13H30N)2SnCl6, we performed thermogravimetric analysis on the sample (C13H30N)2SnCl6:20%Sb. The TGA curve (Fig. S19, ESI†) shows that the sample begins to decompose when the temperature reaches 253 °C, indicating that the synthesized sample has good high temperature stability. We measured its PL spectrum after exposing (C13H30N)2SnCl6:20%Sb to the air environment for 30 days (Fig. S20, ESI†), and the results showed that the emission intensity of the sample decreased by only 3%, remaining almost unchanged after 30 days. In addition, the measured XRD pattern (Fig. S21, ESI†) showed that the structure did not change after being placed in the air environment for 30 days, indicating its excellent structural stability. The above results show that Sb3+ doped (C13H30N)2SnCl6 has good emission and environmental stability.
Metal halide perovskites have been widely studied in the fields of energy,65 information encryption,66 and lighting67 due to their excellent properties. Based on the good environmental stability of Sb3+ doped (C13H30N)2SnCl6, we explored its potential as a single-component high-efficiency white light emitting material in the field of solid-state lighting. We fabricated a single component WLED (Fig. S22, ESI†) by coating (C13H30N)2SnCl6:20%Sb powder on a commercial 310 nm UV chip. Fig. 5(a) shows the PL spectrum of the WLED device at a current of 90 mA; side bands at about 400 nm and 800 nm have also been seen besides the dominant dual bands at 510 nm and 678 nm. Its color rendering index is as high as 96.7, which surpasses most of the reported WLEDs.6,67,68 This shows that this compound has excellent properties and possesses large lighting application prospects. The above illustration shows that it can highly reproduce the color of the object itself. The correlated color temperature (CCT) of the WLED device is 5064 K, which belongs to neutral white light, and the corresponding color coordinates of (0.346, 0.380) are shown in Fig. 5(c). Fig. 5(b) shows the emission spectra of the WLED device under different forward bias currents (10–110 mA), and its emission intensity increases with increasing current. Its lumen efficiency is 0.13 lm W−1 at 10 mA. These results indicate the potential application of lead-free 0D perovskite Sb3+ doped (C13H30N)2SnCl6 in the field of solid-state lighting.
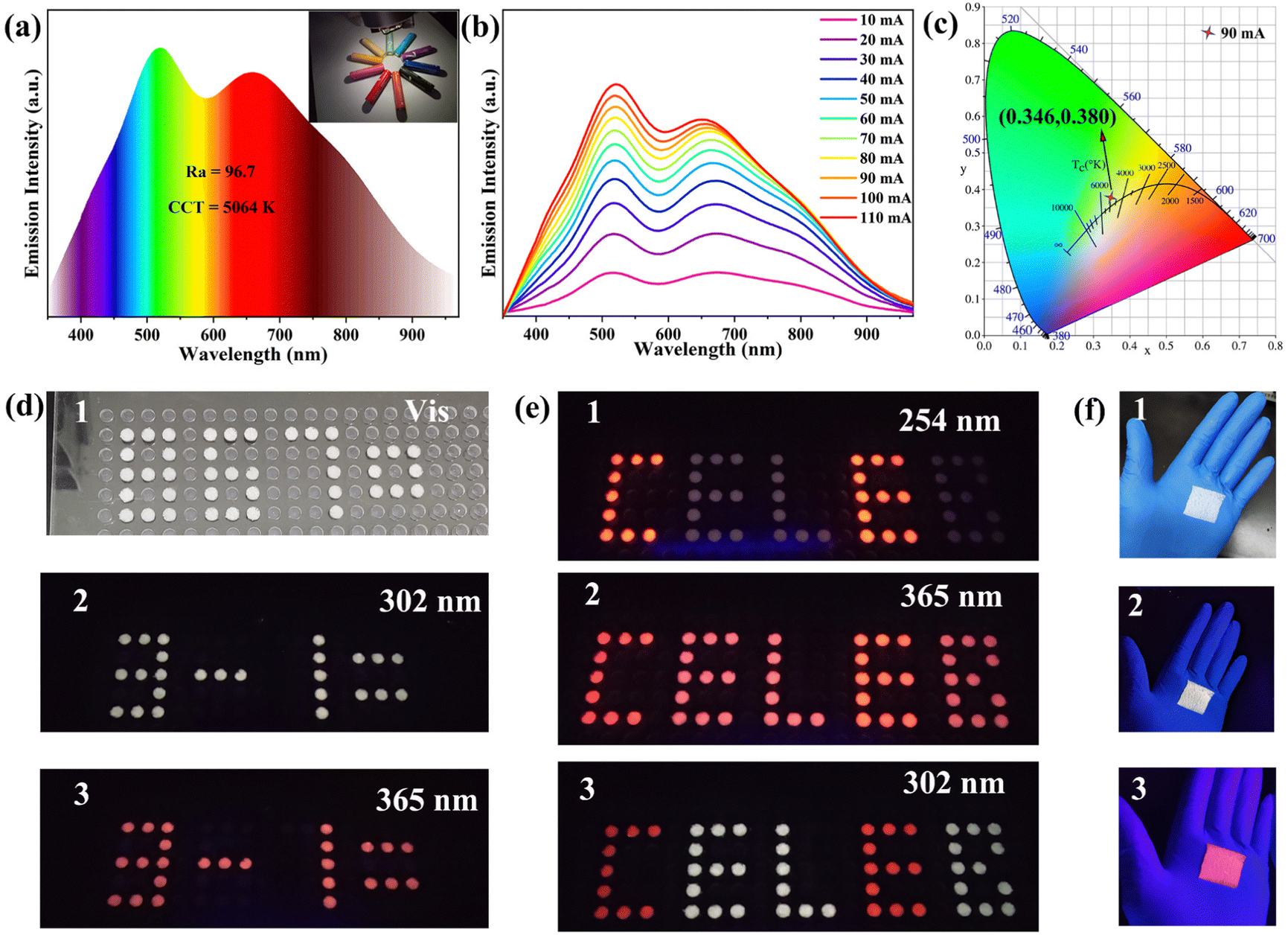 | ||
| Fig. 5 Anti-counterfeiting encryption and lighting display applications of (C13H30N)2SnCl6:20%Sb. (a) PL emission spectra of LED lamps fabricated by combining commercial 310 nm chips and (C13H30N)2SnCl6:20%Sb samples at a driving current of 90 mA. The inset shows a photo of the color rendering of the fabricated LED. (b) Drive current-dependent emission spectra of a fabricated LED. (c) The color coordinate diagram corresponding to the LED device at 90 mA driven current. (d) Photos of modular arithmetic pattern “3–1 = ” under the excitation of (1) visible light, (2) 302 nm UV light on, (3) 365 nm UV light on. (e) Photos of pattern “CELEB” modularized with sample (C13H30N)2SnCl6:20%Sb under (1) visible light, (2) 302 nm UV light, (3) 365 nm UV light excitation. (f) Images of square (C13H30N)2SnCl6:20%Sb-PDMS composites under (1) visible light, (2) 302 nm UV light and (3) 365 nm UV light. | ||
In addition, the wavelength-dependent emission characteristics of Sb3+ doped (C13H30N)2SnCl6 imply that it has two emission centers and varied color, which make it have great application prospects in the fields of information encryption and anti-counterfeiting. As shown in Fig. 5(d), (C13H30N)2SnCl6:20%Sb powder is modularized into a “3–1 = ” formula pattern, and the formula result represents “2”. It appears as a misleading “8670” pattern under visible light, and as a white and red arithmetic pattern “3–1 = ” under 302 and 365 nm UV lamp irradiation, respectively. Reasonable pattern design and wavelength-tunable color as shown make it have potential application prospects in double anti-counterfeiting. Furthermore, we composed a more camouflaged pattern by combining (C13H30N)2SnCl6:20%Sb with C4H12NMnCl3 (Fig. 5(e)). It presents red letters “CE” under 254 nm excitation, red letters “CELEB” under 365 nm excitation, and then evolves into white “ELB” and red “CE” under 302 nm excitation. Different combination patterns under different excitations provide different information, which improves the security of information encryption. As shown in Fig. 5(f), the as-fabricated flexible (C13H30N)2SnCl6:20%Sb-PDMS (polydimethylsiloxane) patterned composites exhibit red and white emission colors when excited by 365 and 302 nm UV light, respectively. It further proves that it is suitable in solid-state lighting and flexible display applications.
Conclusions
In summary, we successfully synthesized Sb3+-doped 0D organic–inorganic hybrid halide perovskite (C13H30N)2SnCl6:Sb by a simple low-temperature solvent evaporation method. The (C13H30N)2SnCl6:Sb crystal exhibits high white (PLQY = 99.32%) and red-light emission (PLQY = 80.98%) under 325 nm and 380 nm excitation, respectively, which indicates that it has very high energy conversion efficiency for emission. Correlative temperature-dependent PL spectroscopy studies reveal that the dual emission bands of (C13H30N)2SnCl6:Sb are mainly related to the excitation energy at different sites. Its unique dual emission bands span the temperature range from 80 K to 320 K under high-energy excitation. The green emission band of (C13H30N)2SnCl6:Sb at 510 nm originates from the confined STE out of 3P1 to 1S0 at 0-d SbCl6 cluster; while the ultrabroad red emission band at 678 nm originates from different types of emission centers, respectively STE radiative recombination out of 3P1 to 1S0 at SbCl5 clusters, and DAP transition between Sn and Sb. In addition, (C13H30N)2SnCl6:Sb has good air environment stability. Single component LED devices based on (C13H30N)2SnCl6:20%Sb have a high color rendering index of 96.7, and we demonstrate the exhibition of this novel 0D lead-free metal halide for solid-state lighting displays. At the same time, it also has excellent anti-counterfeiting and information encryption functions. This work achieves highly efficient white and red-light emission in Sn (IV)-based metal halides at different excitation, which provides a new design strategy for designing high-performance white-emitting hybrid metal halides and possibilities in different optoelectric devices.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by Guangxi NSF grant no. 2020GXNSFDA238004 and the “Guangxi Bagui Scholars” fund and the Scientific and Technological Bases and Talents of Guangxi (Guike AD21238027).References
- C. Zhou, Y. Tian, O. Khabou, M. Worku, Y. Zhou, J. Hurley, H. Lin and B. Ma, ACS Appl. Mater. Interfaces, 2017, 9, 40446–40451 CrossRef CAS PubMed
.
- H. Yuan, F. Massuyeau, N. Gautier, A. B. Kama, E. Faulques, F. Chen, Q. Shen, L. Zhang, M. Paris and R. Gautier, Angew. Chem., Int. Ed., 2020, 59, 2802–2807 CrossRef CAS PubMed
- F. Liu, T. Zhang, D. Mondal, S. Teng, Y. Zhang, K. Huang, D. Wang, W. Yang, P. Mahadevan, Y. S. Zhao, R. Xie and N. Pradhan, Angew. Chem., Int. Ed., 2021, 60, 13548–13553 CrossRef CAS PubMed
- Q. Mo, Q. Qian, Y. Shi, W. Cai, S. Zhao and Z. Zang, Adv. Opt. Mater., 2022, 10, 2201509 CrossRef CAS
- T. Huang, Q. Wei, W. Lin, H. Peng, S. Yao and B. Zou, Mater. Today Phys., 2022, 25, 100703 CrossRef CAS
- H. Peng, Y. Tian, X. Wang, T. Huang, Y. Xiao, T. Dong, J. Hu, J. Wang and B. Zou, J. Mater. Chem. C, 2021, 9, 12184–12190 RSC
- S. Yan, W. Tian, H. Chen, K. Tang, T. Lin, G. Zhong, L. Qiu, X. Pan and W. Wang, Adv. Funct. Mater., 2021, 31, 2100855 CrossRef CAS
- G. Zhou, X. Jiang, M. Molokeev, Z. Lin, J. Zhao, J. Wang and Z. Xia, Chem. Mater., 2019, 31, 5788–5795 CrossRef CAS
- Z. Yuan, C. Zhou, Y. Tian, Y. Shu, J. Messier, J. C. Wang, L. J. van de Burgt, K. Kountouriotis, Y. Xin, E. Holt, K. Schanze, R. Clark, T. Siegrist and B. Ma, Nat. Commun., 2017, 8, 14051 CrossRef CAS PubMed
- M. Zhang, L. Zhao, J. Xie, Q. Zhang, X. Wang, N. Yaqoob, Z. Yin, P. Kaghazchi, S. Zhang, H. Li, C. Zhang, L. Wang, L. Zhang, W. Xu and J. Xing, Nat. Commun., 2021, 12, 4890 CrossRef CAS PubMed
- C. Zhou, S. Lee, H. Lin, J. Neu, M. Chaaban, L.-J. Xu, A. Arcidiacono, Q. He, M. Worku, L. Ledbetter, X. Lin, J. A. Schlueter, T. Siegrist and B. Ma, ACS Mater. Lett., 2020, 2, 376–380 CrossRef CAS
- Y. Liu, C. Wang, Y. Guo, L. Ma, C. Zhou, Y. Liu, L. Zhu, X. Li, M. Zhang and G. Zhao, J. Mater. Chem. C, 2020, 8, 5673–5680 RSC
- R. Gautier, F. Massuyeau, G. Galnon and M. Paris, Adv. Mater., 2019, 31, 1807383 CrossRef PubMed
- B. B. Cui, Y. Han, B. Huang, Y. Zhao, X. Wu, L. Liu, G. Cao, Q. Du, N. Liu, W. Zou, M. Sun, L. Wang, X. Liu, J. Wang, H. Zhou and Q. Chen, Nat. Commun., 2019, 10, 5190 CrossRef PubMed
- L. Mao, P. Guo, M. Kepenekian, I. Hadar, C. Katan, J. Even, R. D. Schaller, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 13078–13088 CrossRef CAS PubMed
- E. R. Dohner, A. Jaffe, L. R. Bradshaw and H. I. Karunadasa, J. Am. Chem. Soc., 2014, 136, 13154–13157 CrossRef CAS PubMed
- T. Huang, H. Peng, Q. Wei, C. Peng, Y. Tian, S. Yao, X. Han and B. Zou, Nano Energy, 2022, 93, 106863 CrossRef CAS
- H. Peng, Y. Tian, X. Wang, S. Yao, T. Huang, Y. Guo, L. Shi and B. Zou, New J. Chem., 2021, 45, 17247–17257 RSC
- D.-Y. Li, Y. Cheng, Y.-H. Hou, J.-H. Song, C.-J. Sun, C.-Y. Yue, Z.-H. Jing and X.-W. Lei, J. Mater. Chem. C, 2022, 10, 3746–3755 RSC
- T. Jiang, W. Ma, H. Zhang, Y. Tian, G. Lin, W. Xiao, X. Yu, J. Qiu, X. Xu, Y. Yang and D. Ju, Adv. Funct. Mater., 2021, 31, 2009973 CrossRef CAS
- Y.-Y. Ma, Y.-R. Song, W.-J. Xu, Q.-Q. Zhong, H.-Q. Fu, X.-L. Liu, C.-Y. Yue and X.-W. Lei, J. Mater. Chem. C, 2021, 9, 9952–9961 RSC
- Y. Liu, A. Wang, J. Wu, C. Wang, Z. Li, G. Hu, S. Sui, J.-X. She, W. Meng, W. Li and Z. Deng, Mater. Adv., 2021, 2, 1320–1327 RSC
- P. Fu, M. Huang, Y. Shang, N. Yu, H. L. Zhou, Y. B. Zhang, S. Chen, J. Gong and Z. Ning, ACS Appl. Mater. Interfaces, 2018, 10, 34363–34369 CrossRef CAS PubMed
- C. Zhou, H. Lin, H. Shi, Y. Tian, C. Pak, M. Shatruk, Y. Zhou, P. Djurovich, M. H. Du and B. Ma, Angew. Chem., Int. Ed., 2018, 57, 1021–1024 CrossRef CAS PubMed
- Y. Jing, Y. Liu, J. Zhao and Z. Xia, J. Phys. Chem. Lett., 2019, 10, 7439–7444 CrossRef CAS PubMed
- G. Song, Z. Li, P. Gong, R. J. Xie and Z. Lin, Adv. Opt. Mater., 2021, 9, 2002246 CrossRef CAS
- H. Shi, D. Han, S. Chen and M.-H. Du, Phys. Rev. Mater., 2019, 3, 034604 CrossRef
- Z. Wang, Z. Zhang, L. Tao, L. Shen, B. Hu, L. Gong, J. Li, X. Chen and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 9974–9978 CrossRef CAS PubMed
- H. Peng, Y. Tian, Z. Yu, X. Wang, B. Ke, Y. Zhao, T. Dong, J. Wang and B. Zou, Sci. China Mater., 2022, 65, 1594–1600 CrossRef CAS
- R. Zeng, L. Zhang, Y. Xue, B. Ke, Z. Zhao, D. Huang, Q. Wei, W. Zhou and B. Zou, J. Phys. Chem. Lett., 2020, 11, 2053–2061 CrossRef CAS PubMed
- C. Li, Z. Luo, Y. Liu, Y. Wei, X. He, Z. Chen, L. Zhang, Y. Chen, W. Wang, Y. Liu, X. Chang and Z. Quan, Adv. Opt. Mater., 2022, 10, 2102746 CrossRef CAS
- J. Jin, Y. P. Peng, Y. Xu, K. Han, A. Zhang, X.-B. Yang and Z. Xia, Chem. Mater., 2022, 34, 5717–5725 CrossRef CAS
- J. Zhou, M. Li, M. S. Molokeev, J. Sun, D. Xu and Z. Xia, J. Mater. Chem. C, 2020, 8, 5058–5063 RSC
- A. Noculak, V. Morad, K. M. McCall, S. Yakunin, Y. Shynkarenko, M. Worle and M. V. Kovalenko, Chem. Mater., 2020, 32, 5118–5124 CrossRef CAS PubMed
- P. Han, C. Luo, S. Yang, Y. Yang, W. Deng and K. Han, Angew. Chem., Int. Ed., 2020, 59, 12709–12713 CrossRef CAS PubMed
- J. Huang, T. Chang, R. Zeng, J. Yan, Q. Wei, W. Zhou, S. Cao and B. Zou, Adv. Opt. Mater., 2021, 9, 2002267 CrossRef CAS
- Y. Jing, Y. Liu, X. Jiang, M. S. Molokeev, Z. Lin and Z. Xia, Chem. Mater., 2020, 32, 5327–5334 CrossRef CAS
- J.-H. Wei, J.-F. Liao, L. Zhou, J.-B. Luo, X.-D. Wang and D.-B. Kuang, Sci. Adv., 2021, 7, eabg3989 CrossRef CAS PubMed
- C.-M. Shi, J.-L. Li, L.-J. Xu, Y. Wu, H.-L. Xuan, J.-Y. Wang and Z.-N. Chen, Sci. China Mater., 2022, 65, 1876–1881 CrossRef CAS
- X. Meng, Q. Wei, W. Lin, T. Huang, S. Ge, Z. Yu and B. Zou, Inorg. Chem., 2022, 61, 7143–7152 CrossRef CAS PubMed
- L. Zhou, L. Zhang, H. Li, W. Shen, M. Li and R. He, Adv. Funct. Mater., 2021, 31, 2108561 CrossRef CAS
- G. Zhang, P. Dang, H. Xiao, H. Lian, S. Liang, L. Yang, Z. Cheng, G. Li and J. Lin, Adv. Opt. Mater., 2021, 9, 2101637 CrossRef CAS
- B. Su, M. Li, E. Song and Z. Xia, Adv. Funct. Mater., 2021, 31, 2105316 CrossRef CAS
- J. P. Perdew and M. Levy, Phys. Rev. Lett., 1983, 51, 1884–1887 CrossRef CAS
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef PubMed
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed
- Z. Yu, H. Peng, Q. Wei, T. Huang, S. Yao, Y. Tian, C. Peng and B. Zou, Mater. Today Chem., 2022, 24, 100781 CrossRef CAS
- R. Zeng, K. Bai, Q. Wei, T. Chang, J. Yan, B. Ke, J. Huang, L. Wang, W. Zhou, S. Cao, J. Zhao and B. Zou, Nano Res., 2020, 14, 1551–1558 CrossRef
- F. Lin, H. Wang, H. Lin, W. Liu and J. Li, Chem. Commun., 2021, 57, 1754–1757 RSC
- J. D. Majher, M. B. Gray, T. Liu, N. P. Holzapfel and P. M. Woodward, Inorg. Chem., 2020, 59, 14478–14485 CrossRef CAS PubMed
- C. Sun, J.-P. Zang, Y.-Q. Liu, Q.-Q. Zhong, X.-X. Xing, J.-P. Li, C.-Y. Yue and X.-W. Lei, CCS Chem., 2021, 4, 3106–3121 CrossRef
- Q. Wei, X. Meng, W. Lin, S. Ge, X. Han, L. Chen, R. Zeng and B. Zou, J. Phys. Chem. Lett., 2022, 13, 8436–8446 CrossRef CAS PubMed
- Y. Dai, Q. Wei, T. Chang, J. Zhao, S. Cao, B. Zou and R. Zeng, J. Phys. Chem. C, 2022, 126, 11238–11245 CrossRef CAS
- F. Lin, H. Tong, H. Lin and W. Liu, Chem. Commun., 2022, 58, 12596–12599 RSC
- H. Lin, Q. Wei, B. Ke, W. Lin, H. Zhao and B. Zou, J. Phys. Chem. Lett., 2023, 14, 1460–1469 CrossRef CAS PubMed
- B. P. Picket and R. L. Benoit, Inorg. Chem., 1967, 6, 1505–1509 CrossRef
- Y. Jing, Y. Liu, M. Li and Z. Xia, Adv. Opt. Mater., 2021, 9, 2002213 CrossRef CAS
- P. Cheng, L. Feng, Y. Liu, D. Zheng, Y. Sang, W. Zhao, Y. Yang, S. Yang, D. Wei, G. Wang and K. Han, Angew. Chem., Int. Ed., 2020, 59, 21414–21418 CrossRef CAS PubMed
- W. Jia, Q. Wei, S. Ge, C. Peng, T. Huang, S. Yao, Y. Tian, T. Chang, R. Zeng and B. Zou, J. Phys. Chem. C, 2021, 125, 18031–18039 CrossRef CAS
- T. L. Brown, W. G. McDugle Jr and L. G. Kent, J. Am. Chem. Soc., 1970, 92, 3645–3653 CrossRef CAS
- M. M. S. Karim, A. M. Ganose, L. Pieters, W. W. Winnie Leung, J. Wade, L. Zhang, D. O. Scanlon and R. G. Palgrave, Chem. Mater., 2019, 31, 9430–9444 CrossRef CAS PubMed
- H. Yin, J. Chen, P. Guan, D. Zheng, Q. Kong, S. Yang, P. Zhou, B. Yang, T. Pullerits and K. Han, Angew. Chem., Int. Ed., 2021, 60, 22693–22699 CrossRef CAS PubMed
- F. J. Amaya Suazo, S. Shaji, D. A. Avellaneda, J. A. Aguilar-Martínez and B. Krishnan, Mater. Sci. Semicond. Process., 2020, 115, 105115 CrossRef CAS
- S. Yao, L. Yang, S. Shi, Y. Zhou, M. Long, W. Zhang, S. Cai, C. Huang, T. Liu and B. Zou, Chin. J. Chem., 2022, 41, 672–678 CrossRef
- C. Wang, Y. Li, Q. Lv, H. Zheng, G. Zhu, X. Xu and Y. Wang, Chem. Eng. J., 2022, 431, 134135 CrossRef CAS
- S. Wang, X. Han, T. Kou, Y. Zhou, Y. Liang, Z. Wu, J. Huang, T. Chang, C. Peng, Q. Wei and B. Zou, J. Mater. Chem. C, 2021, 9, 4895–4902 RSC
- C. Ji, S. Wang, L. Li, Z. Sun, M. Hong and J. Luo, Adv. Funct. Mater., 2018, 29, 1805038 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc00497j |
| This journal is © The Royal Society of Chemistry 2023 |