
Two-dimensional nanosheets of bimetallic chalcogenide-tagged nitrogen-doped carbon as a cathode for high-performance and durable zinc-ion capacitors†
Swati J.
Patil
 *ab,
Nilesh R.
Chodankar
a,
Seung-Kyu
Hwang
cd,
Pragati A.
Shinde
e,
Ganji Seeta
Rama Raju
a,
Kugalur Shanmugam
Ranjith
a,
Smita V.
Karekar
cd,
Yun-Suk
Huh
*cd and
Young-Kyu
Han
*a
*ab,
Nilesh R.
Chodankar
a,
Seung-Kyu
Hwang
cd,
Pragati A.
Shinde
e,
Ganji Seeta
Rama Raju
a,
Kugalur Shanmugam
Ranjith
a,
Smita V.
Karekar
cd,
Yun-Suk
Huh
*cd and
Young-Kyu
Han
*a
aDepartment of Energy and Material Engineering, Dongguk University, Seoul 04620, Republic of Korea. E-mail: swatipatil39@gmail.com; ykenergy@dongguk.edu
bDepartment of Biochemistry and Biophysics, Texas A&M University, College Station, TX 77843, USA
cDepartment of Biological Engineering, NanoBio High-Tech Materials Research Center, Inha University, Incheon 22212, Republic of Korea. E-mail: yunsuk.huh@inha.ac.kr
dDepartment of Biological Sciences and Bioengineering, Inha University, Incheon 22212, Republic of Korea
eDepartment of Sustainable and Renewable Energy Engineering, University of Sharjah, Sharjah 27272, United Arab Emirates
First published on 23rd December 2022
Abstract
Zn-ion capacitors (ZICs) are thought to be potential electrochemical energy storage devices due to their complementary energy density, which is comparable to that of batteries, and superior power characteristics when compared to capacitors. However, the electrochemical stability of current ZICs is inadequate due to the structural instability of the Zn anode associated with the dissolution of conventional cathode materials in conventional aqueous electrolytes. Given the rapid growth of metal chalcogenides in electrochemical energy storage applications, the use of bimetallic chalcogenide-based cathodes in ZICs are desirable. Two-dimensional (2D) bimetallic chalcogenide-tagged nitrogen-doped carbon (NbMo6S8/NC) was prepared as a cathode for use with modified conventional electrolytes in ZICs, affording a maximal specific capacity of 167.89 mA h g−1 at 0.25 A g−1 and superior rate capability. The developed NbMo6S8/NC ZIC delivered a battery-like specific energy of 188.87 and 50.22 W h kg−1 at supercapacitor-like power densities of 250 and 2500 W kg−1, respectively. In a flexible ZIC, the resulting NbMo6S8/NC nanosheets (∼10 nm) could withstand 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 charge–discharge cycles with an initial capacitive retention of 87.60%. A pair of flexible NbMo6S8/NC ZICs connected in series successfully powered light-emitting diodes, demonstrating high potential applications. These findings suggest that NbMo6S8/NC ZIC is capable of driving portable and smart electronics, demonstrating its functionality in real-world applications.
000 charge–discharge cycles with an initial capacitive retention of 87.60%. A pair of flexible NbMo6S8/NC ZICs connected in series successfully powered light-emitting diodes, demonstrating high potential applications. These findings suggest that NbMo6S8/NC ZIC is capable of driving portable and smart electronics, demonstrating its functionality in real-world applications.
Introduction
Despite the high energy-storage capacity of organic-media-based batteries (Li-, Na-, and K-ion batteries) and capacitors (Li-, Na-, and K-ion capacitors), safety remains a pervasive issue that has impeded their widespread application.1–3 Aqueous batteries and capacitors have recently been proposed as alternatives to conventional energy storage systems (EESs) that employ organic media owing to the high safety, low cost, environmental friendliness, and fast charging capability of the former.4–7 Metal-ion hybrid capacitors have gradually gained popularity for clean energy storage using a variety of cations, including monovalent (e.g., Li+, Na+, and K+) and multivalent (e.g., Mg2+, Zn2+, Ni2+, Ca2+, and Al3+) ions.8–11 Specifically, in Zn-ion capacitors (ZICs), the Zn metal anode undergoes fast surface stripping/plating, and ZICs possess a high theoretical energy storage capacity of 823 mA h g−1.12,13 Furthermore, zinc metal is highly compatible with water, and its relatively low redox potential (−0.76 vs. the standard hydrogen electrode) broadens the operating voltage window of aqueous EES systems, with extremely high safety, thereby increasing the energy and power density. Furthermore, Zn ions are more abundant and have relatively weak activity compared to monovalent Li, Na, K, and other ions. These features have fueled the development of mature techniques for producing ZICs with high energy density, where these devices are economic and environmentally friendly.14Mild aqueous electrolytes are non-flammable and “green,” and their ionic conductivity is two orders of magnitude greater than that of organic electrolytes, resulting in significantly improved round-trip efficiency and energy density. However, in aqueous zinc-ion electrolytes, Zn electrodes have a limited lifetime because of dendrite growth and the formation of insulating zinc oxide/hydroxide during use.15 These features are associated with the poor reversibility of the Zn metal anode, where the side reactions include the hydrogen evolution reaction (HER), corrosion, and uneven Zn plating.16 Although conventional aqueous electrolytes contain a large number of free water molecules, several issues must be addressed to meet the anticipated challenges of energy storage. To achieve fast ameliorative transport of the electrolyte ions while reducing the activity of water to mitigate some water-related side reactions and effectively increase the efficiency and operating stability of the electrochemical system, a class of super-concentrated aqueous-based electrolytes, with Zn salt as the main component, has been proposed to overcome the limitations associated with conventional aqueous electrolytes; however, these electrolytes have not been investigated in detail.17,18
An additional critical issue for Zn-based EESs is the cathode material and the dissolution and structural degradation of these materials in traditional Zn-based electrolytes. The most utilized cathode materials, such as MnO2 and V2O5, undergo strong dissolution in Zn-based electrolytes. Consequently, after a few hundred cycles (300–500 cycles), the Zn-based EES loses its electrochemical properties such as energy efficiency and charge-storage capacity.19–22 Hence, it is essential to identify highly efficient yet stable cathode materials to assemble high-performance Zn-based EES cells. Recently, Nb-based materials, such as oxides and chalcogenides, have been reported for energy storage and energy conversion applications owing to their specific features, including high conductivity, layered structure, and excellent redox activity. However, the majority of successfully grown Nb-based oxides and chalcogenides, including nanosheets, nanoparticles, and nanoplates, have been fabricated using a top-down approach involving volatile precursors and toxic environments to facilitate the synthesis process.23–26 One of the difficult tasks in preparing Nb chalcogenides for energy and environmental applications is achieving green chemistry using a bottom-up approach. Layered transition metal dichalcogenides have recently attracted significant research interest owing to their unique physical and chemical properties, which make them promising candidates for energy storage and conversion.27–29 Molybdenum disulfide (MoS2) is a typical transition metal dichalcogenide with a unique layered structure analogous to that of graphene connected via weak van der Waals forces, making it an ideal insertion host for metal cations. MoS2 has been widely studied for rechargeable batteries such as Li-, Na-, and Zn-ion batteries.29–33 In the case of Zn-ion EES systems, the hydrated Zn2+ ions (4.3 Å) in aqueous electrolytes must overcome the high desolvation energy barrier when inserted into the interlayers of MoS2. Remarkably, bivalent Zn2+ interacts strongly with the host framework, resulting in slow intercalation kinetics. Several design strategies have been proposed to alleviate these difficulties, including structural design, defect engineering, phase variation, and interlayer engineering.
In this study, we demonstrate the Chevrel phase of NbMo6S8@nitrogen-doped carbon (NbMo6S8/NC) nanosheets as cathodes for aqueous ZICs. N-doping generates numerous active sites comprising pyridine-N and graphitic-N in the framework of the carbon species. N-doped carbon is recognized as the most appropriate material for energy storage, with a high storage capacity.34 Nitrogen doping also effectively prevents nanoflower aggregation of NbMo6S8, suggesting that nitrogen doping can increase the interlayer spacing and generate a more disordered structure, providing active sites for the target reactants.35,36 Furthermore, N-doped carbon materials with porous structures have a higher specific surface area than the parent carbon material, allowing for more access to the active sites, thereby facilitating superior conductivity, high electron transfer efficiency, and ultimately increasing the charge-storage activity of NbMo6S8.37 The advantages of Zn include its abundant resources, compatibility with safe aqueous liquid electrolytes, and lack of significant risks. Coupled with an aqueous Zn-ion electrolyte, the NbMo6S8/NC ZIC system designed herein exhibits excellent electrochemical performance, with excellent cyclic stability and energy efficiency. Electrochemical analysis of the NbMo6S8/NC electrode based ZIC cell reveals superior electrochemical capacitive properties with a high specific energy (188.87 W h kg−1) and long cycle life of over 15000 cycles, and a discharge capacity decay rate of only 0.00082% per cycle. A pair of flexible NbMo6S8/NC ZICs connected in series successfully powers light-emitting diodes, demonstrating high potential applications.
Results and discussion
A schematic of the formation of the NbMo6S8/NC nanoflowers is shown in Fig. 1(a). Chitosan (CS) has multifunctional properties with multiple functional groups, as well as good biocompatibility and biodegradability. One of the advantages of CS is that it can be converted into N-doped carbon using a standard hydrothermal carbonization method. CS is also a positively charged polyelectrolyte because its many amino groups (–NH2) can be protonated to NH3+ and readily undergo electrostatic interactions with anionic groups in an acidic environment. A simple wet-chemical approach using a hydrothermal method was used to design nanoflower-shaped NbMo6S8 for the first time. The chemical sources of Mo, Nb, and S ions are the ammonium molybdate, niobium chloride, and thiourea precursors, respectively. First, CS was dissolved in an acidic solution and vigorously stirred for 1 h to form a CS solution containing NH3+. Niobium chloride and ammonium molybdate were added to the solution and stirred until the solution became clear. The resultant solution contained negatively charged HNbO32− and MoO42− ions that could be adsorbed on the CS molecular chains through electrostatic interactions with NH3+. Conversely, the CS chains containing NH3+ could be cross-linked to form a network structure by forming ionic bonds with Nb2+ and MoO42−. The cross-linked chitosan chains were then hydrothermally converted into 2D colloidal carbon nanosheets by nitrogen doping. Simultaneously, the Nb2+ and MoO42− ions adsorbed on the CS molecular chain interacted with the H2S released by thiourea to form a Chevrel phase of NbMo6S8, and the local and electronic structure of discrete Mo6S8 cluster units within the extended NbMo6S8 framework grew in situ on the surface to form nanoflower-shaped NbMo6S8. During the hydrothermal process, a few CS chains were embedded in the NbMo6S8 layer, effectively constraining stacking of the NbMo6S8 layers and increasing the interlayer distance. NbMo6S8@nitrogen-doped carbon (NbMo6S8/NC) and NbMo6S8 were further characterized.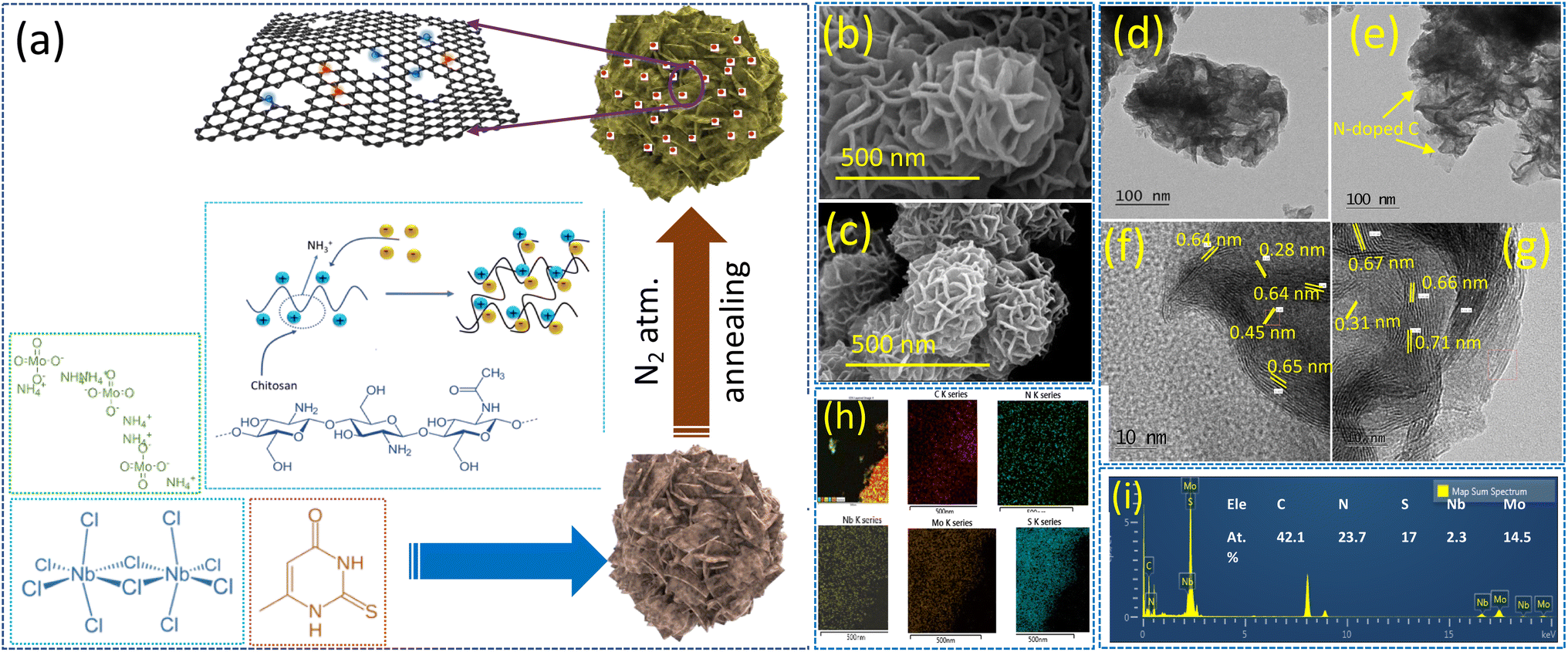 | ||
| Fig. 1 (a) Preparation of NbMo6S8/NC via the hydrothermal process. Morphological characterization of NbMo6S8 and NbMo6S8/NC: SEM images of NbMo6S8 (b) and NbMo6S8/NC (c). TEM images of NbMo6S8 (d) and (f) and NbMo6S8/NC (e) and (g). Elemental mapping (h) and EDS pattern (i) of NbMo6S8/NC. | ||
Field-emission scanning electron microscopy (FE-SEM) and field-emission transmission electron microscopy (FE-TEM) were used to track the morphological growth of the prepared electrode materials. Fig. 1(b) and (c) show the surface micrographs of hydrothermally grown NbMo6S8 and NbMo6S8/NC, respectively. The imaging data show that NbMo6S8 has a nanoflower-like surface structure, whereas NbMo6S8/NC has a nanosheet (∼10 nm) morphology comprising nanoflower bunches with diameters of 200–250 nm. The nanoflower morphology is retained in NbMo6S8 but it is delicate and thus broken owing to a slight decrease in the nanosheet thickness. The low-magnification SEM images of NbMo6S8 also demonstrate well-maintained nanoflower bunches, as shown in the ESI (Fig. S1(a) and (b)†). TEM characterization revealed that the materials had nanoflower-like structures, which is consistent with the SEM analysis (Fig. 1(d) and (e)). It is clear that the number of stacked layers in NbMo6S8/NC is lower than that in NbMo6S8. Fig. 1(e) shows a transparent N-doped carbon coating layer around the nanoflowers, indicating that NbMo6S8 with N-doped carbon was successfully formed. The N-doped carbon coating alters the surface but does not appear to affect the shape of the nanoflowers. The results indicate that the combination of N-doped carbon expanded the interlayer spacing and hindered aggregation of the NbMo6S8 nanosheets. Low-magnification SEM images of the NbMo6S8/NC materials are shown in Fig. S1(c) and (d).† To clearly demonstrate the expanded interspace, the distance between neighboring NbMo6S8 planes was determined using Digital Micrograph software. The highly magnified TEM images in Fig. 2(f) reveal a polycrystalline structure with fringe spacings of 0.64, 0.45, and 0.28 nm, which are consistent with the (101), (110), and (211) lattice planes of NbMo6S8, respectively. Furthermore, the spacings of the NbMo6S8/NC sample increased to 0.71, 0.67, and 0.66 nm, attributed to the (101) plane and 0.31 nm related to the (122) plane, as shown in Fig. 2(g). The interplanar distance was expanded in the layered few-atom nanostructures of the 2D materials, allowing for faster intercalation/deintercalation of the electrolyte metal ions. Fig. 2(h) presents the STEM elemental map of NbMo6S8/NC, which confirms that C, N, Nb, Mo, and S were uniformly grown on the sample surface. The corresponding EDS patterns and atomic percentages in tabular form are presented in Fig. 1(i).
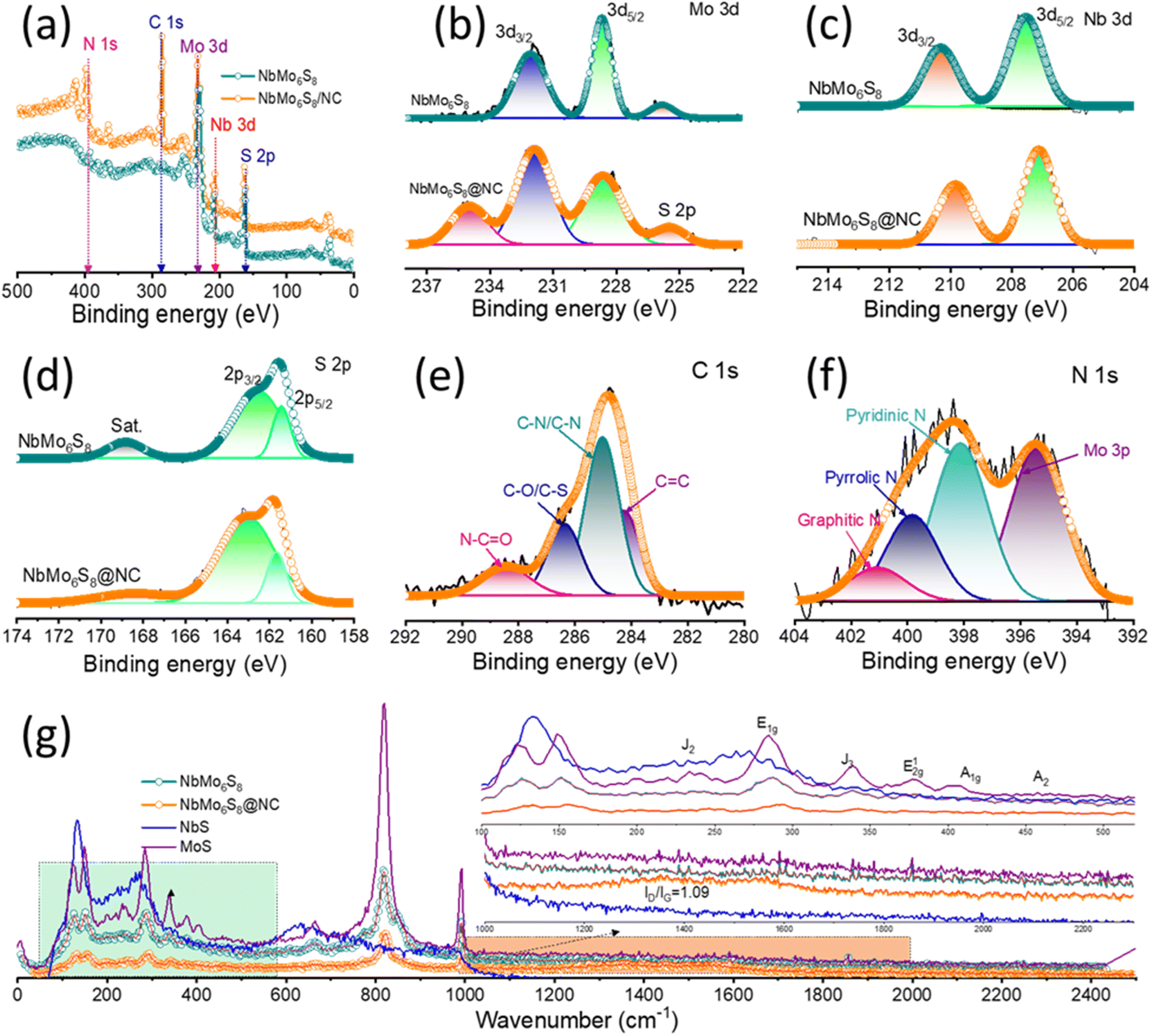 | ||
| Fig. 2 Structural characterization studies: (a) full scan XPS profiles of NbMo6S8 and NbMo6S8/NC. Core-level (b) Mo 3d, (c) Nb 3d, and (d) S 2p XPS profiles of NbMo6S8 and NbMo6S8/NC. Core-level (e) C 1s and (f) N 1s XPS profiles of NbMo6S8/NC. (g) Raman spectra of NbMo6S8 and NbMo6S8/NC. | ||
X-ray photoelectron spectroscopy (XPS) was utilized herein to assess the chemical composition and elemental valence states of the NbMo6S8 and NbMo6S8/NC materials. Fig. 2(a) shows the wide-scan XPS profile, which confirms N-doping in C, along with the presence of Mo, Nb, and S elements in the structure of the NbMo6S8/NC material. The core-level XPS profiles of Nb, Mo, and S for NbMo6S8/NC and NbMo6S8 are shown in Fig. 2(b)–(d), respectively. The Mo 3d peaks at binding energies (BEs) of 232.02 and 228.58 eV are assigned to the 3d3/2 and 3d5/2 states of Mo (as MoS2) in pure NbMo6S8.38 The nearby S 2s peak was fitted to a peak at 226.16 eV, corresponding to S species bound to Mo ions (Mo–S).39 For NbMo6S8 and NbMo6S8/NC, the core-level Nb 3d (Fig. 2(c)) profile displays peaks at 210.31 eV (209.88 eV) and 207.67 eV (207.13 eV) that can be assigned to Nb 3d3/2 and 3d5/2, respectively.40 The S 2p core-level spectrum of NbMo6S8 (Fig. 2(d)) shows two major characteristic peaks at approximately 161.58 and 162.53 eV, corresponding to S 2p3/2 and S 2p1/2, respectively, consistent with the S2− oxidation state.41 The accompanying satellite peak at 168.98 eV for sulfur is attributed to the partial oxidation of NbMo6S8. The C 1s spectrum (Fig. 2(e)) could be deconvoluted into four peaks. The high-intensity peak at 284.26 eV is related to sp2 hybridized carbon (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond) and the 285.05 eV peak represents combined contributions from sp3 carbon in C–C and C–N bonds at the defect site, indicating that NbMo6S8 was indeed in a nitrogen-doped carbon matrix.42–44 The low-intensity peaks at approximately 286.37 and 288.53 eV are attributed to carbon in C–O/C–S and N–CO bonds, respectively. The three major characteristic peaks of N 1s in Fig. 2(f) were assigned to pyridinic-N (398.12 eV), pyrrolic-N (399.83 eV), and graphitic-N (401.07 eV), with pyridinic-N being dominant.45,46 An additional peak appeared at 395.39 eV in the Mo 3p spectrum. The atomic percentage of N in NbMo6S8/NC is 19.8%, which is consistent with the EDS results. Furthermore, the Mo 3d, Nb 3d, and S 2p peaks in the XPS profile of NbMo6S8/NC were negatively shifted compared to those of NbMo6S8, demonstrating strong electron interactions after N atom doping within the carbon.
C bond) and the 285.05 eV peak represents combined contributions from sp3 carbon in C–C and C–N bonds at the defect site, indicating that NbMo6S8 was indeed in a nitrogen-doped carbon matrix.42–44 The low-intensity peaks at approximately 286.37 and 288.53 eV are attributed to carbon in C–O/C–S and N–CO bonds, respectively. The three major characteristic peaks of N 1s in Fig. 2(f) were assigned to pyridinic-N (398.12 eV), pyrrolic-N (399.83 eV), and graphitic-N (401.07 eV), with pyridinic-N being dominant.45,46 An additional peak appeared at 395.39 eV in the Mo 3p spectrum. The atomic percentage of N in NbMo6S8/NC is 19.8%, which is consistent with the EDS results. Furthermore, the Mo 3d, Nb 3d, and S 2p peaks in the XPS profile of NbMo6S8/NC were negatively shifted compared to those of NbMo6S8, demonstrating strong electron interactions after N atom doping within the carbon.
The crystal structure of the prepared materials were characterized using XRD technique. As shown in Fig. S3,† the XRD peaks of NbMo6S8 are consistent with the rhombohedral phase (JCPDS PDF No. 00-041-0794). The peaks appeared at 13.71, 23.55, 34.10, 46.70, and 58.45°, corresponding to the (101), (003), (021), (223), and (315) planes, respectively. To further corroborate the XRD structural features, Raman spectra of the NbMo6S8 and NbMo6S8/NC samples were acquired (Fig. 2(g)) and compared. Prominent peaks appeared at 100–200 cm−2, which served as a unique identifier of the layered nanostructure of the material.47 The peaks at 152.00, 231.01, and 338.55 cm−1 were assigned to the J1, J2, and J3 modes of MoS2, respectively.48 The strong Raman peaks at approximately 283.20, 354.66, and 460.00 cm−1 corresponding to the E1g, E12g, and A2 modes, respectively, are assigned to NbS2 and attributed to the in-plane vibrations of Nb and S atoms.24,47 The Raman spectrum showed a peak at 404.40 cm−1, corresponding to the A1g vibrational mode within the S–Mo–S layer in MoS2.49 The observed peaks at approximately 653.80, 821.04, and 991.76 cm−1 correspond to the NbMo-oxide groups and are associated with the stretching vibration of the longitudinal optic mode of edge-shared O–Mo–NbO.47,50 The spectrum of NbMo6S8/NC shows two distinct peaks at approximately 1357.52 and 1565.30 cm−1, for which the peak intensity ratio (ID/IG) is 1.09; these peaks are attributed to the D-band and G-band of the carbon-based matrix, respectively.
Brunauer–Emmett–Teller (BET) measurements were used to examine the specific surface areas of the NbMo6S8 and NbMo6S8/NC materials. Fig. S7† depicts the N2 adsorption–desorption isotherms of NbMo6S8 and NbMo6S8/NC materials. The specific surface areas of NbMo6S8 and NbMo6S8/NC materials are 96.19 and 154.86 m2 g−1, respectively. The higher electrochemical storage activities of NbMo6S8 and NbMo6S8/NC material nanosheets can be attributed to their larger specific surface area.
To achieve electrochemical stability and resolve the irreversibility issue associated with Zn anodes in aqueous electrolytes, a modified highly concentrated Zn-based electrolyte (MHCZE) that enables dendrite-free Zn plating/stripping and achieves 100% coulombic efficiency is introduced herein. To optimize the electrolyte concentration, the electrochemical properties of MHCZE were investigated using various characterization techniques. First, the viscosity of the electrolyte solutions was measured using a rheometer (Discovery Hybrid Rheometer 2), and the conductivity was measured by electrochemical impedance spectroscopy. The ionic conductivity and shear viscosity plots for the electrolyte solutions at different concentrations are shown in Fig. 3(a) and (d). As expected, the shear viscosity increased linearly with the molar concentration. When the electrolyte concentration was increased from 1 to 17 M, the viscosity increased by 85%. The increase in viscosity may be related to the shift in the frequency of the stretching band of ClO4− (ν1), which suggests the formation of ionic pairs and ionic aggregates in solution, as confirmed by Raman analyses. In addition, the ionic conductivities of the electrolytes increased with increasing MHCZE concentration (Fig. 3(d)). These results suggest that highly concentrated NaClO4 electrolytes have high ionic conductivity, which improves the electrochemical features of the electrochemical energy storage system.
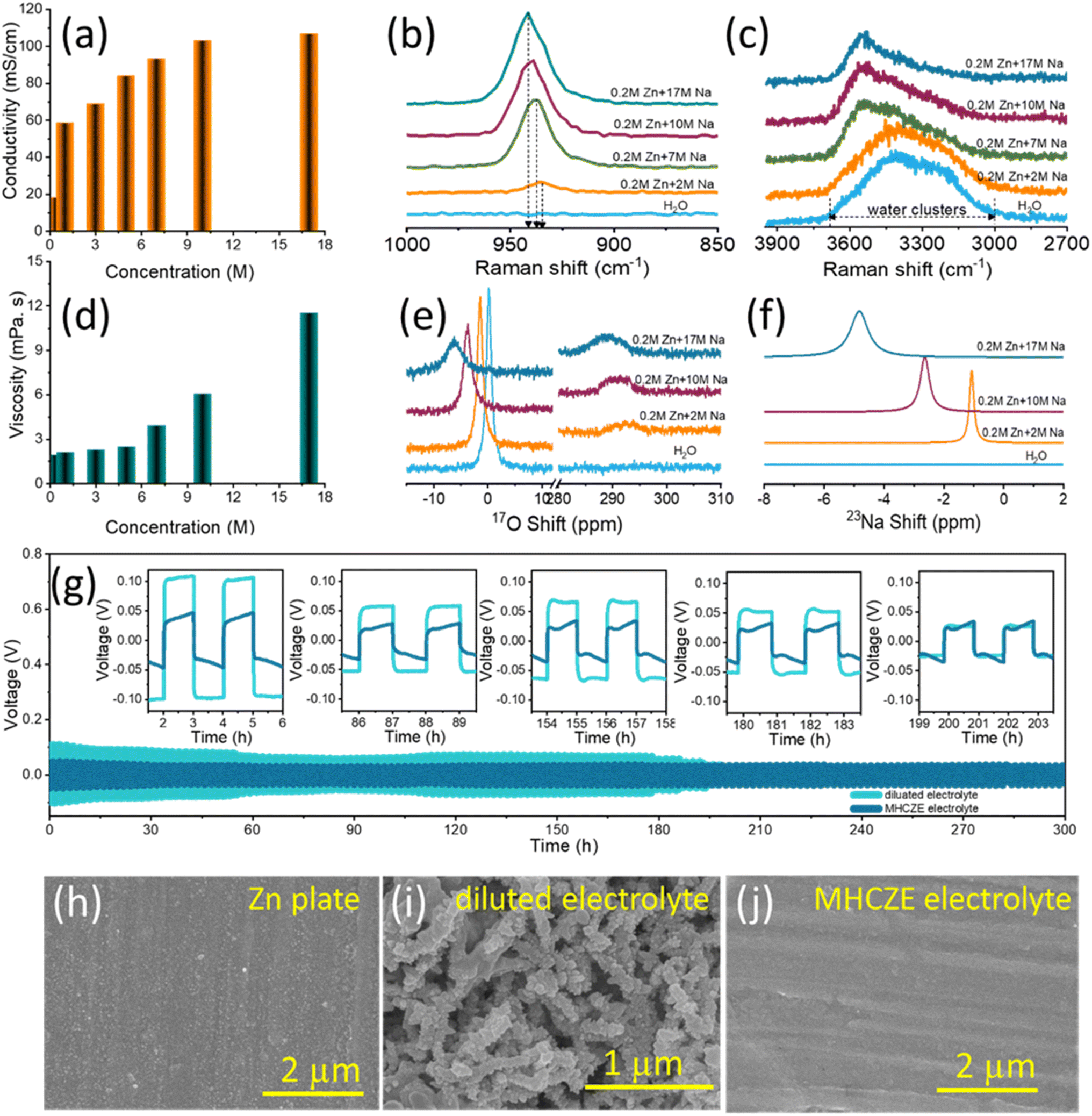 | ||
| Fig. 3 Cation–anion and ion–water interactions in the MHCZE electrolyte solutions. (a) Ionic conductivity and (d) viscosity plots for NaClO4-based MHCZE electrolyte with different concentrations. Raman spectra of (b) Cl–O and (c) O–H stretching vibrations for NaClO4 solutions with varying ZnClO4/NaClO4 molar ratios. (e) 17O, and (f) 23Na NMR chemical shifts with different salt molar ratios. (g) Stripping/plating analysis of a Zn/Zn symmetric cell in dilute and MHCZE electrolytes. Surface morphology of the (h) Zn plate, and after the platting/stripping from (i) ZnClO4- and (j) MHCZE-based cells. | ||
Raman and nuclear magnetic resonance (NMR) measurements were performed to better understand the primary electron transport and cation–anion interactions in the electrolyte. When NaClO4 salt was added to a 0.2 M ZnClO4 aqueous solution, the activity of free water was effectively inhibited, as shown by the Raman spectra (Fig. 3(b)) of the electrolyte solutions at various concentrations. The broadening of the Raman peaks in the range of 3000–3700 cm−1, which correspond to the O–H stretching vibration of clustered water molecules, is associated with diverse hydrogen-bonding modes. The water clusters are usually composed of an ice-like component, ice-like liquid component, and amorphous phase with peaks at 3230, 3450, and 3531 cm−1, respectively.37 As the salt concentration increased, the strong peak related to the O–H stretching vibration (hydrogen–water molecules) shifted to a higher wavenumber, indicating no or weak hydrogen bonds due to the coordination of the water molecules with Na+ ions. The appearance of a sharp and distinct peak at approximately 3551 cm−1 and the disappearance of the original shoulder peaks are attributed to cation-solvated water molecules that are free of hydrogen bonding. These results prove that the strong hydrogen bonds between the free water molecules were significantly shattered, which is consistent with the reduced water activity compared to that of dilute NaClO4 electrolytes. Furthermore, aggregation of NaClO4 in aqueous electrolytes of different concentrations was monitored based on changes in the Cl–O vibration mode of the ClO4− anions, where peaks were apparent in the 915–965 cm−1 wavenumber range (Fig. 3(c)). For dilute NaClO4 solution, the Cl–O peak corresponding to the free anions appeared at 934.25 cm−1. With increasing NaClO4 concentrations, the peak became broader and shifted to a higher wavenumber of 939.6 cm−1 (solvent-separated pairs), whereas the peak at 941.55 cm−1 (contact ion pairs/aggregated cation–anion pairs) became sharper. This sharp change is attributed to the transformation of the water-separated ion pairs into contact ion pairs of Na+ and ClO4− ions, while the electrolyte became more structured.
NMR aids in understanding the interactions between ions and water molecules. Zn2+ has a stronger affinity for water and ClO4− as compared to Na+. The Zn2+ solvation shell in the concentrated electrolyte is primarily occupied by ClO4−, and the interaction between the solitary-pair electrons on the oxygen of water and Zn2+ can be effectively suppressed, resulting in a shielding effect of the 17O signal.51Fig. 3(e) shows the 17O NMR spectra, which are useful because oxygen exists in both water and the salt molecules. A pair of oxygen peaks is anticipated to occur at approximately 0 and 288 ppm from the oxygen in water and ClO4−, respectively. The 17O signal of water in the range of −10 to 10 ppm is more sensitive to the salt concentration, as observed in the Raman spectra, where the peak moved to a lower chemical shift with increasing electrolyte salt concentration. Water and ClO4− in the coordination shell of Na+ in the MHCZE electrolyte cause the electron density around Na+ to increase, resulting in a lower chemical shift for 23Na (Fig. 3(f)). The narrow chemical shift of the 23Na NMR signal is related to the fast exchange of Na+ and ClO4−/water in the solvation shell, suggesting that the MHCZEs should have high ionic conductivity. The electrolyte structure inhibited the movement of water molecules and led to reduced water activity. ClO4− salts have a strong ability to destabilize the overall water structure, suggesting that the solvation-sheath structure of ionic aggregates can be adjusted based on the Hofmeister series. Water-breaking ClO4− ions in the MHCZE electrolyte can form a complex ionic network with water molecules and cations, which generates a unique solvation structure that prevents the formation of hydrogen-bonding networks.52
Fig. 3(g) shows the galvanostatic polarization data for Zn/Zn symmetric cells with dilute and MHCZE electrolytes at a current density of 0.5 mA cm−2. The voltage of the cells with the dilute electrolyte was close to 0 V, implying an internal short-circuit induced by dendrite growth on the Zn plate. Throughout the 10 day test, the voltage in the MHCZE electrolyte cells maintained a smooth profile. A comparatively dilute electrolyte, very low overpotential in the MHCZE electrolyte, and high coulombic efficiency are guaranteed to alleviate Zn stripping/plating, thereby leading to a longer cycle life. The improved stability performance is derived from the formation of a conductive and stable solid interface layer on the Zn plate surface. HR-SEM was used to examine the surface morphologies of the Zn plate in the dilute and MHCZE electrolytes to explore the changes in the surface microstructure before and after long-term plating and stripping (Fig. 3(h)–(j)). A smooth surface was observed for the pristine Zn plate. Notably, after a few cycles, the morphology of the Zn plate in the MHC-ZE electrolyte remained dense and dendrite-free. However, in the dilute electrolyte, a large number of Zn dendrites formed on the surface of the Zn plate. It is evident that the concentrated electrolyte aids in alleviating the effects of dendrite formation on the Zn plate. As a result, stabilized capacity was observed during long-term cycling due to the self-passivation effect that blocked further zinc dendrite formation.53
The prepared samples were used as cathodes in ZICs in the second step of the electrochemical analysis to build full cells; the electrochemical performance of the cells with dilute and MHCZE electrolytes was analyzed. The optimized MHCZE electrolyte composition (0.2 M ZnClO4 and 17 M NaClO4) was then used for electrode analyses. The prices of the selected NaClO4 salt (0.56 $ per g) and ZnClO4 salt (0.34 $ per g) are far cheaper than those of the NaOTf salt (11.2 $ per g), LiClO4 salt (6.75 $ per g), and LiNO3 salt (8.32 $ per g), as shown in Fig. 4(a). The charge-storage behavior of the MHCZE electrolyte was analyzed to determine the charge-storage capacity of the electrode material. The experimental details of the electrochemical analysis are provided in the ESI.† The electrochemical features are compared in Fig. 4(b) to investigate the charge-storage mechanism of the NbMo6S8 and NbMo6S8/NC electrodes in the dilute and MHCZE electrolytes. Fig. 4(b) presents cyclic voltammograms (CVs) of the NbMo6S8 and NbMo6S8/NC electrodes in the voltage range of 0–1.5 V at a scan rate of 20 mV s−1 for comparison of the Zn2+ intercalation/deintercalation electrochemical reaction process. Remarkably, the NbMo6S8/NC electrode displays a pair of redox peaks at 1.21 and 0.59 V vs. Zn2+/Zn that can be ascribed to the insertion/extraction of Zn2+ into/from the NbMo6S8/NC structure. In the dilute electrolyte system, the area under the CV curves, which corresponds to charge storage, is lower than of the NbMo6S8 and NbMo6S8/NC electrodes in the MHCZE electrolyte. These significant differences in the CV curves indicate the vital importance of the MHCZE electrolyte system in enhancing the intercalation kinetics of Zn2+, which was also confirmed by the galvanostatic charge/discharge (GCD) studies (Fig. 4(c)). Evidently, the NbMo6S8/NC electrode has a longer discharge time than NbMo6S8, providing a higher specific capacity. In addition, the area under the discharge curve of the NbMo6S8/NC electrode in MHCZE was much larger than that of NbMo6S8 in the dilute electrolyte, and the former exhibits better energy storage capability, which is attributed to the unique structure of NbMo6S8/NC with a larger surface area and introduced heteroatom content. The N-doped carbon coating contains pyridinic-N and pyrrolic-N, which increase the number of active sites where zinc ions can move because of their defect structures and large interplanar distances, thus improving the conductivity of the electrode material. In addition to structure tuning, adequate surface functionality aids in enhancing the performance of electrode materials. The rapid capacity fading, on the other hand, is caused by a severe metal sulfide pulverization issue caused by the significant volume change throughout Zn-ion insertion/extraction.54 Incorporating nitrogen doped carbon with NbMo6S8 improves electrical transport and reliability, increasing conductivity and effectively expanding the materials' overall electrochemical performance.55 As a result of the pseudocapacitive interactions between electrolyte ions and electrode materials, the capacitance of the electrodes is significantly increased.
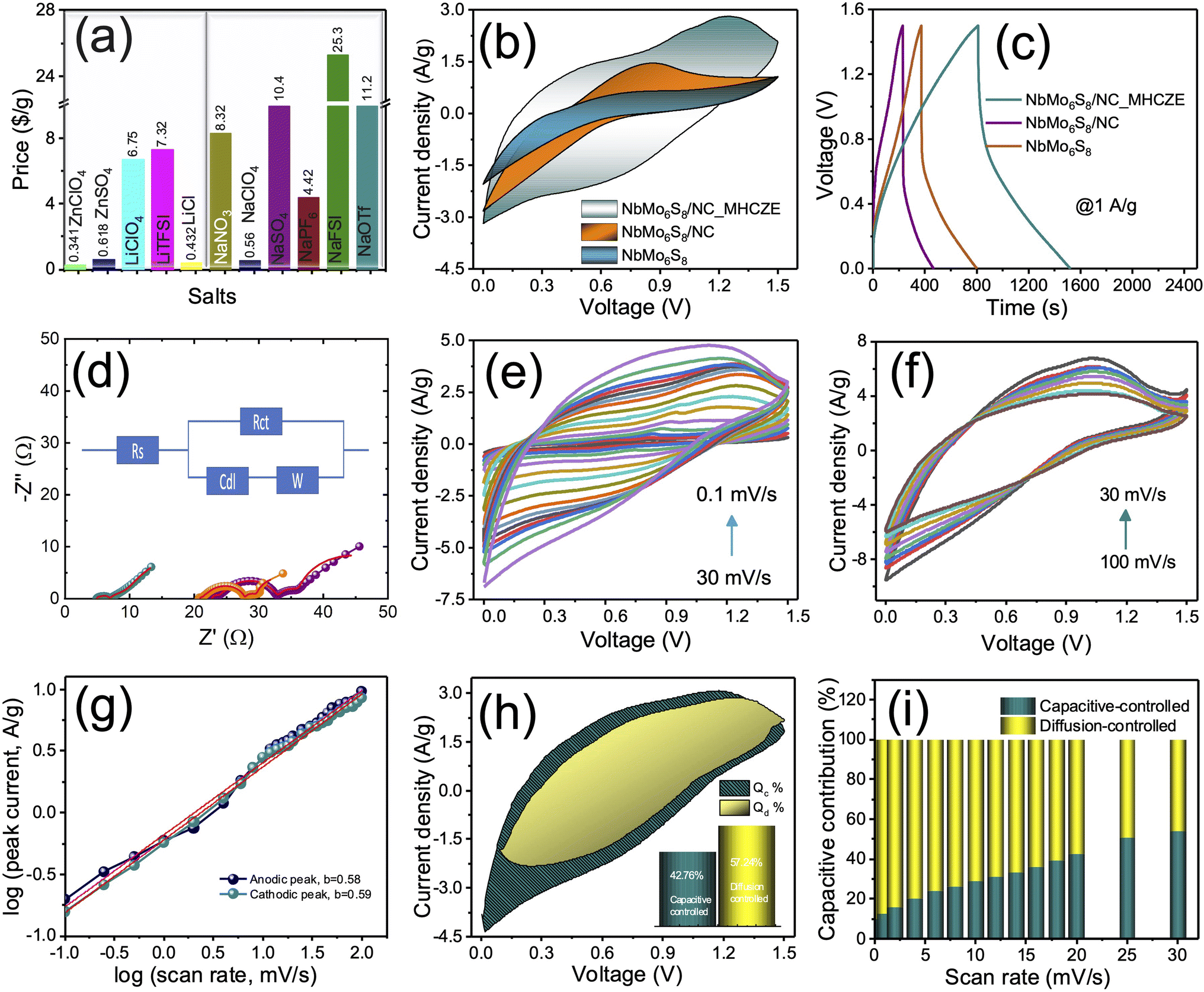 | ||
| Fig. 4 (a) Price comparison of various salts typically used. Electrochemical characterization studies of NbMo6S8 and NbMo6S8/NC in ZICs within the range of 0–1.5 V (vs. Zn2+/Zn). (b) Cyclic voltammograms (CVs) acquired at a lower scan rate of 20 mV s−1, (c) galvanostatic charge–discharge voltage profiles at 1 A g−1 current density, and (d) electrochemical impedance analysis with best-fitted Nyquist data for NbMo6S8 and NbMo6S8/NC cathodes in ZICs. The inset in (d) shows the best fitted equivalent circuit for Nyquist plots. CVs acquired at (e) lower scan rates of 0.1–20 and (f) higher scan rates of 25–100 mV s−1 for the NbMo6S8/NC ZIC in MHCZE electrolyte. (g) Kinetics analysis for calculating b from the plot of log peak current versus scan rate. (h) Characteristic CV profiles representing capacitive (Qc) and diffusion-controlled contributions (Qd) at a scan rate of 20 mV s−1. (i) Charge-storage contributions of the NbMo6S8/NC electrode at different scan rates. | ||
The vital importance of the MHCZE electrolyte system was further confirmed by electrochemical impedance spectroscopy (EIS) analyses (Fig. 4(d)), where the resistance values, which affect the charge-storage kinetics, were significantly different from those in the dilute electrolyte. The Nyquist plot shows a semicircle in the high-frequency region, indicating a charge-transfer process, and a straight line in the low-frequency region, indicating a diffusion process. The typical equivalent circuit model is displayed in the inset of Fig. 4(d) and the corresponding parameter values are provided in Table S1.† The lower solution and charge-transfer resistance (Rct) of the NbMo6S8/NC cathode in the MHCZE electrolyte indicates good conductivity, which effectively improved the kinetics of Zn2+ intercalation by suppressing side reactions. In contrast, the aqueous systems exhibit significantly large Rct and diffusion resistance values, indicating impeded charge-storage dynamics and effective reduction of the charge-storage kinetics of the cathode material.
The CV curves of the NbMo6S8/NC electrode in the MHCZE electrolyte are presented in Fig. 4(e) and (f), indicating the surface adsorption and desorption of Zn2+. The CV curve of the NbMo6S8/NC electrode in the range of 0–1.5 V did not show either a pronounced rectangle or a significant redox peak, indicating that the material exhibits both EDLC and pseudocapacitive behaviors. When the scan rate was increased from 0.1 to 20 mV s−1, the CV profile of NbMo6S8/NC ZIC was not significantly deformed, indicating excellent rate performance and rapid reaction kinetics. The CVs of NbMo6S8/NC ZIC were recorded at higher scan rates (Fig. 4(f)), and the specific capacity was calculated based on the mass of the active materials in the corresponding metallic Zn cells. The relationship between the current (i) and low scanning sweep rates (ν) was fitted to the power-law rule for quantitative analysis.15
| i = aνb, | (1) |
| i(V) = k1ν + k2ν1/2, | (2) |
Fig. 5(a) shows a schematic of an assembled ZIC full device with NbMo6S8/NC as the cathode, a Zn plate as the anode, and MHCZE solution as the electrolyte. To demonstrate the effect of the MHCZE electrolyte on the electrode performance, the capacities and specific energy contributed by the discharge voltage platform were calculated using the area of the discharge region in the GCD curve. Fig. 5(b) shows that the GCD curve was quasi-linear at various current densities, which is consistent with the CV results. The charge and discharge profiles of NbMo6S8/NC confirmed the reduced diffusion resistance and improved charge-storage kinetics of the system. The electrochemical performance of the NbMo6S8/NC and NbMo6S8 electrodes was quantified by calculating the specific capacities from the plateau in the GCD curve. The specific capacities of the NbMo6S8 and NbMo6S8/NC electrodes were 25.64 and 36.22 mA h g−1, respectively, at a current density of 0.16 A g−1. Modification of the electrolyte significantly improved the electrochemical properties of the NbMo6S8/NC electrode, which not only promoted the kinetics of the ion intercalation/deintercalation reaction but also improved the structural stability of the electrode material. This modification did not produce obvious distortions at different current densities, indicating excellent performance. From Fig. 5(c), specific capacities of 188.67, 167.89, 110.49, 76.50, 52.75, 48.68, and 47.25 mA h g−1 were calculated for the NbMo6S8/NC electrode in the MHCZE electrolyte at current densities of 0.16, 0.25, 0.5, 1, 1.5, 2.0, and 2.5 A g−1, respectively. The specific capacities of the fabricated ZICs decreased with increasing current density, particularly at high current densities. It is well known that at high current densities, limited utilization of the electrode material occurs because the electrolyte ions are not capable of penetrating the inner surface of the active material, and charge storage occurs only at the outer surface during repeated charge and discharge processes.37 It is also noted that at higher current densities, the electrolyte and electrode materials interact ineffectively due to diffusion limitations of the internal structure of the electrode materials, resulting in a decrease in specific capacitance.
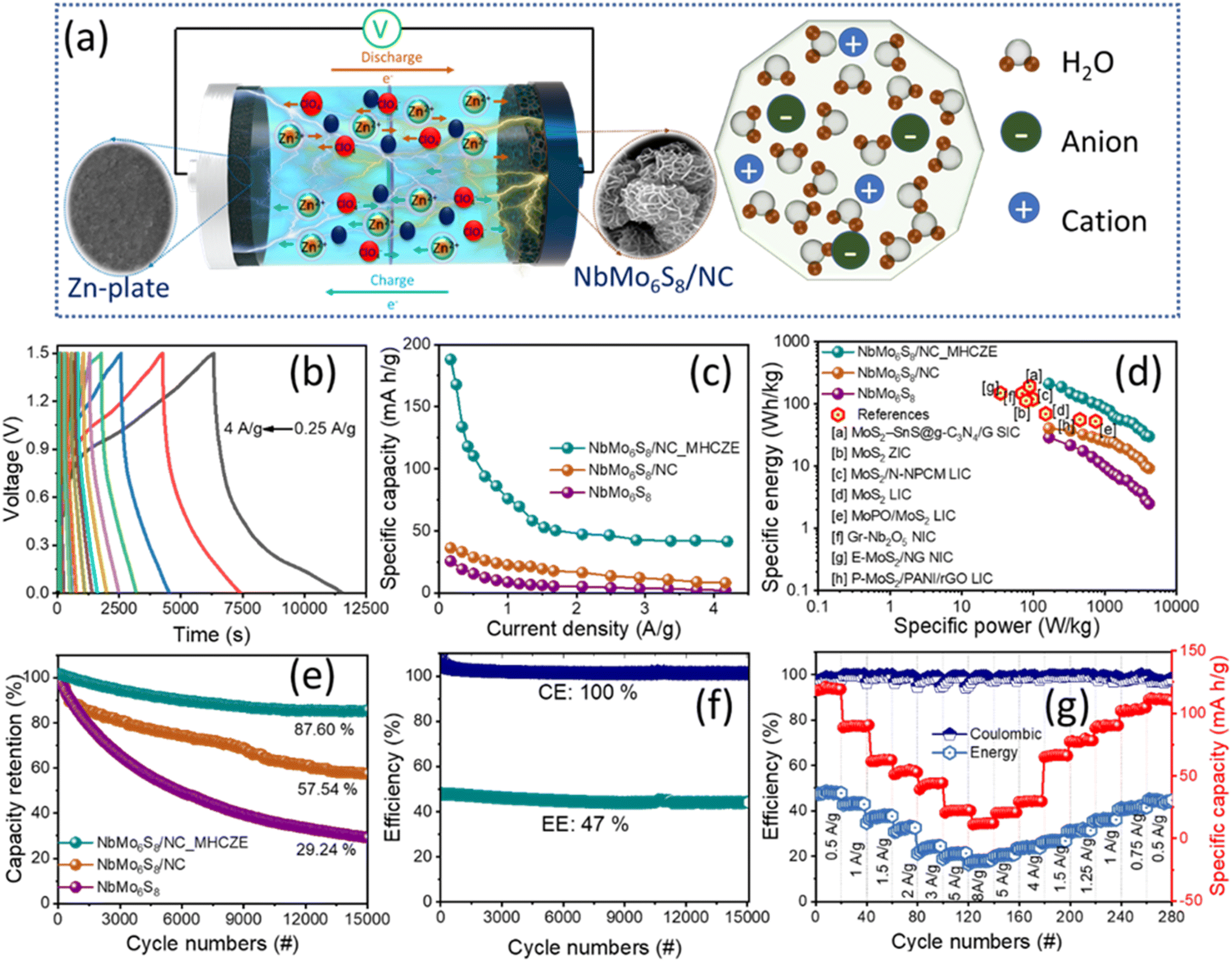 | ||
| Fig. 5 (a) Schematic of the NbMo6S8/NC ZIC construction and working mechanism. (b) Charge–discharge profiles of the NbMo6S8/NC electrode in MHCZE electrolyte. (c) Specific capacity, (d) Ragone plot, and (e) long-term cycling capacity retention at a current density of 2.5 A g−1 for NbMo6S8 and NbMo6S8/NC ZICs. (f) Coulombic and energy efficiencies, and (g) a capability test at different current densities for the NbMo6S8/NC electrode in MHCZE electrolyte. | ||
Fig. 5(d) shows the Ragone plot for the NbMo6S8/NC-based ZICs, where the specific energy and power were calculated from each discharge area platform. The specific energy and power were obtained in accordance with the specific capacity. As seen in the Ragone plot, the as-prepared NbMo6S8/NC ZIC had a high specific energy of 188.87 W h kg−1 and a specific power of 250 W kg−1, and still maintained an impressive energy density of 50.22 W h kg−1 when the specific power was increased by more than ten-fold to 2500 W kg−1. This performance is superior to that of many previously reported metal sulfide-based electrodes, and even when compared to metal oxides.56–70 The cycling stability of the ZIC is also an important and meaningful parameter to consider when evaluating the charge-storage performance. The capacitance retention of the NbMo6S8/NC electrode was evaluated over 15000 cycles at 2.5 A g−1 current density. Fig. 5(e) illustrates that the capacity retention rates of the NbMo6S8 and NbMo6S8/NC electrodes in the dilute electrolyte were 29.24 and 57.54%, respectively, and that the NbMo6S8/NC electrode exhibited excellent long-term cycling performance (87.60% capacity retention) in the MHCZE electrolyte over 15000 cycles. The energy and coulombic efficiency of the electrodes were further evaluated over 15000 charge–discharge cycles (Fig. 5(f)). Fig. 5(g) shows the rate capabilities of the NbMo6S8/NC electrode when the current density was increased stepwise. The NbMo6S8/NC ZIC was first cycled at 0.5 A g−1 for 20 cycles, followed by a stepwise increase in the charging–discharging current density rate to 8 A g−1. Even at high current densities, the NbMo6S8/NC electrode could achieve extraordinary discharge capacity and good efficiency, demonstrating the benefits of the heteroatoms in facilitating electrolyte diffusion, as well as improving the redox reaction kinetics. When the current density was reduced to 0.5 A g−1, the capacity returned to the original level, highlighting the excellent cycle stability.
The NbMo6S8/NC cathode was operated against the Zn anode in the MHCZE electrolyte during charging and discharging and then analyzed by ex situ characterization (SEM, XPS, and XRD). Fig. 6(a) shows the GCD profile of the NbMo6S8/NC cathode, and the inset points show the selected half-discharge/charge (a./c.), full discharge/charge (b./d.), and initial state (int.). As shown in the inset, the micrographs of the cathode in the selected states were similar. Charge and discharge plateau-like regions at −0.58 and 0.75 V, respectively, were clearly visible, corresponding to the insertion/extraction of Zn2+ ions from NbMo6S8/NC. Because the cathode was prepared by grinding NbMo6S8/NC with activated carbon, the surface morphology of the cathode in the initial state was slightly compacted and dense. The morphology of the NbMo6S8/NC cathode did not change significantly (1.5 V) during the charging process from state c (0.75 V) to state d. Woolly nanoparticles appeared on the NbMo6S8/NC surface during discharging (state a), indicating that Zn-ion intercalation began. After the cathode was fully discharged at state b (0 V), nanoflakes formed on the surface, and some woolly nanoparticles appeared in the SEM images of the NbMo6S8/NC surface and then disappeared during the charging process.
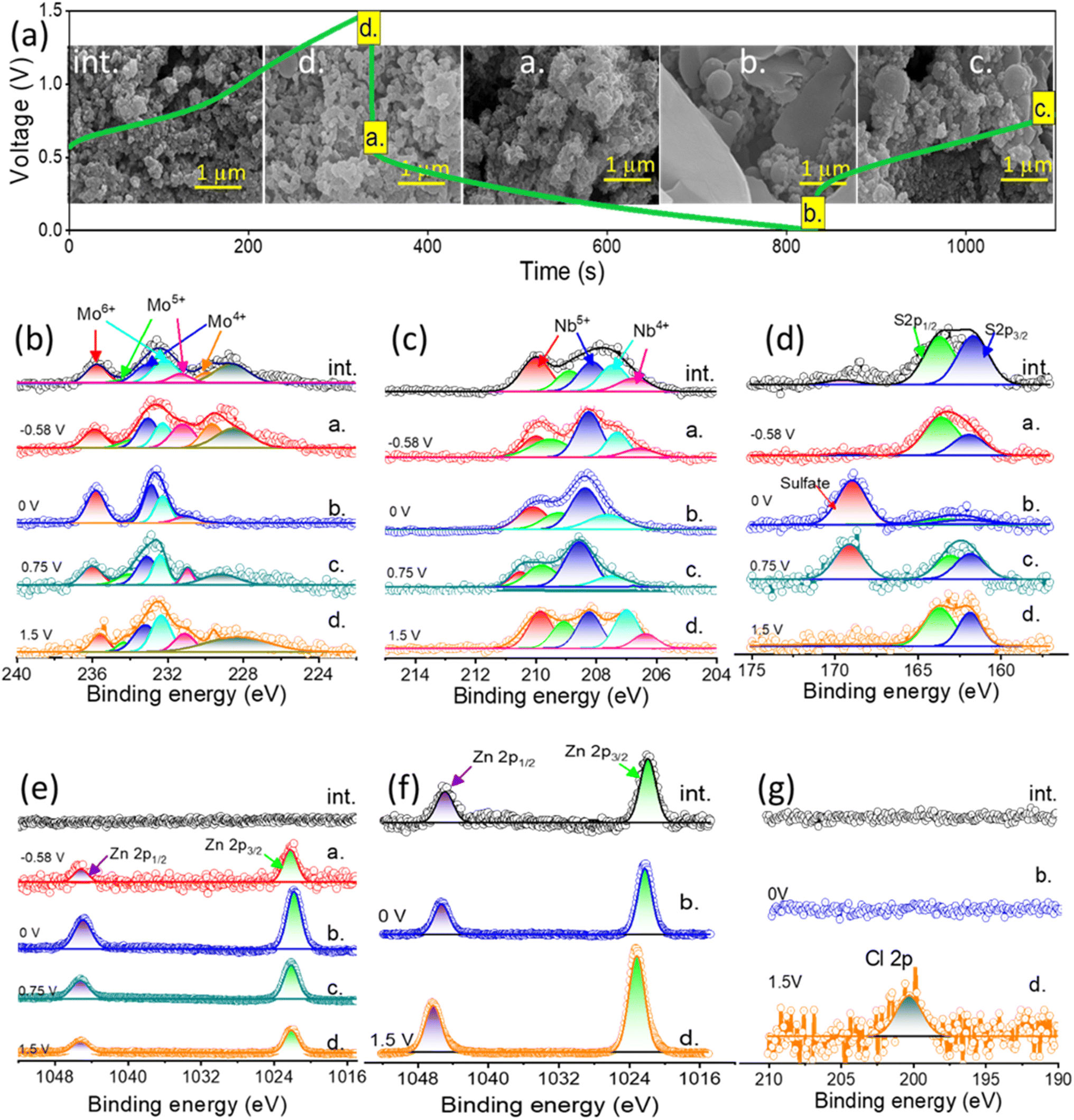 | ||
| Fig. 6 Ex situ characterization of the NbMo6S8/NC cathode and Zn anode in MHCZE. (a) Charge–discharge plots at 1 A g−1. The inset shows the SEM micrographs of the NbMo6S8/NC cathode at selected states. Ex situ XPS profiles of (b) Mo 3d, (c) Nb 3d, (d) S 2p, and (e) Zn 2p for the NbMo6S8/NC cathode. XPS (f) Zn 2p and (g) Cl 2p core-level spectra of the Zn anode in half-charge/discharge, full charge/discharge, and original states. | ||
XPS measurements were further performed to examine the valence state change of Mo, Nb, and S, in the NbMo6S8/NC cathode. Fig. 6(b)–(e) shows the XPS core-level spectrum during the charge/discharge process and compares it with the initial state of NbMo6S8/NC. No traces of Zn were detected in the Zn 2p XPS spectrum of the initial state of the NbMo6S8/NC electrode. Despite the use of a high concentration of NaClO4 as a supporting salt during the charge/discharge process, dominant features related to Zn2+ were observed, whereas no features related to Na+ insertion/removal were observed, as evidenced by the ex situ Zn 2p and Na 1s core-level spectra, respectively (Fig. 6(e) and S5(d)†). Furthermore, the Na 1s core-level spectrum remained unchanged in the fully discharged and charged states, suggesting the presence of stable and immobile Na+ ions in the layered structures of the NbMo6S8/NC cathode during charge/discharge. In contrast, Zn2+ was prominently mobilized during discharging, as shown by the adsorbed Zn2+ on the cathode surface and intercalated Zn2+ in the layered structures of the NbMo6S8/NC cathode in Fig. 6(e). The intense pair of peaks at 1022.14 and 1044.89 eV is attributed to Zn 2p3/2 and 2p1/2, respectively. The fully charged state of the cathode exhibited a weak Zn 2p signal owing to adsorption of the electrolyte on the electrode surface. The lack of impact of Na+ intercalation/deintercalation on the chemical composition of the electrode is related to the larger size of the Na+ cation (0.102 nm) compared to Zn2+ (0.074 nm). XPS was also used to investigate the change in the valence state of Mo. As shown in Fig. 6(b), the best fit after deconvolution produced three doublets in the Mo 3d spectra. The peak at lower binding energy is attributed to the Mo 3d5/2 (228.6 eV) state in NbMo6S8. The XPS peak centered at 226.35 eV is attributed to S 2s. The three peaks in the initial-state Mo 3d core-level spectrum were quantitatively deconvoluted into peaks of the Mo2+ (3d3/2: 232.45 eV and 3d5/2: 228.52 eV), Mo4+ (3d3/2: 233.29 eV and 3d5/2: 229.78 eV), Mo5+ (3d3/2: 234.26 eV and 3d5/2: 231.57 eV), and Mo6+ (3d3/2: 235.00 eV) valence states.71 When the NbMo6S8 electrode is discharged to state a during Zn-ion intercalation, the prominent characteristic peaks of Mo6+ are suppressed, and the characteristic peaks of Mo5+ disappear. The presence of dominant signals of Mo4+ and Mo2+ emerged in the fully discharged state b, corresponding to Zn-ion intercalation and the reduction of Mo5+. The peaks associated with Mo5+ that appeared in the Mo 3d spectrum during the reverse charging from states b to c are related to the oxidation of Mo4+, and these peaks returned to the original state when the cell was fully charged during the Zn-ion deintercalation process. The XPS core-level spectra of Nb 3d are shown in Fig. 6(b). Access of the guest Zn2+ ions to available intercalation sites on the surface of the NbMo6S8/NC cathode is improved when the electrode is discharged to state b (−0.58 V). Deconvolution of the Nb 3d spectrum yielded two doublet peaks at 207.28 and 206.62 eV, which correspond to Nb4+ 3d5/2 and 3d3/2, respectively (Fig. 6(b)).72 During the discharging process, the Nb4+ content decreased significantly, and a prominent peak corresponding to Nb5+ was observed, demonstrating the phase transition of NbMo6S8 triggered by Zn2+ intercalation. During charging, Zn ions are deintercalated from the cathode, resulting in partial reduction of Nb5+. The data collected from the NbMo6S8/NC cathode in the cell with the MHCZE electrolyte show clear differences in the initial, discharging, and charging states of S. The S 2p spectrum was deconvoluted into four peaks (Fig. 6(d)). The BEs of the most intense S 2p doublets were determined to be 161.05 (2p3/2) and 162.04 eV (2p1/2) for S2− species, and 163.14 and 164.40 eV (2p3/2) for bridging sulfur S42− and Sx2−, respectively.73 In addition, a minor S 2p peak was present at 168.78 eV, associated with the thiosulfate (S2O32− or SO32−) species formed on the metal surface.74 Furthermore, when the NbMo6S8/NC electrode was discharged, a new peak appeared and was deconvoluted into doublets at approximately 168.64 and 169.86 eV, attributed to the polysulfides of ZnxNbxS–O and ZnxMoSx–O generated by Zn2+ intercalation into NbMo6S8/NC. On reversing the charging process, due to Zn-ion deintercalation, the strong XPS peaks of S 2p gradually returned to their initial state, while the most intense XPS peaks of the polysulfides gradually diminished until they disappeared near the end of the charging process, indicating that Zn2+ insertion/extraction into/from the cathode is a reversible process. In addition, it is clear that the discharge/charge process of the NbMo6S8 cathode in MHCZE involves a transition from S2− to polysulfides and the reverse reaction forms sulfur; this process is mostly observed in Li–S batteries.75 These observations confirm that the NbMo6S8 cathode is well suited to host Zn2+ and allows fast and reversible Zn2+ intercalation/deintercalation. XRD studies were conducted for detailed structural analysis, and the results are shown in Fig. S7(a)–(e).† In the fully discharged state, Zn2+ is intercalated with the cathode material, forming additional mixed structural phases of Zn0.67Nb11.33O0.67 and ZnMoO4. The obtained results demonstrate that Nb and Mo ions participate in Zn intercalation/deintercalation at the NbMo6S8/NC surface. The contracted crystal structure, in contrast, recovered to its initial state following Zn2+ ion deintercalation during the charging process. These results are consistent with the XPS findings that NbMo6S8/NC is an appropriate cathode material for stable Zn-ion charge storage. The structural properties of the Zn anode during the charge and discharge processes were characterized by XPS and XRD. Plating and stripping alternated at the anode surface (Fig. 6(f)). Owing to ClO4− reduction, a Cl-containing layer of ZnCl2 could form on the surface of the Zn anode, ensuring a potential gradient across this surface layer and allowing Zn plating beneath this layer (Fig. 6(g)).
Developing miniaturized energy storage devices is critical for utilizing renewable clean energy, particularly for easy access to natural energy. Furthermore, because of the excellent flexibility of the NbMo6S8/NC electrode, a flexible ZIC (FZIC) device set was assembled. The electrode's active loading mass is 11 mg cm−2 and it is acceptable for commercial use.76 The CVs of the designed NbMo6S8/NC FZICs at different scan rates are shown in Fig. S6(a).† The CV shape was well-maintained even at a high scan rate of 100 mV s−1, revealing the fast charge and discharge properties of the NbMo6S8/NC FZICs. Unfortunately, electronic components that are manufactured typically have a wide range of operating voltages. Weakening the actual output voltage of the energy storage units in series and parallel is the appropriate path for their practical use. The designed capacitor device has a working voltage of 1.5 V, which can be increased by connecting the two devices in series (Fig. 7(a)–(c)). In series connections, the NbMo6S8/NC FZICs were capable of lighting up a red and green light-emitting diode (LED) (Fig. 7(f)). The total capacity could be doubled by connecting the two devices in parallel. The CV profiles exhibit an elevated level of integration. The superior Zn-ion kinetics of the NbMo6S8/NC FZIC was further supported by the Nyquist plot obtained by impedance analyses of single, parallel, and series combinations with the best-fitted equivalent circuit model, as illustrated in Fig. 7(d).
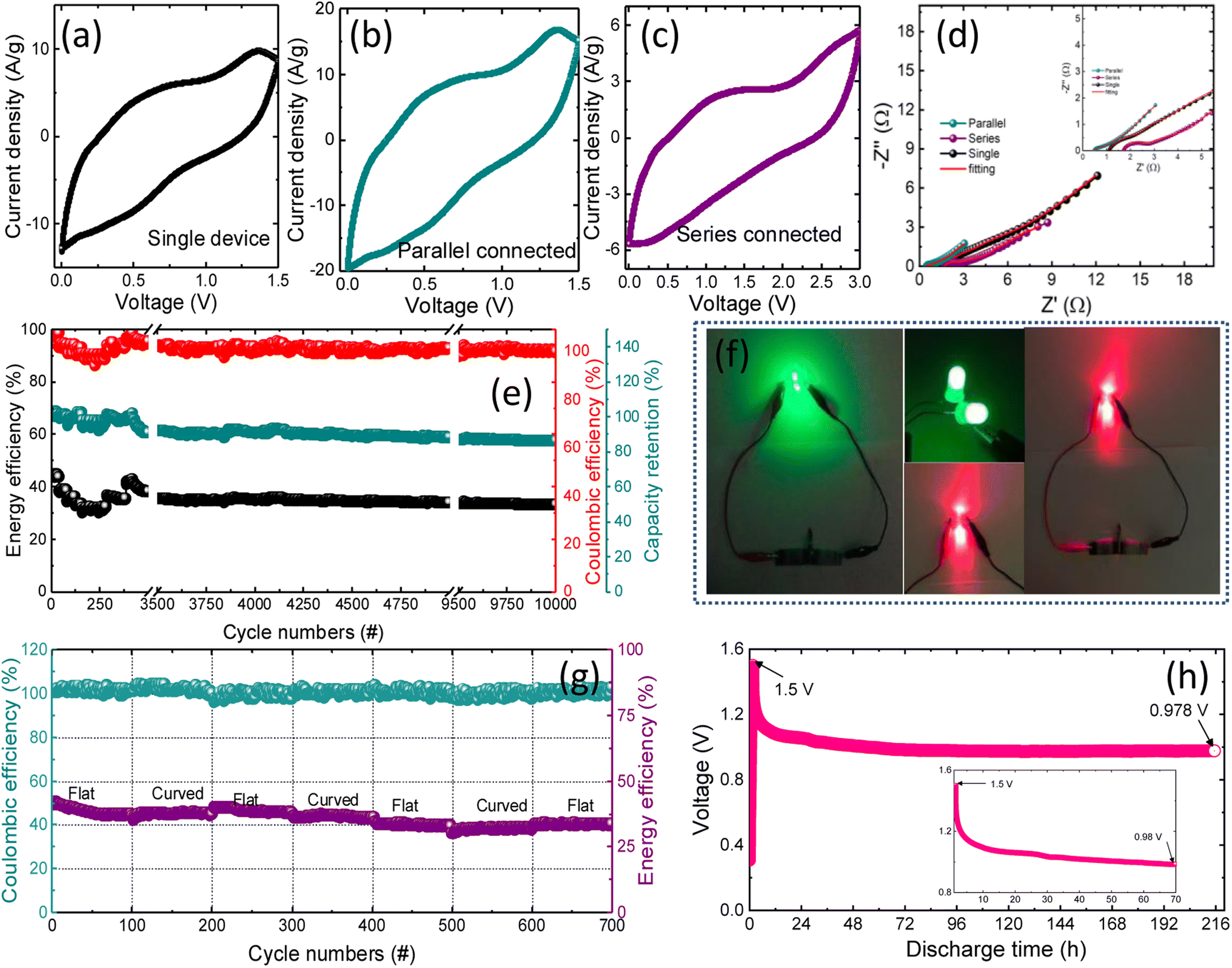 | ||
| Fig. 7 Electrochemical characterization of the NbMo6S8/NC FZIC device. CV profiles of (a) single, and two FZICs in (b) parallel and in (c) series combination and the corresponding Nyquist plots (d). (e) Rate performance of NbMo6S8/NC FZICs from 2.5 to 7.5 A g−1 showing stability over 10000 cycles. (f) Digital photographs of NbMo6S8/NC FZICs powering two red and green LED devices. (g) Flexible test under flat and bent conditions over 100 cycles each. (h) Self-discharge profiles of NbMo6S8/NC FZICs after the devices were charged to 1.5 V at 0.125 A g−1. | ||
The coulombic and energy efficiencies of NbMo6S8/NC FZICs were obtained over 10000 charge–discharge cycles, with respective values of 2.05 and 6.51% for decay in the initial cycles (Fig. 7(e)). The charge–discharge curves of the designed FZIC in the flat and curved states, as shown in Fig. 7(g), reveal that the flexible NbMo6S8/NC ZIC device can work reliably under high-stress conditions, proving that the FZIC has wide industrial applicability and potential implementation. In terms of cycling performance, the fabricated NbMo6S8/NC FZICs retained more than 80% of the energy efficiency after one hundred cycles each of continuous bending–flat trials. The illumination of green and red LEDs illustrates that the NbMo6S8/NC FZICs have enormous potential for power applications. The self-discharge profile (Fig. 7(h)) was obtained by evaluating the open-circuit voltage change after fully charging the devices to 1.5 V at 0.125 A g−1 and allowing them to undergo self-discharge for 216 h (9 days). The voltage at the FZIC terminals was continually monitored. The potential of the NbMo6S8/NC FZICs dropped to 0.52 V after 216 h, equivalent to 34.8% voltage loss in the FZIC. When the device is fully charged, the solvent is removed from the cathode side. This decrease can continue even after the current is cut off, which causes the cathode potential to rapidly decrease. This process is affected by the surface characteristics of the electrode and is not controlled by diffusion owing to current leakage.77 Finally, the results show that the NbMo6S8/NC FZIC has a high bearable current, allowing it to meet the current commercial electronics benchmark and power high-energy appliances.
Conclusion
In summary, NbMo6S8/NC was prepared as a cathode material through hydrothermal synthesis. The synthesized NbMo6S8/NC nanosheets show outstanding electrochemical performance in zinc-ion capacitors. The obtained NbMo6S8/NC nanosheets display a specific capacity of 167.89 mA h g−1 at 0.25 A g−1 as a cathode for ZICs. Furthermore, the ZIC affords a competitive specific energy of 188.87 W h kg−1 at a specific power of 250 W kg−1, demonstrating high-rate capability and retaining 87.60% of the initial capacitance over 15000 charge–discharge cycles. The designed NbMo6S8/NC FZIC devices connected in series are capable of powering light-emitting diodes. This novel NbMo6S8/NC cathode demonstrates the use of a modified high-concentration aqueous zinc-ion electrolyte with lower cost and higher ionic conductivity to achieve high-energy electrochemical performance, which can be beneficial for efficient, eco-friendly, and high-performance energy storage systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIT) (2022R1A2C2008968). This research was performed by the Industrial Strategic Technology Development Program-Strategic Core Material Independent Technology Development Project (20010193, development of electrode fabrication technology using pitch for an MCDI system applied to lithium recovery from low grade brine and waste solution and manufacturing technology of lithium compound for anode material of lithium secondary batteries) funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea).References
- J. Zheng and L. A. Archer, Sci. Adv., 2021, 7, eabe0219 CrossRef CAS PubMed
.
- Q. Liu, Y. Wang, X. Yang, D. Zhou, X. Wang, P. Jaumaux, F. Kang, B. Li, X. Ji and G. Wang, Chem, 2021, 7, 1993–2021 CAS
- Z. Pan, X. Liu, J. Yang, X. Li, Z. Liu, X. J. Loh and J. Wang, Adv. Energy Mater., 2021, 11, 1–24 Search PubMed
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S. Z. Qiao, Sci. Adv., 2020, 21 DOI:10.1126/sciadv.aba4098
- Y. Li, B. Huang, X. Zhao, Z. Luo, S. Liang, H. Qin and L. Chen, J. Power Sources, 2022, 527, 231149 CrossRef CAS
- L. Xuan, L. Chen, Q. Yang, W. Chen, X. Hou, Y. Jiang, Q. Zhang and Y. Yuan, J. Mater. Chem. A, 2015, 3, 17525–17533 RSC
- W. Xu, X. Zhao, F. Zhan, Q. He, H. Wang, J. Chen, H. Wang, X. Ren and L. Chen, Energy Storage Mater., 2022, 53, 79–135 CrossRef
- A. Ponrouch, C. Frontera, F. Bardé and M. R. Palacín, Nat. Mater., 2016, 15, 169–172 CrossRef CAS PubMed
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 1–7 Search PubMed
- S. Chen, D. Zhao, L. Chen, G. Liu, Y. Ding, Y. Cao and Z. Chen, Small Struct., 2021, 2, 2100082 CrossRef CAS
- M. Arnaiz, D. Shanmukaraj, D. Carriazo, D. Bhattacharjya, A. Villaverde, M. Armand and J. Ajuria, Energy Environ. Sci., 2020, 13, 2441–2449 RSC
- D. Selvakumaran, A. Pan, S. Liang and G. Cao, J. Mater. Chem. A, 2019, 7, 18209–18236 RSC
- H. Cui, H. Mi, C. Ji, F. Guo, Y. Chen, D. Wu, J. Qiu and H. Xie, J. Mater. Chem. A, 2021, 9, 23941–23954 RSC
- Y. P. Deng, R. Liang, G. Jiang, Y. Jiang, A. Yu and Z. Chen, ACS Energy Lett., 2020, 5, 1665–1675 CrossRef CAS
- S. J. Patil, N. R. Chodankar, S. K. Hwang, G. S. R. Raju, K. S. Ranjith, Y. S. Huh and Y. K. Han, Energy Storage Mater., 2022, 45, 1040–1051 CrossRef
- G. Liang, F. Mo, H. Li, Z. Tang, Z. Liu, D. Wang, Q. Yang, L. Ma and C. Zhi, Adv. Energy Mater., 2019, 9, 1–12 Search PubMed
- H. Gao, K. Tang, J. Xiao, X. Guo, W. Chen, H. Liu and G. Wang, J. Energy Chem., 2022, 69, 84–99 CrossRef CAS
- X. Ji, eScience, 2021, 1, 99–107 CrossRef
- Y. Ma, M. Xu, R. Liu, H. Xiao, Y. Liu, X. Wang, Y. Huang and G. Yuan, Energy Storage Mater., 2022, 48, 212–222 CrossRef
- Z. Chen, Y. Qin, D. Weng, Q. Xiao, Y. Peng, X. Wang, H. Li, F. Wei and Y. Lu, Adv. Funct. Mater., 2009, 19, 3420–3426 CrossRef CAS
- Y. Lu, T. Zhu, W. van den Bergh, M. Stefik and K. Huang, Angew. Chem., Int. Ed., 2020, 59, 17004–17011 CrossRef CAS PubMed
- S. Khamsanga, R. Pornprasertsuk, T. Yonezawa, A. A. Mohamad and S. Kheawhom, Sci. Rep., 2019, 9, 1–9 CrossRef CAS PubMed
- C. Witteveen, K. Górnicka, J. Chang, M. Månsson, T. Klimczuk and F. O. von Rohr, Dalton Trans., 2021, 50, 3216–3223 RSC
- X. Wang, J. Lin, Y. Zhu, C. Luo, K. Suenaga, C. Cai and L. Xie, Nanoscale, 2017, 9, 16607–16611 RSC
- Y. Zhou, Z. Wang, P. Yang, X. Zu, L. Yang, X. Sun and F. Gao, ACS Nano, 2012, 6, 9727–9736 CrossRef CAS PubMed
- L. Najafi, S. Bellani, R. Oropesa-Nuñez, B. Martín-García, M. Prato, V. Mazánek, D. Debellis, S. Lauciello, R. Brescia, Z. Sofer and F. Bonaccorso, J. Mater. Chem. A, 2019, 7, 25593–25608 RSC
- L. Lin, W. Lei, S. Zhang, Y. Liu, G. G. Wallace and J. Chen, Energy Storage Mater., 2019, 19, 408–423 CrossRef
- H. Z. Qinbai Yun, L. Li, Z. Hu, Q. Lu and B. Chen, Adv. Mater., 2019, 32, 1903826 Search PubMed
- N. Alinejadian, L. Kollo and I. Odnevall, Mater. Sci. Semicond. Process., 2022, 139, 106331 CrossRef CAS
- M. A. H. Xiaolei Wang, G. Li, M. H. Seo, F. M. Hassan and Z. Chen, Adv. Energy Mater., 2015, 5, 1501106 CrossRef
- L. bo Tang, B. Zhang, T. Peng, Z. jiang He, C. Yan, J. Mao, K. Dai, X. wen Wu and J. chao Zheng, Nano Energy, 2021, 90, 106568 CrossRef
- B. D. Boruah, B. Wen and M. De Volder, ACS Nano, 2021, 15, 16616–16624 CrossRef CAS PubMed
- G. Pan, J. Li, L. Han, W. Peng, X. Xu, T. Lu, M. A. Amin, Y. Yamauchi, M. Xu and L. Pan, Inorg. Chem. Front., 2022, 9, 1666–1673 RSC
- R. Yuksel, O. Buyukcakir, P. K. Panda, S. H. Lee, Y. Jiang, D. Singh, S. Hansen, R. Adelung, Y. K. Mishra, R. Ahuja and R. S. Ruoff, Adv. Funct. Mater., 2020, 30 Search PubMed
- H. Lim, H. Kim, S. O. Kim and W. Choi, Chem. Eng. J., 2020, 387, 124144 CrossRef CAS
- Z. Yang, L. Zhu, C. Lv, R. Zhang, H. Wang, J. Wang and Q. Zhang, Mater. Chem. Front., 2021, 5, 5880–5896 RSC
- L. F. Chen, Y. Lu, L. Yu and X. W. Lou, Energy Environ. Sci., 2017, 10, 1777–1783 RSC
- L. Kendall, A. Chamaani, Z. Piontkowski, T. E. Beechem, M. Ridley, E. J. Opila, G. Zangari and S. J. McDonnell, ACS Appl. Energy Mater., 2021, 4, 13676–13683 CrossRef CAS
- Y. Tong, A. Gao, Q. Zhang, T. Gao, J. Yue, F. Meng, Y. Gong, S. Xi, Z. Lin, M. Mao, S. Peng, X. Wang, D. Xiao, D. Su, Y. Luo, H. Li, L. Chen, L. Suo and L. Gu, Energy Storage Mater., 2021, 37, 87–93 CrossRef
- X. Cai, H. Yan, R. Zheng, H. Yu, Z. Yang, X. Zhang, M. Xia, W. Chen, Y. Cui and J. Shu, Inorg. Chem. Front., 2021, 8, 444–451 RSC
- S. J. Patil, J. H. Kim and D. W. Lee, J. Power Sources, 2017, 342, 652–665 CrossRef CAS
- X. Han, G. He, Y. He, J. Zhang, X. Zheng, L. Li, C. Zhong, W. Hu, Y. Deng and T. Y. Ma, Adv. Energy Mater., 2018, 8, 1–13 Search PubMed
- S. A. Shah, X. Shen, M. Xie, G. Zhu, Z. Ji, H. Zhou, K. Xu, X. Yue, A. Yuan, J. Zhu and Y. Chen, Small, 2019, 15, 1804545 CrossRef PubMed
- P. Wu, K. Wang, S. Yu, M. Feng, Y. Chen, S. Liu and J. Fu, ACS Appl. Energy Mater., 2022, 5, 137–148 CrossRef CAS
- J. Zhang, L. Qu, G. Shi, J. Liu, J. Chen and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 2230–2234 CrossRef CAS PubMed
- L. N. Lu, Y. L. Luo, H. J. Liu, Y. X. Chen, K. Xiao and Z. Q. Liu, Chem. Eng. J., 2022, 427, 132041 CrossRef CAS
- N. Usha, R. Sivakumar, C. Sanjeeviraja and Y. Kuroki, J. Alloys Compd., 2015, 649, 112–121 CrossRef CAS
- X. Fan, P. Xu, D. Zhou, Y. Sun, Y. C. Li, M. A. T. Nguyen, M. Terrones and T. E. Mallouk, Nano Lett., 2015, 15, 5956–5960 CrossRef CAS PubMed
- L. Yu, L. Lu, L. Zeng, X. Yan, X. Ren and J. Z. Wu, J. Phys. Chem. C, 2021, 125, 1940–1946 CrossRef CAS
- X. Xi, F. Zeng, H. Cao, C. Cannilla, T. Bisswanger, S. de Graaf, Y. Pei, F. Frusteri, C. Stampfer, R. Palkovits and H. J. Heeres, Appl. Catal., B, 2020, 272, 118950 CrossRef CAS
- X. Bu, L. Su, Q. Dou, S. Lei and X. Yan, J. Mater. Chem. A, 2019, 7, 7541–7547 RSC
- Y. Zhu, J. Yin, X. Zheng, A. H. Emwas, Y. Lei, O. F. Mohammed, Y. Cui and H. N. Alshareef, Energy Environ. Sci., 2021, 14, 4463–4473 RSC
- R. Fei, H. Wang, Q. Wang, R. Qiu, S. Tang, R. Wang, B. He, Y. Gong and H. J. Fan, Adv. Energy Mater., 2020, 10, 2002741 CrossRef CAS
- X. Wei, Y. Zhang, B. Zhang, Z. Lin, X. Wang, P. Hu, S. Li, X. Tan, X. Cai, W. Yang and L. Mai, Nano Energy, 2019, 64, 103899 CrossRef CAS
- A. Ilnicka, M. Skorupska, M. Szkoda, Z. Zarach, P. Kamedulski, W. Zielinski and J. P. Lukaszewicz, Sci. Rep., 2021, 11, 1–11 CrossRef PubMed
- Z. Qin, X. Pan, G. Qiu, Z. Zhang, Y. Chen, S. Nasir Khisro, Y. Zhang, L. Zhao and X. Chen, Chem. Eng. J., 2022, 428, 131179 CrossRef CAS
- S. Li, Y. Liu, X. Zhao, Q. Shen, W. Zhao, Q. Tan, N. Zhang, P. Li, L. Jiao and X. Qu, Adv. Mater., 2021, 33, 2007480 CrossRef CAS PubMed
- H. J. Zhang, Q. C. Jia and L. Bin Kong, Appl. Surf. Sci., 2020, 531, 147222 CrossRef CAS
- Z. Sheng, P. Qi, Y. Lu, G. Liu, M. Chen, X. Gan, Y. Qin, K. Hao and Y. Tang, ACS Appl. Mater. Interfaces, 2021, 13, 34495–34506 CrossRef CAS PubMed
- S. Li, Y. Liu, X. Zhao, K. Cui, Q. Shen, P. Li, X. Qu and L. Jiao, Angew. Chem., 2021, 133, 20448–20455 CrossRef
- J. Ju, L. Zhang, H. Shi, Z. Li, W. Kang and B. Cheng, Appl. Surf. Sci., 2019, 484, 392–402 CrossRef CAS
- X. Yuan, S. Qiu and X. Zhao, ACS Appl. Mater. Interfaces, 2021, 13, 34238–34247 CrossRef CAS PubMed
- J. Liu, P. Xu, J. Liang, H. Liu, W. Peng, Y. Li, F. Zhang and X. Fan, Chem. Eng. J., 2020, 389, 124405 CrossRef CAS
- C. Zhan, W. Liu, M. Hu, Q. Liang, X. Yu, Y. Shen, R. Lv, F. Kang and Z. H. Huang, NPG Asia Mater., 2018, 10, 775–787 CrossRef CAS
- H. Li, Q. Yang, F. Mo, G. Liang, Z. Liu, Z. Tang, L. Ma, J. Liu, Z. Shi and C. Zhi, Energy Storage Mater., 2019, 19, 94–101 CrossRef
- W. Xu, C. Sun, K. Zhao, X. Cheng, S. Rawal, Y. Xu and Y. Wang, Energy Storage Mater., 2019, 16, 527–534 CrossRef
- J. Jiang, Y. Zhang, Y. An, L. Wu, Q. Zhu, H. Dou and X. Zhang, Small Methods, 2019, 3, 1900081 CrossRef
- X. Wang, Q. Li, L. Zhang, Z. Hu, L. Yu, T. Jiang, C. Lu, C. Yan, J. Sun and Z. Liu, Adv. Mater., 2018, 30, 1800963 CrossRef PubMed
- S. Liang, S. Zhang, Z. Liu, J. Feng, Z. Jiang, M. Shi, L. Chen, T. Wei and Z. Fan, Adv. Energy Mater., 2021, 11, 1–11 Search PubMed
- J. Chao, L. Yang, H. Zhang, J. Liu, R. Hu and M. Zhu, J. Power Sources, 2022, 450, 227680 CrossRef
- S. Li, Y. Liu, X. Zhao, K. Cui, Q. Shen, P. Li, X. Qu and L. Jiao, Angew. Chem., Int. Ed., 2021, 60, 20286–20293 CrossRef CAS PubMed
- S. J. Patil, N. R. Chodankar, S. K. Hwang, G. S. R. Raju, K. S. Ranjith, Y. S. Huh and Y. K. Han, Energy Storage Mater., 2022, 45, 1040–1051 CrossRef
- Q. Zhu, J. Wang, X. Liu, N. Ebejer, D. Rambabu and A. Vlad, Angew. Chem., 2020, 16579–16586 CrossRef CAS PubMed
- V. Shutthanandan, M. Nandasiri, J. Zheng, M. H. Engelhard and W. Xu, J. Electron Spectrosc. Relat. Phenom., 2019, 231, 2–10 CrossRef CAS
- S. Zhou, S. Yang, D. Cai, C. Liang, S. Yu, Y. Hu, H. Nie and Z. Yang, Adv. Sci., 2022, 9, 2104205 CrossRef CAS PubMed
- Q. Chen, J. Jin, Z. Kou, C. Liao, Z. Liu, L. Zhou, J. Wang and L. Mai, Small, 2022, 16(14), 2000091 CrossRef PubMed
- F. Barzegar, A. A. Khaleed, F. U. Ugbo, K. O. Oyeniran, D. Y. Momodu, A. Bello, J. K. Dangbegnon and N. Manyala, AIP Adv., 2016, 6, 115306 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta07524e |
| This journal is © The Royal Society of Chemistry 2023 |