
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fundamental insight into enhanced activity of Pd/CeO2 thin films in hydrogen oxidation reaction in alkaline media†
Mathilde
Luneau
 *ab,
Linnéa
Strandberg
b,
Gerard
Montserrat-Sisó
b,
Victor
Shokhen
b,
Roopathy
Mohan
b,
Henrik
Grönbeck
b and
Björn
Wickman
b
*ab,
Linnéa
Strandberg
b,
Gerard
Montserrat-Sisó
b,
Victor
Shokhen
b,
Roopathy
Mohan
b,
Henrik
Grönbeck
b and
Björn
Wickman
b
aDepartment of Chemistry and Chemical Engineering, Competence Centre for Catalysis, Chalmers University of Technology, Gothenburg, SE 41296, Sweden. E-mail: mathilde.luneau@chalmers.se
bDepartment of Physics, Competence Centre for Catalysis, Chalmers University of Technology, Gothenburg, SE 41296, Sweden
First published on 19th July 2023
Abstract
Palladium supported on ceria (Pd/CeO2) has recently raised strong interest as an alternative catalyst to platinum on the anode electrode in anion exchange membrane fuel cells. Herein, we provide new insight into the enhanced activity of Pd/CeO2 in hydrogen oxidation reaction (HOR) in alkaline media. Using well-defined model thin films, we show that Pd/CeO2 thin films lead to enhanced activity in HOR compared to pure Pd thin films. In situ characterization using electrochemical quartz crystal microbalance provide in-depth understanding of the role of CeO2. CeO2 leads to fundamental differences in adsorption and absorption of key reaction intermediates during HOR. In combination with characterization and theoretical calculations, Pd atoms embedded in CeO2 are shown to be present on the prepared thin films and active for hydrogen activation but are not able to bind CO during CO-stripping characterization. Finally, an estimation of the source of hydroxyl intermediates provided by CeO2 – which could be directly participating in the reaction – is presented herein. Fundamental understanding of the Pd–CeO2 interface in HOR opens new ways to reduce the amount of noble metals in alkaline fuel cells.
 Mathilde Luneau | Dr Mathilde Luneau is an Assistant Professor at the Department of Chemistry and Chemical Engineering at Chalmers University of Technology in Gothenburg, Sweden. Before joining Chalmers University of Technology, she was a postdoctoral fellow at Harvard University, USA in the group of Prof. Cynthia Friend. Mathilde obtained her PhD from the University of Lyon 1, France in 2016 where she worked at the Institute of Research on Catalysis and Environment (IRCELyon). Her research focuses on the design and understanding of novel catalytic materials for applications in electrocatalytic and thermocatalytic reactions. |
Introduction
Demand for non-fossil fuel-based electricity is growing rapidly and conversion of hydrogen into electricity in fuel cells has received increasing interest in the past decades. The most common fuel cell is presently the proton-exchange membrane fuel cell (PEMFC). One drawback with PEMFCs is, however, the use of Pt-based catalysts. Pt-based catalysts are required to achieve a high activity for the fuel cell reactions and to withstand the harsh acidic conditions and varying electric potential over long periods of operation. The high cost associated with scarce Pt-based catalysts is limiting the use of fuel cells. To remedy this problem, anion-exchange membrane fuel cells (AEMFCs) have gained interest in recent years.1 The high pH in AEMFCs allows the use of alternative Pt-free catalysts although the reaction rate of the hydrogen oxidation reaction (HOR) in alkaline media is orders of magnitude lower than in acidic conditions.2 Catalytic materials with enhanced HOR activity in alkaline media must thus be designed. A promising approach is to design catalysts with strong metal–support interfaces.Recently, CeO2 has been used as a support for Pd in AEMFC applications and it was proposed that the Pd/CeO2 metal–support interface plays a prominent role in the improved activity.3,4 While Pd–PdO catalysts have shown good performance in various electrochemical reactions such as CO2 electroreduction and methanol electrooxidation, they have poor activity for HOR.5–7 The promotion of Pd catalysts by supports such as ceria has however led to significant enhancement in HOR. The metal-support interface dynamics have been shown to affect performance in heterogeneous catalysis.8 Their understanding and modulation can thus lead to important enhancement in activity. These interactions have been shown to drive catalytic activity and to be most impactful for dispersed single metal atoms and nanoparticles smaller than ca. 4 nm.9,10 In recent years, most efforts have been put on establishing strategies to best utilize, tune and maximize these interactions to enhance activity. Due to the diversity of possible metal–support combinations and interactions, the interface dynamics have been investigated for a wide range of applications.
In particular, the special synergy between precious metals and ceria (CeO2) has raised a lot of attention in the past decades.11,12 Ceria is known to act as an “oxygen storage” material with the ability to gain and release oxygen as Ce oxidation state changes between Ce4+ and Ce3+. In industrial applications, this oxygen storage capability has been utilized in the automotive three way catalysts where CeO2 is used as a promoter to remove pollutants originating from the combustion of gasoline.13 The rare earth metal Ce, either used as a support in CeO2 or as atomically dispersed species, has also led to significant enhancement in electrocatalytic applications and reactions, such as in oxygen evolution reaction, oxygen reduction reaction and in solid oxide fuel cells.11,14–16 These beneficial interactions between precious metals and ceria have mainly been understood in terms of strong metal–support interaction, unique redox properties of CeO2, electronic interaction between the metal and the metal oxide, and hydrogen spillover.17
In alkaline media, HOR has been suggested to proceed via two main mechanisms: (i) the Tafel–Volmer pathway (combining eqn (1) and (3)) or (ii) the Heyrovsky–Volmer pathway (combining eqn (2) and (3)):
| H2 + 2* ⇌ 2H*, Tafel reaction | (1) |
| H2 + OH− +* ⇌ H* + H2O + e−, Heyrovsky reaction | (2) |
| H* + OH− ⇌ * + H2O + e−, Volmer reaction | (3) |
The hydrogen binding energy is often used as a descriptor for hydrogen evolution and oxidation reactions.18 In a so-called volcano plot, the logarithm of the exchange current density in alkaline media was correlated with the calculated hydrogen binding energy (H-BE) on metal surfaces, with Pt near the top.19 In this interpretation, weak hydrogen binding results in a slow Volmer step, limiting the overall rate. Strong hydrogen binding (such as on Pd) leads to a competition between adsorption and desorption which limits the Tafel step. Following the Sabatier principle, an ideal catalyst must bind the hydrogen atoms not too strongly and not too weakly.
In alkaline media, adsorbed hydroxyl groups can also play an important role in the reaction.20,21 Adsorbed OH groups are traditionally considered as spectators or inhibiting species.22 OH adsorption (OH− + * ⇌ OH* + e−) can inhibit H2 adsorption and dissociation if OH binds too strongly and if they compete for the same active sites. However, in alkaline media, OH groups have also been shown to play a promoting role in HOR.20,21 Notably, a bifunctional effect was suggested to occur on PtRu catalyst and led to enhanced HOR activity compared to pure Pt.20 The authors proposed that adsorbed OH groups on oxyphilic Ru could react with hydrogen intermediates adsorbed on nearby Pt sites. Therefore, both H and OH adsorption strengths must be considered in alkaline media.23
In addition to its oxygen storage capability, CeO2 also has the ability to interact with hydrogen, in two different ways: one through the hydroxylation of the surface oxide and the second through the incorporation of hydrogen into the ceria lattice.24 Surface hydroxyl groups are thus expected to play an important role in the reaction. In different applications such as steam reforming of ethanol, studies have shown that in the presence of water, CeO2 participated in reactions by dissociating water and providing hydroxyl groups that can react.25 Modified hydrogen interactions and hydroxyl groups participation in the reaction mechanism are thus important parameters that can influence the reactivity of Pd/CeO2 catalysts.
A recent theoretical study investigated the Tafel–Volmer pathway for HOR on Pd/CeO2 in alkaline media.26 The authors attributed the catalytic improvement to a balance between the H and OH adsorption strengths and the strong interaction between Pd and the ceria support. Importantly, isolated Pd atoms on CeO2 were suggested to lead to better performances than Pd clusters and were suggested to be the active sites. Looking at the Tafel step, isolated Pd atoms on CeO2 were calculated to have comparable dissociation energy (DE) than Pd(111) (H2-DE = 0.05 vs. 0.03 eV, respectively). The authors reported a stronger binding of hydrogen – i.e. more negative binding energy (BE) – on isolated Pd atoms on CeO2 compared to extended Pd(111) surface (H-BE = −1.29 eV vs. −0.69 eV, respectively). In the Volmer step, the hydroxyl intermediate formed by adsorbing OH− reacts with the dissociated hydrogen. On isolated Pd atoms on CeO2, OH groups were found to bind less strongly than on extended Pd(111) surfaces (OH-BE = −3.25 eV vs. −3.33 eV). One important finding is that OH* was calculated to be able to bind to cerium, with H adsorbed on isolated Pd atoms, leading to an interface between Pd and Ce. Further understanding is still needed to elucidate the role of CeO2 in the reaction.
Here we study model Pd/CeO2 thin films with well-defined compositions and structures to gain fundamental understanding of the metal–metal oxide interface and its influence on HOR activity in alkaline media. Model thin films are ideal systems to study the fundamental metal/ceria interactions and have not yet been employed for HOR on Pd/CeO2 in alkaline media. We develop an approach combining electrochemical measurements, in situ electrochemical characterization, ex situ microscopy and spectroscopy, and theoretical calculations to describe the interactions between Pd and CeO2.
A key aspect of our study is the use of electrochemical quartz crystal microbalance (E-QCM) analysis. E-QCM is a powerful in situ technique generally used to evaluate the stability of materials.27 It is also a powerful method to investigate and quantify mass changes occurring on surfaces due to phenomena of dissolution, adsorption, absorption, physisorption and desorption, while the reaction is occurring.28 Using E-QCM, we provide fundamental understanding of the adsorption, absorption and desorption phenomena occurring on Pd/CeO2 thin films during HOR. We show that on pure Pd thin films, mass changes are dominated by the absorption of hydrogen while on Pd/CeO2 they are dominated by the oxidation/reduction of ceria and adsorption of hydroxyl groups. One of the hypotheses for enhanced reactivity of Pd/CeO2 in HOR is that CeO2 – at the interface with Pd – serves as a source of OH groups which react with adsorbed H to form water.29 In this study, we provide quantitative evidence that this hypothesis is valid using an in situ technique on model thin films.
Methods
Thin films were synthesized via electron-beam physical vapor deposition (AVAC HVC600) on glassy-carbon electrodes. Prior to deposition, the electrodes (5 mm diameter, Sigradur® disks from HTW GmbH) were cleaned by sequential sonication in acetone (Sigma-Aldrich), isopropanol (VWR) and Milli-Q water (18.2 MΩ cm) for 10 minutes each, and dried with N2 (Grade N6.0, Strandmöllen) followed by plasma cleaning in O2 atmosphere at 100 W for 1 minute. After cleaning, a 20 nm Ti underlayer was evaporated first to improve adhesion properties of Pd films to the electrodes. So-called pure Pd samples were prepared by evaporating 40 nm Pd thin films on the Ti adhesion layer. Pd/CeO2 samples were prepared by evaporating a 20 nm CeO2 film followed by a 2 nm Pd film. The electrodes have a geometrical surface area of 0.196 cm2.Electrochemical activity was studied using cyclic voltammetry (CV) in a standard three-electrode electrochemical cell in a rotating disk electrode (RDE) configuration (Pine Research Instrumentation). CVs were measured in 0.1 M KOH electrolyte in a polytetrafluoroethylene (PTFE) cell at room temperature using a graphite rod as counter electrode and a Hg/HgO reference electrode. All potentials in this study are reported versus the reversible hydrogen electrode (RHE) which were determined using a Pt counter electrode and a Pt working electrode while flowing H2. Conditioning was performed in Ar by cycling at 100 mV s−1 for 20 cycles followed by 10 cycles at 50 mV s−1. Hydrogen oxidation reaction was performed in H2 by cycling at 5 mV s−1 and rotating at 1600 rpm. In another set of experiments, the rotation rate was varied between 400, 900, 1600 and 2500 rpm while cycling at 50 mV s−1 in H2 after conditioning to obtain Koutecky–Levich plots. The cells were cleaned thoroughly with a piranha solution (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of concentrated sulfuric acid to 30% hydrogen peroxide solution) and rinsed with Milli-Q water between experiments. To further limit contamination, two cells were used: one for Pd-based samples and one for CeO2 only.
:1 ratio of concentrated sulfuric acid to 30% hydrogen peroxide solution) and rinsed with Milli-Q water between experiments. To further limit contamination, two cells were used: one for Pd-based samples and one for CeO2 only.
The Koutecky–Levich equation was used to estimate the number of transferred electrons (n):
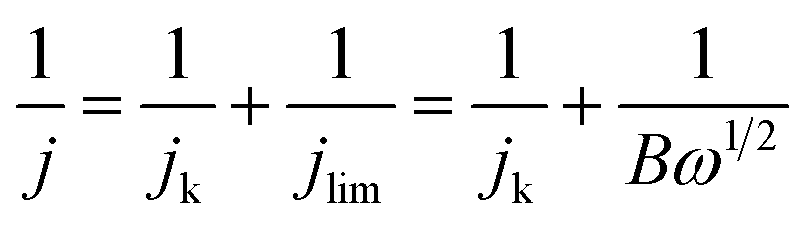 | (4) |
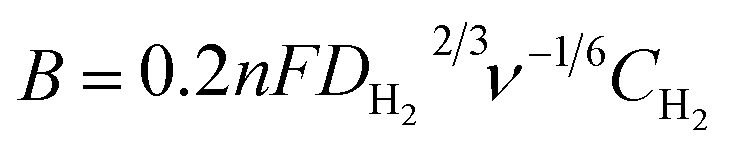 , and where j is the current density, jk is the kinetic current density, jlim is the diffusion-limited current density, ω is the rotation rate in rpm, F is the Faraday constant (96485 C mol−1), DH2 is the diffusion coefficient of H2 in water (4.58 × 10−5 cm2 s−1 at 20 °C),30ν is the kinematic viscosity (0.01 cm2 s−1)30 and CH2 is the bulk concentration of H2 in the solution (1 × 10−6 mol cm−3).31 The constant 0.2 is used as the rotation rate is in rpm.23 The Koutecky–Levich plots were plotted using current densities measured at 0.8 VRHE. This potential was chosen as it corresponds to the diffusion-limited region on both samples. At lower potentials, samples were not limited by the same phenomena (i.e. kinetic vs. diffusion).
, and where j is the current density, jk is the kinetic current density, jlim is the diffusion-limited current density, ω is the rotation rate in rpm, F is the Faraday constant (96485 C mol−1), DH2 is the diffusion coefficient of H2 in water (4.58 × 10−5 cm2 s−1 at 20 °C),30ν is the kinematic viscosity (0.01 cm2 s−1)30 and CH2 is the bulk concentration of H2 in the solution (1 × 10−6 mol cm−3).31 The constant 0.2 is used as the rotation rate is in rpm.23 The Koutecky–Levich plots were plotted using current densities measured at 0.8 VRHE. This potential was chosen as it corresponds to the diffusion-limited region on both samples. At lower potentials, samples were not limited by the same phenomena (i.e. kinetic vs. diffusion).
Mass changes related to adsorption, absorption, physisorption or desorption phenomena were evaluated in situ with electrochemical-quartz crystal microbalance (Fig. 1). E-QCM measurements were performed on quartz crystals. The backside of the quartz crystals was precoated with Au on a Ti adhesion layer (Biolin Scientific). The crystals were first cleaned by sequential sonication in acetone, isopropanol and Milli-Q water for 10 minutes each, and dried in N2 (Grade N6.0, Strandmöllen) as a final step. Thin films of Pd and Pd/CeO2 were evaporated on the crystals: (i) 20 nm Pd thin film on a 20 nm Ti adhesion layer for pure Pd samples, (ii) 2 nm Pd on 20 nm CeO2 on a 20 nm Ti adhesion layer for the Pd/CeO2 samples (Fig. 1b and c). Pd/CeO2 was deposited on a Au thin layer to improve conductivity. The pure Pd thin films were conductive enough and did not require the use of a Au layer. The Pd and Pd/CeO2 crystals have a geometrical surface area of 0.227 cm2. In addition to these crystals, two control samples were prepared: (i) 20 nm CeO2 on a Ti adhesion layer and (ii) Pd 2 nm on a Ti adhesion layer.
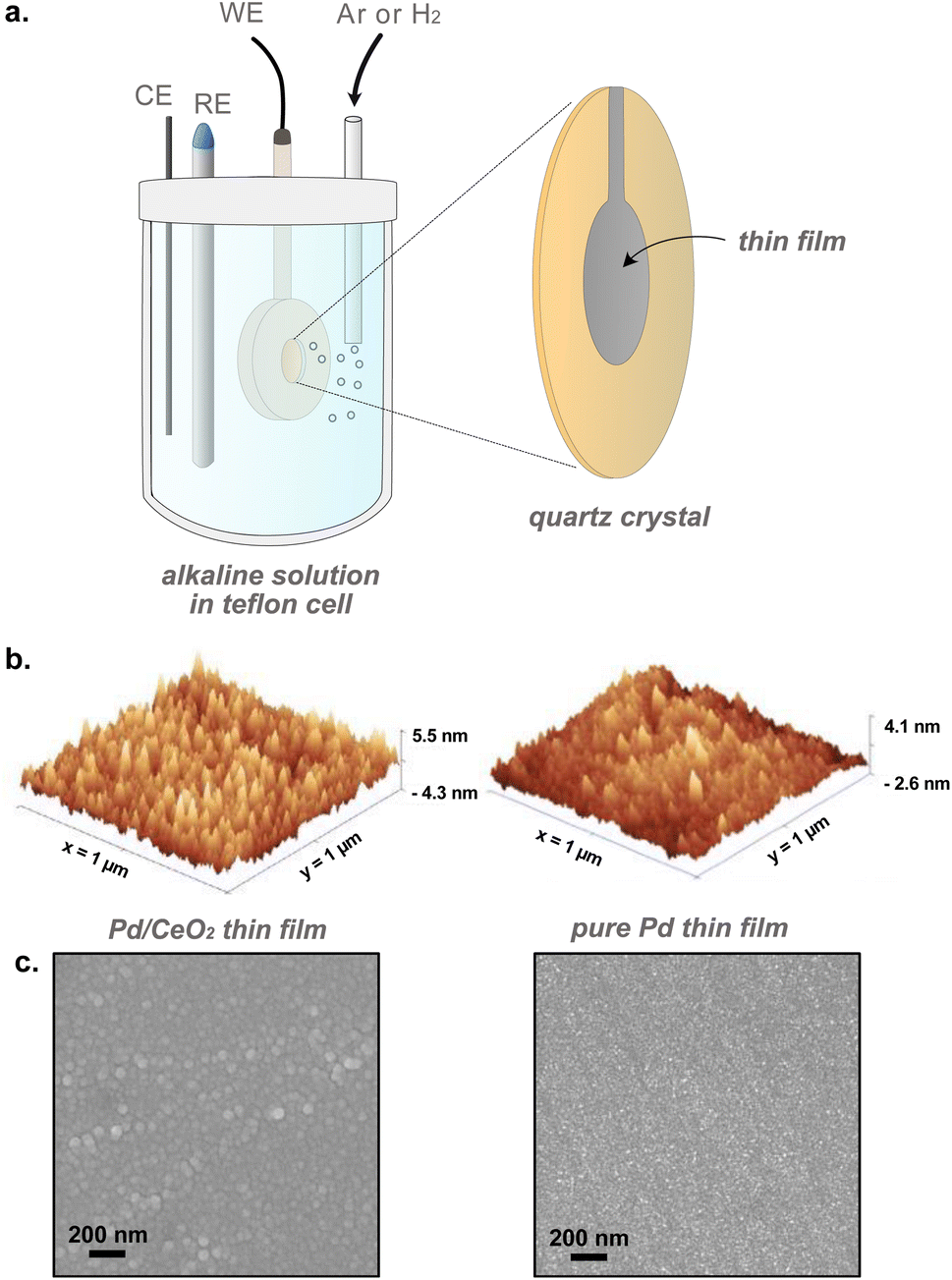 | ||
| Fig. 1 (a) Schematic showing the setup used for in situ electrochemical characterization of thin films using E-QCM. RE, CE and WE refer to reference, counter and working electrodes, respectively. (b) Atomic force microscopy and (c) scanning electron microscopy images of Pd/CeO2 and pure Pd thin films prepared by evaporation. | ||
The E-QCM measurements were performed using a dip holder from MicroVacuum connected to a QSense Explorer frequency response analyzer (Biolin Scientific) with an in-house built adapter. The QCM data was recorded with the QSoft401 software (Biolin Scientific). The dip holder was made of PEEK, sealed using FFPM O-rings and assembled using nylon screws. The CV and frequency response were measured simultaneously on the same crystal.
The quartz crystal resonant frequency change Δfn at different harmonic or overtone can be related to the mass change Δm using the Sauerbrey equation, assuming a spatially uniform film32 (eqn (5)):
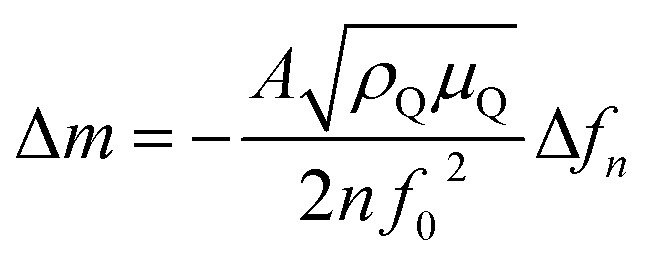 | (5) |
In our system, a 1 Hz change in frequency corresponds to around ∼2 ng cmgeo−2 change in mass. Previous work done on 200 nm Pd film has however shown that the mass of hydrogen absorbed inside pure Pd bulk cannot be estimated with high accuracy due to additional frequency shifts attributed to the stresses generated when H-atoms diffuses into Pd.35 Therefore, we plot Δf instead of Δm for clarity purposes, and make quantitative estimations related to mass changes only when away from the hydrogen absorption region (ERHE > 0.3 V).
Electrochemically active surface area (ECSA) was assessed to provide a comparison between the very well defined pure Pd and Pd/CeO2 films. Two different methods were used here to estimate their ECSAs: one derived from the charge required to oxidize a monolayer of adsorbed CO (CO-stripping method) and one derived from the charge resulting from the reduction of Pd oxide and/or hydroxide. Estimations using the charge related to hydrogen underpotential deposition is not appropriate in the case of Pd films as the H adsorption process is also coupled with an absorption of H into the Pd lattice, rendering estimations inaccurate.36
CO-stripping was performed by dosing CO in the electrolyte at 0.1 VRHE for 10 min and re-saturating with Ar for 30 min. The potential was then increased to 1.2 VRHE with a scan rate of 20 mV s−1 and cycled again to subtract the oxidation charge in the first positive-going scan associated with CO adsorbed on Pd, by the second scan corresponding to a CO-free surface. The ECSA was estimated by using a conversion charge of 330 μC cmPd−2.36 Estimations using the Pd oxide reduction method were done by integrating the charge in the last CV performed during conditioning, right before the HOR reaction, at a scan rate of 50 mV s−1 and with an inversion potential Einversion = 1.25 VRHE. The corresponding ECSA was estimated using a conversion charge of 375 μC cmPd−2.36
The structure of the thin films and sensors were probed using scanning electron microscopy (SEM – ZEISS Ultra-55 Scanning Electron Microscope). High-resolution transmission electron microscopy (HRTEM – FEI Titan operated at 300 kV) images were acquired on TEM grids (lacey carbon golden grids, Ted Pella, Inc.) onto which 20 nm of CeO2 and 2 nm of Pd were evaporated. Atomic force microscopy (AFM) images were acquired using a Bruker dimension 3100 scanning probe microscope (SPM). Chemical composition of the surface species was assessed with X-ray photoelectron spectroscopy (XPS) with a PHI 5000 VersaProbe III spectrometer equipped with a monochromatic AlKα source. Narrow scan measurements were acquired with dual charge compensation and aligned with the adventitious carbon peak (C 1s) at 284.8 eV before analysis. Wide-angle X-ray scattering (WAXS) patterns in grazing incidence were acquired using the SAXSLAB Mat:Nordic instrument equipped with a micro-focus Cu X-ray source and a Dectris Pilatus 300 K R and 100 R detectors. The entire beam path was evacuated before each measurement to 0.5 mbar to minimize air scattering. The sample to detector distance was calibrated before each series of measurements using LaB6. Exposure times were set to 6 hours per sample. Incidence angle of 0.4° was chosen to maximize the signal from the top layer. WAXS data was collected on the 100 R detector mounted on a goniometer inside of the evacuated chamber. Data reduction was performed using the SAXSgui software. Phase matching was performed using Bruker EVA software with the PDF + 2023 database.
Density functional theory (DFT) calculations were performed to gain insight into the oxidation state and structure of Pd on prepared Pd/CeO2 thin films. The calculations were done using the Vienna Ab initio Simulation Package37–40 using an approach described elsewhere.41 The core–valence interaction was described with the plane augmented wave (PAW) method,42 the Perdew, Burke and Ernzerhof (PBE) exchange–correlation functional43 was used together with a Hubbard-U correction.44 The Kohn–Sham orbitals are expanded with plane waves using a 450 eV cut-off. The atomic structures were optimized with the conjugate gradient method. A 3 × 2√3 surface cell was used for CeO2(111) and the Brillouin zone sampling was restricted to the gamma point. The slabs are in all cases separated by at least 15 Å of vacuum. The core level shifts were calculated accounting for complete screening of the core hole.41 The use of periodic boundary conditions requires a neutral supercell and we have compensated the presence of core holes by a compensating jellium background.45
Results and discussion
Electrochemical characterization on glassy-carbon electrodes
Electrochemical characterization of Pd and Pd/CeO2 films on glassy-carbon electrodes in the RDE set-up shows that the two systems have fundamentally different interactions with hydrogen. The CV profile performed in Ar-saturated 0.1 M KOH solutions on pure Pd is consistent with previously reported Pd thin films in alkaline solutions (Fig. 2a).23 Pd and Pd/CeO2 show very distinct potentials for hydrogen desorption. Hydrogen desorption/oxidation occurs from 0.0 to 0.4 VRHE (anodic peak) on Pd while it occurs from 0.0 to ca. 0.15 VRHE on Pd/CeO2. Hydrogen absorption seems more pronounced on Pd compared to Pd/CeO2. On Pd, there is a sharp cathodic peak starting from 0.2 until 0.0 VRHE which can be associated with hydrogen absorption into the Pd lattice to form Pd hydride. On Pd/CeO2, this process only occurs from ca. 0.05 VRHE.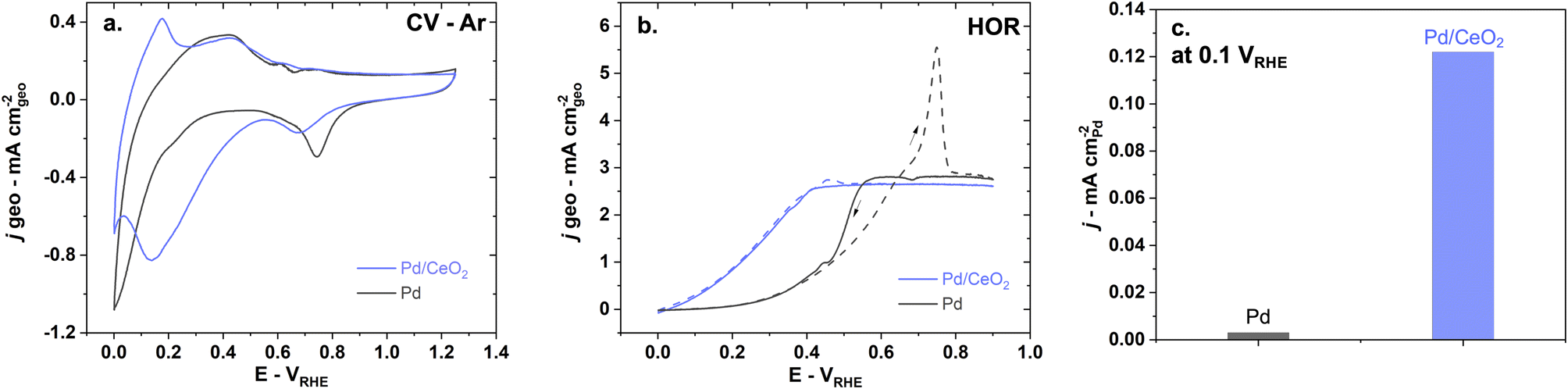 | ||
| Fig. 2 The presence of CeO2 has a significant impact on hydrogen and oxygen interactions with Pd and leads to better HOR performance as shown in (a) CV at a 50 mV s−1 scan rate in Ar-saturated 0.1 M KOH, (b) HOR performed in H2-saturated 0.1 M KOH at 5 mV s−1 and 1600 rpm and (c) comparison of specific activity at 0.1 VRHE on pure Pd (black) and Pd/CeO2 (blue). | ||
Pd oxide species appear to be different on pure Pd compared to Pd/CeO2. This is deduced from the difference in Pd surface oxides/hydroxides reduction potentials (Fig. 2a) with cathodic peaks at 0.75 VRHE and 0.67 VRHE on Pd and Pd/CeO2 respectively. On pure Pd, the reduction of the Pd oxide can proceed via: Pd–O + H2O + 2e− → Pd + 2OH−. However, this reaction will be affected by the distribution and local coordination of Pd. Extended Pd surfaces on pure Pd thin films are thus expected to show different reduction peaks compared with Pd at the interface with ceria (including embedded Pd). This suggests that ceria not only impacts hydrogen interactions but also oxygen species.
Hydrogen oxidation is significantly enhanced on Pd/CeO2 compared to pure Pd (Fig. 2b and c). In H2-saturated KOH solution, the diffusion-limited current plateau is reached at 0.6 VRHE on pure Pd which is consistent with previous literature.7,46 On Pd/CeO2 that plateau is reached at a much lower potential of 0.4 VRHE. The slightly lower diffusion-limited current density on Pd/CeO2 could be due to limited leaching/pealing off of small amounts of Pd/CeO2 active sites during rotation at 1600 rpm. Moreover, from 0 to 0.15 VRHE, no measurable current is observed on pure Pd which could indicate that the Tafel step (step that does not involve charge transfer) is dominating in this potential range. The enhancement is demonstrated by a ca. 40-fold increase in specific activity at 0.1 VRHE on Pd/CeO2 compared to Pd (Fig. 2c) which is consistent with other published work (Table S2†).46 It should also be noted that there is an important anodic peak at 0.75 VRHE on pure Pd with a current significantly above the diffusion-limited current (Fig. 2b). This suggests that another reaction is occurring, notably the oxidation of hydrogen atoms absorbed in the Pd bulk.36 Such high current peak is however not present on Pd/CeO2 confirming that the presence of CeO2 and the lower film thickness (2 nm vs. 20 nm for Pd/CeO2 and pure Pd thin films respectively) lead to significant changes in the interaction with hydrogen (Fig. 2b).
Pd and Pd/CeO2 present similar Tafel slopes (Fig. 3a). The Tafel slopes are 139 ± 5 and 135 ± 5 mV dec−1 respectively, close to 120 mV dec−1. A value close to 120 mV dec−1 can be attributed to various reaction mechanisms (notably Tafel–Volmer) but it cannot be solely used to identify the rate determining step.47 These values are however consistent with previously reported values for Pd/CeO2-based catalysts in KOH solution.3
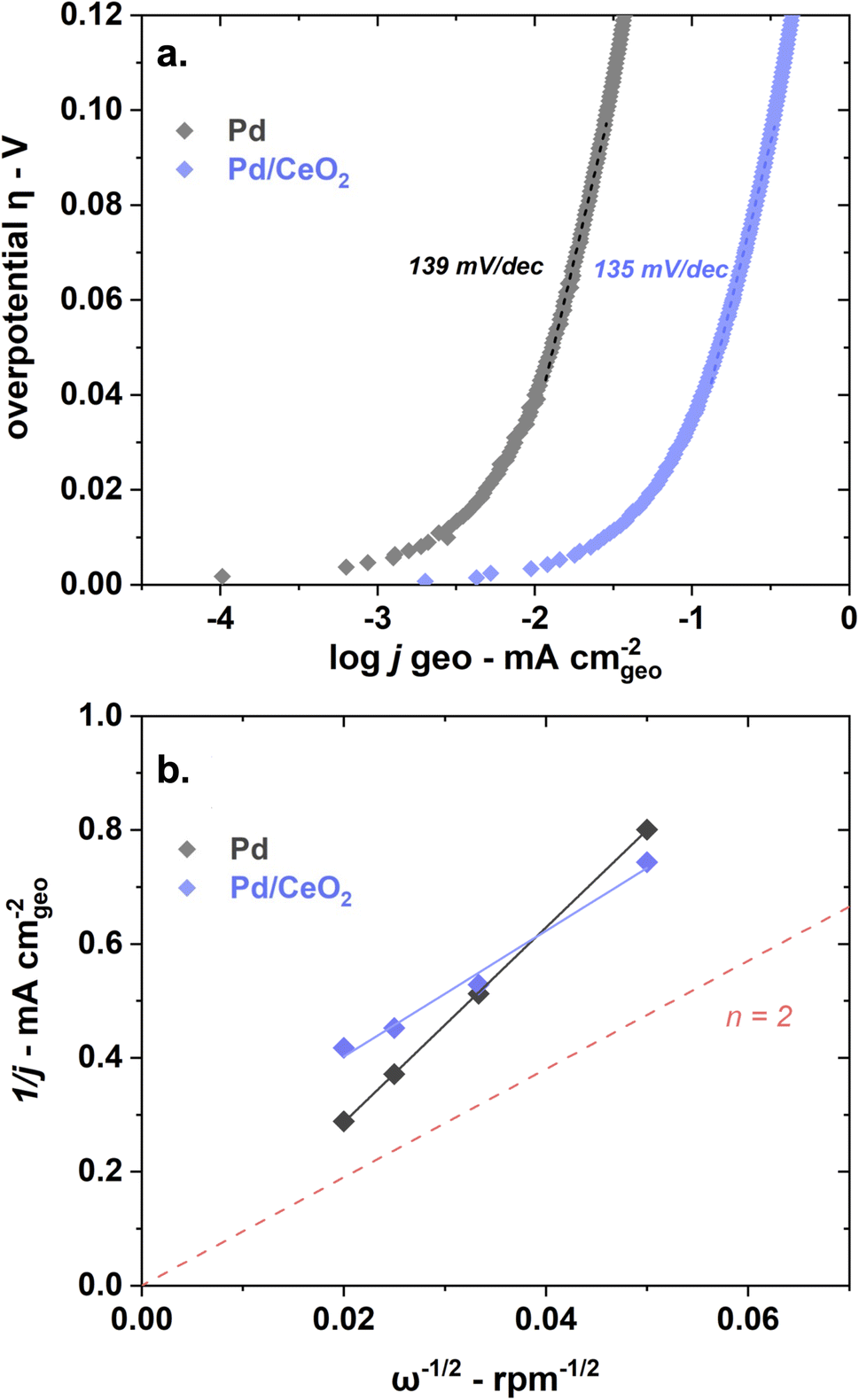 | ||
| Fig. 3 Pd and Pd/CeO2 show similar Tafel slopes but different estimated number of transferred electrons as shown in (a) a Tafel plot obtained at 5 mV s−1 and 1600 rpm and (b) Koutecky–Levich plot obtained at 0.8 VRHE with measurements at 400, 900, 1600 and 2500 rpm at 50 mV s−1. Both reactions were performed in H2-saturated 0.1 M KOH. | ||
Both samples appear to present first order kinetics with respect to hydrogen as shown by the Koutecky–Levich plots (Fig. 3b) but present important dissimilarities. Pd/CeO2 displays a linear behavior but does not intercept at the origin. This means that at this potential (0.8 VRHE), the process is not purely diffusion-controlled. The number of transferred electron is estimated to be 2 which is expected for this reaction. Pure Pd, in turn, shows a close to zero intercept which means that the current is almost exclusively diffusion-limited at this potential. However, the number of transferred electrons is closer to 1. This deviation could be related to the interference of hydrogen absorbed into the Pd bulk, rendering the accurate estimation of transferred electrons difficult.36
New insights from E-QCM during conditioning
E-QCM measurements were performed to gain new insight into the role of CeO2 at the interface with Pd. The CV obtained with a scan rate of 50 mV s−1 during conditioning in Ar saturated solution while measuring the frequency changes are consistent with the one obtained on the glassy-carbon electrodes (Fig. S1a†). The scan rate was decreased to 5 mV s−1 in the following experiments to better capture mass changes. The ECSA obtained from the integration of charge of Pd oxide reduction before reaction is very similar for Pd and Pd/CeO2 thin films (Table S1†). The frequency changes were thus not corrected for ECSA and were plotted as measured for clarity purposes.Importantly, the dissipation is the same at the beginning and at the end of the CVs for both Pd and Pd/CeO2 during conditioning and HOR (Fig. S2†). During conditioning in Ar and during HOR, small variations in the dissipation was noticed during the cycles, but the start and end points were similar. This further establishes that (i) the observed changes are solely related to mass changes and (ii) the surface roughness is not significantly affected during cycles.
No measurable metal dissolution occurs on pure Pd thin films in alkaline media in the potential window studied here. The frequency changes were measured as a function of the potential in an Ar-saturated 0.1 M KOH solution (Fig. 4a). These frequency changes appear reversible as the frequency at the beginning of the cycle at ∼0 VRHE is the same after the cycling is completed. This is indicative of a stable thin film with no Pd dissolution. The reversibility of the changes also demonstrate that the entire oxygen–hydrogen potential region is encompassed in this measurement. This contrasts with previous QCM studies showing significant Pd dissolution in acidic solution.48
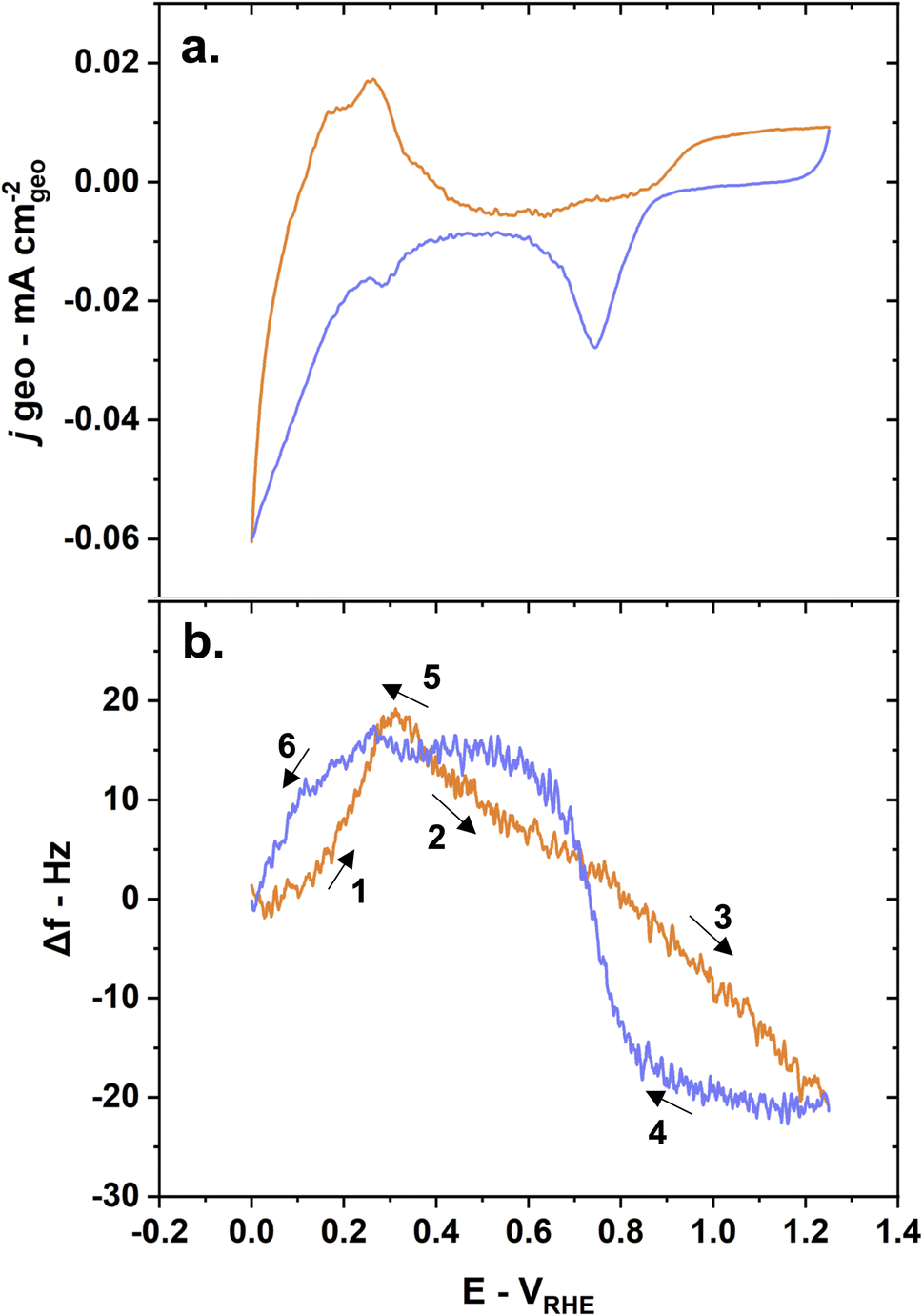 | ||
| Fig. 4 Mass changes due to hydrogen adsorption, absorption, and desorption behaviors induce frequency changes on pure Pd crystals during conditioning as shown by (a) CV and (b) E-QCM measurements with anodic scans in orange and cathodic scans in blue. Reaction conditions: 5 mV s−1 scan rate, Ar-saturated 0.1 M KOH solution. | ||
The changes in frequency during the CV can be described in several steps (Fig. 4b):
(1) Hydrogen desorption/oxidation processes lead to an increase in frequency – corresponding to a mass loss – during the anodic scan until ∼0.3 VRHE.
(2) Water physisorption/adsorption processes starting at ∼0.3 VRHE until ∼0.75 VRHE lead to a decrease in frequency – corresponding to a mass gain.49 This is also commonly referred to as the double layer region.
(3) OH adsorption and subsequent chemisorbed oxide layer formation leads to a continuing decrease in frequency – corresponding to a mass again – until the inversion potential (1.25 VRHE).
(4) Pd oxide layer reduction leads to a drastic increase in frequency – corresponding to a mass loss – at ∼0.9 VRHE during the cathodic scan.
(5) Hydrogen adsorption leads to a small increase in frequency – corresponding to a mass loss – at around 0.3 VRHE. This counter intuitive mass loss is due to the displacement of adsorbed water and hydroxyl groups by lighter hydrogen atoms.
(6) Hydrogen absorption leads a drastic decrease in frequency – corresponding to a mass gain and the formation of Pd hydride – starting from 0.3 VRHE until 0 VRHE.
Interestingly, the frequency changes on Pd/CeO2 as a function of the potential show drastic differences compared to pure Pd. With CeO2 as a support for Pd, the changes in frequency are slightly irreversible (Fig. 5b). The difference in frequency at the beginning and end of the cycle is however only less than 5 Hz – corresponding to <10 ng cm−2 (using eqn (5)). Possible dissolution of Pd could be an explanation for this small change. A complementary study comparing E-QCM measurements with varying inversion potentials would give further insight into the effect of Pd distribution and oxidation states on stability, but is beyond the scope of this work.28 It should be noted here that the control sample made of 2 nm of Pd on a 20 nm Ti film (without CeO2) were not stable and degraded during CV, making a comparison impossible.
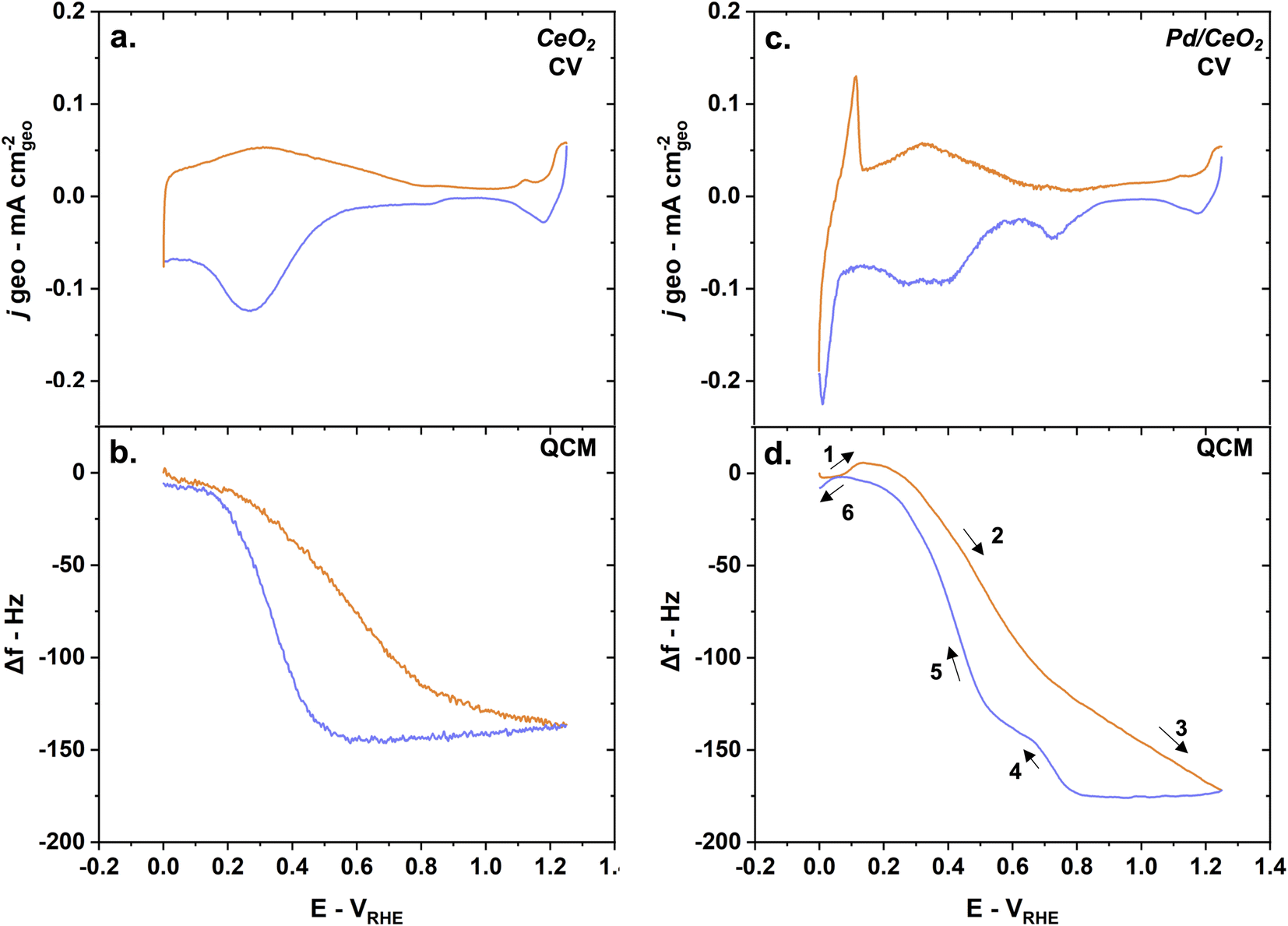 | ||
| Fig. 5 The mass changes measured on Pd/CeO2 are dominated by adsorption and desorption phenomena occurring on CeO2 during conditioning as shown by (a) CV on CeO2 and simultaneous (b) QCM measurements and (c) CV on Pd/CeO2 and simultaneous (d) QCM measurements. The anodic scans are represented in orange while the cathodic scans are in blue. Reaction conditions: 5 mV s−1 scan rate, Ar-saturated 0.1 M KOH solution. | ||
On CeO2 alone, the frequency changes can be largely attributed to adsorption and desorption of hydroxyl groups as well as oxidation-reduction of ceria (Fig. 5a and b). The frequency decreases drastically from 0.2 VRHE until 0.8 VRHE. This could correspond to a significant mass increase related to possible oxidation to Ce4+ and OH uptake. The small changes in current densities observed at higher potentials are contributions from the Au layer used to promote conductivity. When the potential is decreased, the frequency is constant until ∼0.5 VRHE. From 0.5 VRHE until 0.2 VRHE, the frequency increases back to its starting point. This would correspond to a mass loss, related to the possible reduction to Ce3+ and desorption of hydroxyl groups from ceria.
The mass changes measured on Pd/CeO2 are dominated by the oxidation/reduction of ceria and adsorption/desorption of hydroxyl groups (Fig. 5c and d). The various steps occurring on pure Pd can also be observed on Pd/CeO2 but there are both minor and major differences:
(1) Hydrogen desorption/oxidation processes lead to an increase in frequency – corresponding to a mass loss – during the anodic scan until ∼0.1 VRHE. That represents a ∼0.2 VRHE negative shift compared to pure Pd. This is consistent with the shift in the desorption/oxidation current peak observed on the CV obtained on both the glassy carbon electrode (Fig. 2a) and quartz crystal (Fig. 5c). The increase in frequency is only 6–7 Hz compared to 16–17 Hz on pure Pd, meaning that there is less absorbed hydrogen on Pd/CeO2 compared to Pd, which is to be expected due to the difference in thicknesses in Pd (2 nm vs. 20 nm).
(2) Oxidation of ceria to Ce4+ and adsorption of OH groups lead to a decrease in frequency starting at ∼0.1 VRHE corresponding to a significant mass gain (Fig. 5d).
(3) Chemisorbed oxide layer formation on Pd leads to a continuing decrease in frequency – corresponding to a mass again – until the inversion potential (1.25 VRHE), as was observed on pure Pd. This significant mass gain is not observed on pure CeO2 (Fig. 5d) which confirms that this additional mass gain is related to the Pd oxide species.
(4) Pd oxide layer reduction leads to a drastic increase in frequency – corresponding to a mass loss – at ∼0.8 VRHE during the cathodic scan. This is consistent with the negative shift in Pd oxide reduction potentials observed on the glassy-carbon electrodes (Fig. 2a) and quartz crystals (Fig. 5c).
(5) Desorption and reduction processes lead to a significant increase in frequency – corresponding to a mass loss – starting at around 0.6 VRHE.
(6) Hydrogen absorption leads a small decrease in frequency – corresponding to a small mass gain – starting from 0.05 VRHE until 0 VRHE. The difference is only ∼5–6 Hz which is consistent with step 1 and corresponds to Pd hydride formation.
An important observation is that during conditioning, the increase in frequency associated with Pd oxides reduction in pure Pd is 35 Hz which corresponds to a mass loss of ∼69 ng cmgeo−2. This is very similar to the frequency changes associated with the reduction of Pd oxides on Pd/CeO2 of 32 Hz which corresponds to a mass loss of ∼63 ng cmgeo−2. This is consistent with the fact that the two thin films have similar ECSAPd (Table S1†).
One of the other major observations from these sets of measurements during conditioning in Ar is that in an inert gas environment, OH adsorption processes and oxidation/reduction on CeO2 are not affected by the presence of Pd. The increase in frequency starting at 0.6 VRHE on negative scans is approximately the same on Pd/CeO2 and CeO2. The frequency increases of about 145 Hz in both cases. Considering a surface roughness of ca. 1.5 (derived from the ECSA in Table S1†), a Δf of 145 Hz would correspond to a Δm of 285 ng cmgeo−2. With a calculated Ce surface density of 7.7 × 1014 of Ce atoms per cm2, this would give a O/Ce ratio of 9, supporting the hypothesis that a bulk process could also be occurring (i.e. reduction). This increase is thus mostly related to OH desorption from the CeO2 support and reduction of ceria to Ce3+.
New insights from E-QCM during HOR
E-QCM measurements give critical insights into the major differences between Pd and Pd/CeO2 during HOR. The CV obtained with a scan rate of 5 mV s−1 during HOR in H2-saturated solution while measuring the frequency changes are consistent with the one obtained on glassy-carbon electrodes (Fig. 2a, b, 6a and c). Pure Pd thin films show poor HOR activity while Pd/CeO2 shows significant enhancement in activity (Fig. 6a and c).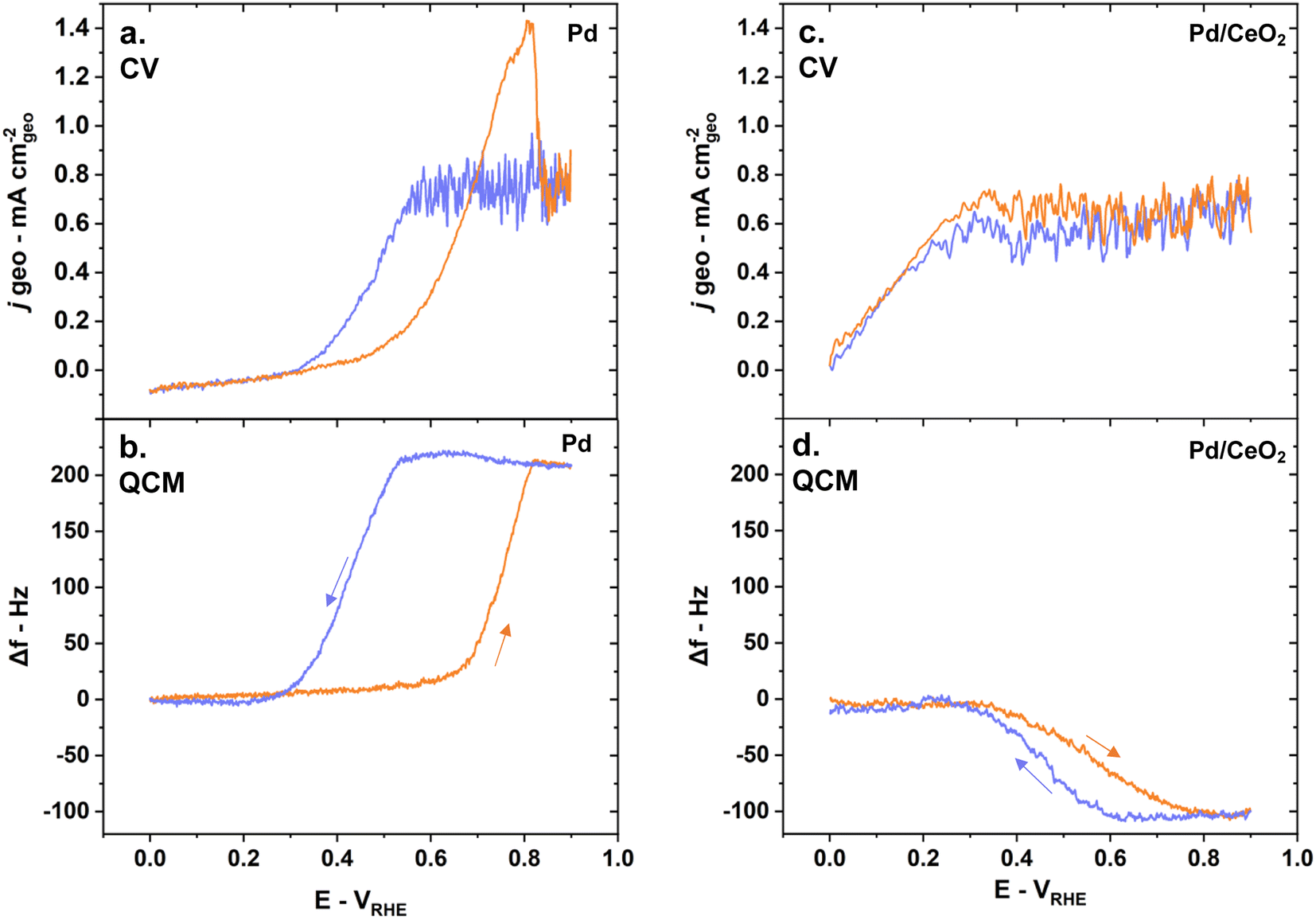 | ||
| Fig. 6 E-QCM reveals fundamental differences during HOR on Pd and Pd/CeO2 as shown by (a) the measured current density during CV and (b) simultaneous E-QCM measurements on pure Pd; (c) measured current density during CV and (d) simultaneous E-QCM measurements on Pd/CeO2. The anodic scans are represented in orange and the cathodic scans in blue. Reaction conditions: 5 mV s−1 scan rate, H2-saturated 0.1 M KOH solution. | ||
On pure Pd thin films, mass changes are dominated by the presence of absorbed hydrogen while on Pd/CeO2 they are dominated by the oxidation/reduction of ceria and adsorption of hydroxyl groups. Indeed, the frequency changes as a function of the potential vary greatly between the two samples during HOR (Fig. 6b and d). On Pd thin films, the frequency increases as the potential increases in the anodic scan (Fig. 6b). This corresponds to a mass loss due to the desorption of absorbed hydrogen in the Pd bulk and its subsequent oxidation to form water. It should be noted here that pure Pd thin films deposited on a Au layer gave similar E-QCM response (Fig. S4†). On Pd/CeO2 the frequency decreases as the potential increases in the anodic scan (Fig. 6d). This corresponds to a mass gain due to the desorption/oxidation of adsorbed hydrogen on the Pd surface, hydroxyl adsorption and oxidation of ceria to Ce4+. During the cathodic scans, the processes are reversed to reach their initial state. A decrease in frequency is observed on pure Pd (Fig. 6b), corresponding to a mass gain as hydrogen atoms are re-incorporated and absorbed into Pd bulk. In contrast, an increase in frequency is observed on Pd/CeO2 (Fig. 6d), corresponding to a mass loss due to the displacement of adsorbed hydroxyl groups, reduction of ceria to Ce3+ and adsorption of hydrogen atoms on the Pd surface.
E-QCM measurement on CeO2 during HOR suggests that the adsorbed OH on CeO2 in Pd/CeO2 could indeed participate in the HOR (Fig. 7). When comparing the frequency changes on CeO2 and Pd/CeO2, there is a difference of ∼55 Hz corresponding to ∼108 ng cmgeo−2 or ∼73 ng cmPd−2 during HOR (Fig. 7). We propose that this unaccounted mass could correspond to the reaction of OH intermediates on CeO2 in close contact with adsorbed H on Pd which supports the hypothesis that Pd at the interface with CeO2 is the active site. We argue that if CeO2 played no part in the reaction, the mass increase associated with OH adsorption processes and oxidation of ceria during HOR would be similar on both CeO2 and Pd/CeO2 just like during conditioning (Fig. 5b and d).
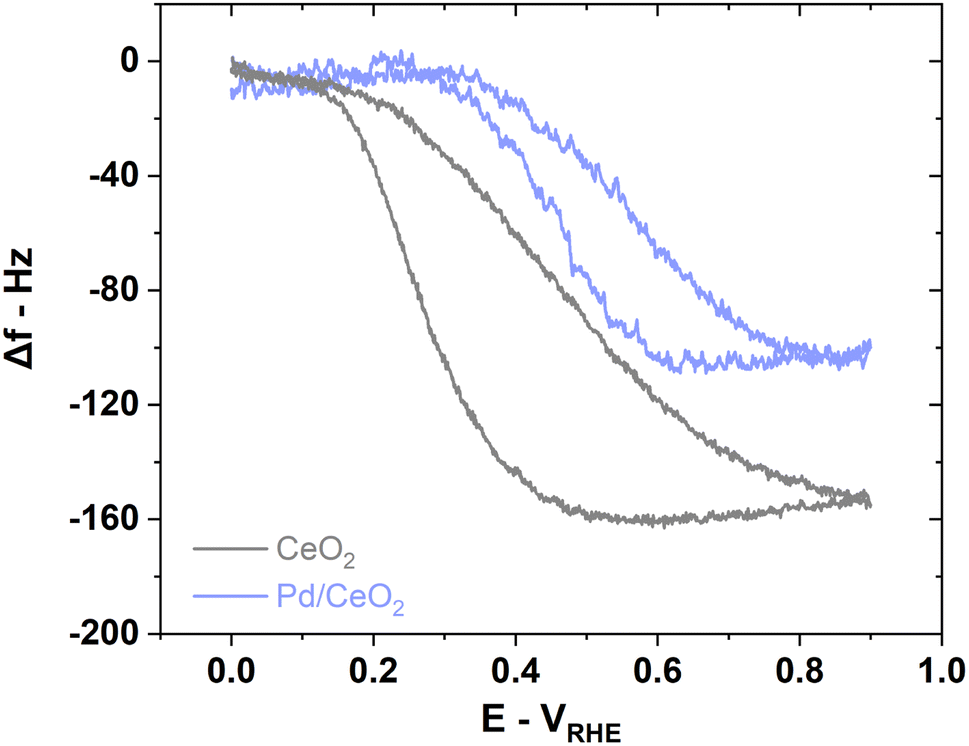 | ||
| Fig. 7 EQCM measurements on pure CeO2 reveals the role played by the support on Pd/CeO2 represented by the difference in frequency during HOR. Reaction conditions: 5 mV s−1 scan rate, H2-saturated 0.1 M KOH solution. | ||
Pd is well dispersed on CeO2 as shown by HR-TEM
Strong interactions between Pd and CeO2 on Pd/CeO2 are observed with TEM (Fig. 8). The Pd thickness (2 nm) was chosen to yield a good dispersion and close interaction of Pd while avoiding a full coverage of the CeO2 film. The TEM images confirm that such close interaction was achieved. The surface appears homogeneous and presents two types of regions – both containing Pd and CeO2 – but in different ratios: darker regions are associated with Ce-rich domains and lighter regions with Pd-rich domains. The presence of Pd and CeO2 phases is confirmed with the fast Fourier transform (FFT) analysis (Fig. 8c). The semi-oriented diffraction pattern rings are consistent with Pd (2.3 Å (111), 1.9 Å (200)) and CeO2 (3 Å (111), 1.9 Å (220), 1.6 Å (311)). The presence of the CeO2 phase on Pd/CeO2 before and after reaction is also confirmed by WAXS (Fig. S5†). | ||
| Fig. 8 Close interactions between Pd and CeO2 are observed on TEM images with lighter areas corresponding to Pd rich domains while darker areas can be attributed to Ce-rich domains as shown on (a) and (b) TEM images and (c) FFT analysis. | ||
Strong metal–support interactions are identified with ex situ XPS
On pure Pd thin films, surface Pd is in its metallic state before reaction even though Pd is prone to oxidation when exposed to air and ambient conditions (Fig. 9a). The binding energy of Pd 3d5/2 is 335 ± 0.1 eV as determined by ex situ XPS before reaction. This is consistent with metallic Pd binding energies reported in previous surface studies.50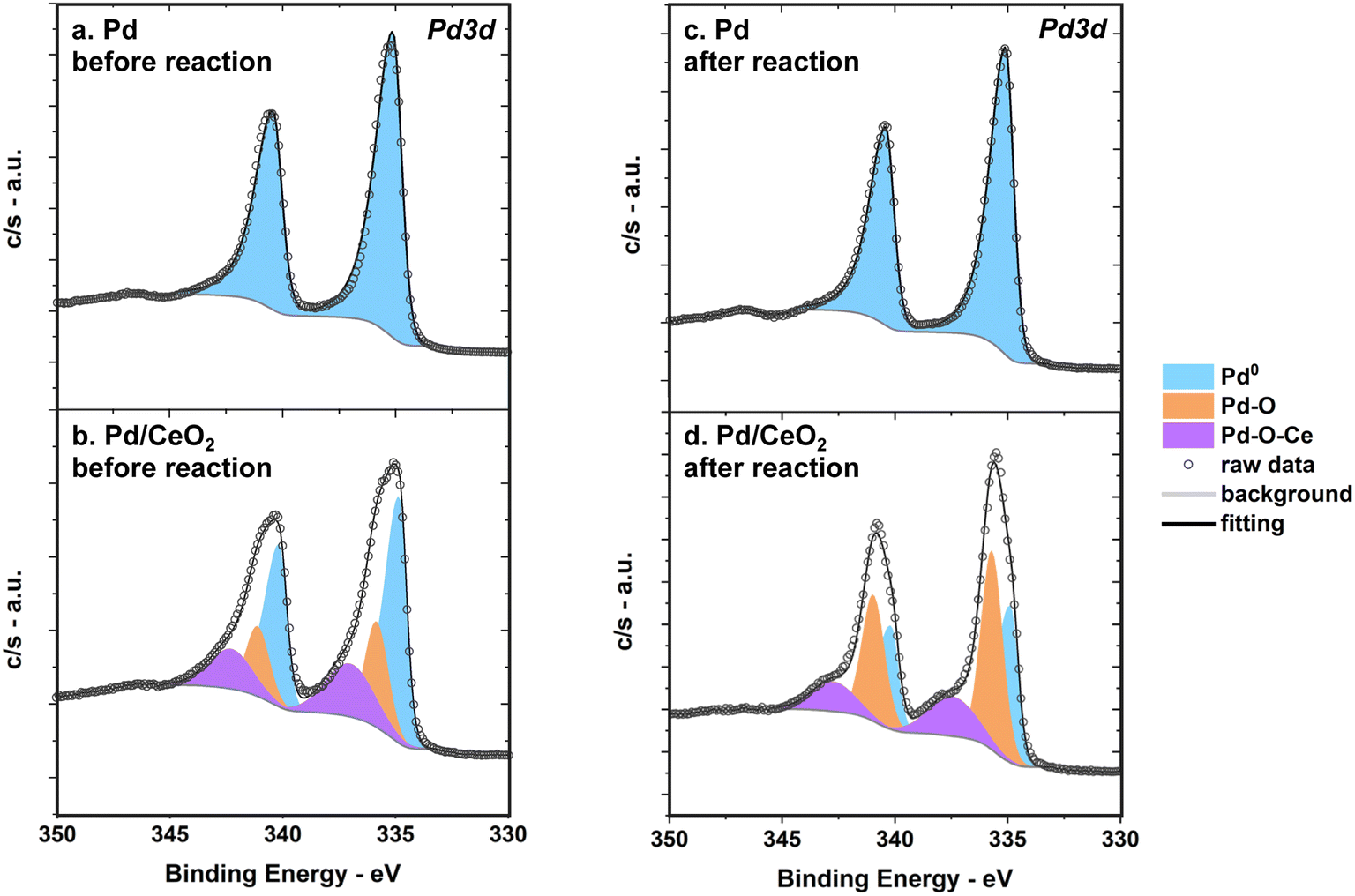 | ||
| Fig. 9 XPS spectra measured before and after reaction show that Pd remains metallic on pure Pd thin films while Pd is partly oxidized on Pd/CeO2 as shown by the Pd 3d region of pure Pd (a) before reaction and (c) after reaction, and the Pd 3d region of Pd/CeO2 (b) before reaction and (d) after reaction. | ||
The Pd 3d region of Pd/CeO2 measured by XPS indicates that there are multiple Pd species with different oxidation states before reaction (Fig. 9b). At least three different contributions could be identified after fitting: a first species at 334.8 ± 0.1 eV (referred to as Pd0), a second at 336 ± 0.1 eV (Pd–O), and a third at 337.5 ± 0.1 eV (Pd–O–Ce). This is consistent with previous studies of Pd single atoms dispersed and covalently bonded to CeO2.8,51 This confirms that an important fraction of Pd is in close interaction with Ce. XPS measurements alone can however not fully describe all possible Pd species and theoretical insight from calculations of Pd 3d core level shifts are needed (see section below).
After reaction, Pd remains metallic in pure Pd while Pd/CeO2 still presents at least three main species but with different ratios compared to the fresh sample (Fig. 9c and d). There is a decrease in the metallic Pd0 fraction after reaction on Pd/CeO2: from 55% on the fresh sample to 38% on the used sample (Fig. 9d, Table S3†).
As expected from E-QCM, CV and HOR conditions appear to influence the oxidation state of Ce in Pd/CeO2 thin films. The ceria support consists mostly of Ce4+ species before reaction as measured by XPS (Fig. S3†). The uncertainty in the background and noise of the Ce 3d region renders a quantitative analysis of the fresh sample difficult. After reaction, a clear contribution from Ce3+ can be measured. This suggests that reduction/oxidation of ceria indeed occur during cycling in HOR. The elemental composition of the Pd/CeO2 sample was also investigated with XPS. It should be noted that the Pd 3d5/2/Ce 3d area ratio decreases after reaction (Table S3†), which is consistent with the slight irreversibility of the frequency changes observed during E-QCM (Fig. 4). This would further support the hypothesis that slight dissolution of Pd occurs on Pd/CeO2 during experiment.
Calculated Pd 3d core level shifts
A set of DFT calculations was performed to facilitate the interpretation of the measured Pd 3d binding energies. The considered structures on CeO2(111) are (a) a Pd atom embedded in the top layer of the surface, (b) a Pd ad-atom on the surface, (c) a small Pd7O8 cluster adsorbed on the surface and (d) a small Pd4 cluster with one atom embedded in the surface (Fig. 10). The structures are chosen to represent different possibilities that have been discussed in the literature for single atoms and small clusters on CeO2(111).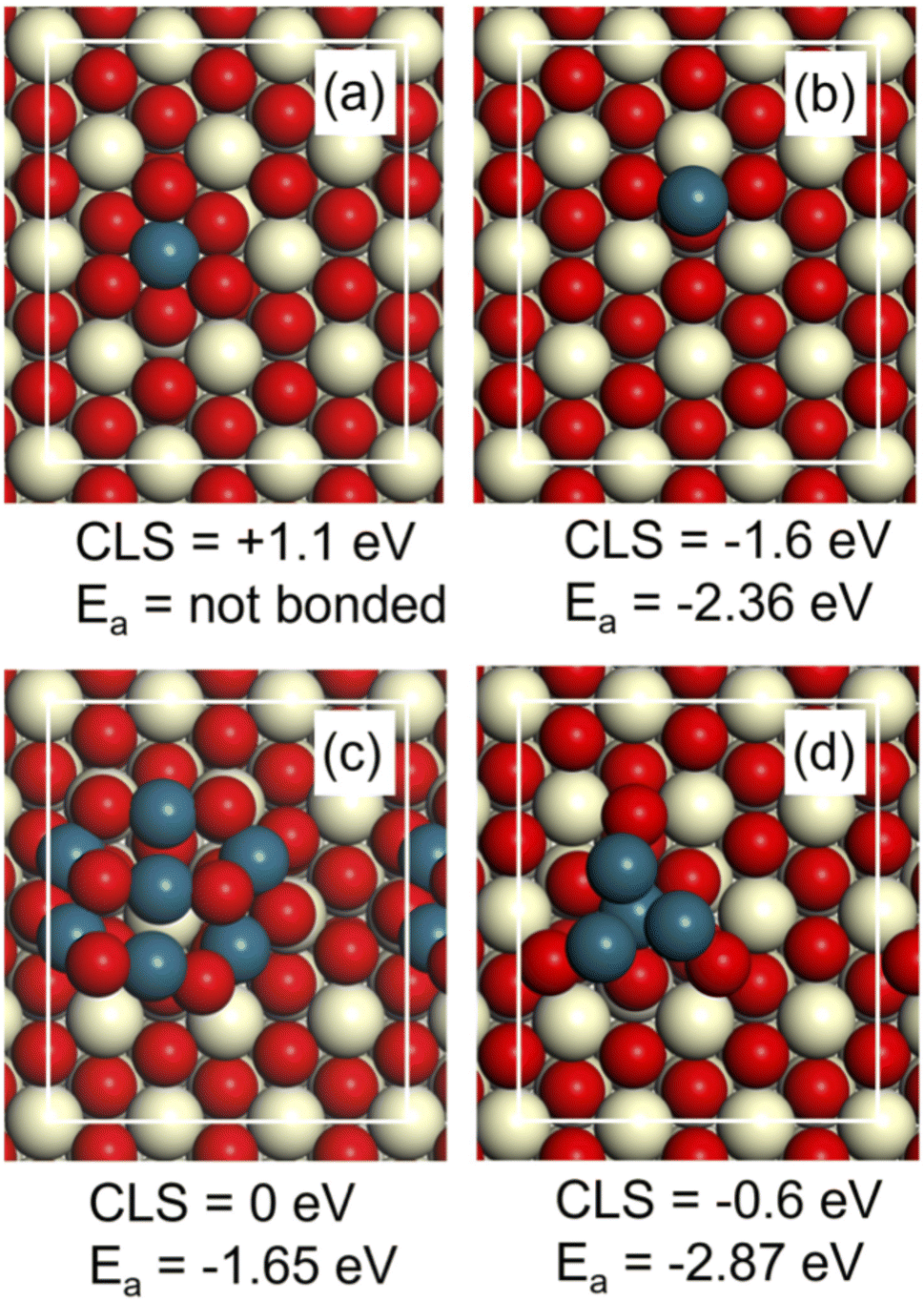 | ||
| Fig. 10 The considered Pd-structures on CeO2(111) have significant differences in Pd 3d core level shifts (CLS) and CO adsorption energy (Ea). (a) Pd atom embedded in the top layer, (b) Pd atom adsorbed on the surface, (c) Pd7O8 cluster, (d) Pd4 cluster with one atom embedded in the surface. The Pd 3d CLS are calculated with respect to PdO. Atomic color code: oxygen (red), palladium (blue) and cerium (beige). | ||
The Pd 3d binding energy is sensitive to the chemical state of the metal. The shift in binding energy is calculated here with respect to the binding energy in PdO by considering a unit cell with three layers of CeO2(111) including the explored Pd structure and three layers of PdO(101). A four-fold Pd atom in the top layer of PdO(101) is used as the reference atom. The computed shift is in this approach converged within about 0.1 eV as compared to bulk PdO.52 The size of the surface cell is determined by the computed lattice constant for CeO2, which is 5.49 Å. Using the lattice constant of CeO2, implies that the PdO slab is strained by 8% tensile strain in one direction and 7% compressive strain in the other direction. Test for structure (a) using instead the lattice constant of PdO for the surface cell altered the core level shift by less than 0.1 eV.
The Pd atom embedded in the top layer of the surface (a) has a shift of +1.1 eV with respect to PdO (Fig. 10). A Pd ad-atom (b) has instead a shift of −1.6 eV with respect to PdO. The oxide cluster (c) has on average a shift similar to PdO and the small Pd4 cluster has a negative shift of −0.6 eV (Fig. 10). In this case, it is the three atoms not embedded in the surface that have a negative shift, whereas the embedded atom has zero shift with respect to PdO. The calculated shifts indicate that the measured value at 337.5 eV in Fig. 9 could be attributed to Pd embedded in CeO2 (a). It is only embedded Pd that has a substantial positive shift with respect to PdO. The large shift is consistent with a higher charging of Pd as corroborated by a Bader charge analysis. The initial state charge of the embedded Pd is calculated to be 8.63e, whereas the average charge of Pd in PdO is calculated to be 9.11e. The clear negative shift of a Pd ad-atom on CeO2(111) (b) is also consistent with the Bader analysis, which yields the initial state charge of 9.76e.
CO-stripping experiments
CO-stripping voltammetry was carried out three consecutive times on both Pd and Pd/CeO2 quartz crystals. The first runs were performed immediately after HOR and describe the catalyst post-reaction. Interestingly, the first runs vary greatly compared to the second and third runs on both samples. For the first runs, Pd shows a 40 mV shift to lower potential compared to Pd/CeO2, indicating more facile CO oxidation (Fig. S7†). This is counter intuitive as ceria is expected to facilitate oxidation of CO.53 The other runs however present similar onsets and potential peaks for both Pd and Pd/CeO2, indicating that potential cycling and/or exposure to CO have changed the surface. In the literature, the same stripping onset and peak potential have been observed for nanostructured Pd–CeO2/C and Pd/C in 2 M KOH54 and 0.1 M KOH.3Mass loss associated with the oxidation of CO and desorption of CO2 are consistent with the ECSA calculations on pure Pd thin films. As expected, the mass loss (or increase in frequency) measured with E-QCM occurs at lower potential on the first run for Pd compared to the other runs which is consistent with more facile oxidation (Fig. S7†). Moreover, there is a difference in mass loss of 29.3 and 26.7 ng cm−2 between the first and the second run which is consistent with different calculated ECSA by cmPd2 (Table S1†). This mass change can be related to a number of CO molecules adsorbed per cmPd2. Considering a surface density of ∼1.3 × 1015 Pd atoms per cm2 (averaged value of Pd(100) and Pd(111)), this gives a Pd to CO ratio of ∼2.2. This ratio would translate into a preferred bridged CO adsorption mode, on average. This is consistent with a recent FTIR study showing that after purging CO from the cell with Ar, the main adsorbed species are water through OH bonds and bridged CO.55 No atop CO was observed after purging in Ar. It should be noted that this FTIR study was performed at 0.4 VRHE and in acidic media which could also affect the binding conformation of CO.
In the case of Pd/CeO2, the mass loss associated with the oxidation of CO and desorption of CO2 is lower than expected. The mass loss in the first and second runs is only ∼13 and ∼14.5 ng cmPd−2 respectively. Using the same atom density as for extended Pd surfaces gives a Pd to CO ratio of ∼4.3. This ratio is higher than expected. This is partly due to an overestimation of the surface of density of Pd atoms as the surface density of Pd atoms in Pd/CeO2 is expected to be lower than on close-packed surfaces. Most importantly, this discrepancy could be due to different adsorption energies of CO on Pd/CeO2.
The CO adsorption energy was calculated on the different model structures (Fig. 10). The adsorption energy (Ea) on the adatom (b) and the cluster (d) is high, which is consistent with the well-known high reactivity of undercoordinated metal sites such as small clusters. The adsorption energy on the Pd7O8 cluster is consistent with the adsorption energy on PdO(101).52 Importantly, we find that CO does not adsorb on the embedded Pd atom (Fig. 10a). This is consistent with the low adsorption energy of CO on Pd four-fold coordinated to oxygen, such as in the PdO(100) surface.56 Thus, embedded Pd has a clear signature in XPS but should not be visible in CO-stripping. It should be noted that the embedded site is, however, active for H2 dissociation. We find that H2 dissociates without barrier on embedded Pd in a heterolytic fashion forming PdH and an OH group. The subsequent formation of two OH group is strongly preferred thermodynamically by 3 eV. The results highlight the fact that sites that may be relevant for the considered reaction may not be visible in CO-stripping. Additional research involving calculations of the entire potential energy surface of the reactions is, however, required to make conclusions regarding the activity of the considered sites.
Conclusion
The investigation of well-defined model thin films revealed fundamental differences between Pd and Pd/CeO2 during hydrogen oxidation reaction in alkaline media. Pd/CeO2 led to an expected enhancement in HOR performance compared to pure Pd thin films. By combining electrochemical measurements on glassy carbon electrodes, in situ electrochemical characterization with quartz crystal microbalance, microscopy and spectroscopy techniques and theoretical calculations, we were able to give a new insight into the enhanced reactivity of Pd/CeO2 in HOR in alkaline media. Strong interaction between Pd and Ce was confirmed by spectroscopy and theoretical calculations where an important fraction of Pd was found to be embedded in the ceria support. E-QCM measurements enabled an estimation of the large source of hydroxyl intermediates provided by CeO2, supporting the hypothesis that these intermediates could be directly participating in the reaction. This work indicates that the interface between Pd and ceria must be carefully designed to provide synergies and to lead to enhanced reactivity. Finally, this study underlines the importance of model thin films as appropriate tools to study the interface between metal and supports in hydrogen oxidation reaction in alkaline media.Author contributions
ML and BW designed the study, ML wrote the paper, HG performed the DFT calculations and participated in discussions, LS and GMS prepared the materials, ML ran the electrochemical tests, the E-QCM analyses, performed the XPS study and collected the SEM and AFM images, VS performed the HR-TEM analysis, RM provided help with the preliminary results.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Swedish Research Council (Project No. 2018-03927) and the Swedish Foundation for Strategic Research (Project No. EM16-0060 and ARC19-0026). The Competence Centre for Catalysis is hosted by Chalmers University of Technology and financially supported by the Swedish Energy Agency (Project No. 52689-1) and the member companies Johnson Matthey, Perstorp, Powercell, Preem, Scania CV, Umicore, and Volvo Group. We acknowledge the staff of Myfab Chalmers and the Chalmers Materials Analysis Laboratory (CMAL) where sample preparation and physical characterization were carried out. The calculations were performed at PDC (Stockholm) via a SNIC grant.References
- L. Osmieri, L. Pezzolato and S. Specchia, Curr. Opin. Electrochem., 2018, 9, 240–256 CrossRef CAS.
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. Yan, P. Zelenay and Y. S. Kim, J. Power Sources, 2018, 375, 170–184 CrossRef CAS.
- R. K. Singh, E. S. Davydova, J. Douglin, A. O. Godoy, H. Tan, M. Bellini, B. J. Allen, J. Jankovic, H. A. Miller, A. C. Alba-Rubio and D. R. Dekel, Adv. Funct. Mater., 2020, 30, 2002087 CrossRef CAS.
- M. Bellini, M. V. Pagliaro, A. Lenarda, P. Fornasiero, M. Marelli, C. Evangelisti, M. Innocenti, Q. Jia, S. Mukerjee, J. Jankovic, L. Wang, J. R. Varcoe, C. B. Krishnamurthy, I. Grinberg, E. Davydova, D. R. Dekel, H. A. Miller and F. Vizza, ACS Appl. Energy Mater., 2019, 2, 4999–5008 CrossRef CAS.
- T.-J. Wang, F.-M. Li, H. Huang, S.-W. Yin, P. Chen, P.-J. Jin and Y. Chen, Adv. Funct. Mater., 2020, 30, 2000534 CrossRef CAS.
- T.-J. Wang, W.-S. Fang, Y.-M. Liu, F.-M. Li, P. Chen and Y. Chen, J. Energy Chem., 2022, 70, 407–413 CrossRef CAS.
- H. A. Miller, A. Lavacchi, F. Vizza, M. Marelli, F. D. Benedetto, F. D'Acapito, Y. Paska, M. Page and D. R. Dekel, Angew. Chem., Int. Ed., 2016, 55, 6004–6007 CrossRef CAS.
- V. Muravev, G. Spezzati, Y.-Q. Su, A. Parastaev, F.-K. Chiang, A. Longo, C. Escudero, N. Kosinov and E. J. M. Hensen, Nat. Catal., 2021, 4, 469–478 CrossRef CAS.
- T. W. v. Deelen, C. H. Mejía and K. P. d. Jong, Nat. Catal., 2019, 2, 955–970 CrossRef.
- J. Yang, W. Li, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2003300 CrossRef CAS PubMed.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- Q. Wang, K. L. Yeung and M. A. Bañares, Catal. Today, 2020, 356, 141–154 CrossRef CAS.
- R. J. Gorte, AIChE J., 2010, 56, 1126–1135 CAS.
- M. Li, X. Wang, K. Liu, H. Sun, D. Sun, K. Huang, Y. Tang, W. Xing, H. Li and G. Fu, 2023, 2302462.
- X. Wang, Y. Tang, J.-M. Lee and G. Fu, Chem Catal., 2022, 2, 967–1008 CrossRef CAS.
- X. Wang, Y. Zhu, H. Li, J.-M. Lee, Y. Tang and G. Fu, Small Methods, 2022, 6, 2200413 CrossRef CAS.
- N. Acerbi, S. C. E. Tsang, G. Jones, S. Golunski and P. Collier, Angew. Chem., Int. Ed., 2013, 125, 7891–7895 CrossRef.
- C. A. Campos-Roldán and N. Alonso-Vante, Electrochem. Energy Rev., 2019, 2, 312–331 CrossRef.
- W. Sheng, M. Myint, J. G. Chen and Y. Yan, Energy Environ. Sci., 2013, 6, 1509 RSC.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. v. d. Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 CrossRef CAS PubMed.
- J. Li, S. Ghoshal, M. K. Bates, T. E. Miller, V. Davies, E. Stavitski, K. Attenkofer, S. Mukerjee, Z.-F. Ma and Q. Jia, Angew. Chem., Int. Ed., 2017, 56, 15594 CrossRef CAS PubMed.
- N. M. Markovic and P. N. R. Jr, Surf. Sci. Rep., 2002, 45, 117–229 CrossRef CAS.
- G. Montserrat-Siso and B. Wickman, Electrochim. Acta, 2022, 420, 140425 CrossRef CAS.
- K. Sohlberg, S. T. Pantelides and S. J. Pennycook, J. Am. Chem. Soc., 2001, 132, 6609–6611 CrossRef.
- W. Xu, Z. Liu, A. C. Johnston-Peck, S. D. Senanayake, G. Zhou, D. Stacchiola, E. A. Stach and J. A. Rodriguez, ACS Catal., 2013, 3, 975–984 CrossRef CAS.
- S. Sahoo, D. R. Dekel, R. Maric and S. P. Alpay, ACS Catal., 2021, 11, 2561–2571 CrossRef CAS.
- P. C. K. Vesborg, B. Seger and I. Chorkendorff, J. Phys. Chem. Lett., 2015, 6, 951–957 CrossRef CAS PubMed.
- L. Strandberg, V. Shokhen, M. Luneau, G. Lindbergh, C. Lagergren and B. Wickman, ChemElectroChem, 2022, 9, e202200591 CAS.
- F. D. Speck, F. S. M. Ali, M. T. Y. Paul, R. K. Singh, T. Böhm, A. Hofer, O. Kasian, S. Thiele, J. Bachmann, D. R. Dekel, T. Kallio and S. Cherevko, Chem. Mater., 2020, 32, 7716–7724 CrossRef CAS.
- W. Haynes, in Handbook of Chemistry and Physics, CRC Press, 95 edn, 2014 Search PubMed.
- S. A. Tschupp, S. E. Temmel, N. P. Salguero, J. Herranz and T. J. Schmidt, J. Electrochem. Soc., 2017, 164, E3448–E3456 CrossRef CAS.
- J. Kankare, Langmuir, 2002, 18, 7092–7094 CrossRef CAS.
- I. Reviakine, A. N. Morozov and F. F. Rossetti, J. Appl. Phys., 2004, 95, 7712 CrossRef CAS.
- B. Wickman, H. Grönbeck, P. Hanarp and B. Kasemo, J. Electrochem. Soc., 2010, 157, B592 CrossRef CAS.
- M. Grden, J. Kotowski and A. Czerwinski, J. Solid State Electrochem., 2000, 273–278 CAS.
- S. Henning, J. Herranz and H. A. Gasteiger, J. Electrochem. Soc., 2015, 162, F178–F189 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 49, 14251 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- A. Posada-Borbon, N. Bosio and H. Grönbeck, Surf. Sci., 2021, 705, 121761 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- M. V. d. Bossche, N. M. Martin, J. Gustafson, C. Hakanoglu, J. Weaver, E. Lundgren and H. Grönbeck, J. Chem. Phys., 2014, 141, 034706 CrossRef PubMed.
- V. Yarmiayev, M. Alesker, A. Muzikansky, M. Zysler and D. Zitoun, J. Electrochem. Soc., 2019, 166, F3234 CrossRef CAS.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- M. Grden, J. Kotowski and A. Czerwinski, J. Solid State Electrochem., 1999, 3, 348–351 CrossRef CAS.
- K. Shimazu and H. Kita, J. Electroanal. Chem., 1992, 341, 361–367 CrossRef CAS.
- J. Andersen, D. Hennig, E. Lundgren, M. Methfessel, R. Nyholm and M. Scheffler, Phys. Rev. B, 1994, 50, 17525 CrossRef CAS PubMed.
- K. R. Priolkar, P. Bera, P. R. Sarode, M. S. Hegde, S. Emura, R. Kumashiro and N. P. Lalla, Chem. Mater., 2002, 14, 2120–2128 CrossRef CAS.
- R. Westerström, M. E. Messing, S. Blomberg, A. Hellman, H. Grönbeck, J. Gustafson, N. M. Martin, O. Balmes, R. v. Rijn, J. N. Andersen, K. Deppert, H. Bluhm, Z. Liu, M. E. Grass, M. Hävecker and E. Lundgren, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 115440 CrossRef.
- G. Spezzati, Y. Su, J. P. Hofmann, A. D. Benavidez, A. T. DeLaRiva, J. McCabe, A. K. Datye and E. J. M. Hensen, ACS Catal., 2017, 7, 6887–6891 CrossRef CAS.
- H. A. Miller, M. V. Pagliaro, M. Bellini, F. Bartoli, L. Wang, I. Salam, J. R. Varcoe and F. Vizza, ACS Appl. Energy Mater., 2020, 3, 10209–10214 CrossRef CAS.
- A. Winiwarter, M. J. Boyd, S. B. Scott, D. C. Higgins, B. Seger, I. Chorkendorff and T. F. Jaramillo, ChemElectroChem, 2021, 8, 250–256 CrossRef CAS.
- K. Zorn, S. Giorgio, E. Halwax, C. R. Henry, H. Grönbeck and G. Rupprechter, J. Phys. Chem. C, 2011, 115, 1103–1111 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01879b |
| This journal is © The Royal Society of Chemistry 2023 |