
Enhancing the cycle-life of initial-anode-free lithium-metal batteries by pre-lithiation in Mn-based Li-rich spinel cathodes†
Leiyu
Chen‡
a,
Chao-Lung
Chiang‡
 b,
Guifan
Zeng
a,
Yonglin
Tang
a,
Xiaohong
Wu
a,
Shiyuan
Zhou
a,
Baodan
Zhang
a,
Haitang
Zhang
a,
Yawen
Yan
a,
Tingting
Liu
d,
Hong-Gang
Liao
a,
Chuanwei
Wang
*e,
Xiaoxiao
Kuai
*ac,
Yan-Gu
Lin
*b,
Yu
Qiao
*ac and
Shi-Gang
Sun
a
b,
Guifan
Zeng
a,
Yonglin
Tang
a,
Xiaohong
Wu
a,
Shiyuan
Zhou
a,
Baodan
Zhang
a,
Haitang
Zhang
a,
Yawen
Yan
a,
Tingting
Liu
d,
Hong-Gang
Liao
a,
Chuanwei
Wang
*e,
Xiaoxiao
Kuai
*ac,
Yan-Gu
Lin
*b,
Yu
Qiao
*ac and
Shi-Gang
Sun
a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, PR China. E-mail: kuaixiaoxiao@xmu.edu.cn; yuqiao@xmu.edu.cn
bNational Synchrotron Radiation Research Center, Hsinchu, 30076 Taiwan, Republic of China. E-mail: lin.yg@nsrrc.org.tw
cFujian Science & Technology Innovation Laboratory for Energy Materials of China (Tan Kah Kee Innovation Laboratory), Xiamen, 361005, PR China
dSchool of Environmental Science and Engineering, Suzhou University of Science and Technology, Suzhou 215009, China
eCell Research Department, Amperex Technology Limited, Ningde, 352100, China. E-mail: Wangcw2@atlbattery.com
First published on 3rd May 2023
Abstract
Anode-free lithium metal batteries (AF-LMBs) are attracting growing attention due to the burgeoning pursuit of high energy density. However, the poor reversibility of lithium-ion plating/stripping on the Cu anode triggers multiple issues, including rapid capacity loss, active lithium loss, and short lifetime. Herein, a cathode pre-lithiation strategy combined with a fluorine-containing electrolyte is introduced to achieve long-lifetime AF-LMBs. Furthermore, contact pre-lithiation, a more advanced and convenient strategy, has been introduced to precisely control the lithiation degree by adjusting the contact time. It has been demonstrated that over-lithiated Li1.3Mn2O4 can deliver additional lithium ions during the charging process, thus exhibiting 94.4% capacity retention after 40 cycles.
 Yu Qiao | Yu Qiao is currently a professor in the College of Chemistry and Chemical Engineering at Xiamen University. He earned his B.S. degree from the University of Science and Technology of China (USTC) in 2013. He received his Ph.D. degree from Tsukuba University (Japan) in 2019, and he worked as a postdoctoral fellow at the National Institute of Advanced Industrial Science and Technology (AIST, Japan). His research interests are focused on the development of electrochemical energy storage devices, surface/interfacial electrochemistry, and operando spectroscopic characterization. |
Introduction
Rechargeable Li-ion batteries (LIBs) have developed into the most commonly used electrochemical energy storage devices.1–4 Nowadays, with the urgent requirement of achieving maximum energy density, lithium metal batteries (LMBs) have been spotlighted as next-generation energy storage devices.5–8 Compared with traditional LMBs, anode-free lithium metal batteries (AF-LMBs) have a higher theoretical energy density.9–12 However, owing to the lack of lithium compensation, the poor reversibility of lithium-ion plating/stripping, and non-uniform lithium deposition on the Cu anode, AF-LMBs face the tremendous challenge of rapid capacity fading, which hinders their practical implementation.9,13–15 In the last few years, numerous strategies (such as optimizing electrolytes, modifying current collectors, constructing artificial solid electrolyte interphase (SEI) layers, and pre-lithiation strategies) have been proposed to extend the lifetime of AF-LMBs.16–18 Among these strategies, pre-lithiation strategies can effectively compensate for the irreversible lithium loss during the first few cycles, improve the initial coulombic efficiency (ICE), and thus obtain better cycle performance. However, anode pre-lithiation strategies cannot apply to AF-LMBs because Li-ions cannot intercalate into the Cu anode, different from other anodes (i.e., carbon-, Si- and graphite-based anodes).19,20Currently, efforts have also been made to pre-install additional lithium sources into the cathode which can be classified as adding Li-containing additives and forming an over-lithiated cathode.21,22 Li-containing additives in the cathode can effectually mitigate the lithium loss, however, sacrificial salts (Li2C4O4, Li2C2O4, and Li3N, etc.) would generate gases (i.e., CO, CO2, or N2) at the specific voltage after the first charge, which would affect the electrode stability, the energy density of the system, and the whole cycling environment, hindering the practical application.23,24 The over-lithiated cathode can release additional Li-ions during the first charge, which are stored in unoccupied crystallographic sites, to offset the active lithium consumed to form the SEI and the lithium loss in the subsequent cycles.25–28 Notably, the over-lithiated cathode strategy does not introduce inactive ingredients, making it more convenient for practical applications than additives.
LiMn2O4 (LMO) is a non-toxic and environmentally friendly cathode material with low cost and outstanding rate performance. Meanwhile, over-lithiated Li1+xMn2O4 (L1+xMO, 0 ≤ x ≤ 1) obtained by over-lithiation of LiMn2O4 was proposed as a lithium reservoir compound to provide an additional lithium source to overcome the capacity loss, and considered as a promising cathode material for AF-LMBs.29 Over-lithiated cathodes on AF-LMBs can improve the ICE and prolong the lifetime, but the lifetime has still been threatened by the undesirable average coulombic efficiency (ACE). To achieve desirable ACE, the strategy of optimizing electrolytes has been applied. Electrolyte engineering will modify the interface chemistry of AF-LMBs by introducing fluorine-containing additives to obtain high capacity retention (CR). Therefore, combining over-lithiated cathodes with optimized electrolytes can extend the lifespan of AF-LMBs to the best.
In this work, the Cu||L1.3MO AF-LMB was constructed using the Cu anode and over-lithiated Li1.3Mn2O4 (L1.3MO) cathode obtained from conventional LMO (Fig. S1 and Table S1†), combined with a fluorine-containing electrolyte to prolong the AF-LMB lifetime. Additional lithium can be provided by the I41/amd type Li2Mn2O4 (L2MO) during the first charge, and thus offset the lithium consumption. The crystal structure of the cathode material after pre-lithiation has been studied by in situ X-ray diffraction (XRD). In addition, the transition between LMO and L2MO is reversible, which has been demonstrated by ex situ X-ray absorption structure (XAS) analysis. Considering that electrochemical pre-lithiation cannot be used in practical applications, we propose a contact pre-lithiation method, which can precisely control the lithiation degree by adjusting the contact time. At the same time, benefiting from the over-lithiated cathode and optimized electrolyte, a Cu||L1.3MO anode-free cell with a high ICE of 99% can still achieve a capacity retention of 94.4% after 40 cycles at 0.5C discharge.
Results and discussion
The electrochemical performance of the Cu||LMO anode-free coin-cell shows a rapid capacity loss during different cycles without excess lithium (Fig. 1a). As for the typical anode-free cell, disappointing initial coulombic efficiency (ICE) is attributed to the lithiophilic organic–inorganic solid electrolyte interphase (SEI) and Li dendrite formation on the Cu anode, and undesirable average coulombic efficiency (ACE) which is triggered by non-uniform Li plating/stripping on the lithiophobic Cu anode (Fig. S2†).5,25,30 However, the ICE and ACE of the Li||LMO half-cell (Fig. S3†) upon cycling with different numbers reached superior values, about 96.3% and 99%, respectively, indicating that the poor cycling stability of the Cu||LMO AF-LMB results from the Cu anode. To date, various strategies for optimizing anode-free cells have been reported in several literature reports. Electrolyte optimization is an effective strategy to increase the lifetime because it can improve the ACE by promoting lithium morphology and lithium plating/stripping on the Cu anode.16,31 In addition, to improve the ICE of anode-free cells, pre-lithiation strategies have been applied to extend the lifespan, which can provide additional lithium-ions to compensate for the irreversible Li loss during the initial cycles. Herein, we use a strategy gained from the aforementioned considerations by combining optimized electrolytes with cathode pre-lithiation to reduce active lithium loss and supplement the irreversible capacity loss, extending the lifetime of anode-free cells.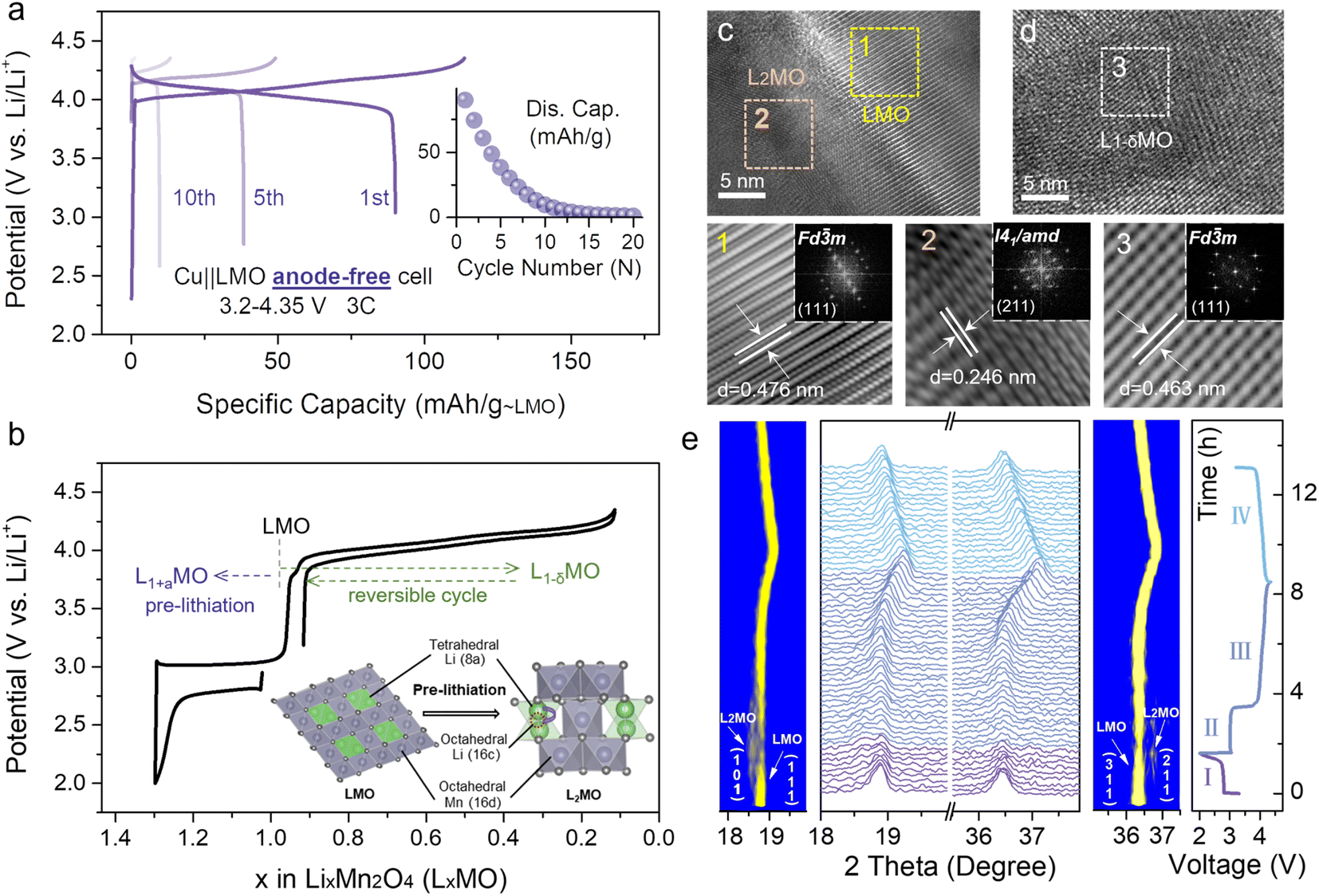 | ||
| Fig. 1 (a) Charge–discharge curves of the Cu||LMO anode-free cell in the voltage range from 3.2 to 4.35 V at 3C rate. (b) The charge/discharge profiles of the Li||LMO half-cell (LxMO (0 ≤ x ≤ 2)). Insets are the schematic illustration of the reversible phase transition between LMO and L2MO. High-resolution transmission electron microscopy (HRTEM) images of the LMO (c) discharged to 2 V and (d) recharged to 4.35 V, and the corresponding (inverse) fast Fourier transformation (FFT) patterns. (e) Two-dimensional contour map of in situ X-ray diffraction (XRD) diffraction patterns for the (101), (111), (311), and (211) reflections, and the corresponding galvanostatic charge–discharge curve of the Li||LMO half-cell. | ||
From the point-of-view of specific energy, environmental pollution (toxicity), and cost, a spinel LMO cathode is one of the most promising cathodes for lithium-ion batteries (LIBs) owing to the low toxicity of Mn sources, abundant reserves, high energy density, and excellent rate performance. LMO compounds have the unoccupied octahedral site (16c) and the formula can be denoted as [Li+]8a[Mn3+–Mn4+]16d[□]16c[O4]32e (□ represents the octahedral vacancy).32 The [Mn2]16d[□]16c[O4]32e skeleton is a three-dimensional network of tetrahedral and octahedral coplanar structures, which benefit fast lithium-ion diffusion. Additional lithium ions can intercalate into the empty 16c octahedral sites in the LMO structure, allowing LMO to transform into Li2Mn2O4 (L2MO) and enabling the application of pre-lithiation strategies.
Fig. 1b displays the charge–discharge curves of the Li||LMO half-cell, related to the pre-lithiation process, and the schematic illustration of the reversible phase transition between LMO and L2MO. Li-ions at the 8a site continuously insert into empty 16c octahedral sites during the first discharge process, and then the cubic spinel converts to a tetragonal spinel L2MO.1,32 Besides, high-resolution transmission electron microscopy (HRTEM) and in situ X-ray diffraction (XRD) were carried out to visually demonstrate the phase transition between LMO and L2MO during the whole charge–discharge cycling process (Fig. 1c–e). Fig. 1e shows the evolution of XRD patterns and the corresponding zoom in the contour plot of the main peak (111) and (311) throughout the entire four regions (the specific reaction equations are shown in Fig. S4†), clearly demonstrating the two-phase transition process.33 In region I, during the first discharge process, the characteristic peaks (111) and (311) of LMO gradually divide into two peaks, and new peaks (101) and (211) which are related to the L2MO form, respectively, indicating one Li-ion is intercalated into the empty octahedral site of cubic LMO to transform partial LMO into tetragonal L2MO, resulting in the coexistence of LMO and L2MO.34 Region II (recharge to 3.9 V) is the reverse process of region I, involving a phase transition from L2MO (tetragonal) to LMO (cubic), and all the characteristic reflections of L2MO gradually disappear. In region III (recharge to 4.35 V), on further de-intercalation of Li-ions from the transformed LMO (cubic), the (111) and (311) peaks of LMO are observed to shift to higher degrees, inducing a solid–solution reaction to form cubic-spinel-like Li-deficient Li1−δMn2O4 (0 ≤ δ ≤ 1).33 When re-discharged to 3.2 V (region IV), all the characteristic reflections of LMO shifted back to the initial position, indicating that L2MO reversibly transforms into LMO. In Fig. 1c, the lattice fringes of over-lithiated LMO reveal the (111) plane of LMO and (211) plane of L2MO with interlayer spacings of 0.476 nm (region 1, yellow) and 0.246 nm (region 2, orange), respectively, indicating the coexistence of LMO and L2MO. Furthermore, the Li-deficient LMO after charge to 4.35 V remains a cubic-spinel structure (Fig. 1d, region 3, white), which is similar to the initial cubic-spinel LMO (Fig. S5†), implying that partial pre-lithiation will not affect the LMO structure. HRTEM results are consistent with in situ XRD. Meanwhile, L2MO, formed by first discharge to 2 V, will release additional Li-ions to the anode during the subsequent cycles. By controlling the re-discharge cut-off voltage, partial Li-ions can be stored on the anode to prolong the lifetime of anode-free cells without sacrificing the energy density (Fig. 1b).
Intercalation of too many Li-ions into the LMO structure will affect the structural stability, and therefore, it is necessary to precisely control the lithiation degree of cathodes. When the lithiation degree exceeds 30%, the structure of the LMO will be damaged (Fig. S6†). Based on the above considerations, the molar percentage of L2MO in the L1+xMO cathode was kept below 30% in the subsequent tests. When the initial discharge cut-off voltage is set to 2 V, L1.3MO is obtained (Fig. S7, S8 and Table S2†). Fig. 2a (gray line) displays the whole electrochemical lithiation charge–discharge curves of the Li||L1.3MO half-cell (denoted as cell-B). To explore the effect of structural stability after over-lithiation, the electrochemical performance of the Li||L1.3MO half-cell was further compared with that of the typical Li||LMO half-cell (denoted as cell-A, Fig. S9†) shown in Fig. 2b (3.2–4.35 V, at 1C (1C = 148 mA g−1)). Notably, cell-B released an outstanding discharge capacity of 115.6 mA h g−1 with 99.46% ICE, ascribed to the electrochemical pre-lithiation in the initial discharge process. Furthermore, cell-B exhibits superior capacity retention and cycling stability after 500 cycles, consistent with or even better than that of cell-A, indicating that partial pre-lithiation will not affect the structural stability of LMO and the reversible phase transformation between LMO and L2MO (cyclic voltammetry curves confirmed this conclusion again, Fig. S10†). However, due to the typical Jahn–Teller distortion and manganese disproportionation reaction, the LMO cathodes exhibit unstable long-term stability in the voltage range from 2 V to 4.35 V (Fig. S11†).
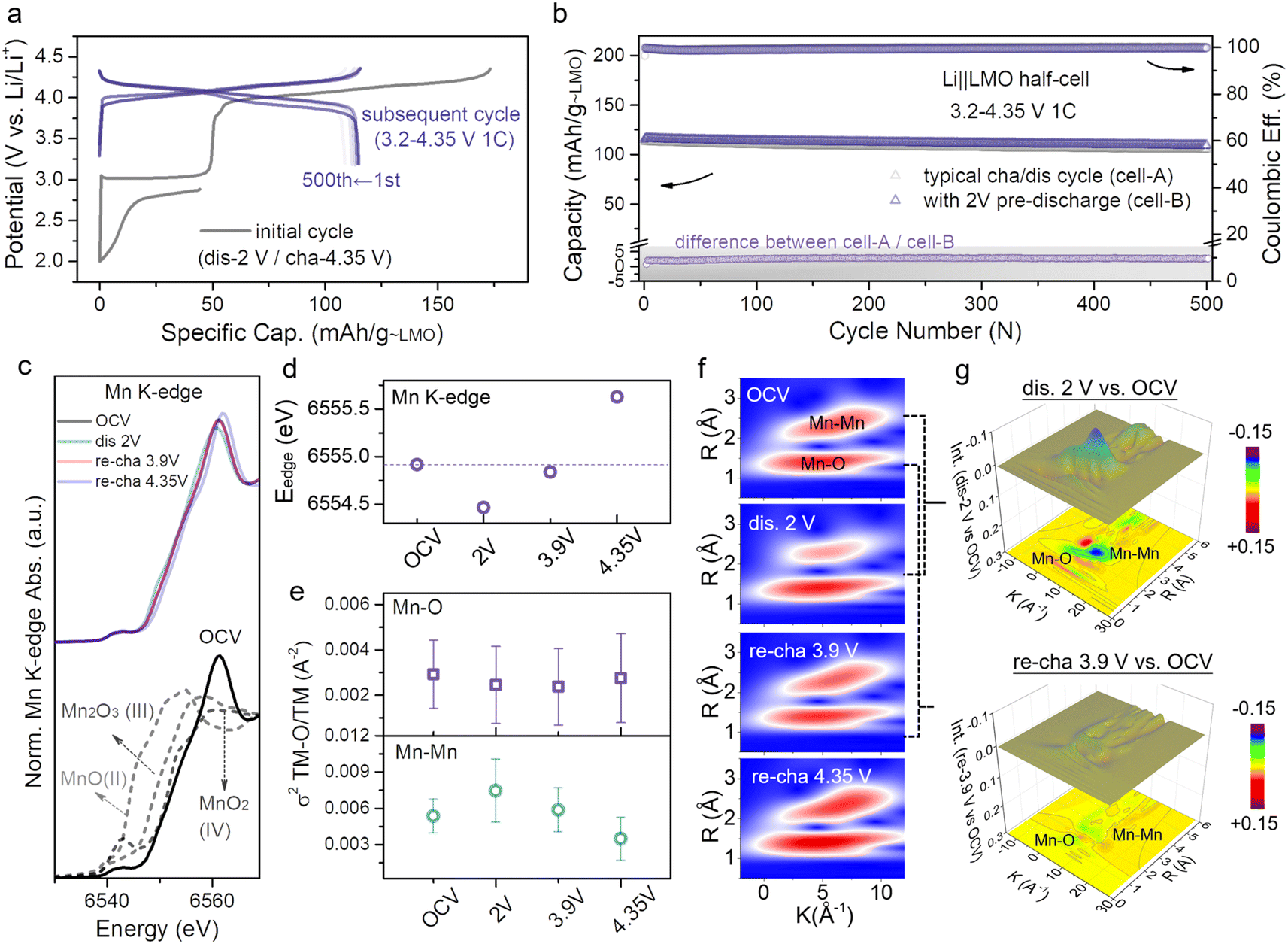 | ||
| Fig. 2 (a) Voltage profiles of the Li||L1.3MO half-cell that was initially discharged to 2 V and then cycled in the voltage range of 3.2–4.35 V (denoted as cell-B). (b) Cycling performances of cell-A (Fig. S9†) and cell-B, along with the difference of discharge capacity between cell-A and cell-B. (c) XANES spectra, and (d) edge positions at the Mn K-edge of the LMO electrodes at different voltage states. (e) Debye–Waller factor σ2, and (f) WT-EXAFS spectra of the Mn–O/Mn bond of the LMO at different states. (g) Color 3D WT-EXAFS spectra showing the differences between WT-EXAFS spectra of the Mn–O bond and Mn–Mn bond (first discharge to 2 V vs. OCV (top) and re-charge to 3.9 V vs. OCV (bottom)). | ||
The analysis of the cathode structure will deliver valuable information for further comprehending the prominent electrochemical performance.35 The X-ray absorption structure (XAS) technique, especially X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS), is suitable for obtaining information regarding the electronic and local structure of cathode materials.36 The ex situ XAS spectra at the Mn K-edge have been photon-energy-calibrated, absorbance-normalized, and then used to capture the internal structural evolution of LMO after pre-lithiation. Fig. 2c and d provide information about the valence state variation of Mn during the whole charge–discharge process at four specific potentials (open circuit voltage (OCV), dis 2 V, re-cha 3.9 V and re-cha 4.35 V; dis: discharge, re-cha: re-charge), and the corresponding point position in the charge–discharge curve is shown in Fig. S12.† The XANES spectra at the OCV state by contrast with several manganese oxides standards (MnO, Mn2O3, and MnO2) are stacked in the Fig. 2c bottom for reference. The edge position of the OCV state discloses the coexistence of Mn3+ and Mn4+ in LMO. With the continual intercalation of Li-ions, upon discharge to 2 V, the edge position of Mn shifted from 6654.9 eV to lower energy (6554.5 eV) as a result of the reduction of Mn4+ to lower valence manganese, inducing the formation of a new tetragonal phase L2MO to result in the coexistence of LMO and L2MO. The Mn edge position backward shifted to the initial position while recharging to 3.9 V. When reaching a higher voltage (re-cha 4.35 V) owing to excessive deintercalation of Li-ions, the edge position shifted to higher energy (6555.6 eV), resulting in a solid–solution reaction accompanied by the oxidation of Mn3+ to Mn4+. The changes in the Mn K-edge position indicated the reversibility of the LMO structure after pre-lithiation.
In addition, EXAFS spectra (Fig. S13†) provide information about bond length, coordination number, and the Debye–Waller factor (σ2) of the coordination atoms for the Mn atom, such as O and Mn. Fig. S14† presents Fourier transform (FT) magnitude and the relevant fitting result of Mn K-edge EXAFS spectra in the OCV state, corresponding to a model of dealing EXAFS data. The two shells at ≈1.5 and 2.5 Å correspond to the Mn–O and Mn–Mn interactions. The σ2 provides a measure of the structural disorder in a material, as shown in Fig. 2e. The σ2 value of Mn–Mn increased and the bond strength (R) of Mn–Mn weakened (Fig. S13†) while discharging to 2 V, suggesting that the intercalation of Li-ions in octahedral vacancies results in the transformation of the LiO4 tetrahedron into LiO6 octahedra, which affects the Mn–Mn interaction so that the Mn–Mn coordination and system structure disorder become intensified, giving rise to the two-phase transition from LMO to L2MO. During the process of recharging to 3.9 V accompanied by the deintercalation of Li-ions, the Mn–Mn σ2 value recovered to the initial value along with a slight deviation leading to the local structure being ordered, indicating the high reversibility of the Li-ion intercalation/deintercalation and LMO structure after pre-lithiation, which was consistent with the results of XANES. With the further deintercalation of Li-ions, the σ2 value of Mn–Mn decreased and the Mn–Mn distance became shorter (Fig. S13†), suggesting that the LMO structure became more ordered. In the LMO structure, the Mn–O distance does not vary much with the complete discharge–charge process due to the Mn atoms being coordinated by 6 adjacent O atoms at equal distances. To further explore more intuitive changes of the surrounding coordination environments of transition metal after a whole Li-ion (de)intercalation process, wavelet transform (WT) analysis of Mn K-edge spectra is further performed. Fig. 2f exhibits two different regions: Mn–O (first shell) and Mn–Mn (second shell). The strength of Mn–Mn at dis 2 V is weakened compared with the OCV state, owing to the covalent effect of Li-ions intercalating into vacancies on the two-phase transition. Notably, the strength difference between dis 2 V and OCV is quite obvious (blue) as seen from the color 3D WT-EXAFS spectra in Fig. 2g. In addition, along with the lithium-ions deintercalated from the octahedral sites, when re-charging to 3.9 V, the strength of Mn–Mn goes back to the strength of Mn–Mn at the initial OCV state with a slight difference, consistent with the results in Fig. 2g (yellow). The strength variability of Mn–Mn suggests that the covalent effect has no effect on structural reversibility.
Based on the aforementioned results, the cathode pre-lithiation strategy has been applied to anode-free cells. However, only utilizing the cathode pre-lithiation strategy to improve ICE (Fig. S15a†) or using optimized electrolytes to improve ACE (Fig. S15b†) still results in rapid capacity loss, and the lifetime of the anode-free cell cannot extend to the best. To get the maximum energy density and long-term stable anode-free devices, the cathode pre-lithiation strategy combined with a fluorine-containing electrolyte was applied to achieve long-lifetime anode-free cells. Depending on the structural properties of LMO, extra Li-ions can be stored in the LMO structure. The lithiation degree can be determined by the cut-off potential. An anode-free cell consisting of an over-lithiated L1.3MO cathode and a Cu anode outperforms the Cu||LMO anode-free cell (Fig. 3a and b) by delivering a higher initial discharge specific capacity of 112 mA h g−1, with an outstanding capacity retention of 93.1% after 40 cycles (Fig. 3c, S16 and S17†). The ICE of the anode-free cell increases from 84.4% to 98.8%, due to the pre-lithiation strategy, which stores extra Li-ions in the LMO structure, which can offset the irreversible loss of active lithium in the subsequent cycles. Meanwhile, electrolyte optimization promotes the reversibility of lithium plating/stripping, which contributes to the long lifespan of anode-free cells. More importantly, L1.3MO is outstanding in rate performance (Fig. S18†), which makes it possible to realize anode-free cells with high current density. When discharged at 1C rate, the ICE of the Cu||L1.3MO anode-free cell (Fig. S19b†) is 98%, with a high capacity retention of 83.3% after 40 cycles (Fig. 3d), certainly surpassing the Cu||LMO anode-free cell (Fig. S19a†). Therefore, the outstanding electrochemical performance of the L1.3MO cathode undoubtedly demonstrates its great potential for anode-free cells.
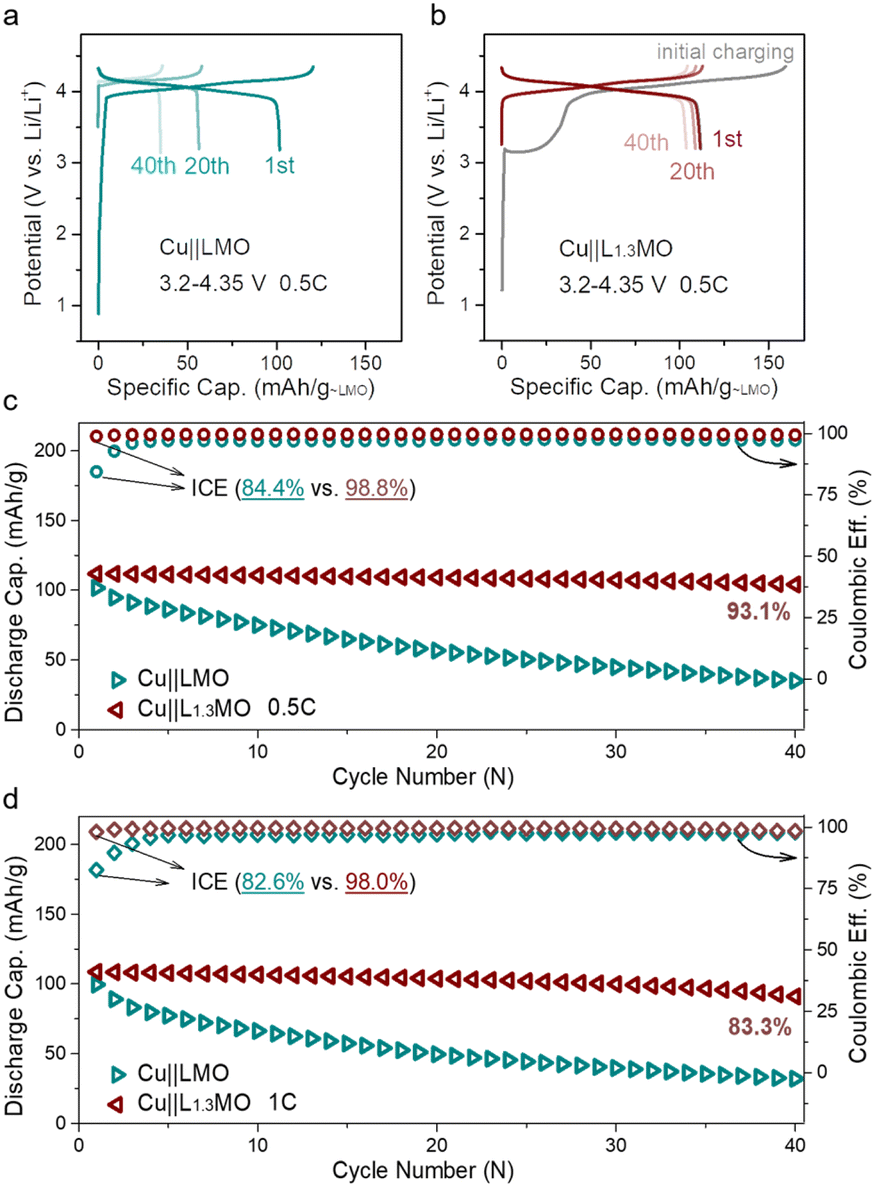 | ||
| Fig. 3 Charge–discharge curves of (a) Cu||LMO and (b) Cu||L1.3MO anode-free coin cells, and (c) corresponding cycling performance and CE of (a) and (b) at 0.5C discharge rate. (d) Cycling performance and CE of Cu||LMO (Fig. S19a†) and Cu||L1.3MO (Fig. S19b†) at 1C discharge rate. (L1.3MO cathodes were prepared via the electrochemical pre-lithiation method). | ||
Electrochemical pre-lithiation has been widely used in laboratories, however, the complex assembly/disassembly process hinders its practical applications. To date, some alternative strategies have been proposed. First, lithium metal powder can be mixed into the electrode for pre-lithiation; nevertheless, this strategy is relatively risky to cause short-circuit.21 Second, the lithiation degree of ultra-thin lithium films can be controlled by their thickness, but their cost makes them impractical for large-scale practical applications.26 Moreover, there is a piece of bad news, massively reported strategies have been applied to common anodes rather than Cu anodes, and most of these strategies cannot be applied to Cu anodes. In this work, contact pre-lithiation, a more advanced and convenient strategy, has been introduced for application to the cathode, and the schematic is displayed in Fig. 4a. When the unlithiated LMO cathode comes into direct contact with lithium metal through the pre-prepared electrolyte, the spontaneous pre-lithiation process will be initiated due to the potential difference between LMO and lithium metal (lithium metal has a much lower potential than LMO). In the pre-lithiation process, the lithium metal transforms into Li-ions, and then gradually intercalates into LMO octahedral vacancies. To inspect the feasibility of contact pre-lithiation and electrochemical pre-lithiation, in situ XRD was conducted (Fig. 4b and c). For electrochemical pre-lithiation, during the discharge process of the plateau around 2.7 V, the main peak (111) gradually divided into two, and a new diffraction peak (101) related to L2MO appeared, indicating the formation of L2MO. A similar tendency was observed in the in situ XRD patterns of contact pre-lithiation (Fig. 4c), and as the contact time increased, the (101) peak also appeared, confirming that contact pre-lithiation is reliable. More importantly, the lithiation degree (x value, (0 ≤ x ≤ 2)) can be precisely controlled by controlling the contact time (Fig. S20†), and the integrity of the cathode can be maintained during the pre-lithiation process. Notably, by calculating the proportion of the (101) peak area, when the contact time extended to 2 min, the same over-lithiation degree was achieved using contact pre-lithiation and electrochemical pre-lithiation (Fig. 4d). Furthermore, over-lithiated L1.3MO was achieved via contact pre-lithiation of LMO for 2 min, and then a Cu||L1.3MO anode-free cell was constructed (Fig. 4e). The electrochemical performance of the Cu||L1.3MO anode-free cell prepared by contact pre-lithiation is tantamount to the electrode prepared by electrochemical pre-lithiation, confirming that contact pre-lithiation has no effect on the LMO structure. In addition, the Cu||L1.3MO anode-free cell with a high ICE of 99% exhibits a high capacity retention of 94.4% after 40 cycles (Fig. 4e), exhibiting its great potential for practical applications.
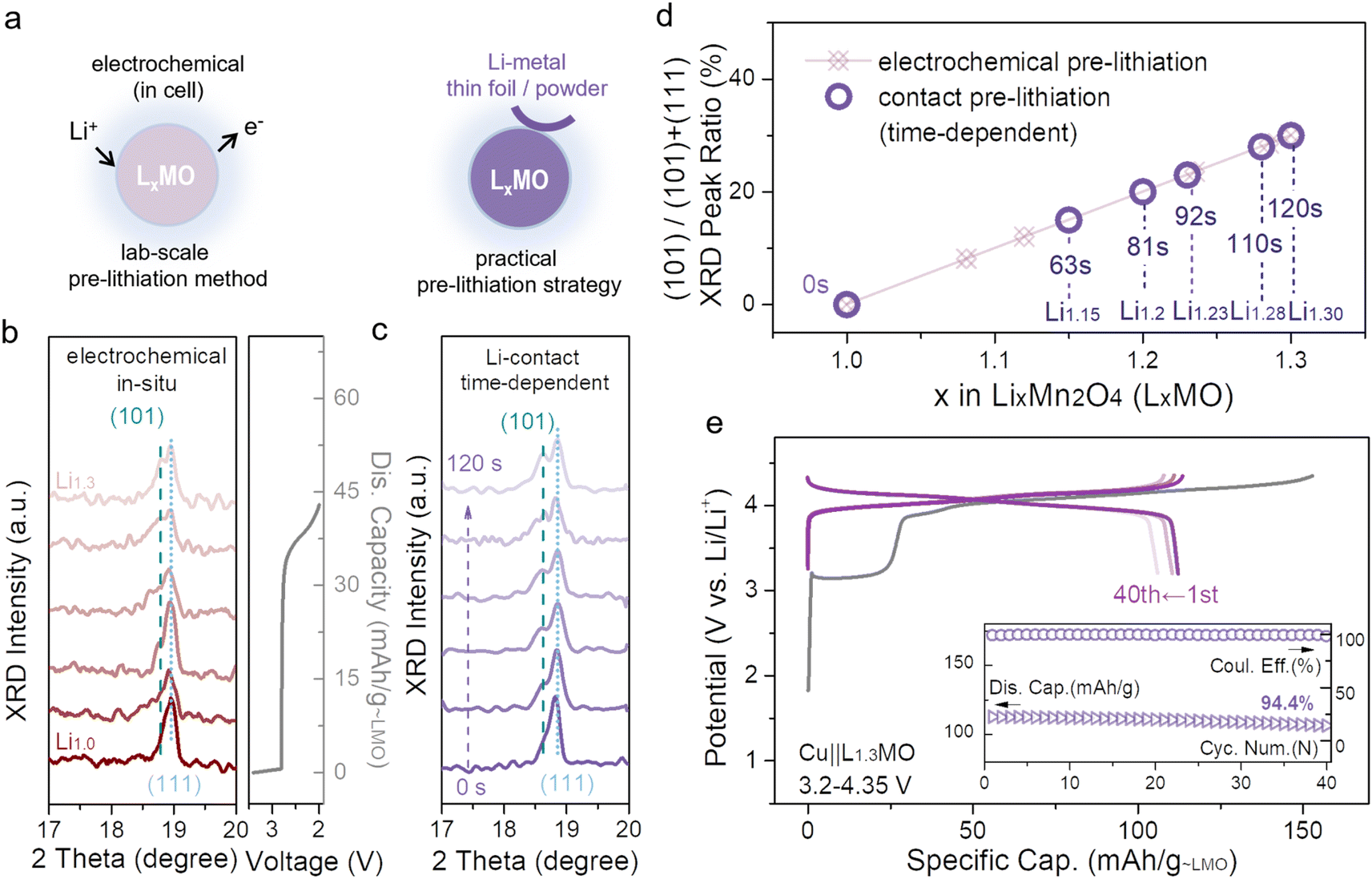 | ||
| Fig. 4 (a) Schematic illustration of electrochemical pre-lithiation (left) and contact pre-lithiation (right). (b) In situ XRD patterns of the Li||LMO half-cell discharged to 2 V, and the corresponding galvanostatic charge–discharge curve (according to electrochemical pre-lithiation). (c) In situ XRD patterns of contact pre-lithiation which depends on contact time were obtained to observe the change of the main peak (111). (d) The ratio of the (101) peak area corresponds to different x values in LxMO (0 ≤ x ≤ 2) (according to in situ XRD patterns in (b) and (c)). (e) Typical galvanostatic charge/discharge curves of the Cu||L1.3MO (2 min) anode-free coin cell at 0.5C discharge rate, and the inset represents the corresponding cycling performance. | ||
Conclusions
In summary, a cathode strategy combined with optimized electrolytes has been successfully proposed to achieve a long life-span of AF-LMBs. The variation of the crystal structure after pre-lithiation and the reversible phase transition between conventional Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m type LiMn2O4 and I41/amd type Li2Mn2O4 have been demonstrated by in situ XRD, HRTEM, and ex situ XAS. Additional lithium ions are intercalated into the vacancies in the LiMn2O4 structure to form over-lithiated Li2Mn2O4, and then Li2Mn2O4 delivers additional lithium ions during the first charge process to offset active lithium loss and prolong the life-span of AF-LMBs. Furthermore, contact pre-lithiation, a more advanced and convenient strategy at the engineering level, has been introduced to precisely control the lithiation degree by controlling the contact time. Cu|| Li1.3Mn2O4 AF-LMBs have been constructed by the application of contact pre-lithiation combined with a fluorine-containing electrolyte and exhibit a high capacity retention of 94.4% after 40 cycles. We believe that this could spark a storm of related studies for the design of AMF-LMBs.
m type LiMn2O4 and I41/amd type Li2Mn2O4 have been demonstrated by in situ XRD, HRTEM, and ex situ XAS. Additional lithium ions are intercalated into the vacancies in the LiMn2O4 structure to form over-lithiated Li2Mn2O4, and then Li2Mn2O4 delivers additional lithium ions during the first charge process to offset active lithium loss and prolong the life-span of AF-LMBs. Furthermore, contact pre-lithiation, a more advanced and convenient strategy at the engineering level, has been introduced to precisely control the lithiation degree by controlling the contact time. Cu|| Li1.3Mn2O4 AF-LMBs have been constructed by the application of contact pre-lithiation combined with a fluorine-containing electrolyte and exhibit a high capacity retention of 94.4% after 40 cycles. We believe that this could spark a storm of related studies for the design of AMF-LMBs.
Author contributions
L. C., C. W., X. K. and Y. Q. contributed to the design of the research and performed the experimental data analysis. L. C. conducted the materials synthesis, electrochemical studies and cell performance studies. C. C. and Y. L. conducted the XAS experiments and the analysis of XAS results. G. Z., Y. T., and B. Z. helped to conduct the XRD experiment and FAULTS simulation. X. W. and T. L. conducted the LSV and SEM experiments. S. Z. conducted the HR-TEM experiment. H. Z. and Y. Y. drew the schematics. X. K., Y. L. and Y. Q. supervised the work. All authors discussed the results, co-wrote and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Natural Science Foundation of China (grant no. 22179111 and 22021001), the Ministry of Science and Technology of China (grant no. 2021YFA1201900), the Basic Research Program of Tan Kah Kee Innovation Laboratory (grant no. RD2021070401), the Principal Fund from Xiamen University (grant no. 20720210015), and the Fundamental Research Funds for the Central Universities (grant no. 20720220010). This research also employed the resources of the National Synchrotron Radiation Research Center (NSRRC) in Hsinchu, Taiwan, R.O.C. and Beijing Synchrotron Radiation Laboratory (under contract no. 2021-BEPC-PT-005771, 2021-BEPC-PT-005765, 2021-BEPC-PT-005760, and 2022-BEPC-PT-006478).Notes and references
- Y. Huang, Y. Dong, S. Li, J. Lee, C. Wang, Z. Zhu, W. Xue, Y. Li and J. Li, Adv. Energy Mater., 2021, 11, 2000997 CrossRef CAS.
- S. Zhang, N. S. Andreas, R. Li, N. Zhang, C. Sun, D. Lu, T. Gao, L. Chen and X. Fan, Energy Storage Mater., 2022, 48, 44–73 CrossRef.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- B. D. Adams, J. Zheng, X. Ren, W. Xu and J. G. Zhang, Adv. Energy Mater., 2018, 8, 1702097 CrossRef.
- Y. Qiao, H. Deng, P. He and H. Zhou, Joule, 2020, 4, 1445–1458 CrossRef CAS.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- X. Fan, L. Chen, O. Borodin, X. Ji, J. Chen, S. Hou, T. Deng, J. Zheng, C. Yang, S. C. Liou, K. Amine, K. Xu and C. Wang, Nat. Nanotechnol., 2018, 13, 715–722 CrossRef CAS PubMed.
- S. Nanda, A. Gupta and A. Manthiram, Adv. Energy Mater., 2020, 11, 2000804 CrossRef.
- C. Fang, X. Wang and Y. S. Meng, Trends Chem., 2019, 1, 152–158 CrossRef CAS.
- L. Lin, K. Qin, Y. S. Hu, H. Li, X. Huang, L. Suo and L. Chen, Adv. Mater., 2022, 34, e2110323 CrossRef PubMed.
- H. Wang, Y. Liu, Y. Li and Y. Cui, Electrochem. Energy Rev., 2019, 2, 509–517 CrossRef CAS.
- Z. Tong, B. Bazri, S.-F. Hu and R.-S. Liu, J. Mater. Chem. A, 2021, 9, 7396–7406 RSC.
- J. Xiang, L. Yang, L. Yuan, K. Yuan, Y. Zhang, Y. Huang, J. Lin, F. Pan and Y. Huang, Joule, 2019, 3, 2334–2363 CrossRef CAS.
- Y. Tian, Y. An, C. Wei, H. Jiang, S. Xiong, J. Feng and Y. Qian, Nano Energy, 2020, 78, 105344 CrossRef CAS.
- A. J. Louli, A. Eldesoky, R. Weber, M. Genovese, M. Coon, J. deGooyer, Z. Deng, R. T. White, J. Lee, T. Rodgers, R. Petibon, S. Hy, S. J. H. Cheng and J. R. Dahn, Nat. Energy, 2020, 5, 693–702 CrossRef CAS.
- J. Chen, Q. Li, T. P. Pollard, X. Fan, O. Borodin and C. Wang, Mater. Today, 2020, 39, 118–126 CrossRef.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J. G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed.
- H. J. Kim, S. Choi, S. J. Lee, M. W. Seo, J. G. Lee, E. Deniz, Y. J. Lee, E. K. Kim and J. W. Choi, Nano Lett., 2016, 16, 282–288 CrossRef CAS PubMed.
- Z. Cui, F. Zou, H. Celio and A. Manthiram, Adv. Funct. Mater., 2022, 32, 2203779 CrossRef CAS.
- L. Jin, C. Shen, Q. Wu, A. Shellikeri, J. Zheng, C. Zhang and J. P. Zheng, Adv. Sci., 2021, 8, e2005031 CrossRef PubMed.
- Z. Song, K. Feng, H. Zhang, P. Guo, L. Jiang, Q. Wang, H. Zhang and X. Li, Nano Energy, 2019, 66, 104175 CrossRef CAS.
- Y. Qiao, H. Yang, Z. Chang, H. Deng, X. Li and H. Zhou, Nat. Energy, 2021, 6, 653–662 CrossRef CAS.
- K. Park, B.-C. Yu and J. B. Goodenough, Adv. Energy Mater., 2016, 6, 1502534 CrossRef.
- L. Chen, C. L. Chiang, X. Wu, Y. Tang, G. Zeng, S. Zhou, B. Zhang, H. Zhang, Y. Yan, T. Liu, H. G. Liao, X. Kuai, Y. G. Lin, Y. Qiao and S. G. Sun, Chem. Sci., 2023, 14, 2183–2191 RSC.
- L. Lin, K. Qin, Q. Zhang, L. Gu, L. Suo, Y. S. Hu, H. Li, X. Huang and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 8289–8296 CrossRef CAS PubMed.
- L. Lin, K. Qin, M. Li, Y.-s. Hu, H. Li, X. Huang, L. Chen and L. Suo, Energy Storage Mater., 2022, 45, 821–827 CrossRef.
- F. Wang, B. Wang, J. Li, B. Wang, Y. Zhou, D. Wang, H. Liu and S. Dou, ACS Nano, 2021, 15, 2197–2218 CrossRef CAS PubMed.
- V. Aravindan, S. Nan, M. Keppeler and S. Madhavi, Electrochim. Acta, 2016, 208, 225–230 CrossRef CAS.
- M. Mao, X. Ji, Q. Wang, Z. Lin, M. Li, T. Liu, C. Wang, Y. S. Hu, H. Li, X. Huang, L. Chen and L. Suo, Nat. Commun., 2023, 14, 1082 CrossRef CAS PubMed.
- R. Weber, M. Genovese, A. J. Louli, S. Hames, C. Martin, I. G. Hill and J. R. Dahn, Nat. Energy, 2019, 4, 683–689 CrossRef CAS.
- X. Hou, X. Liu, H. Wang, X. Zhang, J. Zhou and M. Wang, Energy Storage Mater., 2023, 57, 577–606 CrossRef.
- B. Song, G. M. Veith, J. Park, M. Yoon, P. S. Whitfield, M. J. Kirkham, J. Liu and A. Huq, Chem. Mater., 2018, 31, 124–134 CrossRef.
- F. Luo, C. Wei, C. Zhang, H. Gao, J. Niu, W. Ma, Z. Peng, Y. Bai and Z. Zhang, J. Energy Chem., 2020, 44, 138–146 CrossRef.
- Y. Shiraishi, I. Nakai, T. Tsubata, T. Himeda and F. Nishikawa, J. Soild State Chem., 1997, 133, 587–590 CrossRef CAS.
- Y. Terada, K. Yasaka, F. Nishikawa, T. Konishi, M. Yoshio and I. Nakai, J. Solid State Chem., 2001, 156, 286–291 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01823g |
| ‡ L. Chen and C.-L. Chiang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |