
Halide perovskite quantum dots for photocatalytic CO2 reduction
Wentao
Song
,
Guobin
Qi
and
Bin
Liu
 *
*
Department of Chemical and Biomolecular Engineering, National University of Singapore, Singapore 117585, Singapore. E-mail: cheliub@nus.edu.sg
First published on 16th February 2023
Abstract
Halide perovskite quantum dots have recently attracted increasing research interest in photocatalytic CO2 reduction due to their high light absorption coefficient, tunable bandgap, and long charge diffusion length. However, the intrinsic high radiative recombination and instability of perovskite quantum dots limit the development of perovskite-based photocatalysis, which is under investigation by different research groups. This review is focused on the current progress and challenges of halide perovskite quantum dots for photocatalytic CO2 reduction. The structures and properties of perovskite quantum dots as well as the mechanism and obstacles for photocatalytic CO2 reduction are first summarized in detail. Subsequently, composition optimization, surface modification, and heterojunction construction of perovskite-based photocatalysts are introduced to enhance photogenerated carrier separation and photocatalytic CO2 activity. Finally, we discuss the challenges and opportunities of perovskite quantum dots for photocatalytic CO2 reduction and further propose several prospective directions for future research.
10th anniversary statementSince the Journal of Materials Chemistry was split into three independent journals (A, B, and C) in 2013, the Journal of Materials Chemistry A has developed rapidly into one of the leading scientific journals in the energy and sustainability fields. Over the past 15 years, our group and co-workers have published 16 papers in the Journal of Materials Chemistry (A, B, and C), which include various topics, such as dye-sensitized solar cells, perovskite solar cells, water splitting, and CO2 reduction. With the strong support from the materials and energy community, we wish JMCA great success and hope that it will grow to be one of the most influential journals in the near future. |
1. Introduction
Photocatalytic CO2 reduction is one of the very effective and environmentally friendly strategies to convert CO2 to valuable chemical fuels.1,2 In pursuit of high CO2 conversion efficiency, many semiconductor photocatalysts with appropriate energy bands have been utilized for photocatalytic CO2 reduction.3–5 Nonetheless, the solar-to-fuel efficiency is far lower than 10%, making it less suitable for large-scale industrial production. The high recombination rate and low efficiency of photocatalysts prevent their widespread applications.6–8Since the pioneer work of halide perovskite materials in solar cells by Miyasaka in 2009,9 halide perovskite materials have attracted much research interest in optoelectronics and photoelectrical conversion.10,11 In 2015, Kovalenko and colleagues synthesized perovskite quantum dots (QDs) or nanocrystals (NCs) through the hot-injection method for the first time.12 The relatively stable perovskite QDs have great potential for practical applications in various photoelectrical fields, including solar cells, light-emitting diodes (LEDs), photodetectors, and so on.13–17 Recently, perovskite QDs have been explored for photocatalytic CO2 reduction due to their high light absorption coefficient, tunable bandgap, and long carrier transport distance.18,19 Photocatalysis generally involves several key processes: light absorption, photogenerated carrier separation and migration, surface catalytic reaction, and product desorption. The high light absorption coefficient means more light can be captured for photocatalytic CO2 reduction. The tunable bandgap allows perovskite to easily optimize the bandgap and redox potential, and the long carrier diffusion distance shows great opportunity for carriers to migrate to the active site. These unique properties could make perovskite QDs promising photocatalysts for CO2 reduction. However, the radiative recombination induced by high photoluminescence quantum yield (PLQY) and low stability against light, heat, oxygen, and moisture of perovskite QDs have an adverse effect on photocatalytic CO2 reduction.20,21 To overcome these obstacles, some methods have been developed to improve the photocatalytic CO2 performance of perovskite QDs.22 For example, various heterojunction composites were constructed to enhance the photogenerated carrier separation and photocatalytic stability for highly efficient CO2 reduction.23 A series of composition optimization and surface modification measures have been performed to improve the stability and efficiency of perovskite-based photocatalysts. Although several reviews have summarized the current progress of different photocatalytic processes and stability issues, nevertheless, very few reports were focused on perovskite QDs for photocatalytic CO2 reduction or discussed the difficulties of photocatalytic CO2 reduction from the perspective of perovskite fundamentals and the photocatalytic mechanism.24,25 In addition, despite the considerable progress made for CO2 photoreduction, it is noteworthy that the superior photocatalytic activity and long-term stability of perovskite photocatalysts are yet to be realized.26 It is thus important to understand the photocatalytic mechanism, summarize the current progress, and provide insightful perspectives into further development of perovskite QDs for photocatalytic CO2 reduction.27,28
This review highlights the recent success and reliable measures of perovskite-based photocatalysts for CO2 photoreduction, and proposes appropriate strategies for future design, as shown in Fig. 1. First, the fundamentals of perovskite QDs, as well as the mechanism and obstacles for photocatalytic CO2 reduction are summarized in detail. Afterwards, current successful strategies, including composition optimization, surface modification and heterojunction construction, are introduced to enhance the photocatalytic CO2 activity and stability of perovskite-based photocatalysts. Finally, we discuss the challenges and opportunities of perovskite QDs for photocatalytic CO2 reduction and further propose several prospective directions for future research.29
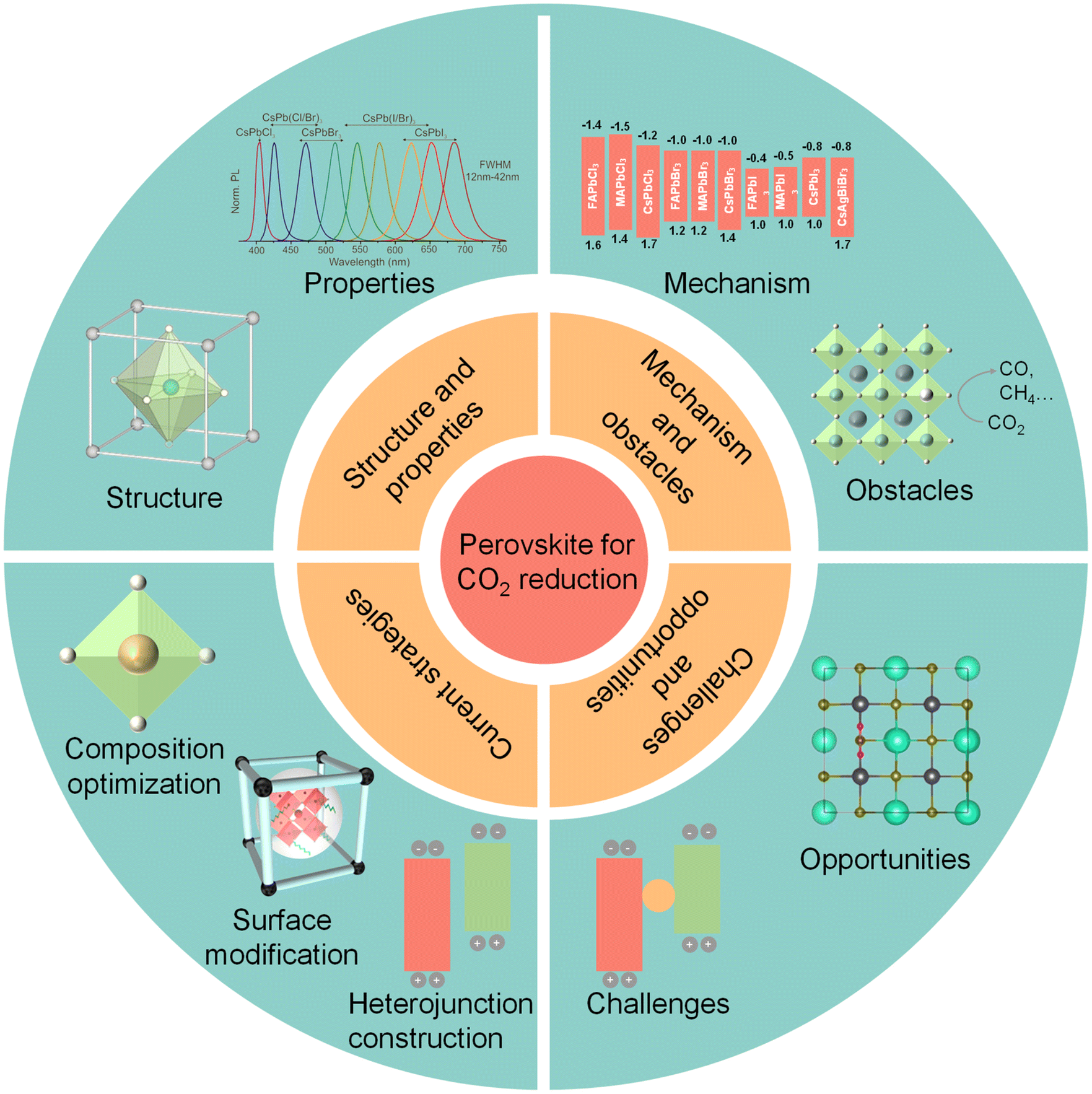 | ||
| Fig. 1 Schematic illustrations of halide perovskite QDs for CO2 reduction: structure and properties, photocatalytic mechanism and obstacles, current strategies, challenges, and opportunities. Reproduced from ref. 17 with permission from American Chemical Society, copyright 2015; reproduced from ref. 91 with permission from Wiley-VCH, copyright 2022; reproduced from ref. 105 with permission from Royal Society of Chemistry, copyright 2022. | ||
2. Halide perovskite fundamentals, photocatalytic mechanism, and obstacles for CO2 reduction
2.1 Crystal structure and properties
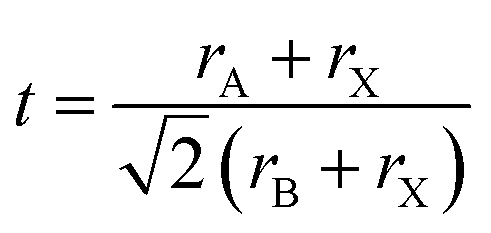 , rA, rB, and rX represent the radii of A, B, and X, respectively) is used to measure the structural stability of perovskite materials.31 When t ranges from 0.8–1, it fits structural constraints and constructs stable three-dimensional perovskite. Nonetheless, when the radius of the A cation is too large, it would cause t to be larger than 1 and usually form lower-dimensional phases, such as two-dimensional (2D) perovskites (e.g., A2PbI4), whereas when t is smaller than 0.8, other structures, such as perovskite oxide (ABO3), are stable.32
, rA, rB, and rX represent the radii of A, B, and X, respectively) is used to measure the structural stability of perovskite materials.31 When t ranges from 0.8–1, it fits structural constraints and constructs stable three-dimensional perovskite. Nonetheless, when the radius of the A cation is too large, it would cause t to be larger than 1 and usually form lower-dimensional phases, such as two-dimensional (2D) perovskites (e.g., A2PbI4), whereas when t is smaller than 0.8, other structures, such as perovskite oxide (ABO3), are stable.32
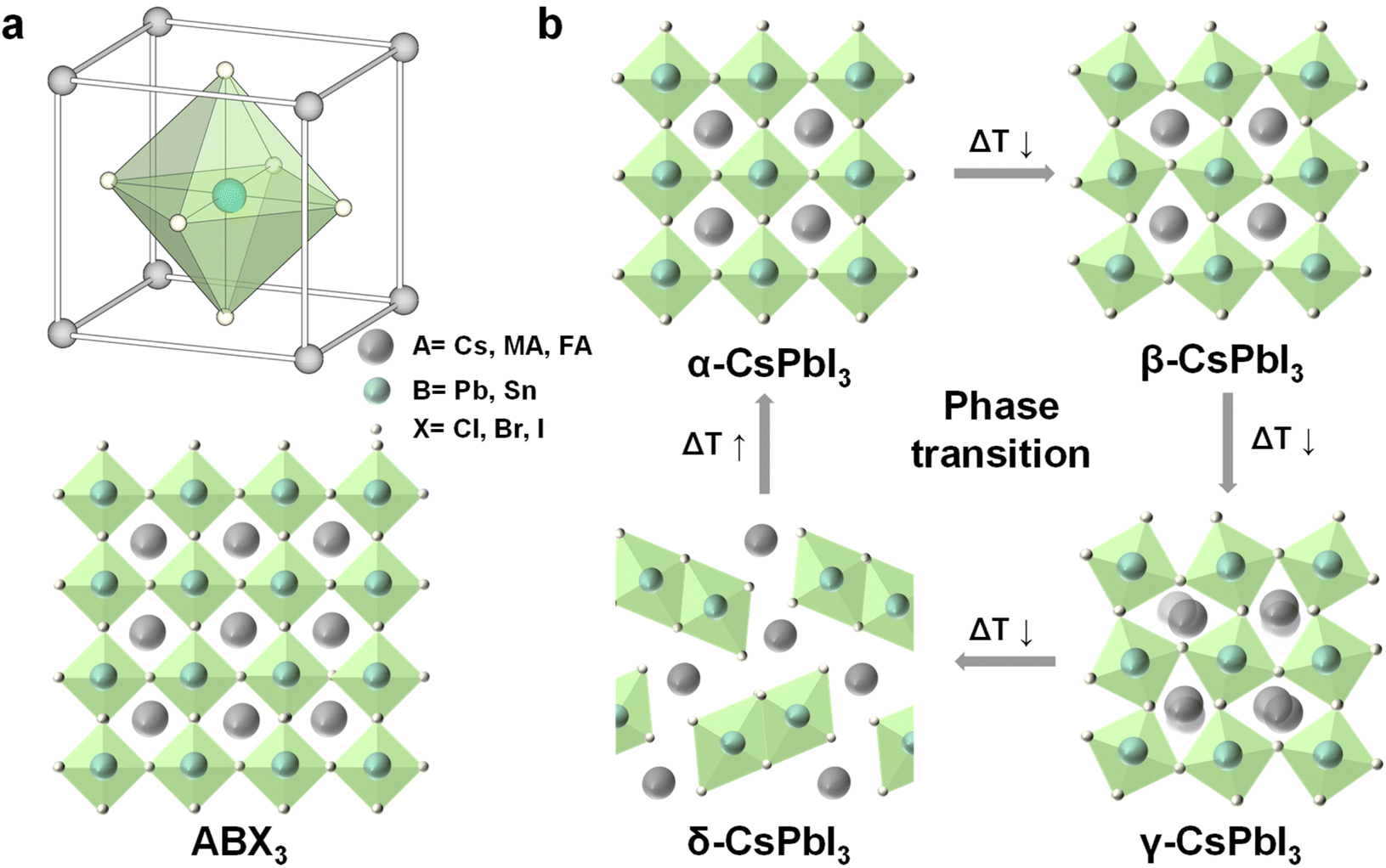 | ||
| Fig. 2 (a) Illustration of the halide perovskite structure ABX3, A = Cs+, CH3NH2+ (MA+), or CH2(NH2)2+ (FA+), B = Pb2+, or Sn2+; X = Cl−, Br−, or I−; (b) crystal structure of the different CsPbI3 phases and relative phase transitions. Reproduced from ref. 33 with permission from American Association for the Advancement of Science, copyright 2019. | ||
In addition, according to the bond angle of Pb-X-Pb, the halide perovskite structure can be divided into optically active black perovskite phases: cubic (α), tetragonal (β), and orthorhombic (γ) phases. The ideal Pb-X-Pb angle is 180°, corresponding to cubic perovskite. The distortion of the cubic structure would change the angle of Pb-X-Pb and reduce the crystal symmetry, which would form low symmetric tetragonal and orthorhombic perovskite.33Fig. 2b depicts the crystal structure of different phases and their relative phase transitions of CsPbI3. At room temperature, the metastable black perovskite phases would undergo phase transformation into the stable non-perovskite phase (δ-CsPbI3), which is non-photoactive and undesirable for optoelectronics.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Br or Br:I. The tunable bandgap makes perovskite QDs good candidates for efficient photocatalysts.
:Br or Br:I. The tunable bandgap makes perovskite QDs good candidates for efficient photocatalysts.
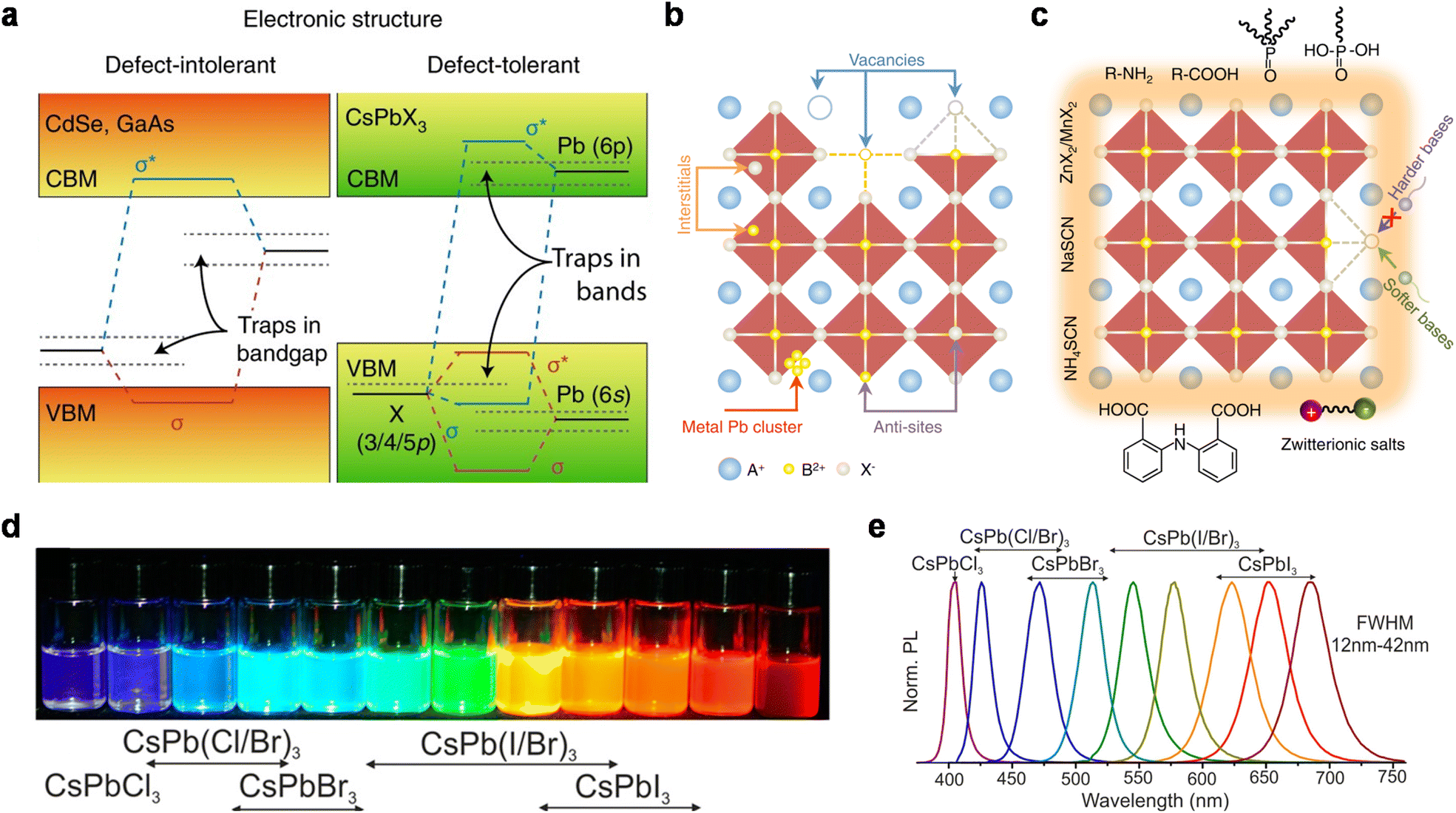 | ||
| Fig. 3 (a) Scheme of the electronic band structure of typical defect-intolerant semiconductors and perovskite QDs. In conventional semiconductors, the bandgap is formed between bonding (σ) and antibonding (σ*) orbitals. Point defects or dangling bonds emerge as weak bonding or non-bonding states within the bandgap. In perovskite QDs, the bandgap is formed between two antibonding orbitals. Only shallow traps are formed within the CB or VB, with little influence on optical properties. Reproduced from ref. 35 with permission from Springer Nature, copyright 2018. (b) Point defects of perovskite QDs, including vacancies, interstitials, anti-sites, and metal Pb clusters. Reproduced from ref. 39 with permission from Springer Nature, copyright 2021. (c) Common passivating ligands for perovskite QDs: zwitterionic salts, R–NH2, R–COOH, etc. Reproduced from ref. 39 with permission from Springer Nature, copyright 2021. (d) Perovskite QDs in toluene under a UV lamp (λ = 365 nm). Reproduced from ref. 17 with permission from American Chemical Society, copyright 2015. (e) Representative PL spectra of mixed-halide perovskite QDs. Reproduced from ref. 17 with permission from American Chemical Society, copyright 2015. | ||
2.2 Photocatalytic mechanism
Photocatalysis refers to a chemical reaction with the aid of light and a photocatalyst, which can absorb light to promote electron excitation from the VB to the CB, and thus leave holes in the VB. The photogenerated electron and hole pairs can be used for two half-reactions: reduction and oxidation reactions. Taking CO2 reduction by H2O as an electron source as an example, the CB minimum of a photocatalyst must be more negative than the electrochemical potential of CO2 reduction and the VB maximum must be more positive than the electrochemical potential of H2O oxidation to ensure that the reaction can occur. That is to say, the bandgap of a photocatalyst should meet the band requirement of both CO2 reduction and H2O oxidation, which limits the selection of a single photocatalyst. Therefore, sacrificial agents with lower oxidation potentials are added to replace H2O and decrease the required bandgap.41 Considering the photocatalytic thermodynamics, a proper energy band plays a vital role in reaction redox potentials. The redox potentials of perovskite QDs together with traditional semiconductors and photocatalytic CO2 reduction to C1 products are depicted in Fig. 4. It could be clearly seen that the CBs of perovskite QDs are more negative than that of traditional semiconductors, which means perovskite QDs are more favourable for CO2 reduction and H2 production. However, when it comes to water oxidation for O2 production and ·OH production, nearly all the perovskite QDs cannot perform this reaction owing to the weak oxidation potentials. Notably, most traditional semiconductors show good potential for O2 production, indicating suitable candidates to form a heterojunction with halide perovskite QDs. From the standpoint of thermodynamics, halide perovskite QDs could yield all the C1 products from photocatalytic CO2 reduction. In fact, photocatalytic CO2 reduction is strongly affected by both thermodynamics and kinetics.42,43 For example, CO2 reduction to CO or HCOOH involves a 2H+/2e− process, and this multiparticle (from bond breaking of the CO2 molecule to bond formation of CO or HCOOH molecules) transfer faces kinetic limitations. For the 6H+/6e− processes of CO2 reduction to CH3OH, the complex multiparticle and multielectron processes are less favourable in a single photocatalytic reaction. Thus, despite all the C1 products meeting thermodynamic conditions, the general products of photocatalytic CO2 reduction on perovskite QDs are CO and CH4. Besides, some similar thermodynamic potentials of products induce several side reactions and low selectivity, such as CO2 reduction to CH3OH and CO, or direct reduction of 2H+ to H2.4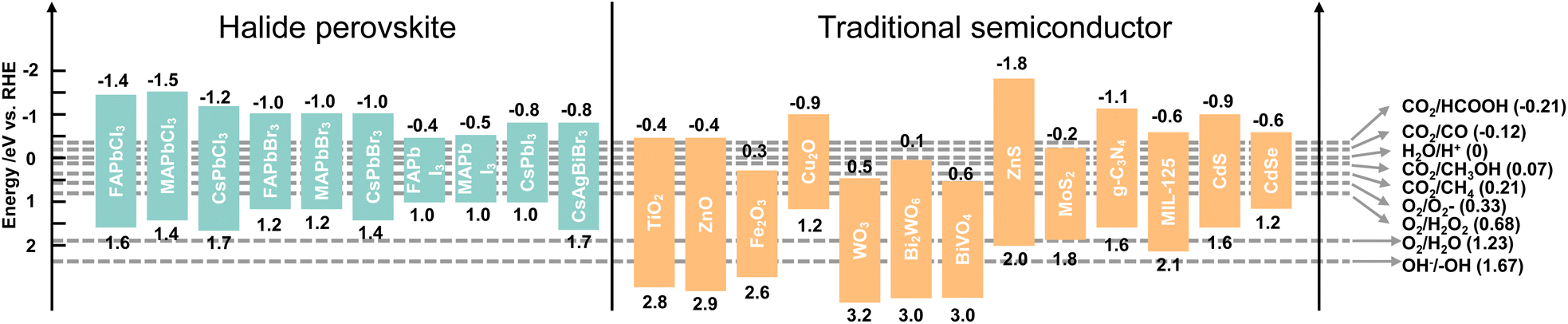 | ||
| Fig. 4 Band edge positions of traditional photocatalysts and different halide perovskites relative to the reversible hydrogen electrode (RHE). Reproduced from ref. 25 and ref. 26 with permission from American Chemical Society, copyright 2022. | ||
2.3 Obstacles for photocatalytic CO2 reduction
Although halide perovskite QDs have unique physical and chemical properties, the intrinsic structural instability and high carrier recombination rate limit their practical application in photocatalytic CO2 reduction.Apart from instability in a polar solvent, perovskite materials would also decompose rapidly when exposed to moisture, heat, light, and oxygen.50Fig. 5 shows the environmental factors and possible degradation mechanisms of perovskite materials.51 The common degradation mechanisms of perovskite materials include polymorphic transformation, hydration, decomposition, and oxidation. Unlike organic–inorganic hybrid perovskite QDs, all-inorganic perovskite QDs do not have hygroscopic organic cations. Hence, all-inorganic perovskite QDs would not be degraded by hydration.52 Long-term light irradiation would cause the desorption of surficial ligands, transportation of halide ions and accumulation of NCs.53 These processes originated from the rapid formation of halide vacancies after light illumination, providing a path for oxygen or water molecules into the perovskite lattice.54 It is also a great challenge for halide perovskite materials to operate at a high temperature. Hybrid organic–inorganic perovskite QDs decompose before melting (melting point: MAPbI3: 150–200 °C; FAPbI3: 290–300 °C), while the melting point of the all-inorganic QDs are higher (CsPbBr3 and CsPbI3: 450–500 °C).35 In addition, perovskite QDs would be stable only for an extremely low oxygen concentration, because high oxygen partial pressures could generate reactive intermediates such as superoxide species under light illumination.55,56
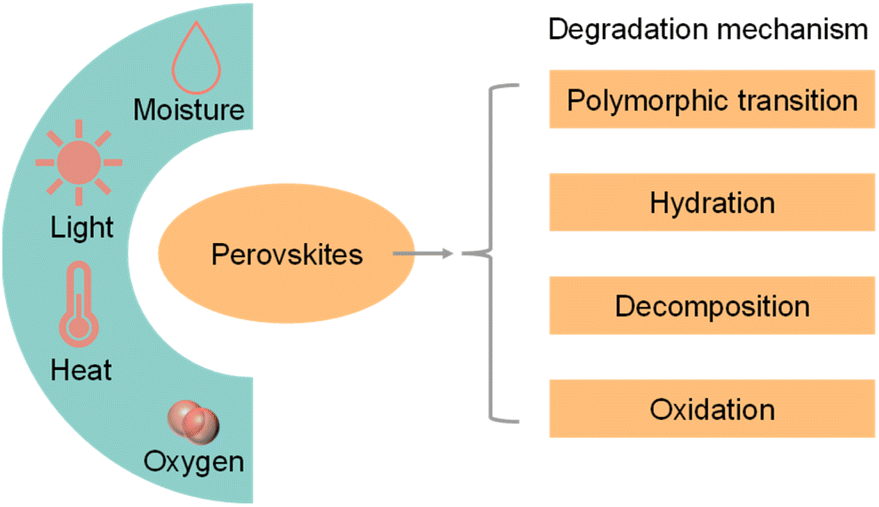 | ||
| Fig. 5 Scheme of the environmental factors (moisture, light, heat, and oxygen) and possible degradation mechanisms of perovskite materials. Reproduced from ref. 51 with permission from Elsevier, copyright 2018. | ||
Another unignorable factor for perovskite instability is the phase transition at room temperature, especially for CsPbI3. The photoactive cubic phase of perovskite (α-CsPbI3) would spontaneously undergo phase transition to the non-perovskite phase (δ-CsPbI3) owing to the lower formation energy of δ-CsPbI3. Only the photoactive perovskite phase can absorb visible light and show good photocatalytic activity for CO2 reduction. Some studies would incorporate the ions with a larger radius into the A-site (such as, FA and MA) or replace I− with Br− to stabilize α-CsPbI3 QDs.44,57
3. Current strategies for perovskite photocatalytic CO2 reduction
Even though halide perovskite QDs have been widely utilized for solar cells,58 research on perovskite QDs for photocatalysis is still in the initial stage. In the former parts, we understood the mechanism and obstacles of perovskite QDs for photocatalytic CO2 reduction from the fundamental and basic properties. Herein, recent progress and successful strategies to improve photocatalytic performance and stability are given in the following sections, including composition optimization (doping, bandgap adjustment, and morphology regulation), surface modification (ligand modification, oxide encapsulation, and MOF encapsulation) and heterostructure construction (Schottky heterojunction, type II heterojunction, and Z-scheme heterojunction).3.1 Composition optimization
The intrinsic physical and chemical properties of a photocatalyst influence its photocatalytic performance. For a photocatalyst, its intrinsic composition involves the band structure, bandgap, charge transfer, crystal face, active site, and so on. Doping, bandgap adjustment, and morphology regulation are general measures to optimize the intrinsic composition and improve the photocatalytic activity for CO2 reduction.59,60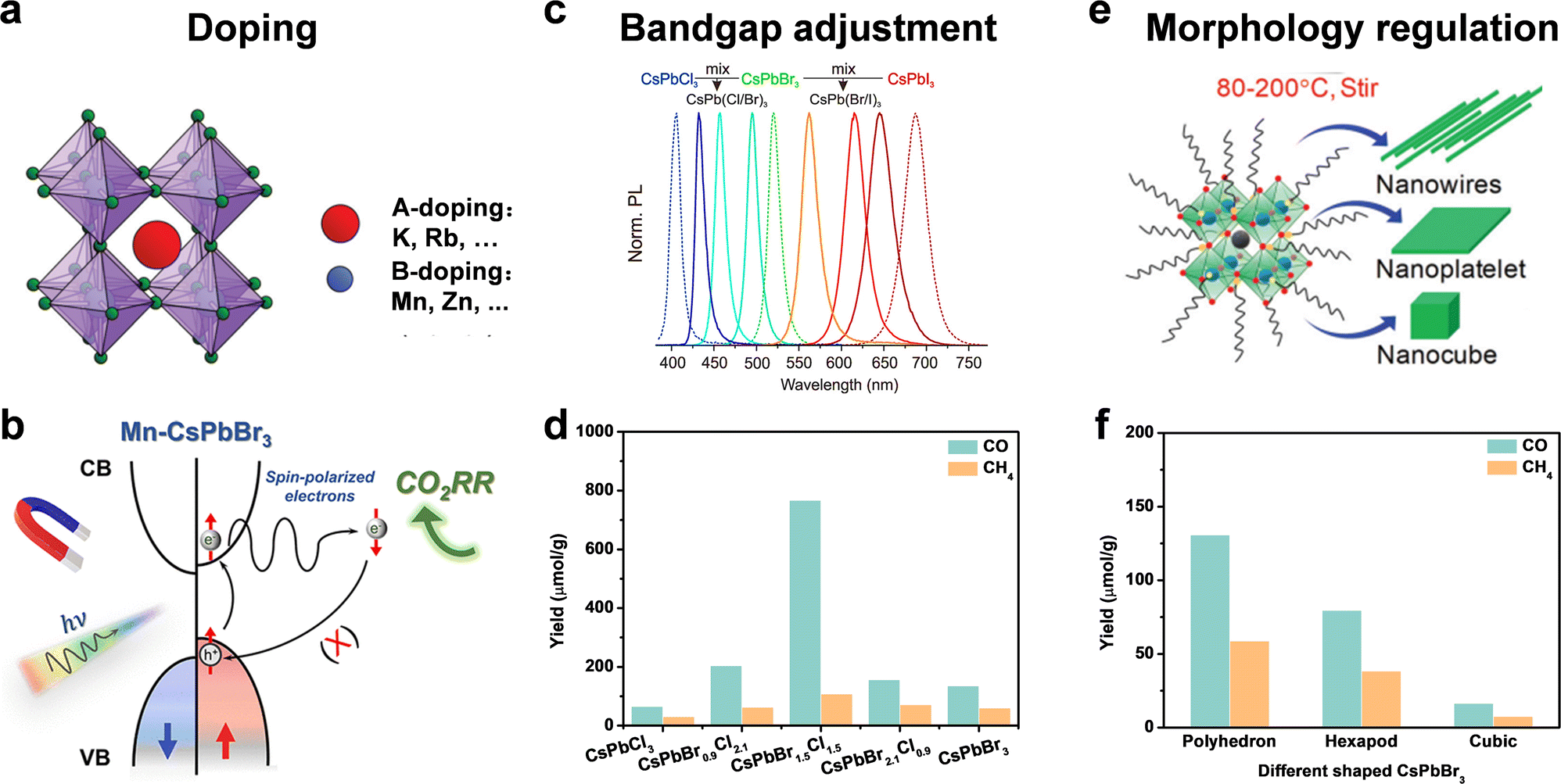 | ||
| Fig. 6 (a) Doping: A-site doping and B-doping; reproduced from ref. 62 with permission from Royal Society of Chemistry, copyright 2020. (b) Suppressed electron spin polarization of Mn doped CsPbBr3; reproduced from ref. 59 with permission from American Chemical Society, copyright 2022. (c) Bandgap adjustment: halide ratio; reproduced from ref. 12 with permission from American Chemical Society, copyright 2015. (d) Yields of CO2 reduction under 9 h Xe lamp irradiation with AM 1.5; reproduced from ref. 74 with permission from Elsevier, copyright 2015. (e) Morphology regulation: nanowires, nanoplatelets, and nanocubes; reproduced from ref. 75 with permission from Royal Society of Chemistry, copyright 2020. (f) Yields of CO and CH4 from CO2 reduction after 4 h using different shaped CsPbBr3 nanostructures; reproduced from ref. 77 with permission from American Chemical Society, copyright 2020. | ||
Although an A-site cation was thought to hardly influence optoelectronic properties of perovskite materials, some recent studies have confirmed that A-site doping (such as K+,67 Rb+,68,69 and so on) could improve stability and facilitate carrier transport. Because X-site anions directly affect the bandgap, we will discuss the X-site dopants in bandgap adjustment.
Another strategy to optimize the bandgap is to change the halide ratio. As shown in Fig. 6c, through different halide compositions, the emission spectra can be easily changed over the full-visible wavelength of 410–700 nm.12 Su et al. synthesized CsPbClxBr3−x NCs with different ratios of Br and Cl by the hot-injection method.74 The yield of photocatalytic CO2 reduction to CO and CH4 varied with the ratio of Br and Cl. CsPbBr1.5Cl1.5 exhibited the highest yield of CO (767 μmol g−1) and CH4 (108 μmol g−1), a nearly 4.5- and 9.1-fold enhancement compared to that of pristine CsPbBr3 and CsPbCl3 under the same conditions for 9 h irradiation (Fig. 6d), attributed to the efficient photogenerated carrier migration and separation of CsPbBr1.5Cl1.5 NCs.
Different facets and crystal orientations have different active sites and surface energy. A good crystal face should enable the efficient adsorption of reactants and the desorption of products. Besides, the photogenerated electrons and holes tend to accumulate on different facets, leading to facet-dependent photocatalytic properties. Pradhan and cooperators explored the polyhedron-shaped nanocubes, hexapods, and cube-shaped CsPbBr3 nanostructure for CO2 photoreduction.77 As shown in Fig. 6f, the polyhedron-shaped nanocubes exhibited the best photocatalytic activity (130.7 μmol g−1 for CO and 58.7 μmol g−1 for CH4 for 4 h) due to the fastest charge transfer for photocatalysis. TEM images showed polyhedron shapes had eight {112}, eight {012}, and four {100} prominent facets, while cube shapes were composed of four {110} and two {002} facets. DFT calculations showed that {112} and {102} facets were more superior for surface adsorption of CO2 and desorption of reduction products compared to the dominant {110} and {002} facets of orthorhombic cube shaped CsPbBr3. As facets affect photocatalytic performance, different facets of perovskite QDs should be further studied for efficient catalytic reaction.
It is noteworthy that the formation process of perovskite QDs is within sub-seconds through hard-to-control ionic metathesis nucleation and the growth process. Kovalenko's group reported a room-temperature method to fabricate monodisperse and isolable perovskite QDs with sizes from 3 to 13 nm.78 The processes of nucleation and growth were slowed down to 30 min by using trioctylphosphine oxide (TOPO) complexing with PbBr2 as the monomer of the Cs[PbBr3] solute and adding a long-chain zwitterion-lecithin at the end of formation. The slower nucleation and growth processes could be observed by in situ optical absorption and emission spectroscopy. The controllable synthesis of perovskite QDs will encourage researchers to obtain desirable facets for photocatalysis, which further helps understand the relationships between different facets of perovskite QDs and catalytic performance.
3.2 Surface modification
The surface states of semiconductors represent termination of the periodic arrangement of crystals. This would generate dangling bonds on the surface and alter the surficial band structure. Hence, the crystal surface state can affect surface energy, coordination number, molecular adsorption, interface carrier transfer, and so on. In this section, we focus on ligand modification and encapsulation to improve the stability and photocatalytic performance of halide perovskite QDs.Although many studies have utilized organic molecules as ligands to improve the stability of perovskite QDs and passivate the surface defects, it seemed that most studies also enhanced radiative recombination and fluorescence intensity.81 As depicted in Fig. 7a, common passivated organic molecules include alkylammonium salts, alkylcarboxylic/alkylphosphonic/alkylsulfonic acids, thiol/thiolate, zwitterionic molecules, and so on.82,83 However, to realize the application in photocatalysis, the specific modified ligands should boost the stability and facilitate carrier transfer simultaneously.84 Li et al. used thionyl bromide (SOBr2) to promote the stripping of original ligands (OA/OAm) and compensate Br vacancies on CsPbBr3 NCs to improve the stability, which could also facilitate the charge transfer and separation from CsPbBr3 to g-C3N4 (Fig. 7b).27 CsPbBr3–SOBr2 showed an enhanced CO2 reduction activity (total electron consumption: 69 μmol g−1 h−1), which was about 6-fold higher than that of pristine CsPbBr3. After further combination with g-C3N4, the photocatalytic CO2 yield reached 190 μmol g−1 h−1. Li et al. selected tetrafluoroborate salts (BF4−) as a defect treatment agent and then loaded Co2+ as a cocatalyst.85 The result showed that CsPbBr3–BF4 had a photocatalytic CO2 performance of 83.8 μmol g−1 h−1 electron consumption, demonstrating that such surface molecules could reduce carrier recombination and facilitate charge separation.
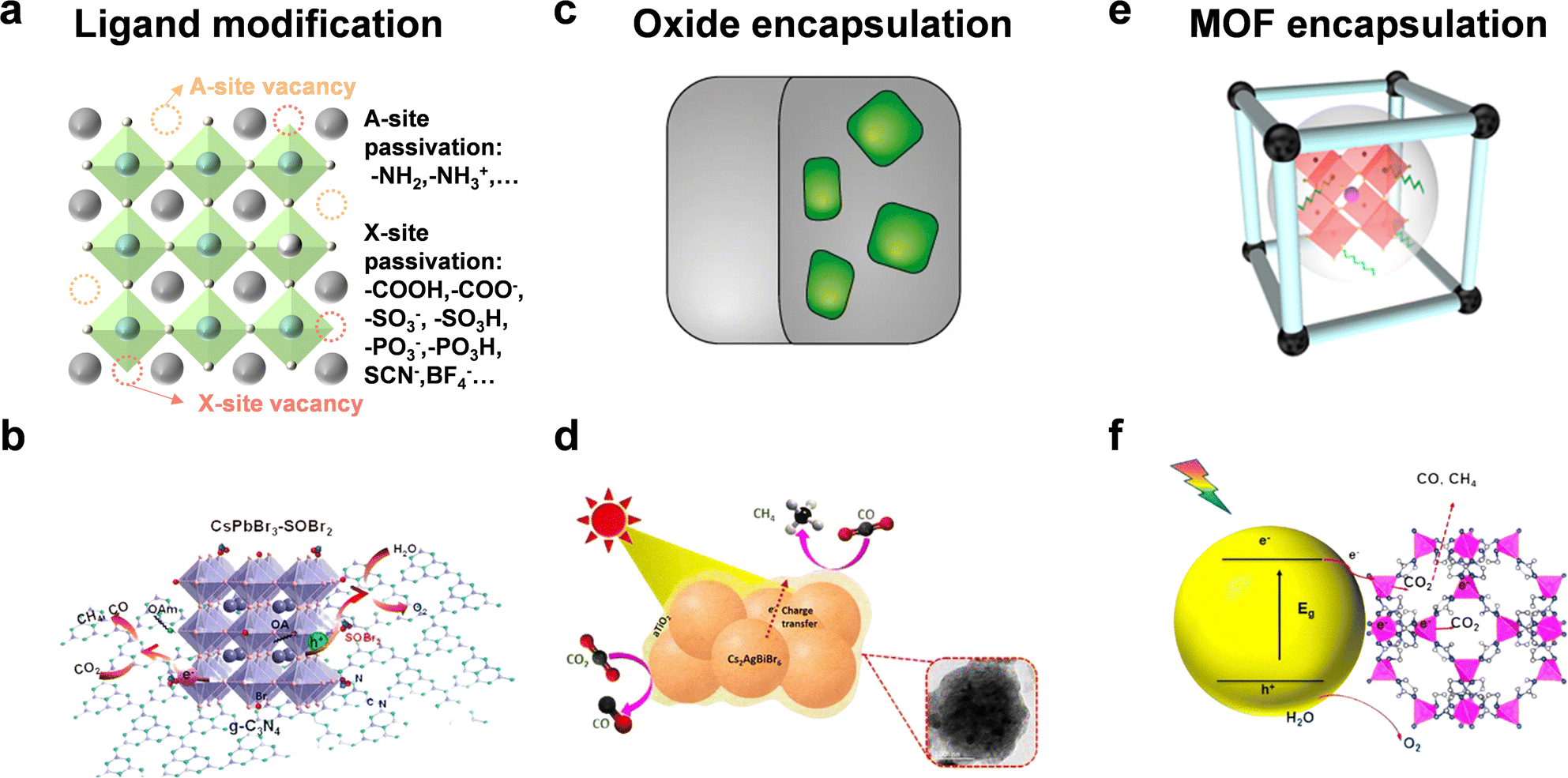 | ||
| Fig. 7 (a) Ligand modification; reproduced from ref. 83 with permission from Springer Nature, copyright 2020. (b) CsPbBr3 with SOBr2 modification for photocatalytic CO2 reduction; reproduced from ref. 27 with permission from American Chemical Society, copyright 2022. (c) Oxide encapsulation; reproduced from ref. 35 with permission from Springer Nature, copyright 2018. (d) Photocatalytic CO2 reduction of TiO2-encapsulated Cs2AgBiBr6 NCs; reproduced from ref. 87 with permission from American Chemical Society, copyright 2022. (e) MOF encapsulation; reproduced from ref. 91 with permission from Wiley-VCH, copyright 2022. (f) CsPbBr3@zeolitic nanocomposite for photocatalytic CO2 reduction. Reproduced from ref. 92 with permission from American Chemical Society, copyright 2018. | ||
A protective layer of metal oxide can isolate perovskite QDs from the external environment and benefit charge transfer (Fig. 7c). As shown in Fig. 7d, Satapathi et al. fabricated amorphous TiO2-coated Cs2AgBiBr6 NCs via the anti-solvent recrystallization method.87 In contrast with pristine Cs2AgBiBr6, the Cs2AgBiBr6-amorphous TiO2 nanocomposite showed an 11-fold enhancement of CO (22.87 μmol g−1) and CH4 (33.86 μmol g−1) production for 2 h reaction because of more efficient charge separation and transfer. Some other oxide semiconductors, such as SnO2,88 and ZnO,89 also allow great potential for photocatalytic CO2 reduction.
Metal organic frameworks (MOFs) are good candidates for photocatalytic CO2 reduction because of the controllable porous structure, high surface area, superior conductivity, and adsorption ability.90 The integration of MOFs and perovskite QDs offers more possibilities for CO2 transformation into valuable products (Fig. 7e).91 Su's group designed cobalt-based zeolitic imidazolate frameworks (ZIFs) to coat on the surface of CsPbBr3 QDs (Fig. 7f).92 The coated ZIFs could promote CO2 activation and improve CO2 photoconversion as a co-catalyst for photocatalysis. After optimization, CsPbBr3@ZIF-8 and CsPbBr3@ZIF-67 displayed total electron consumption rates of 15.498 and 29.630 μmol g−1 h−1, respectively, which were 1.39- and 2.66-fold higher than that of CsPbBr3 QDs.
Besides, some conductive materials have also been used to coat perovskite QDs for photocatalytic CO2 reduction. For example, MXenes, a member of 2D conductive materials, was utilized to encapsulate Cs2AgBiBr6 NCs to facilitate efficient charge separation and transport.93 To conclude, surface modification is an effective measure to maintain structural stability of perovskite QDs. If these nanocomposites are applied for photocatalysis, it is necessary that these ligands or coating materials can facilitate charge migration and separation.
3.3 Heterojunction construction
To promote photogenerated carrier transfer and separation, many scientists have constructed a series of interfacial heterojunctions. Because of the carrier confinement effect of a type-I heterojunction and without synergistic effects of a type-III heterojunction, they are not preferable for photocatalysis and will not be discussed in this section. Here, we mainly focus on these heterojunctions: a Schottky heterojunction, type-II heterojunction, direct Z-scheme heterojunction, and all-solid Z-scheme heterojunction, as shown in Fig. 8. Recently, many reports have fabricated different types of perovskite-based heterostructures for photocatalytic CO2 reduction,94,95 which are further summarized and discussed below.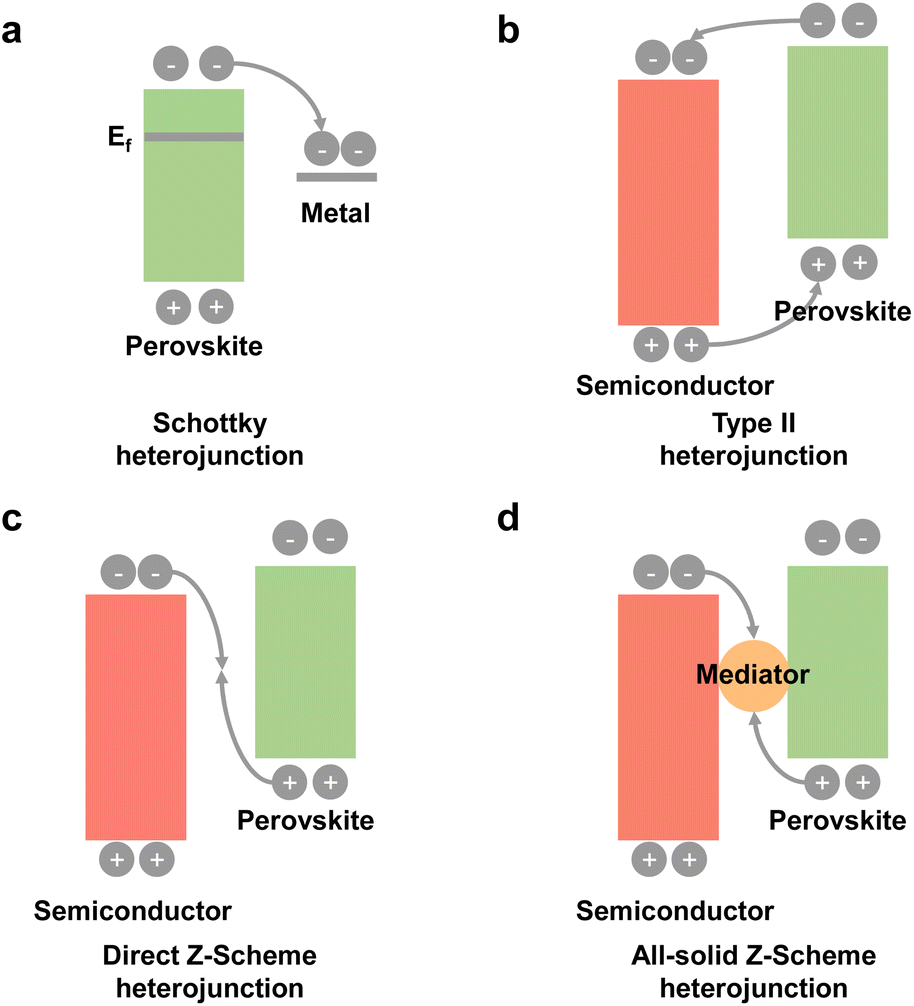 | ||
| Fig. 8 Schematic illustration of heterojunctions under irradiation: a (a) Schottky heterojunction; (b) type II heterojunction; (c) direct Z-scheme heterojunction; (d) all-solid Z-scheme heterojunction. Ef: work function. | ||
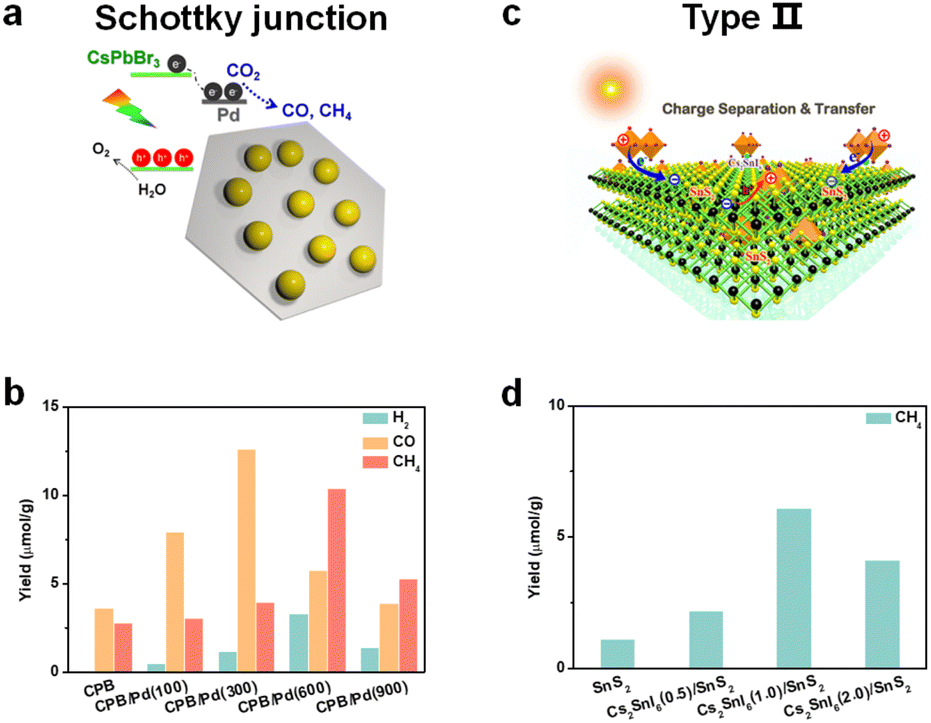 | ||
| Fig. 9 (a) CsPbBr3/Pd nanosheet Schottky junction for CO2 reduction; reproduced from ref. 98 with permission from American Chemical Society, copyright 2018. (b) Photocatalytic CO2 reduction performances after 3 h of constant illumination; 100, 300, 600, and 900 mean the volume (μL) of added Pd nanosheets; CsPbBr3: abbreviated as CPB; reproduced from ref. 98 with permission from American Chemical Society, copyright 2018. (c) Cs2SnI6/SnS2 nanosheet type-II heterojunction for CO2 reduction; reproduced from ref. 106 with permission from American Chemical Society, copyright 2010. (d) Photocatalytic CO2 reduction to CH4 of SnS2, Cs2SnI6(0.5)/SnS2, Cs2SnI6(1.0)/SnS2, and Cs2SnI6(2.0)/SnS2 nanocomposites; 0.5, 1.0, and 2.0 correspond to the concentration (1.0 mM, 2.0 mM, and 4.0 mM) of CsI; reproduced from ref. 106 with permission from American Chemical Society, copyright 2019. | ||
Some reports found that the surface plasmon resonance (SPR) effect caused by the interaction between the incident photons and the conduction electrons of metal nanoparticles also contributed to photocatalysis.99,100 Kuang and cooperators synthesized Au decorated CsPbBr3 nanoparticles.101 Under light illumination (λ > 420 nm), photogenerated electrons could be transferred from CsPbBr3 to Au, but when irradiated with longer wavelength light (λ > 580 nm), the induced hot electrons of Au from the SPR effect could be transferred to CsPbBr3 and further facilitate photocatalytic CO2 reduction. Besides, prolonged stability of the Schottky heterostructure is also a big issue.102 After long-term irradiation, the precious metal nanoparticles would accumulate and induce a drop in photocatalytic activity. Thus, the use of precious metals to construct Schottky heterojunctions deserves more attention.
Gong's group synthesized a 0D/0D type-II CsPbBr3/CdSe heterojunction by an in situ growth method.105 DFT calculations showed that there was a strong electronic coupling of Pb–Se and Br–Cd bonds, which facilitated fast electron transfer and separation. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) found that the intermediate Cd–C2O4− adduct could enable efficient CO2 activation and accelerate CO production. With the help of water to provide electrons and protons, the yield of CO reached 115.26 μmol g−1 under visible light illumination for 3 h, which was about 4.6-fold higher than that of CsPbBr3 QDs. Kuang et al. constructed hybrid Cs2SnI6/SnS2 nanosheets via in situ synthesis for the first time (Fig. 9c).106 Ultraviolet photoelectron spectroscopy (UPS) analysis and theoretical calculations showed that the co-sharing of a Sn atom makes Cs2SnI6/SnS2 come into intimate contact with a type-II heterojunction. Under visible-light illumination, the CH4 yield of Cs2SnI6/SnS2 was 6.09 μmol g−1, nearly 5.4 times higher than that of SnS2 (Fig. 9d), which originated from the improved light absorption and charge separation after incorporation of Cs2SnI6.
Even though it is not easy to construct a Z-scheme heterojunction, some reports have successfully fabricated a Z-scheme heterojunction. Xie et al. synthesized a series of SnS2/CsPbBr3 heterostructures for photocatalytic CO2 reduction (Fig. 10a).107 Interface and surface carrier dynamic investigations confirmed that the SnS2/CsPbBr3 heterojunctions satisfied direct Z-scheme transfer dynamics and could effectively improve carrier separation efficiency. As shown in Fig. 10b, SnS2/CsPbBr3 showed much higher CO production than pristine CsPbBr3 (marked as 0% SnS2 in Fig. 10b). Li's group used Cs2AgBiBr6 and Sr2FeNbO6 to construct a Z-scheme heterostructure.108In situ light-irradiation X-ray photoelectron spectroscopy (XPS) spectra and theoretical calculations confirmed that the Cs2AgBiBr6/Sr2FeNbO6 composites belonged to a direct Z-scheme heterojunction. With the irradiation of visible light (λ ≥ 420 nm), the photocatalytic CO and CH4 yields reached 50.00 and 8.12 μmol g−1 h−1, respectively. Li's group also fabricated a 0D/2D Cs2AgBiBr6/Bi2WO6 heterojunction, and the photocatalytic results showed that the Cs2AgBiBr6/Bi2WO6 heterojunction achieved 7-fold higher activity than pristine Cs2AgBiBr6 under simulated solar light (AM 1.5G).109 Besides, the CsPbBr3/Bi2WO6Z-scheme heterojunctions have also been reported.19,110
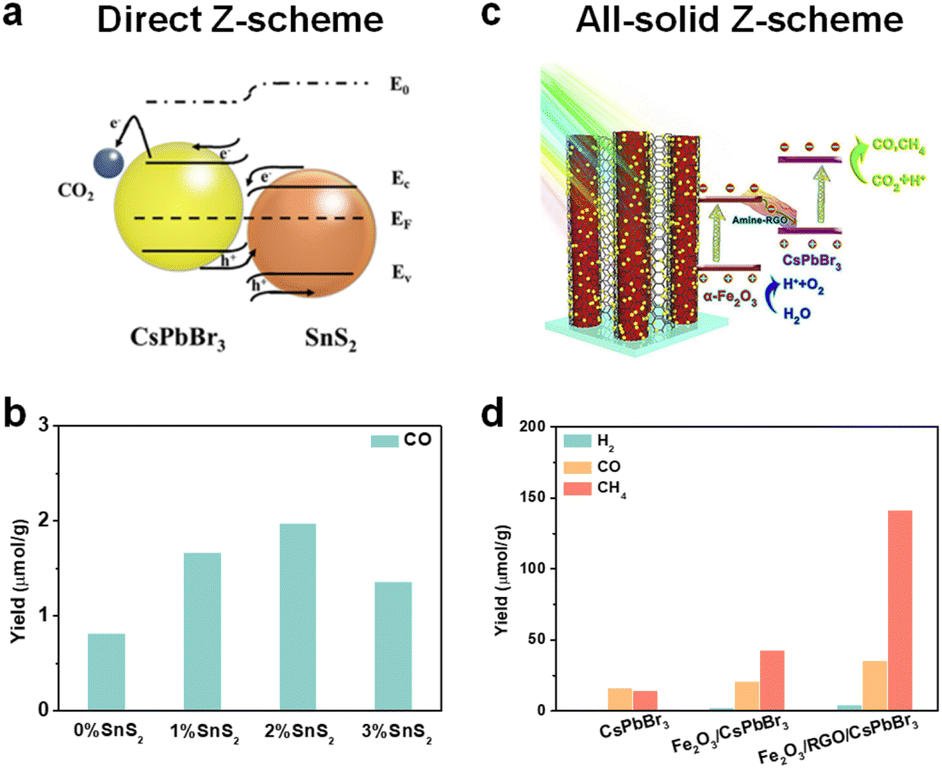 | ||
| Fig. 10 (a) Direct Z-scheme SnS2/CsPbBr3 heterojunction for CO2 reduction; reproduced from ref. 107 with permission from Wiley-VCH, copyright 2022. (b) Photocatalytic CO2 reduction performance of SnS2/CsPbBr3 heterojunctions under illumination; the percentage represents the added SnS2; reproduced from ref. 107 with permission from Wiley-VCH, copyright 2022. (c) All-solid-state Z-scheme α-Fe2O3/Amine-RGO/CsPbBr3 heterojunctions for photocatalytic CO2 reduction; reproduced from ref. 110 with permission from Elsevier, copyright 2020. (d) Photocatalytic CO2 reduction performances of CsPbBr3, α-Fe2O3/CsPbBr3, and α-Fe2O3/Amine-RGO (RGO)/CsPbBr3 after 15 h irradiation; reproduced from ref. 110 with permission from Elsevier, copyright 2020. | ||
In addition, a perovskite-based all-solid Z-scheme heterojunction has also been reported. As shown in Fig. 10c, Kuang et al. constructed a CsPbBr3/reduced graphene oxide (RGO)/Fe2O3Z-scheme heterojunction for photocatalytic CO2 reduction.110 The effective Z-scheme electron transfer path from the CB of Fe2O3 to the VB of CsPbBr3 was confirmed by electron spin resonance (ESR) analysis. Under irradiation, photogenerated electrons and holes could be effectively separated for CO2 reduction at CsPbBr3 and H2O oxidation at Fe2O3, respectively. In contrast with CsPbBr3 and Fe2O3/CsPbBr3, this effective photogenerated carrier separation and transfer of the Z-scheme Fe2O3/Amine-RGO/CsPbBr3 heterojunction enabled highly efficient CO2 photoreduction. As depicted in Fig. 10d, after 15 h irradiation, the Fe2O3/Amine-RGO/CsPbBr3 heterojunction had the highest yield of CO (35.47 μmol g−1) and CH4 (141.81 μmol g−1).
In short, the Z-scheme heterojunction shows tremendous advantages for photocatalysis owing to high redox potential, high-efficiency charge separation, and wide light-harvesting. However, the construction of a Z-scheme heterojunction is very complicated on account of interfacial interaction, the internal electric field, and band energy requirement.
4. Challenges and opportunities
Due to remarkable photophysical and photochemical properties, halide perovskite QDs display great potentials for photocatalytic CO2 reduction, as summarized in Table 1. However, severe instability and charge recombination inhibit the photocatalytic CO2 reduction of perovskite QDs. Although some useful strategies have been carried out and some progress has been made for photocatalytic CO2 reduction, many challenges should also be highlighted and addressed before practical application, such as, long-term stability, Pb-free perovskite, Z-scheme formation scheme, and theoretical calculations (Fig. 11).| Photocatalyst | Light source | Solution | Product (μmol g−1 h−1) | Measured stability | Ref. |
|---|---|---|---|---|---|
| CsPbBr3 | 100 W Xe lamp, AM 1.5G | Ethyl acetate | CO, 4.3 | 8 h | 76 |
| CH4, 1.5 | |||||
| H2, 0.1 | |||||
| CsPbBr3/GO | 100 W Xe lamp AM 1.5G, 150 mW cm−2 | Ethyl acetate | CO, 4.9 | 12 h | 18 |
| CH4, 2.5 | |||||
| H2, 0.13 | |||||
| Cs2AgBiBr6/MXene | 300 W Xe lamp, 420 nm filter | Water vapor | CO, 11.1 | 5 h | 93 |
| CH4, 1.3 | |||||
| H2, 8.9 | |||||
| CsPbBr3@ZIF-67 | 100 W Xe lamp, AM 1.5G, 150 mW cm−2 | Water vapor | CO, 0.77 | 18 h | 92 |
| CH4, 3.51 | |||||
| Cs2SnI6/SnS2 | 150 mW cm−2 visible light, 400 nm filter | Water/methanol vapor | CH4, 6.09 | 9 h | 106 |
| Cd:CsPbBr3 |
450 W Xe lamp, AM 1.5G, 150 mW cm−2 | Ethyl acetate/water | CO, 12.6 | 8 h | 130 |
| CH4, 26.5 | |||||
| CsPbBr3−xClx | 300 W Xe lamp, AM 1.5G, 200 mW cm−2 | Ethyl acetate | CO, 85, | 8 h | 74 |
| CH4, 12 | |||||
| Cs3Bi2Br9/TiO2 | 300 W Xe lamp, AM 1.5G, 70 mW cm−2 | Isopropanol | CO, 4.2 | 7 h | 23 |
| CH4, 32.9 | |||||
| CsPbBr3/CdSe | 300 W Xe lamp, AM 1.5G, 420 nm filter, 150 mW cm−2 | Ethyl acetate/H2O | CO, 38.4 | 9 h | 105 |
| CH4, 8.5 | |||||
| α-Fe2O3/Amine-RGO/CsPbBr3 | 150 W Xe lamp, AM 1.5G, 420 nm filter, 150 mW cm−2 | Water vapor | CO, 2.4 | 40 h | 110 |
| CH4, 9.5 | |||||
| H2, 0.3 |
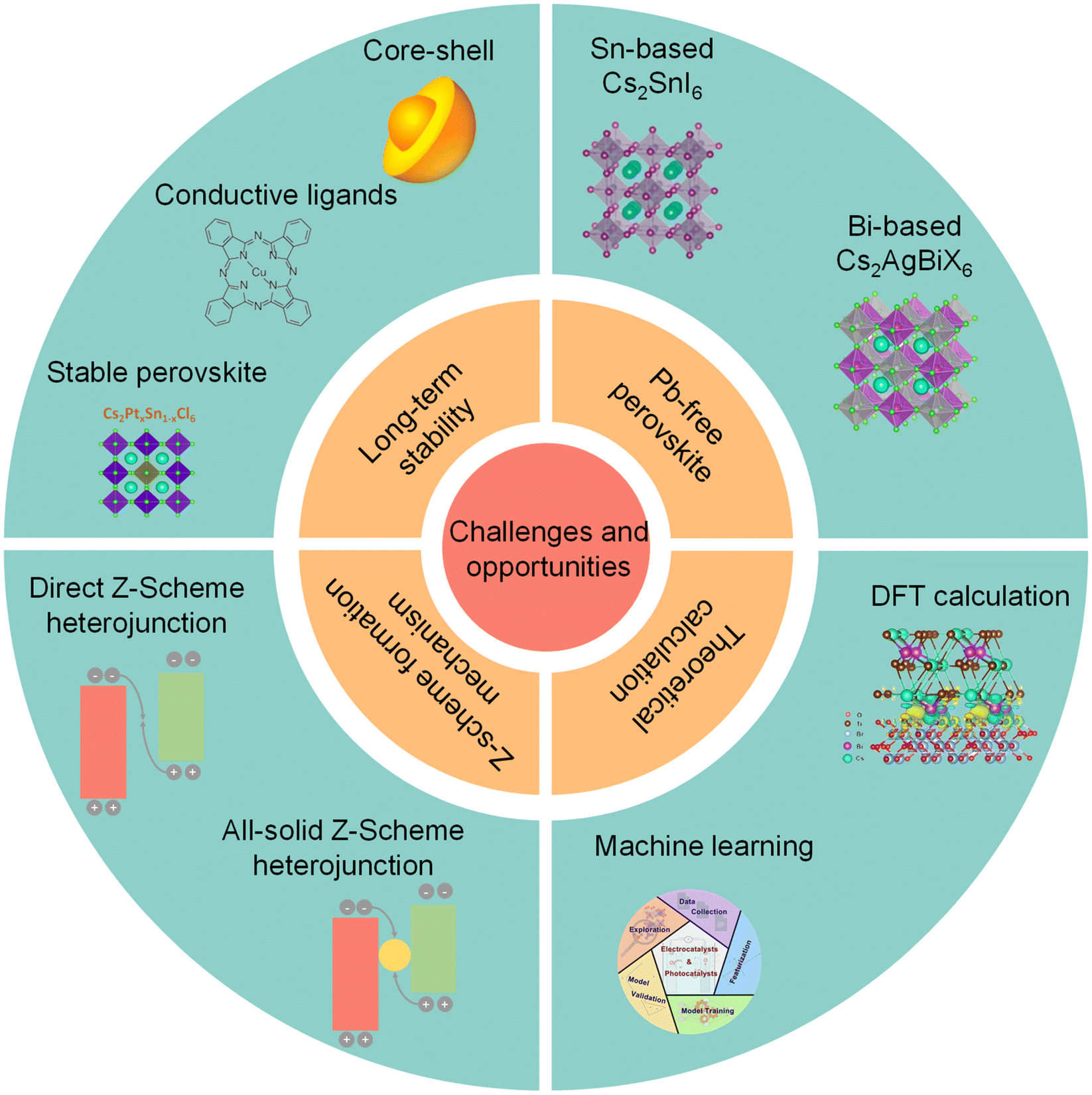 | ||
| Fig. 11 Challenges and opportunities of photocatalytic CO2 reduction on perovskite QDs. Insets are reproduced from ref. 35 with permission from Springer Nature, copyright 2018; reproduced from ref. 114 with permission from Springer Nature, copyright 2020; reproduced from ref. 116 with permission from Wiley-VCH, copyright 2021. Reproduced from ref. 127 with permission from Wiley-VCH, copyright 2021. Reproduced from ref. 128 with permission from American Chemical Society, copyright 2020. Reproduced from ref. 23 with permission from Wiley-VCH, copyright 2022. Reproduced from ref. 133 with permission from American Chemical Society, copyright 2022. | ||
4.1 Long-term stability
The intrinsic ionic properties of halide perovskite materials lead to high instability in a polar solvent.111–113 The labile ligands are easily desorbed from the surface of perovskite QDs. All these problems would affect the long-term stability of a perovskite photocatalyst. To promote structural stability of perovskite QDs, exploiting suitable ligands to substitute OA/OAm is a feasible strategy. However, many reported studies focus on passivating surface defects, reducing non-radiative recombination, and improving stability, which have been applied for LEDs and solar cells. Thus, it is necessary to design a suitable conductive ligand to improve long-term stability and facilitate charge transfer simultaneously.114 For instance, poly[(thiophene)-alt-(6,7-difluoro-2-(2-hexyldecyloxy)-quinoxaline)], a stable and conjugated molecule, has been used to improve the stability and efficiency of perovskite solar cells.115In fact, one of the most effective strategies is to construct a core–shell structure, which can preserve perovskite QDs from outer adverse environments. Unfortunately, recent studies just utilized perovskite-based core–shell nanoparticles to improve water-resistivity. Because of insulation or band misalignment, it seems that these shells cannot realize charge transfer and separation. Furthermore, the synthesis of perovskite-based core–shell nanoscale heterostructures has not been realized. A stable conductive shell can maintain the structural stability of core perovskite QDs and facilitate electron transfer for photocatalytic CO2 reduction. Additionally, existing studies concentrate on the stability of the intrinsic unstable perovskite for photocatalytic CO2 reduction. It should be noted that if we can synthesize perovskite that is intrinsically stableagainst water, it is much better for photocatalytic CO2 reduction, e.g., Cs2PtxSn1−xCl6.116
Besides, low-dimensional halide perovskites allow great possibility for photocatalytic CO2 reduction and solar fuel production.117,118 In contrast with three-dimensional perovskite, two-dimensional (2D) perovskite generally has higher stability due to the hydrophobic bulky organic A-site cations in the structure.119,120 Moreover, 2D perovskites have more surface defects because of their large specific area, rendering them good candidates for photocatalysis.121 Li's group realized the stabilization of 2D hybrid perovskites in aqueous solution and found that the length of A-site organic cations could affect the nanostructures, electron transfer, and photocatalytic activity process significantly.117
Recently, thiocyanates (SCN−) and formates (HCOO−) have been utilized to synthesize stable pseudo-halide perovskite materials for application in solar cells.122 Compared to halide perovskite, these pseudo-halide perovskites have better stability at room temperature because of the SCN− or HCOO− anion in the axial positions of the lead halide octahedron which results in an asymmetric electronic structure to maintain the desired framework.123 The tunable band structure and relatively low exciton binding energy of pseudo-halide perovskites also contribute to obtaining desirable bandgaps and electron transfer. From these perspectives, the pseudo-halide perovskite materials also show promising prospects for photocatalytic CO2 reduction.
4.2 Pb-free perovskite
Due to the high toxicity of Pb, Pb-based perovskite would induce severe environmental pollution, which stops practical industrial application.124,125 Unlike solar cells, whose toxicity could be mitigated by device encapsulation, a perovskite photocatalyst would directly come into contact with the surroundings. To solve this problem, some Pb-free perovskite QDs have been obtained, such as Cs2SnI6,126,127 Cs2AgBiX6,128 and Cs2NaBiCl6.129 Nonetheless, the photocatalytic CO2 activities of these substitutions are lower than that of Pb-based perovskite QDs. As a result, much attention should be paid to finding stable low-toxic or non-toxic perovskite materials with high catalytic performance.1304.3 Z-scheme formation mechanism
In the past few years, a Z-scheme heterojunction has acquired increasing interest because of higher redox potentials than the type-II heterojunction. So far, some studies have constructed perovskite-based Z-scheme heterojunctions.23 Here are two questions to be addressed in the future. First, fabricating a Z-scheme heterojunction needs complicated conditions, including interfacial properties, surface ligands, band structures, and so on. To confirm whether a heterojunction corresponds to the Z-scheme, a series of advanced characterization studies should be carried out.131 For example, in situ XPS can demonstrate the changes in binding energy before and after light illumination. This can directly provide solid information to confirm the emergence of a Z-scheme heterojunction. Secondly, although an internal build-in electric field is a commonly used concept to explain the Z-scheme, the internal electric field also emerges in a type-II heterojunction. Therefore, the real formation mechanism of a Z-scheme heterojunction is still unclear for the whole catalysis field, and deserves more efforts from physicist, chemists, and materials scientists.1324.4 Theoretical calculations and machine learning
DFT calculations can be used to understand the photocatalytic mechanism. For example, Lu, et al. conducted DFT to figure out the high selectivity of Cs3Bi2Br9/mesoporous TiO2 and Cs2AgBiBr6/mesoporous TiO2 for photocatalytic CO2 reduction, and the results showed that the high selectivity was attributed to the Bi-adsorption-mediated hydrogenation of CO (*HCO) rather than CO desorption.23 Li's group used DFT to calculate the work function of CsPbBr3 and CsPbBr3 with Br vacancies to understand the different electron migration processes from CsPbBr3 to g-C3N4.27 Thus, DFT can provide valuable insights into the mechanism of photocatalytic CO2 reduction and heterojunction construction. Besides, machine learning is a suite of data-driven computational methods that uses a range of algorithms to build models based on patterns in data, which can be applied to predict new structures and materials.133 Recently, machine learning has been successfully utilized for the discovery and prediction of functional materials, such as electrocatalysts, photocatalysts, and optoelectronics.134–136 For example, Chan et al. used machine learning and high-throughput computations to design mixed cation halide perovskite alloys.137 Based on ABX3 halide perovskite alloys where mixing occurs at the B-site, ∼18000 novel compounds were proposed and 392 were screened to be stable with suitable band gaps and defect tolerance. Hence, machine learning approaches can also be utilized to identify stable halide perovskite compositions and design new perovskite photocatalysts.
In conclusion, composition optimization, surface modification, and heterojunction construction are effective strategies to promote superior stability and efficient photocatalytic performance, which have been summarized in this review. However, it is still a challenge to realize long-term stability and high catalytic activity for perovskite-based photocatalysts. Construction of core–shell heterostructures and discovery of low-dimensional perovskite materials are promising approaches to overcome these obstacles. Besides, the photocatalytic CO2 reduction path and Z-scheme formation mechanism should be further understood. DFT calculations, machine learning, and operando characterization can provide reliable and detailed information, which may help researchers understand these complicated processes.137 Furthermore, to design and find low-lead or free-lead perovskite photocatalysts holds great promise for environment protection and practical applications. We hope that this review could provide some assistance for rational design of perovskite-based photocatalysts and attract more researchers to this promising research field.
Author contributions
W. S., G. Q, and B. L. conceived the idea and wrote the paper. B. L. supervised the project. All the authors contributed to the overall scientific interpretation and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National University of Singapore (A-0001423-06-00) and the Singapore National Research Foundation (A-000916-01-00).Notes and references
- X. Gao and W. Yang, Chem, 2020, 6, 1041–1042 CAS.
- Y. Wang, E. Chen and J. Tang, ACS Catal., 2022, 12, 7300–7316 CrossRef CAS PubMed.
- G. Q. Zhao, J. Hu, X. Long, J. Zou, J. G. Yu and F. P. Jiao, Small, 2021, 17, 2102155 CrossRef CAS PubMed.
- E. Gong, S. Ali, C. B. Hiragond, H. S. Kim, N. S. Powar, D. Kim, H. Kim and S.-I. In, Energy Environ. Sci., 2022, 15, 880–937 RSC.
- Y. Wei, F. You, D. Zhao, J. Wan, L. Gu and D. Wang, Angew. Chem., Int. Ed. Engl., 2022, 61, e202212049 CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- W. Song, Y. Wang, C. Wang, B. Wang, J. Feng, W. Luo, C. Wu, Y. Yao and Z. Zou, ChemCatChem, 2021, 13, 1711–1716 CrossRef CAS.
- D. Li, K. Yang, J. Lian, J. Yan and S. Liu, Adv. Energy Mater., 2022, 12, 2201070 CrossRef CAS.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- Y. Hou, E. Aydin, M. De Bastiani, C. Xiao, F. H. Isikgor, D. J. Xue, B. Chen, H. Chen, B. Bahrami, A. H. Chowdhury, A. Johnston, S. W. Baek, Z. Huang, M. Wei, Y. Dong, J. Troughton, R. Jalmood, A. J. Mirabelli, T. G. Allen, E. Van Kerschaver, M. I. Saidaminov, D. Baran, Q. Qiao, K. Zhu, S. De Wolf and E. H. Sargent, Science, 2020, 367, 1135–1140 CrossRef CAS PubMed.
- J. Peng, F. Kremer, D. Walter, Y. Wu, Y. Ji, J. Xiang, W. Liu, T. Duong, H. Shen, T. Lu, F. Brink, D. Zhong, L. Li, O. Lee Cheong Lem, Y. Liu, K. J. Weber, T. P. White and K. R. Catchpole, Nature, 2022, 601, 573–578 CrossRef CAS PubMed.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed.
- D. Ma, K. Lin, Y. Dong, H. Choubisa, A. H. Proppe, D. Wu, Y. K. Wang, B. Chen, P. Li, J. Z. Fan, F. Yuan, A. Johnston, Y. Liu, Y. Kang, Z. H. Lu, Z. Wei and E. H. Sargent, Nature, 2021, 599, 594–598 CrossRef CAS PubMed.
- C. R. Kagan, E. Lifshitz, E. H. Sargent and D. V. Talapin, Science, 2016, 353, aac5523 CrossRef PubMed.
- K. Sakhatskyi and M. V. Kovalenko, Light Sci. Appl., 2022, 11, 271 CrossRef CAS PubMed.
- H. Tsai, S. Shrestha, R. A. Vilá, W. Huang, C. Liu, C.-H. Hou, H.-H. Huang, X. Wen, M. Li, G. Wiederrecht, Y. Cui, M. Cotlet, X. Zhang, X. Ma and W. Nie, Nat. Photon., 2021, 15, 843–849 CrossRef.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- Y. F. Xu, M. Z. Yang, B. X. Chen, X. D. Wang, H. Y. Chen, D. B. Kuang and C. Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed.
- Y. Jiang, H. Y. Chen, J. Y. Li, J. F. Liao, H. H. Zhang, X. D. Wang and D. B. Kuang, Adv. Funct. Mater., 2020, 30, 2004293 CrossRef CAS.
- M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Angew. Chem., Int. Ed. Engl., 2018, 130, 13758–13762 CrossRef.
- Z. J. Li, E. Hofman, J. Li, A. H. Davis, C. H. Tung, L. Z. Wu and W. Zheng, Adv. Funct. Mater., 2017, 28, 1704288 CrossRef.
- H. Huang, D. Verhaeghe, B. Weng, B. Ghosh, H. Zhang, J. Hofkens, J. A. Steele and M. B. J. Roeffaers, Angew. Chem., Int. Ed. Engl., 2022, 61, e202203261 CAS.
- Q. M. Sun, J. J. Xu, F. F. Tao, W. Ye, C. Zhou, J. H. He and J. M. Lu, Angew. Chem., Int. Ed. Engl., 2022, 61, e202200872 CAS.
- J. Yuan, H. Liu, S. Wang and X. Li, Nanoscale, 2021, 13, 10281–10304 RSC.
- J. T. DuBose and P. V. Kamat, ACS Energy Lett., 2022, 7, 1994–2011 CrossRef CAS.
- J. Wang, Y. Shi, Y. Wang and Z. Li, ACS Energy Lett., 2022, 7, 2043–2059 CrossRef CAS.
- Q. Zheng, J. Wang, X. Li, Y. Bai, Y. Li, J. Wang, Y. Shi, X. Jiang and Z. Li, ACS Mater. Lett., 2022, 4, 1638–1645 CrossRef CAS.
- R. Das, A. Patra, S. K. Dutta, S. Shyamal and N. Pradhan, J. Am. Chem. Soc., 2022, 144, 18629–18641 CrossRef CAS PubMed.
- S. Chen, H. Yin, P. Liu, Y. Wang and H. Zhao, Adv. Mater., 2022, 2203836 Search PubMed.
- X. Ling, J. Yuan and W. Ma, Acc. Mater. Res., 2022, 3, 866–878 CrossRef CAS.
- Y. Tang, S. Tang, M. Luo, Y. Guo, Y. Zheng, Y. Lou and Y. Zhao, Chem. Commun., 2021, 57, 7465–7479 RSC.
- J. Y. Kim, J. W. Lee, H. S. Jung, H. Shin and N. G. Park, Chem. Rev., 2020, 120, 7867–7918 CrossRef CAS PubMed.
- J. A. Steele, H. Jin, I. Dovgaliuk, R. F. Berger, T. Braeckevelt, H. Yuan, C. Martin, E. Solano, K. Lejaeghere, S. M. J. Rogge, C. Notebaert, W. Vandezande, K. P. F. Janssen, B. Goderis, E. Debroye, Y. K. Wang, Y. Dong, D. Ma, M. Saidaminov, H. Tan, Z. Lu, V. Dyadkin, D. Chernyshov, V. Van Speybroeck, E. H. Sargent, J. Hofkens and M. B. J. Roeffaers, Science, 2019, 365, 679–684 CrossRef CAS PubMed.
- R. X. Yang and L. Z. Tan, J. Chem. Phys., 2020, 152, 034702 CrossRef CAS PubMed.
- Q. A. Akkerman, G. Raino, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS PubMed.
- E. M. Sanehira, A. R. Marshall, J. A. Christians, S. P. Harvey, P. N. Ciesielski, L. M. Wheeler, P. Schulz, L. Y. Lin, M. C. Beard and J. M. Luther, Sci. Adv., 2017, 3, eaao4204 CrossRef PubMed.
- J. W. Lee, S. Tan, S. I. Seok, Y. Yang and N. G. Park, Science, 2022, 375, eabj1186 CrossRef CAS PubMed.
- B. Chen, P. N. Rudd, S. Yang, Y. Yuan and J. Huang, Chem. Soc. Rev., 2019, 48, 3842–3867 RSC.
- X. K. Liu, W. Xu, S. Bai, Y. Jin, J. Wang, R. H. Friend and F. Gao, Nat. Mater., 2021, 20, 10–21 CrossRef CAS PubMed.
- D. P. Nenon, K. Pressler, J. Kang, B. A. Koscher, J. H. Olshansky, W. T. Osowiecki, M. A. Koc, L. W. Wang and A. P. Alivisatos, J. Am. Chem. Soc., 2018, 140, 17760–17772 CrossRef CAS PubMed.
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem., 2018, 2, 17105 Search PubMed.
- S. Navarro-Jaén, M. Virginie, J. Bonin, M. Robert, R. Wojcieszak and A. Y. Khodakov, Nat. Rev. Chem., 2021, 5, 564–579 CrossRef.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- J. Gan, J. He, R. L. Z. Hoye, A. Mavlonov, F. Raziq, J. L. MacManus-Driscoll, X. Wu, S. Li, X. Zu, Y. Zhan, X. Zhang and L. Qiao, ACS Energy Lett., 2019, 4, 1308–1320 CrossRef CAS.
- H. Huang, B. Pradhan, J. Hofkens, M. B. J. Roeffaers and J. A. Steele, ACS Energy Lett., 2020, 5, 1107–1123 CrossRef CAS.
- F. Krieg, S. T. Ochsenbein, S. Yakunin, S. Ten Brinck, P. Aellen, A. Suess, B. Clerc, D. Guggisberg, O. Nazarenko, Y. Shynkarenko, S. Kumar, C. J. Shih, I. Infante and M. V. Kovalenko, ACS Energy Lett., 2018, 3, 641–646 CrossRef CAS PubMed.
- J. Shamsi, Z. Dang, P. Ijaz, A. L. Abdelhady, G. Bertoni, I. Moreels and L. Manna, Chem. Mater., 2018, 30, 79–83 CrossRef CAS PubMed.
- S. Park, W. J. Chang, C. W. Lee, S. Park, H.-Y. Ahn and K. T. Nam, Nat. Energy, 2016, 2, 16185 CrossRef.
- X. Zhang, R. Tang, F. Li, R. Zheng and J. Huang, Sol. RRL, 2022, 6, 202101058 Search PubMed.
- J. A. Christians, P. A. Miranda Herrera and P. V. Kamat, J. Am. Chem. Soc., 2015, 137, 1530–1538 CrossRef CAS PubMed.
- M.-G. Ju, M. Chen, Y. Zhou, J. Dai, L. Ma, N. P. Padture and X. C. Zeng, Joule, 2018, 2, 1231–1241 CrossRef CAS.
- Y. Zhou and Y. Zhao, Energy Environ. Sci., 2019, 12, 1495–1511 RSC.
- N. Li, X. Chen, J. Wang, X. Liang, L. Ma, X. Jing, D. L. Chen and Z. Li, ACS Nano, 2022, 16, 3332–3340 CrossRef CAS PubMed.
- A. Senocrate, T. Acartürk, G. Y. Kim, R. Merkle, U. Starke, M. Grätzel and J. Maier, J. Mater. Chem. A, 2018, 6, 10847–10855 RSC.
- J. Chen, X. Hong, Y. Wang, X. Guan, R. Wang, Y. Wang, H. Du, Y. Zhang and S. Shen, J. Phys. Chem. Lett., 2022, 13, 1806–1824 CrossRef CAS PubMed.
- W. Chi and S. K. Banerjee, Chem. Mater., 2021, 33, 4269–4303 CrossRef CAS.
- T. Zhang, M. I. Dar, G. Li, F. Xu, N. Guo, M. Gratzel and Y. Zhao, Sci. Adv., 2017, 3, e1700841 CrossRef PubMed.
- Y. K. Wang, K. Singh, J. Y. Li, Y. Dong, X. Q. Wang, J. M. Pina, Y. J. Yu, R. Sabatini, Y. Liu, D. Ma, J. Liu, Z. Liu, Y. Gao, O. Voznyy, W. Ma, M. K. Fung, L. S. Liao and E. H. Sargent, Adv. Mater., 2022, 34, 2200854 CrossRef CAS PubMed.
- C. C. Lin, T. R. Liu, S. R. Lin, K. M. Boopathi, C. H. Chiang, W. Y. Tzeng, W. C. Chien, H. S. Hsu, C. W. Luo, H. Y. Tsai, H. A. Chen, P. C. Kuo, J. Shiue, J. W. Chiou, W. F. Pong, C. C. Chen and C. W. Chen, J. Am. Chem. Soc., 2022, 144, 15718–15726 CrossRef CAS PubMed.
- L. Li and Z. Zhang, Chem. Eng. J., 2022, 434, 134811 CrossRef CAS.
- C. Tang, C. Chen, W. Xu and L. Xu, J. Mater. Chem. A, 2019, 7, 6911–6919 RSC.
- C. H. Lu, G. V. Biesold-McGee, Y. Liu, Z. Kang and Z. Lin, Chem. Soc. Rev., 2020, 49, 4953–5007 RSC.
- Y. Gao, Y. Cui, J. Li, Y. Xu, J. Hu and T. He, J. Phys. Chem. C, 2022, 126, 6694–6699 CrossRef CAS.
- H. Bian, T. Liu, D. Li, Z. Xu, J. Lian, M. Chen, J. Yan and S. Frank Liu, Chem. Eng. J., 2022, 435, 135071 CrossRef CAS.
- Y. Guo, J. Su, L. Wang, Z. Lin, Y. Hao and J. Chang, J. Phys. Chem. Lett., 2021, 12, 3393–3400 CrossRef CAS PubMed.
- J. Zhu, L. Zhou, Y. Zhu, J. Huang, L. Hou, J. Shen, S. Dai and C. Li, Small, 2022, 18, 2104399 CrossRef CAS PubMed.
- M. T. Hoang, A. S. Pannu, C. Tang, Y. Yang, N. D. Pham, K. Gui, X. Wang, S. Yambem, P. Sonar, A. Du and H. Wang, Adv. Opt. Mater., 2020, 8, 202000742 Search PubMed.
- D. H. Parmar, J. M. Pina, H. Choubisa, G. Bappi, K. Bertens and E. H. Sargent, Adv. Mater., 2021, 33, 2008690 CrossRef CAS PubMed.
- J. Li, X. Du, G. Niu, H. Xie, Y. Chen, Y. Yuan, Y. Gao, H. Xiao, J. Tang, A. Pan and B. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 989–996 CrossRef PubMed.
- P. Li, Y. Lin, M. Ma, M. Zhang, J. Li, Z. Wang, V. Maheskumar, Z.-Y. Jiang and R. Zhang, ACS Appl. Energy Mater., 2022, 5, 1942–1952 CrossRef CAS.
- Y.-X. Feng, G.-X. Dong, K. Su, Z.-L. Liu, W. Zhang, M. Zhang and T.-B. Lu, J. Energy Chem., 2022, 69, 348–355 CrossRef CAS.
- N. H. Alotaibi, G. M. Mustafa, N. A. Kattan, Q. Mahmood, H. Albalawi, M. Morsi, H. H. Somaily, M. A. Hafez, H. I. Mahmoud and M. A. Amin, J. Solid State Chem., 2022, 313, 123353 CrossRef CAS.
- Z. Liu, H. Yang, J. Wang, Y. Yuan, K. Hills-Kimball, T. Cai, P. Wang, A. Tang and O. Chen, Nano Lett., 2021, 21, 1620–1627 CrossRef CAS PubMed.
- S.-H. Guo, J. Zhou, X. Zhao, C.-Y. Sun, S.-Q. You, X.-L. Wang and Z.-M. Su, J. Catal., 2019, 369, 201–208 CrossRef CAS.
- L. Chouhan, S. Ghimire, C. Subrahmanyam, T. Miyasaka and V. Biju, Chem. Soc. Rev., 2020, 49, 2869–2885 RSC.
- J. Hou, S. Cao, Y. Wu, Z. Gao, F. Liang, Y. Sun, Z. Lin and L. Sun, Eur. J. Chem., 2017, 23, 9481–9485 CrossRef CAS PubMed.
- S. Shyamal, S. K. Dutta, T. Das, S. Sen, S. Chakraborty and N. Pradhan, J. Phys. Chem. Lett., 2020, 11, 3608–3614 CrossRef CAS PubMed.
- Q. A. Akkerman, T. P. T. Nguyen, S. C. Boehme, F. Montanarella, D. N. Dirin, P. Wechsler, F. Beiglbock, G. Raino, R. Erni, C. Katan, J. Even and M. V. Kovalenko, Science, 2022, 377, 1406–1412 CrossRef CAS PubMed.
- W. Fu, A. G. Ricciardulli, Q. A. Akkerman, R. A. John, M. M. Tavakoli, S. Essig, M. V. Kovalenko and M. Saliba, Mater. Today, 2022, 58, 275–296 CrossRef CAS.
- S. Tan, T. Huang, I. Yavuz, R. Wang, M. H. Weber, Y. Zhao, M. Abdelsamie, M. E. Liao, H. C. Wang, K. Huynh, K. H. Wei, J. Xue, F. Babbe, M. S. Goorsky, J. W. Lee, C. M. Sutter-Fella and Y. Yang, J. Am. Chem. Soc., 2021, 143, 6781–6786 CrossRef CAS PubMed.
- Y. F. Lan, J. S. Yao, J. N. Yang, Y. H. Song, X. C. Ru, Q. Zhang, L. Z. Feng, T. Chen, K. H. Song and H. B. Yao, Nano Lett., 2021, 21, 8756–8763 CrossRef CAS PubMed.
- Y. Bai, M. Hao, S. Ding, P. Chen and L. Wang, Adv. Mater., 2022, 34, 2105958 CrossRef CAS PubMed.
- J. Xue, R. Wang and Y. Yang, Nat. Rev. Mater., 2020, 5, 809–827 CrossRef CAS.
- S. Liang, S. He, M. Zhang, Y. Yan, T. Jin, T. Lian and Z. Lin, J. Am. Chem. Soc., 2022, 144, 12901–12914 CrossRef CAS PubMed.
- J.-C. Wang, N. Li, A. M. Idris, J. Wang, X. Du, Z. Pan and Z. Li, Sol. RRL, 2021, 5, 202100154 Search PubMed.
- W. Song, D. Wang, J. Tian, G. Qi, M. Wu, S. Liu, T. Wang, B. Wang, Y. Yao, Z. Zou and B. Liu, Small, 2022, 18, 2204763 CrossRef CAS PubMed.
- S. Purohit, S. Shyamal, S. K. Saini, K. L. Yadav, M. Kumar and S. Satapathi, Energy Fuels, 2022, 36, 12170–12180 CrossRef CAS.
- H. Xiao, J. Fu, X. Wei, B. Wang, Q. Qian, J. Huang, R. Li and Z. Zang, Laser Photonics Rev., 2022, 16, 2200276 CrossRef CAS.
- Y. Jiang, J.-F. Liao, Y.-F. Xu, H.-Y. Chen, X.-D. Wang and D.-B. Kuang, J. Mater. Chem. A, 2019, 7, 13762–13769 RSC.
- Q. J. Wu, J. Liang, Y. B. Huang and R. Cao, Acc. Chem. Res., 2022, 55, 2978–2997 CrossRef CAS PubMed.
- C. Zhang, W. Li and L. Li, Angew. Chem., Int. Ed., 2021, 60, 7488–7501 CrossRef CAS PubMed.
- Z.-C. Kong, J.-F. Liao, Y.-J. Dong, Y.-F. Xu, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS.
- Z. Zhang, B. Wang, H.-B. Zhao, J.-F. Liao, Z.-C. Zhou, T. Liu, B. He, Q. Wei, S. Chen, H.-Y. Chen, D.-B. Kuang, Y. Li and G. Xing, Appl. Catal., B, 2022, 312, 121358 CrossRef CAS.
- Y.-H. Chen, J.-K. Ye, Y.-J. Chang, T.-W. Liu, Y.-H. Chuang, W.-R. Liu, S.-H. Liu and Y.-C. Pu, Appl. Catal., B, 2021, 284, 119751 CrossRef CAS.
- Y. Tang, C. H. Mak, G. Jia, K.-C. Cheng, J.-J. Kai, C.-W. Hsieh, F. Meng, W. Niu, F.-F. Li, H.-H. Shen, X. Zhu, H. M. Chen and H.-Y. Hsu, J. Mater. Chem. A, 2022, 10, 12296–12316 RSC.
- R. Tang, H. Sun, Z. Zhang, L. Liu, F. Meng, X. Zhang, W. Yang, Z. Li, Z. Zhao, R. Zheng and J. Huang, Chem. Eng. J., 2022, 429, 132137 CrossRef CAS.
- J.-N. Huang, Y.-J. Dong, H.-B. Zhao, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Mater. Chem. A, 2022, 10, 25212–25219 RSC.
- Y.-F. Xu, M.-Z. Yang, H.-Y. Chen, J.-F. Liao, X.-D. Wang and D.-B. Kuang, ACS Appl. Energy Mater., 2018, 1, 5083–5089 CrossRef CAS.
- T. Chen, M. Zhou, W. Chen, Y. Zhang, S. Ou and Y. Liu, Sustain. Energy Fuels, 2021, 5, 3598–3605 RSC.
- X. Huang, H. Li, C. Zhang, S. Tan, Z. Chen, L. Chen, Z. Lu, X. Wang and M. Xiao, Nat. Commun., 2019, 10, 1163 CrossRef PubMed.
- J.-F. Liao, Y.-T. Cai, J.-Y. Li, Y. Jiang, X.-D. Wang, H.-Y. Chen and D.-B. Kuang, J. Energy Chem., 2021, 53, 309–315 CrossRef CAS.
- J. Chakkamalayath, G. V. Hartland and P. V. Kamat, J. Phys. Chem. C, 2021, 125, 17881–17889 CrossRef CAS.
- S. R. Xu, J. L. Li, Q. L. Mo, K. Wang, G. Wu, Y. Xiao, X. Z. Ge and F. X. Xiao, Inorg. Chem., 2022, 61, 17828–17837 CrossRef CAS PubMed.
- S. Schunemann, M. van Gastel and H. Tuysuz, ChemSusChem, 2018, 11, 2057–2061 CrossRef PubMed.
- G. Yin, X. Qi, Y. Chen, Q. Peng, X. Jiang, Q. Wang, W. Zhang and X. Gong, J. Mater. Chem. A, 2022, 10, 22468–22476 RSC.
- X. D. Wang, Y. H. Huang, J. F. Liao, Y. Jiang, L. Zhou, X. Y. Zhang, H. Y. Chen and D. B. Kuang, J. Am. Chem. Soc., 2019, 141, 13434–13441 CrossRef CAS PubMed.
- R. Zhang, L. Li, Y. Li, D. Wang, Y. Lin and T. Xie, Sol. RRL, 2022, 6, 2200536 CrossRef CAS.
- A. Mahmoud Idris, S. Zheng, L. Wu, S. Zhou, H. Lin, Z. Chen, L. Xu, J. Wang and Z. Li, Chem. Eng. J., 2022, 446, 137197 CrossRef CAS.
- L. Wu, S. Zheng, H. Lin, S. Zhou, A. Mahmoud Idris, J. Wang, S. Li and Z. Li, J. Colloid Interface Sci., 2023, 629, 233–242 CrossRef CAS PubMed.
- Y. Jiang, J.-F. Liao, H.-Y. Chen, H.-H. Zhang, J.-Y. Li, X.-D. Wang and D.-B. Kuang, Chem, 2020, 6, 766–780 CAS.
- K. Sakhatskyi, R. A. John, A. Guerrero, S. Tsarev, S. Sabisch, T. Das, G. J. Matt, S. Yakunin, I. Cherniukh, M. Kotyrba, Y. Berezovska, M. I. Bodnarchuk, S. Chakraborty, J. Bisquert and M. V. Kovalenko, ACS Energy Lett., 2022, 7, 3401–3414 CrossRef CAS PubMed.
- J. Ma, M. Qin, P. Li, L. Han, Y. Zhang and Y. Song, Energy Environ. Sci., 2022, 15, 413–438 RSC.
- C. Fu, Z. Gu, Y. Tang, Q. Xiao, S. Zhang, Y. Zhang and Y. Song, Angew. Chem., Int. Ed., 2022, 61, e202117067 CAS.
- S. Fratini, M. Nikolka, A. Salleo, G. Schweicher and H. Sirringhaus, Nat. Mater., 2020, 19, 491–502 CrossRef CAS PubMed.
- L. Meng, C. Sun, R. Wang, W. Huang, Z. Zhao, P. Sun, T. Huang, J. Xue, J. W. Lee, C. Zhu, Y. Huang, Y. Li and Y. Yang, J. Am. Chem. Soc., 2018, 140, 17255–17262 CrossRef CAS PubMed.
- H. Yin, J. Chen, P. Guan, D. Zheng, Q. Kong, S. Yang, P. Zhou, B. Yang, T. Pullerits and K. Han, Angew. Chem., Int. Ed., 2021, 60, 22693–22699 CrossRef CAS PubMed.
- H. Wang, H. Zhang, J. Wang, Y. Gao, F. Fan, K. Wu, X. Zong and C. Li, Angew. Chem., Int. Ed., 2021, 60, 7376–7381 CrossRef CAS PubMed.
- L. Yan, J. Ma, P. Li, S. Zang, L. Han, Y. Zhang and Y. Song, Adv. Mater., 2022, 34, 2106822 CrossRef CAS PubMed.
- S. Peng, J. Ma, P. Li, S. Zang, Y. Zhang and Y. Song, Adv. Funct. Mater., 2022, 32, 2205289 CrossRef CAS.
- Q. Cao, P. Li, W. Chen, S. Zang, L. Han, Y. Zhang and Y. Song, Nano Today, 2022, 43, 101394 CrossRef CAS.
- M. Que, W. Cai, Y. Zhao, Y. Yang, B. Zhang, S. Yun, J. Chen and G. Zhu, J. Colloid Interface Sci., 2022, 610, 538–545 CrossRef CAS PubMed.
- P. Y. Lin, A. Loganathan, I. Raifuku, M. H. Li, Y. Y. Chiu, S. T. Chang, A. Fakharuddin, C. F. Lin, T. F. Guo, L. Schmidt-Mende and P. Chen, Adv. Energy Mater., 2021, 11, 2100818 CrossRef CAS.
- C. H. Liao, C. H. Chen, J. Bing, C. Bailey, Y. T. Lin, T. M. Pandit, L. Granados, J. Zheng, S. Tang, B. H. Lin, H. W. Yen, D. R. McCamey, B. J. Kennedy, C. C. Chueh and A. W. Y. Ho-Baillie, Adv. Mater., 2022, 34, 2104782 CrossRef CAS PubMed.
- H. Zhu, J. Ma, P. Li, S. Zang, Y. Zhang and Y. Song, Chem, 2022, 8, 2939–2960 CAS.
- H. Luo, P. Li, J. Ma, L. Han, Y. Zhang and Y. Song, Adv. Energy Mater., 2022, 12, 2201242 CrossRef CAS.
- P. Zhou, H. Chen, Y. Chao, Q. Zhang, W. Zhang, F. Lv, L. Gu, Q. Zhao, N. Wang, J. Wang and S. Guo, Nat. Commun., 2021, 12, 4412 CrossRef CAS PubMed.
- S. Ullah, J. Wang, P. Yang, L. Liu, J. khan, S.-E. Yang, T. Xia, H. Guo and Y. Chen, Sol. RRL, 2021, 5, 2000830 CrossRef CAS.
- D. J. Kubicki, M. Saski, S. MacPherson, K. Gal Kowski, J. Lewinski, D. Prochowicz, J. J. Titman and S. D. Stranks, Chem. Mater., 2020, 32, 8129–8138 CrossRef CAS PubMed.
- J. Pi, X. Jia, Z. Long, S. Yang, H. Wu, D. Zhou, Q. Wang, H. Zheng, Y. Yang, J. Zhang and J. Qiu, Adv. Energy Mater., 2022, 12, 2202074 CrossRef CAS.
- S. Bera, A. Patra, S. Shyamal, D. Nasipuri and N. Pradhan, ACS Energy Lett., 2022, 7, 3015–3023 CrossRef CAS.
- Y. Wang, X. Shang, J. Shen, Z. Zhang, D. Wang, J. Lin, J. C. S. Wu, X. Fu, X. Wang and C. Li, Nat. Commun., 2020, 11, 3043 CrossRef CAS PubMed.
- E. M. Akinoglu, D. A. Hoogeveen, C. Cao, A. N. Simonov and J. J. Jasieniak, ACS Nano, 2021, 15, 7860–7878 CrossRef CAS PubMed.
- H. Mai, T. C. Le, D. Chen, D. A. Winkler and R. A. Caruso, Chem. Rev., 2022, 122, 13478–13515 CrossRef CAS PubMed.
- R. Gomez-Bombarelli, J. Aguilera-Iparraguirre, T. D. Hirzel, D. Duvenaud, D. Maclaurin, M. A. Blood-Forsythe, H. S. Chae, M. Einzinger, D. G. Ha, T. Wu, G. Markopoulos, S. Jeon, H. Kang, H. Miyazaki, M. Numata, S. Kim, W. Huang, S. I. Hong, M. Baldo, R. P. Adams and A. Aspuru-Guzik, Nat. Mater., 2016, 15, 1120–1127 CrossRef CAS PubMed.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Nature, 2018, 559, 547–555 CrossRef CAS PubMed.
- S. Chmiela, H. E. Sauceda, K. R. Muller and A. Tkatchenko, Nat. Commun., 2018, 9, 3887 CrossRef PubMed.
- A. Mannodi-Kanakkithodi and M. K. Y. Chan, Energy Environ. Sci., 2022, 15, 1930–1949 RSC.
| This journal is © The Royal Society of Chemistry 2023 |