
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystallographic evidence for global aromaticity in the di-anion and tetra-anion of a cyclophane hydrocarbon†
Wojciech
Stawski
 ab,
Yikun
Zhu
a,
Zheng
Wei
a,
Marina A.
Petrukhina
*a and
Harry L.
Anderson
*b
ab,
Yikun
Zhu
a,
Zheng
Wei
a,
Marina A.
Petrukhina
*a and
Harry L.
Anderson
*b
aDepartment of Chemistry, University at Albany, State University of New York, Albany, NY 12222, USA. E-mail: mpetrukhina@albany.edu
bDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Oxford OX1 3TA, UK. E-mail: harry.anderson@chem.ox.ac.uk
First published on 20th September 2023
Abstract
[24]Paracyclophanetetraene is a classic example of a macrocyclic hydrocarbon that becomes globally aromatic on reduction to the di-anion, and switches to globally anti-aromatic in the tetra-anion. This redox activity makes it promising as an electrode material for batteries. Here, we report the solid-state structures of the di- and tetra-anions of this cyclophane, in several coordination environments. The changes in bond length on reduction yield insights into the global aromaticity of the di-anion (26π electrons), and anti-aromaticity of the tetra-anion (28π electrons), that were previously deduced from NMR spectra of species generated in situ. The experimental geometries of the aromatic di-anion and anti-aromatic tetra-anion from X-ray crystallographic data match well with gas-phase calculated structures, and reproduce the low symmetry expected in the anti-aromatic ring. Comparison of coordinated and naked anions confirms that metal coordination has little effect on the bond lengths. The UV-vis-NIR absorption spectra show a sharp intense peak at 878 nm for the di-anion, whereas the tetra-anion gives a broad spectrum typical of an anti-aromatic system.
Introduction
Reduction of π-conjugated hydrocarbons with alkali metals can profoundly alter their electronic structures and properties.1 [24]Paracyclophanetetraene 1 (PCT, Fig. 1) is one of the first macrocyclic hydrocarbons reported to exhibit global electronic delocalization upon reduction.2,3 The electronic structure of the nonplanar neutral form is described by four localized benzene Clar sextets, separated by cis-vinylene bridges with purely olefinic character. Adding two or four electrons generates a delocalization pathway leading to global aromaticity (di-anion: 26π e−) or anti-aromaticity (tetra-anion: 28π e−), manifested by large chemical shift differences between the inner and outer core protons in the 1H NMR spectra (Hα and Hβ, Fig. 1).3 It has been questioned whether global aromaticity can be reliably assigned in large macrocycles based purely on NMR data,4 which motivated us to check for structural evidence from global aromaticity. The redox-based aromaticity switching in 1 may make it a suitable material for storing energy in batteries,5 and several derivatives of 1 have been synthesized for this application.6 It is desirable to know the solid-state structure of anions involved in these processes, and to understand their coordination chemistry with alkali metal cations. Although the redox behavior of 1 has been known for over 40 years, the crystal structures of the reduced species have not been reported. We decided to take up this challenge and characterize them in the solid state.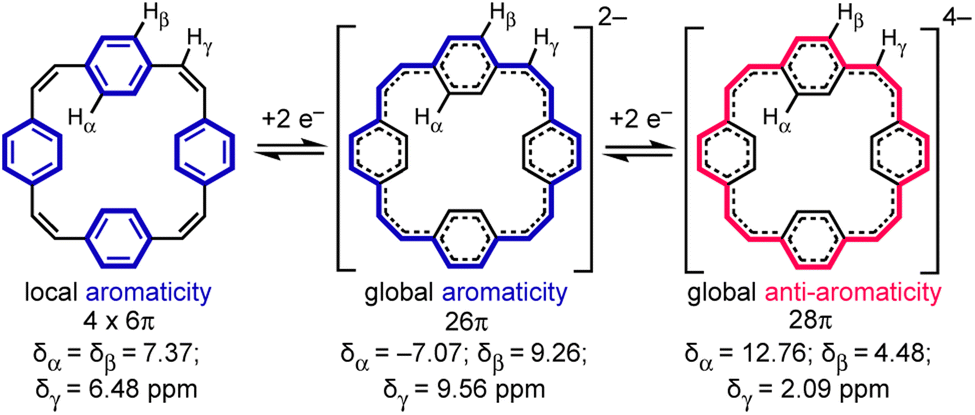 | ||
| Fig. 1 Reduction of neutral 1 (PCT) to the di- and tetra-anion, showing the number of π electrons in the delocalization pathways and 1H NMR chemical shifts in THF-d8, −80 °C (data from ref. 3a). | ||
Results and discussion
Crystals of the PCT salts were grown by reducing the cyclophane with alkali metals in THF, followed by layered addition of hexane. Plate-like crystals of Li and Na salts of the di-anion 12− appeared after few hours. The lithium salt of 12− crystallized with one lithium cation coordinated to a vinylene bridge and the second Li+ solvent-separated (Fig. 2a). The molecular formula of this product can be written as [Li+(THF)4][{Li+(THF)3}(12−)] and we use the shortened Li2-12− (and similarly for other products) throughout the text.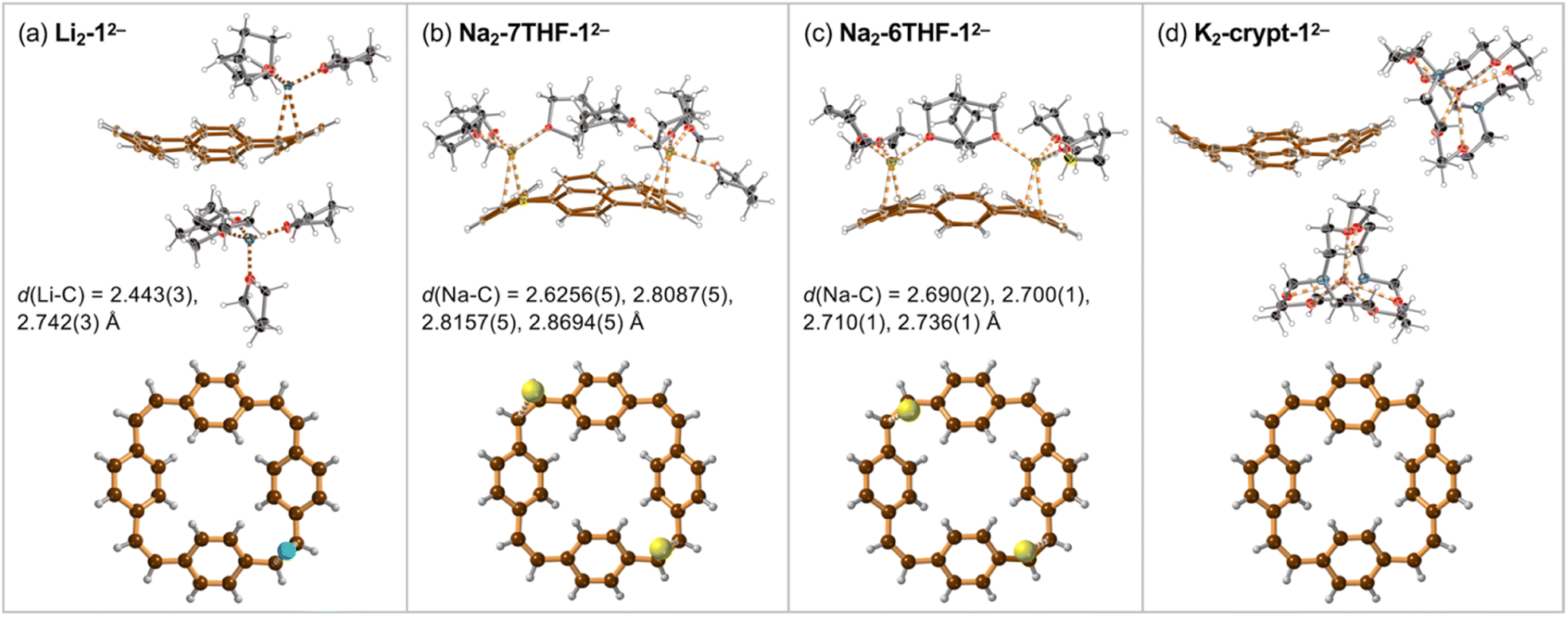 | ||
| Fig. 2 Single crystal X-ray structures of (a) Li2-12−, (b) Na2-7THF-12−, (c) Na2-6THF-12− and (d) K2-crypt-12− (thermal ellipsoids: 40% probability). In each case, the lower image is a projection perpendicular to the mean plane of the PCT di-anion, with coordinated metal cations. The shortest metal-carbon distances are indicated by dashed lines and listed. Solvent molecules and minor disorder components are omitted for clarity. Color code: Li: sky-blue; Na: yellow; K: purple; N: blue; O: red; C: black or brown; H: pale gray. | ||
Reaction of 1 with sodium provided two types of crystals of close–contact complexes, Na2-6THF-12− and Na2-7THF-12− (Fig. 2b and c). In Na2-6THF-12−, described more accurately as [Na+(THF)3]2[12−], both counterions coordinate to three THF molecules and bind to two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bridges. Despite the different numbers of coordinated THF molecules in Na2-7THF-12−, with the more descriptive formula of [Na+(THF)3][Na+(THF)4][12−], both Na+ cations also bind CC bridges, as in Na2-6THF-12−. In both cases, the cations are located on the same side of the hydrocarbon di-anion, on the opposite CC bridges.
C bridges. Despite the different numbers of coordinated THF molecules in Na2-7THF-12−, with the more descriptive formula of [Na+(THF)3][Na+(THF)4][12−], both Na+ cations also bind CC bridges, as in Na2-6THF-12−. In both cases, the cations are located on the same side of the hydrocarbon di-anion, on the opposite CC bridges.
In order to check the impact of metal coordination on electronic delocalization, 18-crown-6 and [2.2.2]cryptand were added to the reaction mixtures with sodium and potassium, respectively. Two solvent-separated products were isolated. [K+(cryptand)]2[12−], abbreviated as K2-crypt-12−, features two cryptand-bound cations, separating them from the hydrocarbon di-anion (Fig. 2d). Similarly, Na2-crown-12−, with the detailed formula of [Na+(18-crown-6)(THF)2]2[12−], contains 12− di-anions solvent-separated from the cations, which coordinate to the crown ether and THF (ESI, Fig. S10†).
The geometries of the 12− di-anions in all five crystal structures are remarkably similar, and they closely resemble the geometry of gas-phase 12− predicted by density functional theory (DFT), as shown by the patterns of bond lengths plotted in Fig. 3 (see also Fig. S17† for comparison of the crystallographic bond lengths in 12− di-anions) and by the overlayed projections in Fig. 4 (solid-state structure in orange; gas-phase structure in blue). Reduction of neutral 1 to 12− results in a global decrease in bond-length alternation around the macrocycle (Fig. 3, black and blue traces). The changes are most striking in the vinylene bridges. For example, in Li2-12−, the single CC bond coordinated to the Li+ cation is most elongated (bond b; 1.402(2) Å) compared with 1.312(5) Å in the neutral 1, but the other three remote CC bonds are almost as elongated (h: 1.386(2) Å; n: 1.374(2) Å, t: 1.383(2) Å) reflecting the global change in electronic structure as the whole system becomes aromatic with a circuit of 26π-electrons. The elongation of the CC double bonds (b, h, n, t) is accompanied by a contraction of the C–C single bonds (a, c, g, i, m, o, s, u), corresponding to a reduction in bond-length alternation.
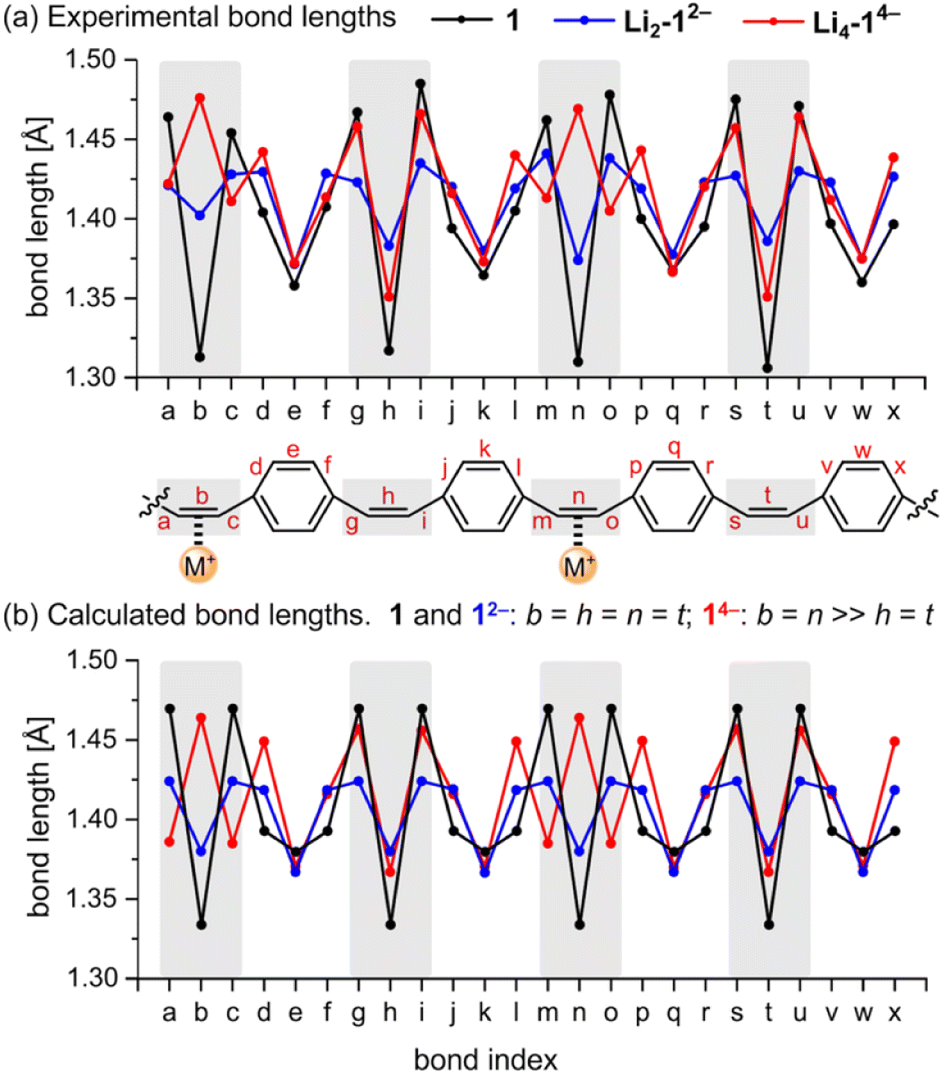 | ||
| Fig. 3 Comparison of (a) crystallographic C–C bond lengths in 1, Li2-12− and Li4-14−, and (b) calculated bond lengths for gas-phase 1, 12− and 14− from DFT (CAM-B3LYP/def2-TZVP). Bonds in the vinylene bridges are marked in gray. Lengths for the para-phenylene units (e.g. d–f) are averages for inner and outer bonds. | ||
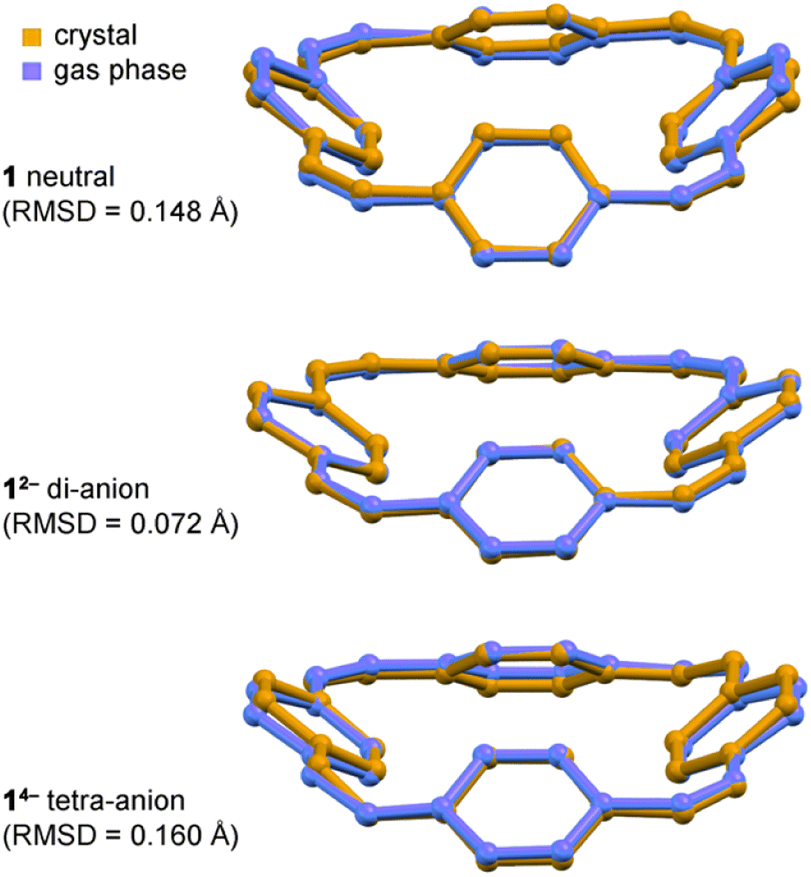 | ||
| Fig. 4 Overlay of crystallographic (orange) and calculated gas-phase (blue) structures of neutral 1, Li2-12− and Li4-14−, with RMSDs between experimental and calculated structures. Note that the Li+ cations were not included in the gas phase DFT calculations. Calculations: CAM-B3LYP def2-TZVP. | ||
We performed DFT calculations on isolated anions in the gas phase in order to compare the experimental bond lengths with the theoretical values (CAM-B3LYP/def2-TZVP; Fig. 3).5,7 In the optimized structure of di-anion, the length of the middle bond in the CC bridge (b) is 1.380 Å whereas the bonds linking it to the para-phenylene rings (a) are 1.424 Å (Fig. 3b). These values are close to the experimentally determined lengths for bridges not involved in metal binding, with the general trend that coordination to metal cations increases the length of the CC bond by ca. 0.01–0.02 Å, depending on the metal. Comparison of the direct-contacted and solvent-separated anions shows that metal coordination only slightly perturbs the effect of electronic delocalization over the whole molecule.
The global electronic delocalization in the di-anion can be evaluated using the harmonic oscillator model of aromaticity (HOMA), which compares bond lengths with those in benzene (see ESI, Section 7†).8 The vinylene bridges of neutral 1 give a HOMA value of −0.66 (non-aromatic), whereas the five crystal structures of 12− salts give HOMA values in the range 0.66–0.73 (compared with an ideal value of 1.00 for fully aromatic).
In all the structures of the 12− di-anion, the torsion angles in the linkers (defined by the coordinates of ethylene bridges and the two carbon atoms directly connected to them) increase substantially upon reduction, consistently with the theoretical predictions and with a reduction in bond order (see ESI, Section 3.2†). The whole C32H24 framework generally becomes flatter on reduction to the di-anion, reflecting greater π-delocalization around the macrocycle. Thus the root-mean-square deviation (RMSD) of the 32 carbon atoms from a plane decreases from 0.527 Å in neutral 1 to 0.395 Å in 12− in the DFT geometries; the experimental RMSDs are 0.411 Å in Li2-12−, 0.501 Å in Na2-7THF-12−, 0.452 Å in Na2-6THF-12− and 0.462 Å in K2-crypt-12−, compared with 0.471 Å in neutral 1.2a
Prolonged contact of 1 with lithium metal in THF provided a tetra-reduced product [Li+(THF)2]4[14−] (Li4-14−, Fig. 5). The lithium cations coordinate to the opposite C2 linkers, two to each vinylene bridge. Each cation is additionally coordinated to two THF molecules. The 14− macrocycle exhibits stronger bond-length alternation than 12− (Fig. 3a, red trace). The C2 bridges that are coordinated to Li+ cations (b and n) are remarkably elongated (1.469(4) and 1.476(4) Å) compared with the CC bond length in neutral 1 (1.312(4) Å). The C2 bridges that are not coordinated by lithium cations (h and t) are slightly elongated (1.350(4) and 1.351(4) Å). Bridges involved in coordination to metal centers are twisted, with torsion angles (48.3(3)/51.5(3)° vs. 5.3(3)/4.4(3)° in 1; see ESI†).
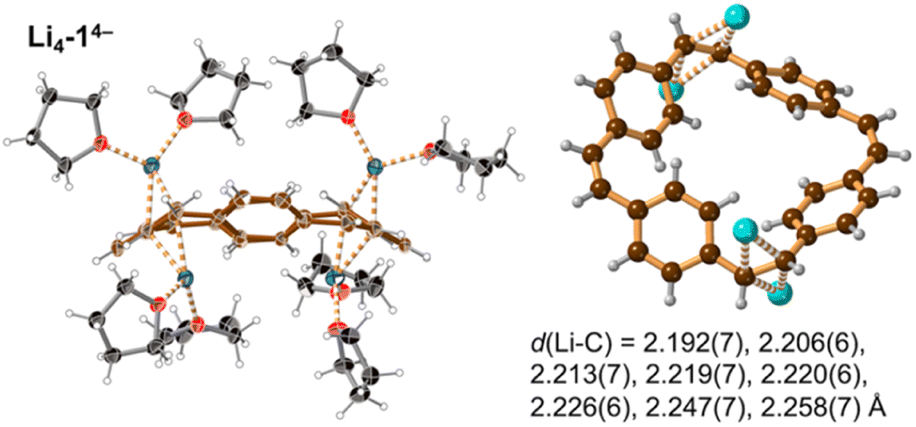 | ||
| Fig. 5 Single crystal X-ray structure of Li4-14−. Thermal ellipsoids at 40% probability level and ball-and-stick view. Minor disorder components are omitted for clarity. Color code as for Fig. 2. The shortest eight Li⋯C distances are indicated by dashed lines and listed. | ||
The pattern of experimental bond lengths and torsion angles in the 14− tetra-anion is similar to that in the gas-phase from DFT calculations (Fig. 3b), revealing that the substantial elongation of the two CC bridges involved in binding to lithium (b and n) is not a result of metal coordination. The gas-phase geometry has two long vinylene bridges (b and n), which have higher negative charge (Fig. S42†). The less symmetrical distribution of bond lengths in 14− is evidently a consequence of its anti-aromatic electronic structure.
Lithium is the only metal which provided a solution of a pure tetra-anion, which parallels results on polycyclic aromatic hydrocarbons.9 Analysis by 1H NMR spectroscopy shows that reduction with sodium yields a mixture of tetra-anion and di-anion (Fig. S25†), and only the di-anion crystallized from this mixture. Prolonged contact with the potassium resulted in a black precipitate, suggesting that the tetra-anion K+ salt is insoluble in THF. Indeed, the 1H NMR spectrum of the potassium di-anion salt was reported by Müllen et al., without commenting on the tetra-anion.3b Contact of a solution of 1 with rubidium or cesium metal causes immediate precipitation, even in the presence of secondary ligands. Addition of crown ethers or [2.2.2]cryptand stops reduction by sodium or potassium at the di-anion stage (for lithium as representative example, see Fig. S37†). When a solution of 1 was exposed to K in the presence of 18-crown-6 or [2.2.2]cryptand, the solution remained intensely orange, without any precipitate, indicating that it also stopped at the di-anion. We can therefore control the maximum available reduction state by adding a secondary ligand.
We recorded 1H NMR spectra of all the crystal samples dissolved in d8-THF. Previously, the only spectra reported were for lithium salts of 12− and 14− (Fig. 1),3a and the potassium salt of 12−, generated in situ.3b All the 1H NMR spectra of the di-anionic species consist of three singlets, two at high chemical shift (outer protons, Hβ and Hγ, 9.2 to 9.6 ppm) and one at negative chemical shift (inner protons, Hα −6.8 to −7.1 ppm). The simplicity of the spectra implies that there is a rapid exchange between metal binding sites. Li2-12− and Na2-12− give sharp signals at −30 °C, whereas it is necessary to decrease the temperature to −80 °C to see partial splitting of two high frequency signals in Na2-crown-12− and K2-crypt-12−, and at this temperature the solubility drops resulting in a poor signal-to-noise ratio. The chemical shifts are summarized in Table 1. As noted previously, the shielded inner proton is sensitive to the counter cation (Hα: δH = −7.10 ppm at −30 °C for Li2-12−, vs. δH = −6.94 ppm for Na2-12−).3b The 7Li NMR spectrum of Li2-12− recorded at −30 °C exhibits a peak at δLi = −3.02 ppm, indicating that binding to Li+ results in shielding of the cation by the π-system (c.f. δLi = 0 ppm for LiCl in d8-THF).
| Compound | T (°C) | δ α (ppm) | δ β (ppm) | δ γ (ppm) |
|---|---|---|---|---|
| a Spectra recorded in THF-d8 at 500 MHz; see Fig. 1 for definition of α, β and γ proton environments. | ||||
| 1 | 25 | 7.32 | 7.32 | 6.42 |
| −30 | 7.44 | 7.44 | 6.39 | |
| Li2-12− | 25 | −6.85 | 9.21 | 9.49 |
| −30 | −7.10 | 9.25 | 9.54 | |
| Na2-12− | −30 | −6.94 | 9.27 | 9.57 |
| Na2-crown-12− | −90 | −7.05 | 9.19 | 9.49 |
| K2-crypt-12− | −80 | −6.76 | 9.23 | 9.46 |
| Li4-14− | 25 | 12.39 | 4.59 | 2.30 |
| −30 | 13.76 | 4.19 | 1.80 | |
In the 1H NMR spectra of Li4-14− recorded at 25 °C, the trend is opposite to that in the di-anion, and the inner protons (Hα) resonate at high chemical shift (12.39 ppm) whereas the outer protons (Hβ and Hγ) appear at low frequencies (2.30 and 4.59 ppm), due to the presence of a paratropic ring current.3,10 The 7Li NMR spectrum shows a broad peak at δLi = 0.25 ppm, implying that the tetra-anion is solvent-separated.
Previous studies of PCT have not mentioned the spectacular color changes that accompany reduction, nor reported the UV-vis-NIR absorption spectra of the reduced species. A solution of neutral PCT in THF exhibits a band at 306 nm, with a shoulder reaching the visible region, resulting in a yellow color (Fig. 6). Reduction with lithium metal gives a bright orange solution, with the most red-shifted band at 878 nm, corresponding to the di-anion. Reduction occurs via a radical anion, with a broad band at 1300 nm, which was previously studied by EPR spectroscopy.11 Further reduction to the tetra-anion is characterized by isosbestic points, indicating that the intermediate trianion does not build up to significant concentrations. The tetra-anion has a rather featureless UV-vis-NIR absorption spectrum, as is characteristic of anti-aromatic species.12 The experimental absorption spectra agree moderately well with simulated spectra from TD-DFT calculations (CAM-B3LYP def2-TZVP) for the free anions in the gas phase (Fig. S43 and S44†). The UV-vis-NIR spectra of anions generated in situ match those recorded upon dissolving crystals (Fig. S22†). The type of counterion (Li+–K+) and the presence of crown ether or cryptand have negligible effect on the optical spectra, in line with the relatively small differences observed in 1H NMR spectra, confirming that the anions are solvent-separated in THF solution.
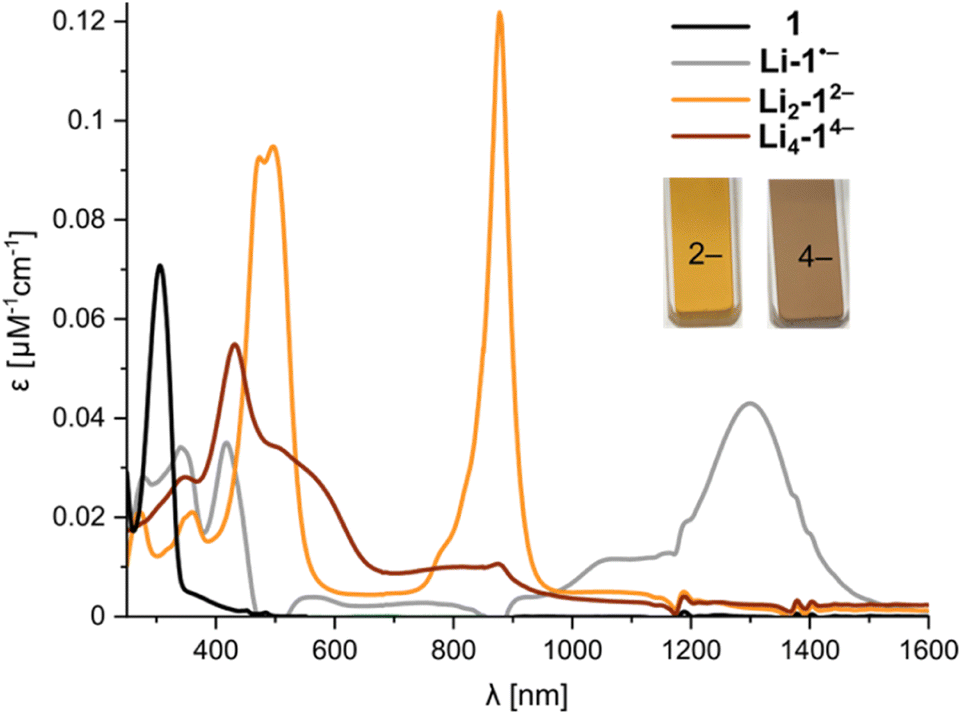 | ||
| Fig. 6 UV-vis-NIR spectra from in situ reduction of 1 with Li metal in THF at 25 °C, corresponding to the neutral macrocycle, radical anion, di-anion and tetra-anion (for details, see Fig. S18†). | ||
Conclusions
Four features of the crystal structures of the 12− and 14− anions are remarkable:(a) Reduced bond-length alternation in the di-anion
Global aromatic delocalization in the di-anion is revealed by the fact that the nominally single bonds (a, c, g, i, m, o, s and u) get shorter, while the nominally double bonds (b, h, n and t) get longer, on reduction of 1 to 12− (Fig. 3), as expected from a simple resonance scheme (Fig. 7). Local aromaticity in the benzene rings could not account for these changes in bond length.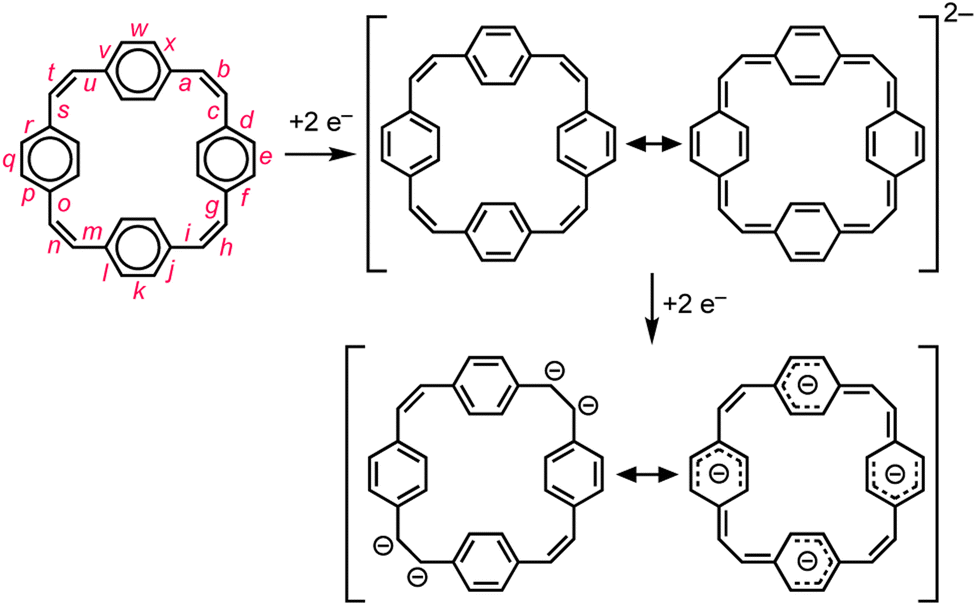 | ||
| Fig. 7 Schematic representation of the changes in delocalization revealed by the crystallographic molecular geometries. Reduction of the neutral cyclophane, with four localized Clar sextets, gives a globally delocalized aromatic di-anion. Further reduction gives a tetra-anion that can be viewed as the combination of four benzyl anions. | ||
(b) Charge localization in the tetra-anion
Symmetry-breaking is a classical signature of anti-aromatic system (as illustrated by the D2h symmetry of cyclobutadiene) and this effect is clearly evident in the crystallographic and calculated geometries of 14−. It is most noticeable from the elongation of bonds b and n, the shortening of bonds t and h, and the elongation of bonds g, i, s and u (Fig. 3 and 6). Charge localization is confirmed experimentally by the locations of the counter ions in the crystal structures (near bonds b and n), and by the by gas-phase DFT Mulliken charge distribution (Fig. S42†). There must be some global anti-aromatic delocalization in 14− because the 1H NMR spectrum indicates a paratropic ring current, but it appears to be less delocalized than the di-anion.(c) Planarization
Text books list the formation of a planar π-system as one of the requirements for aromaticity. Thus it is noteworthy that the crystallographic RMSD of the 32 carbon atoms from a plane decreases from 0.471 Å in neutral 1 to 0.411 Å in Li2-12−. This planarization is achieved through a reduction in the torsion angle between the vinylene and phenylene units, which enhances global delocalization. The corresponding RMSD of 14− is 0.492 Å reflecting its more distorted structure. The 1H NMR spectra of 12− and 14− provide further evidence of planarization. The fact that protons Hα and Hβ give distinct resonances in the 1H NMR spectra of 12− and 14−, but not for neutral 1, shows that reduction increases the barrier for phenylene rotation, which is related to the preference for a planar structure.(d) Match between solid-state and gas-phase geometries
All three features (a)–(c) are observed in the crystal structures and reproduced by gas-phase DFT calculations, without including the counter ions, which confirms that these geometrical effects are caused by the electronic structure, not by interactions with the alkali metal ions. The surprisingly good match between the solid-state and gas-phase geometries is seen by comparing the bond lengths (Fig. 3a and b) and from the overlayed projections in Fig. 4.Taken together, the results presented here strongly support the conclusion that the 12− di-anion is globally aromatic. The diatropic ring current deduced from NMR spectroscopy is not just a magnetic effect; it reports on the global aromatic delocalization, and that delocalization is also expressed through the molecular geometry. The situation with the 14− tetra-anion is more complicated: the molecular structure indicates significant localization, yet the observation of a paratropic ring current indicates that there is some degree of global anti-aromaticity in this 28π system. Paradoxically, the observation of distortion and charge-localization in 14− (which must mitigate against anti-aromaticity) provides strong evidence for anti-aromaticity—it is difficult to identify other possible causes for the observed localization. The results from this work are relevant to understanding electronic delocalization in large π-conjugated macrocycles, and further insights into the structures of alkali metals salts of 1 may be useful for the development of materials for batteries.5
Data availability
The data that support the findings of this study are available in the ESI† of this article. Crystallographic data have been deposited at the Cambridge crystallographic data. Deposition numbers: 2272266 (Li2-12−), 2272267 (Na2-6THF-12−), 2272268 (Na2-7THF-12−), 2272269 (Na2-crown-12−), 2272270 (K2-crypt-12−) and 2272271 (Li4-14−). These data can be obtained free of charge viahttps://www.ccdc.cam.ac.uk/data_request/cif, or by emailing http://data_request@ccdc.cam.ac.uk. Cartesian coordinates of calculated molecular geometries are available from the Zenodo public repository (https://zenodo.org/record/8239309 and https://doi.org/10.5281/zenodo.8239309).Author contributions
H. L. A. initiated the project. W. S. synthesized the neutral macrocycle 1 in Oxford and performed reduction and crystallization experiments under guidance of Y. Z. in Albany. Compounds were characterized by NMR and UV-vis-NIR spectroscopy by W. S. and Y. Z. Glassblowing was done by Y. Z. X-ray diffraction experiments and refinement were performed by Z. W. DFT calculations were done by W. S. The draft manuscript was written by W. S., and edited by H. L. A. and M. A. P. All authors revised the manuscript. Funding was secured by H. L. A. and M. A. P., and they supervised the whole project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European Research Council (grant 885606, ARO-MAT) and U.S. National Science Foundation (grant CHE-2003411) for funding, Oxford Advanced Research Computing (ARC) for computational resources and Keble College, Oxford, for appointing M. A. P. as a Senior Research Visitor. This research used resources of the NSF's ChemMatCARS, Sector 15 at the Advanced Photon Source (APS), Argonne National Laboratory (ANL) supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE-1834750. Use of APS, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by ANL, was supported by the U.S. DOE under contract no. DE-AC02-06CH11357.Notes and references
- (a) A. V. Zabula, S. N. Spisak, A. S. Filatov, A. Y. Rogachev and M. A. Petrukhina, Record alkali metal intercalation by highly charged corannulene, Acc. Chem. Res., 2018, 51, 1541–1549 CrossRef CAS PubMed; (b) Z. Zhou and M. A. Petrukhina, Planar, curved and twisted molecular nanographenes: reduction-induced alkali metal coordination, Coord. Chem. Rev., 2023, 486, 215144 CrossRef CAS; (c) B. Benshafrut, E. Shabtai, M. Rabinovitz and L. T. Scott, π-Conjugated anions: from carbon-rich anions to charged carbon allotropes, Eur. J. Org Chem., 2000, 2000, 1091–1106 CrossRef.
- (a) E. Ljungström, O. Lindqvist and O. Wennerström, The crystal and molecular structure of [24]paracyclophanetetraene, Acta Crystallogr., Sect. B, 1978, 34, 1889–1893 CrossRef; (b) B. Thulin, O. Wennerström, H.-E. Högberg, O. Smidsrød, O. Dahl, O. Buchardt and G. Schroll, A Simple synthesis of [2.2.2.2]-paracyclophane-1,9,17,25-tetraene by a Wittig reaction, Acta Chem. Scand., 1975, 29, 138–139 CrossRef.
- (a) W. Huber, K. Müllen and O. Wennerström, Dianion and tetraanion of [24] paracyclophanetetraene—new unusual perimeter systems, Angew Chem. Int. Ed. Engl., 1980, 19, 624–625 CrossRef; (b) K. Müllen, H. Unterberg, W. Huber, O. Wennerström, U. Norinder and D. Tanner, Diatropic cyclophane dianions, J. Am. Chem. Soc., 1984, 106, 7514–7522 CrossRef; (c) K. Ankner, B. Lamm, B. Thulin and O. Wennerström, Electrochemical reduction of [2(4)]paracyclophanetetraene. A reversible two-electron process, Acta Chem. Scand., 1978, 32, 155–156 CrossRef.
- (a) Z. Badri and C. Foroutan-Nejad, Unification of ground-state aromaticity criteria – structure, electron delocalization, and energy – in light of the quantum chemical topology, Phys. Chem. Chem. Phys., 2016, 18, 11693–11699 RSC; (b) F. Feixas, E. Matito, J. Poater and M. Solà, Quantifying aromaticity with electron delocalisation measures, Chem. Soc. Rev., 2015, 44, 6434–6451 RSC.
- S. Eder, D.-J. Yoo, W. Nogala, M. Pletzer, A. Santana Bonilla, A. J. P. White, K. E. Jelfs, M. Heeney, J. W. Choi and F. Glöcklhofer, Switching between local and global aromaticity in a conjugated macrocycle for high-performance organic sodium-ion battery anodes, Angew. Chem., Int. Ed., 2020, 59, 12958–12964 CrossRef CAS PubMed.
- (a) M. Rimmele, W. Nogala, M. Seif-Eddine, M. M. Roessler, M. Heeney, F. Plasser and F. Glöcklhofer, Functional group introduction and aromatic unit variation in a set of π-conjugated macrocycles: revealing the central role of local and global aromaticity, Org. Chem. Front., 2021, 8, 4730–4745 RSC; (b) S. Eder, B. Ding, D. B. Thornton, D. Sammut, A. J. P. White, F. Plasser, I. E. L. Stephens, M. Heeney, S. Mezzavilla and F. Glöcklhofer, Squarephaneic tetraanhydride: a conjugated square-shaped cyclophane for the synthesis of porous organic materials, Angew. Chem., Int. Ed., 2022, 61, e202212623 CrossRef CAS PubMed.
- Theoretical calculations have been reported previously for these anions, including geometry optimizations, resonance energies and AICD plots, but the coordinates were not published.6 (a) E. Shabtai, O. Segev, R. Beust and M. Rabinovitz, Charged paracyclophanes behave as annulenes with enhanced anisotropy, J. Chem. Soc., Perkin Trans., 2000, 2, 1233–1241 RSC; (b) J. Aihara, Chem. Phys. Lett., 2003, 381, 147–153 CrossRef CAS; (c) F. Plasser and F. Glöcklhofer, Visualisation of chemical shielding tensors (VIST) to elucidate aromaticity and antiaromaticity, Eur. J. Org Chem., 2021, 2021, 2529–2539 CrossRef CAS PubMed.
- (a) T. M. Krygowski and M. K. Cyrański, Structural aspects of aromaticity, Chem. Rev., 2001, 101, 1385–1419 CrossRef CAS PubMed; (b) T. M. Krygowski, H. Szatylowicz, O. A. Stasyuk, J. Dominikowska and M. Palusiak, Aromaticity from the viewpoint of molecular geometry: application to planar systems, Chem. Rev., 2014, 114, 6383–6422 CrossRef CAS PubMed.
- (a) A. Ayalon, A. Sygula, P.-C. Cheng, M. Rabinovitz, P. W. Rabideauv and L. T. Scott, Stable high-order molecular sandwiches: hydrocarbon polyanion pairs with multiple lithium ions inside and out, Science, 1994, 265, 1065–1067 CrossRef CAS PubMed; (b) V. Zabula, A. S. Filatov, S. N. Spisak, A. Yu. Rogachev and M. A. Petrukhina, A main group metal sandwich: five lithium cations jammed between two corannulene tetraanion decks, Science, 2011, 333, 1008–1011 CrossRef PubMed; (c) A. S. Filatov, A. V. Zabula, S. N. Spisak, A. Yu. Rogachev and M. A. Petrukhina, Clamshell opening in the mixed-metal supramolecular aggregates formed by fourfold reduced corannulene for maximizing intercalated metal content, Angew. Chem., Int. Ed., 2014, 53, 140–145 CrossRef CAS PubMed.
- R. Gershoni-Poranne and A. Stanger, Magnetic criteria of aromaticity, Chem. Soc. Rev., 2015, 44, 6597–6615 RSC.
- F. Gerson, W. Huber and O. Wennerström, The radical anions of [2.2.2.2]paracyclophane-1,9,17,25-tetraene and some of its heteroaromatic analogues, Helv. Chim. Acta, 1978, 61, 2763–2772 CrossRef CAS.
- J. Kim, J. Oh, A. Osuka and D. Kim, Porphyrinoids, a unique platform for exploring excited-state aromaticity, Chem. Soc. Rev., 2022, 51, 268–292 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and full characterization for all compounds, spectroscopic data, copies of NMR spectra, MS data. CCDC 2272266–2272271. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04251k |
| This journal is © The Royal Society of Chemistry 2023 |