
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of cyclic peptide monolayers by hydrophobic supramolecular hinges†
Ignacio
Insua‡
 ab,
Annalisa
Cardellini‡
cd,
Sandra
Díaz
a,
Julian
Bergueiro
a,
Riccardo
Capelli
e,
Giovanni M.
Pavan
*cd and
Javier
Montenegro
*a
ab,
Annalisa
Cardellini‡
cd,
Sandra
Díaz
a,
Julian
Bergueiro
a,
Riccardo
Capelli
e,
Giovanni M.
Pavan
*cd and
Javier
Montenegro
*a
aCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS), Departamento de Química Orgánica, Universidade de Santiago de Compostela, 15705, Spain. E-mail: javier.montenegro@usc.es
bI+D Farma Group (GI-1645), Departamento de Farmacoloxía, Farmacia e Tecnoloxía Farmacéutica, Facultade de Farmacia, Universidade de Santiago de Compostela, 15782, Spain
cDepartment of Applied Science and Technology, Politecnico di Torino, 10129 Torino, Italy. E-mail: giovanni.pavan@polito.it
dDepartment of Innovative Technologies, University of Applied Sciences and Arts of Southern Switzerland, Polo Universitario Lugano, 6962 Lugano-Viganello, Switzerland
eDepartment of Biosciences, University of Milan, 20133 Milano, Italy
First published on 27th October 2023
Abstract
Supramolecular polymerisation of two-dimensional (2D) materials requires monomers with non-covalent binding motifs that can control the directionality of both dimensions of growth. A tug of war between these propagation forces can bias polymerisation in either direction, ultimately determining the structure and properties of the final 2D ensemble. Deconvolution of the assembly dynamics of 2D supramolecular systems has been widely overlooked, making monomer design largely empirical. It is thus key to define new design principles for suitable monomers that allow the control of the direction and the dynamics of two-dimensional self-assembled architectures. Here, we investigate the sequential assembly mechanism of new monolayer architectures of cyclic peptide nanotubes by computational simulations and synthesised peptide sequences with selected mutations. Rationally designed cyclic peptide scaffolds are shown to undergo hierarchical self-assembly and afford monolayers of supramolecular nanotubes. The particular geometry, the rigidity and the planar conformation of cyclic peptides of alternating chirality allow the orthogonal orientation of hydrophobic domains that define lateral supramolecular contacts, and ultimately direct the propagation of the monolayers of peptide nanotubes. A flexible ‘tryptophan hinge’ at the hydrophobic interface was found to allow lateral dynamic interactions between cyclic peptides and thus maintain the stability of the tubular monolayer structure. These results unfold the potential of cyclic peptide scaffolds for the rational design of supramolecular polymerisation processes and hierarchical self-assembly across the different dimensions of space.
Introduction
An ever-growing pool of synthetic monomers is opening exciting opportunities in the bottom-up fabrication of two-dimensional (2D) supramolecular materials.1,2 The reversible nature of non-covalent forces endows these assemblies with valuable properties for bio-technological applications such as self-healing,3 stimuli-responsiveness,4 molecular self-sorting5,6 and transient assembly.7 Concepts from current designs contribute to guiding new monomers, architectures and supramolecular interfaces. Despite major advances in this field, the controlled assembly of simple monomers in 2D is still challenging due to the lack of design and mechanistic understanding at the molecular level. For example, monomers are required to elongate in two directions, which often leads to competing assembly pathways that may result in polydisperse and heterogenous samples.8 Yet, the high surface area of 2D assemblies is highly coveted by material scientists and supramolecular chemists alike, finding applications in catalysis,9,10 molecular sieving,11 responsive surfaces12,13 and biosensing,14,15 amongst others.16,17The control of the self-assembly dynamics and structure of 2D supramolecular materials depends on hierarchical stages of propagation.8 Thus, transitions between supramolecular hierarchies determine elongation rates and hence the structure and properties of the resulting 2D material. Artificial intermediate assembly states such as hydrophobic toroids,18,19 DNA origami tiles,20 polymeric rods21,22 or peptoid helices,23 all could transition into a final 2D assembly hierarchically. It is no surprise that nature capitalises on the hierarchical folding of amino acid chains through secondary, tertiary and quaternary structure to reach the structural sophistication of protein architectures. For this reason, peptides are particularly interesting monomers for design-based 2D self-assembly following their natural structure-assembly relationships.24–27 Moreover, synthetic peptide assemblies can perform as artificial enzymes,28,29 protein receptors30 and cytoskeleton-like scaffolds,31,32 providing a powerful building block for functional 2D materials.
While 2D supramolecular systems continue to emerge,8,33 a big gap remains in the mechanistic understanding of their hierarchical self-assembly. The kinetic and thermodynamic constraints that monomers undergo across levels of supramolecular complexity remains overlooked. Computational simulations can be used to tackle this knowledge gap, allowing the modelling of the self-assembling process with single-monomer resolution. Thus, complex polymerisation mechanics such as monomer exchange,34 pathway selection35 or the amplification of packing defects36 can be deconvoluted. However, just a few studies have investigated the mechanism of hierarchical 2D self-assembly, only in the context of solvent exchange-induced polymerisation.23,37,38 A wider scope of monomers and 2D assemblies must be studied to understand the polymerisation process mechanistically and thus distil a general set of rules to guide new hierarchical designs. Despite the peculiarities of each monomer, general mechanistic concepts with broad applicability are key to drive the transition from fundamentally empirical assemblies to rational and predictable molecular designs.
In this article, we investigate the underlying mechanisms driving the hierarchical self-assembly of a new cyclic peptide design equipped with two orthogonal hydrophobic domains for lateral association of new nanotubular assemblies (Fig. 1A). Molecular dynamics (MD) simulations of this peptide monomer revealed a bias towards two-dimensional propagation, which was based on the degree of polymerisation of initially formed nanotubular assemblies. While the polar interaction is barely affected as elongation proceeds, hydrophobic effects are amplified in the tubular state, thus triggering higher order 2D elongation upon a 1D oligomerisation threshold. These results demonstrate that cyclic peptides of alternating chirality constitute an excellent scaffold for the rational design of hierarchical self-assembled systems across different dimensions. The rigid structure of cyclic peptide monomers is exploited here to impose orthogonal supramolecular growth via H-bonding and hydrophobic effects, which direct the elongation in 2D. This strategy is experimentally validated by the assembly of a novel nanotubular monolayer structure. A new supramolecular binding motif, the hydrophobic ‘tryptophan hinge’, provides the required flexibility to the contact points between nanotubes (NTs) in lateral association. MD simulations and synthetic cyclic peptides with selected sequence mutations confirmed the suitability of this versatile scaffold for the rational design of hierarchical self-assembled systems.
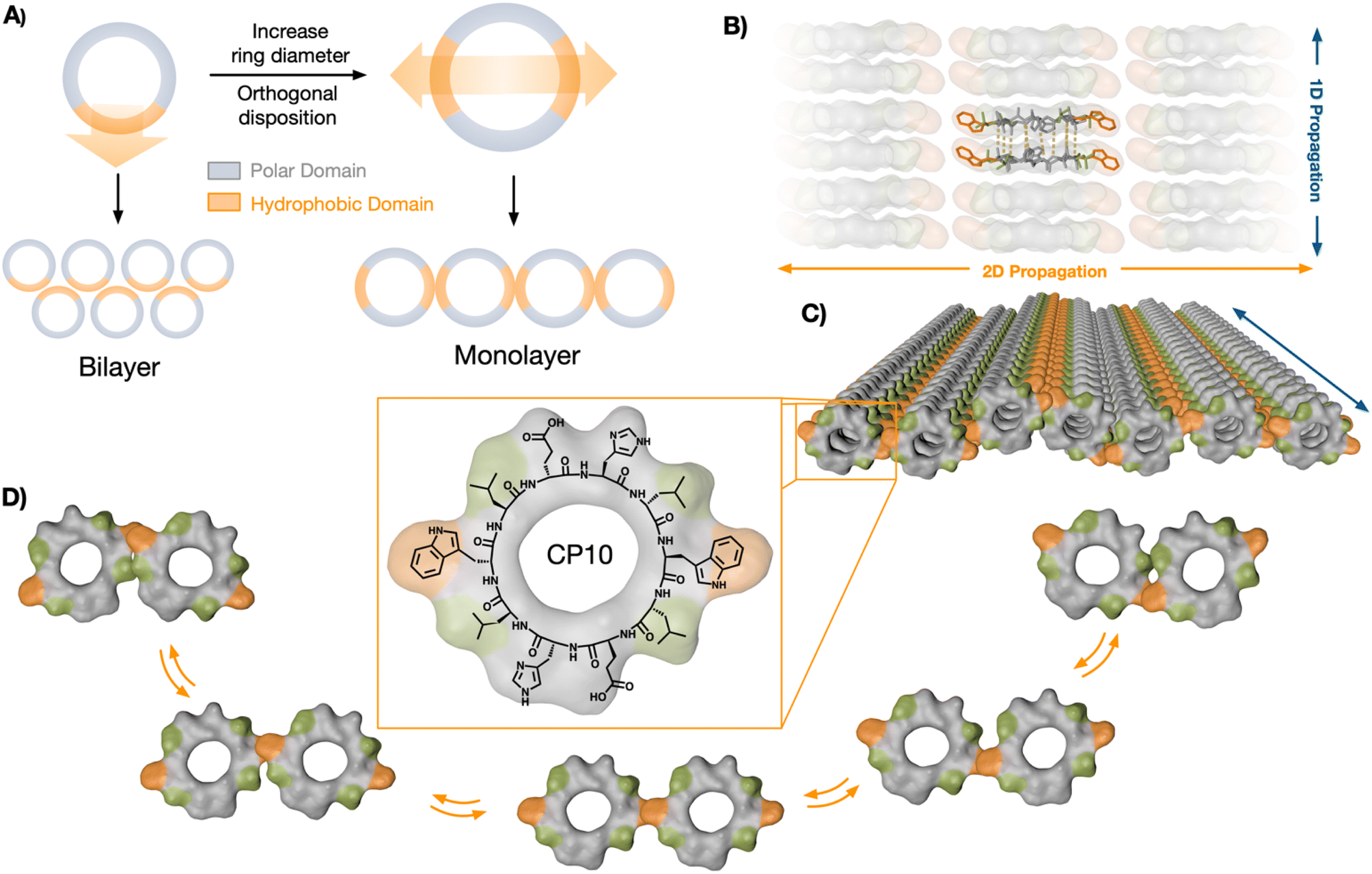 | ||
| Fig. 1 Rational design and structural scheme of self-assembled 2D nanotubular monolayers of cyclic peptide monomers. (A) A cyclic octapeptide (left) with a single hydrophobic domain matches the structural requirements for a staggered bilayer assembly; increasing molecular diameter to a cyclic decapeptide (right) allows the incorporation of two hydrophobic motifs at 180° in a C2 symmetric scaffold for the lateral assembly of nanotubular monolayers. (B) Longitudinal antiparallel β-sheet hydrogen bonded network of the stacked cyclic peptide monomers. (C) Supramolecular structure of a two-dimensional monolayer assembled from nanotubes through lateral hydrophobic contacts between aligned Trp (orange) and Leu (green) residues of CP10. (D) Lateral contact model: ‘Trp hinge’ with the aromatic residue pivoting between the two possible Leu zippers. | ||
Results and discussion
Cyclic peptides (CPs) with alternating D/L chirality are known to undergo longitudinal supramolecular polymerisation to generate nanotubes via inter-backbone H-bonding.39 Recently, we discovered that nanotubes assembled from amphiphilic CP octamers could generate bilayered nanosheets.25 The amphiphilic CPs should segregate polar and hydrophobic domains in the longitudinal axis of the nanotubes to drive their solvophobic association in aqueous media. We rationalised that the axial self-sorting of the peptide's side chains—by polarity-based segregation, electrostatic bridging, etc.—should restrict the rotational freedom of the CP monomers in the nanotubular state.40 Therefore, we hypothesised that monomer fixation along the nanotubular axis should be, a priori, suitable for the rational design of hierarchical self-assembled systems. Under this assumption, the cyclic peptide scaffold would tolerate angle-defined positioning of complementary binding motifs, which can be amplified along the resulting nanotubes to generate new supramolecular architectures (Fig. 1A). Despite the interest in such hierarchical self-assembly models,21,37,41–43 the underlying mechanisms dictating the possible supramolecular stages along the polymerisation process are not yet understood.Therefore, we decided to investigate the relative disposition (i.e. angle) between non-covalent contact points around the rigid CP backbone, aiming to engineer a new hierarchically self-assembled architecture. To incorporate two hydrophobic domains into a CP monomer with C2 symmetry, the diameter of the ring was increased to the decapeptide cyclo-(L-Trp-D-Leu-L-Glu-D-His-L-Leu-D-Trp-L-Leu-D-Glu-L-His-D-Leu) (CP10, Fig. 1C). Nanotubes from CP10 should self-sort their amphiphilic domains and generate confronted hydrophobic faces. As a result, 2D elongation will be induced in a 180° angle, forming a new monolayered structure from peptide nanotubes in lateral association (Fig. 1).
Thus, the hierarchical self-assembly of CP10 was initially modelled using molecular dynamics (MD). To this end, the relative stacking of monomers in parallel or antiparallel configuration was first investigated to unravel the potential internal structure of the resulting nanotubes. All-atoms MD simulations of CP10 nanotubes were run on either configuration, and the energy kinetic profile of the system was monitored in water (Fig. 2A). We found that, while the parallel nanotube disassembled after 100 ns of simulation, the antiparallel analogue retained its full integrity under the same conditions.
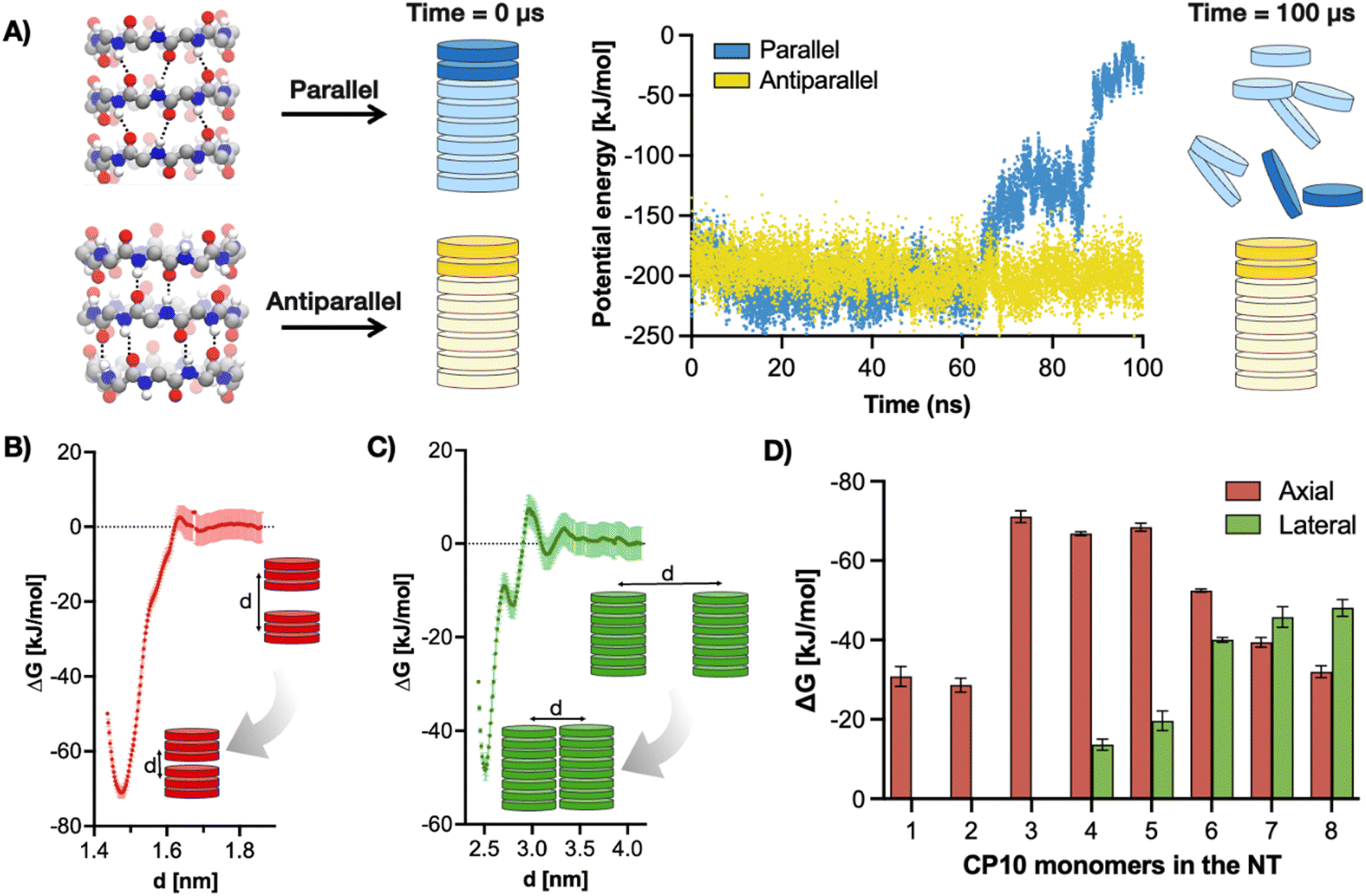 | ||
| Fig. 2 (A) Left: atomistic representation of CP10 in parallel (top) and antiparallel (bottom) configurations. Right: MD simulations of one single nanotube made of eight CP10 units stacked in either parallel (blue) or antiparallel (yellow) configuration. The data show the total interaction energy, including both Lennard-Jones and Coulomb potentials, between the top two CP10 units of the nanotubes (in darker colour) simulated for 100 ns in aqueous solution. (B) Potential of Mean Force (PMF) profile of two 3CP10-NTs in axial assembly and antiparallel configuration. ‘d’ indicates the distance between the centre of mass of the nanotubes. (C) PMF profile of two 8CP10-NTs in lateral assembly and antiparallel configuration. (D) Global free energy (ΔG) minima, i.e. dimerisation free energy, for CP10 nanotubes in axial (red) and radial (green) association (see (B) and (C)) as a function of their oligomerisation degree in antiparallel configuration. Error bars were obtained from the PMF calculation. Because of their low probability to laterally assemble, 1CP10-NT, 2CP10-NT and 3CP10-NT have a negligible lateral (green) dimerisation ΔG. Note: 1D nanotubes are denoted as ‘XCP10-NT’, where ‘X’ indicates the oligomerisation degree [i.e. number of CP10 units constituting the nanotube (NT)]. | ||
To validate this predicted antiparallel stacking, CP10 was synthesised by Fmoc solid-phase strategy44 and analysed by FT-IR, showing frequencies for amide A (3272 cm−1), I (1629 cm−1) and II (1527 cm−1) consistent with those reported in the literature for antiparallel CP nanotubes (Fig. S1†).45 Next, an in silico modelling of the self-assembly mechanism was carried out for CP10 (Fig. 2B–D). Dimerisation energies of CP10 oligomers, both in axial and lateral contact, were calculated as a function of their degree of polymerisation. The comparison of these energies allows the identification of a potential supramolecular bias promoting either dimension of propagation (axial versus lateral) as the polymerisation proceeds. Dimerisation energies were calculated by potential of mean force (PMF) profiling of approaching CP10 oligomers with fixed orientation—i.e. unable to rotate—, where the global minimum of Gibbs free energy correlates to the binding strength between the supramolecular units under study (Fig. 2B and C). Thus, PMF analysis was carried out for axial and lateral interaction of CP blocks with increasing oligomerisation degrees (e.g. monomer–monomer, dimer–dimer, etc.). For axial (i.e. nanotube) elongation, CP10 monomers and dimers appear to be significantly less favoured to associate than larger longitudinal oligomers (Fig. 2D). Short oligomers (3–5mer) display the highest axial association energies, probably due to their increased backbone rigidity and hence better interfacial matching, as compared to mono- and dimeric species. Indeed, our calculations show better interfacial matching between 3mer blocks than in the case of more flexible monomers (Fig. S2 and S3,† see Methods). This cooperative axial elongation progressively drops from pentamers onwards, most likely because of the more defects that can be generated and amplified though longer nanotubular assemblies.35,36 However, regarding lateral 2D association, dimerisation energies could only be extracted from 4mer blocks onwards, showing a gradual increase in association energy up to a 7mer state (Fig. 2D). These phenomena can be rationalised by the enhanced hydrophobicity of longer tubes boosting their solvophobic 2D packing, also limiting the lateral association of small oligomers (3mer an below) to a negligible energy value. In fact, whereas axial elongation seems favoured in short oligomers (<5mers), lateral association energy peaks at a 7mer state, where the preferential dimension of growth is inverted in favour of 2D propagation. Therefore, PMF profiling strongly supports the hierarchical assembly mechanism, where CP10 monomers grow axially until a certain hydrophobicity threshold, which then favours the lateral association of tubular oligomers.
To experimentally test our supramolecular design, the synthesised CP10 was dissolved in phosphate buffer at pH 7.4 and subjected to a heating–cooling cycle to gradually anneal the monomers (see ESI†). The environment-sensitive fluorescent dye thioflavin-T (ThT), which accumulates at β-sheet and hydrophobic interfaces with enhanced fluorescence, was added to CP10 samples to allow their visualisation by epifluorescence microscopy. Nanosheets reaching over 100 μm in lateral dimensions were observed under the microscope from drop cast samples of CP10 (Fig. 3A). It must be noted that micrographs were acquired from dry samples, demonstrating the structural robustness of the supramolecular 2D ensemble even out of solution. The laminar 2D morphology of the afforded assemblies was further confirmed by scanning-transmission electron microscopy (STEM, Fig. 3B). High-resolution TEM revealed two perpendicular diffraction spacings: 4.7 Å corresponds to the reported distance between 1D stacked cyclic peptides,25 while the perpendicular 2.8 Å stems from the lateral contact between tubes in 2D elongation (Fig. 3C). Atomic force microscopy (AFM) revealed a thickness of 2.6 nm (Fig. 3D), which is in range with the diameter of CP10 and thinner than the bilayer structure resulting from cyclic octapeptides.25,43 Fluorescence, electron and atomic force microscopy allowed us to confirm the assembly of tubular monolayers from cyclic peptide monomers bearing confronted hydrophobic domains. pH buffering of the aqueous solution at 7.4 was required to maintain the structural integrity of CP10's 2D monolayers. As previously found for the bilayer peptide assemblies,25 deprotonated glutamic acids are needed to balance the hydrophobic effects with electrostatic repulsion, thus avoiding the uncontrolled aggregation of the supramolecular system observed with acidification (Fig. S4†).
 | ||
| Fig. 3 Dry-state microscopic characterisation of 2D nanosheets composed of CP10 nanotube monolayers: (A) ThT-stained epifluorescence; (B) STEM; (C) electron diffraction pattern by high-resolution TEM and its Fourier transform (inset), revealing two predominant distances: d1 (axial CP spacing) and d2 (lateral 2D spacing); (D) AFM with height profile. | ||
Well-tempered metadynamics simulations46 of two contacting nanotubes (8CP10-NT) were then performed to understand, at the molecular level, the lateral packing within the tubular monolayers (Fig. 4A–C). Free energy landscapes were projected along two geometrical parameters from the 8CP10-NT interface: the number of Trp–Trp contacts established between tubes as an indicator of the efficient packing of the hydrophobic domain, and the sum of distances between NT termini to probe for their relative orientation (i.e. rotation and alignment) (Fig. 4A). In this energy map, long distances (d1 + d2 > 5 nm) represent axially-mismatched nanotubes that cannot fully connect their hydrophobic domains (Fig. S5†). Alternatively, short distances (d1 + d2 ∼ 4 nm) correspond to NTs in parallel and aligned orientation, favourable for 2D elongation (Fig. 4B).
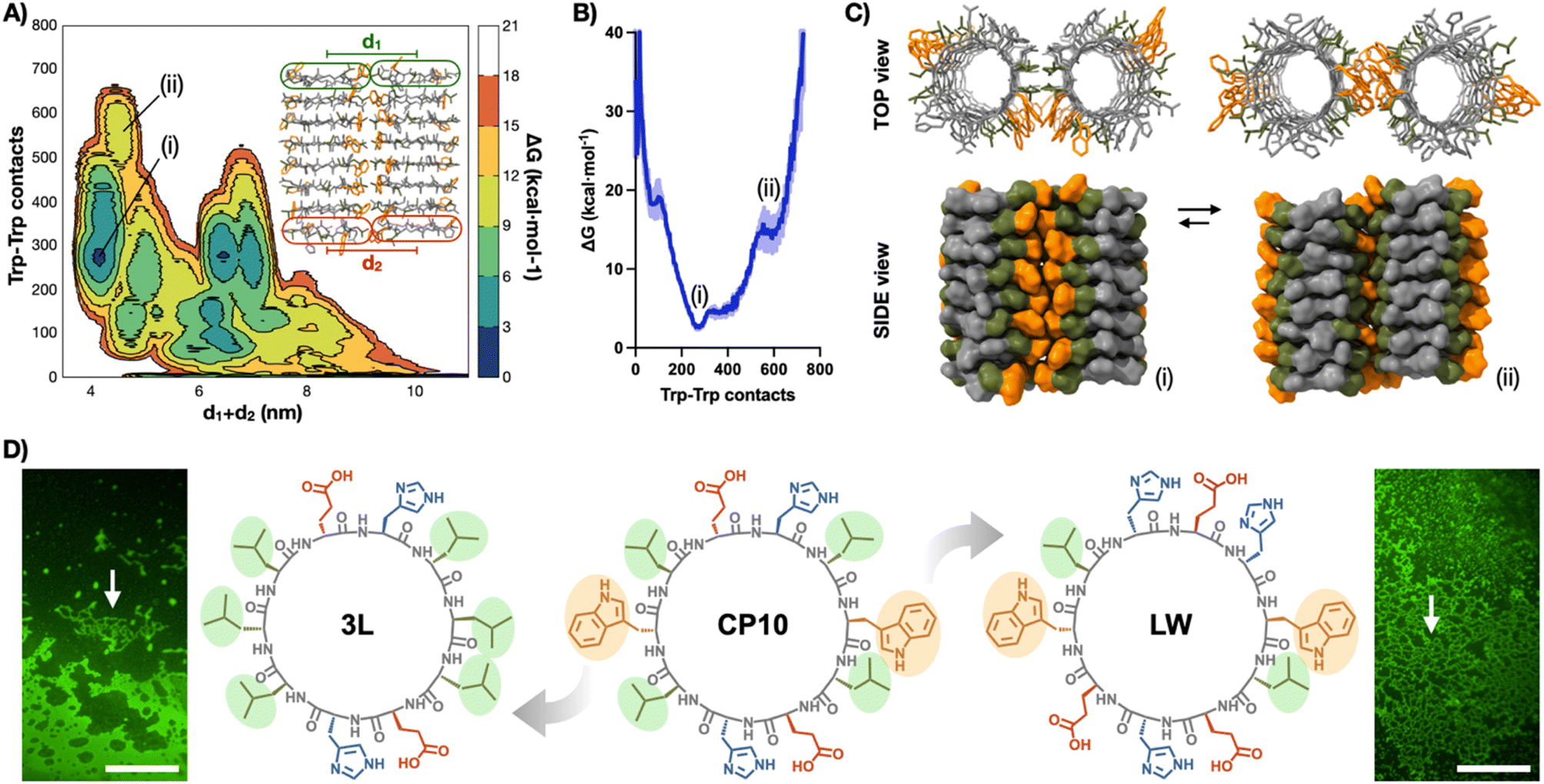 | ||
| Fig. 4 (A) Free energy surface (FES) metadynamics simulation of the spatial evolution of two 8CP10-NTs shown as free energy surface mapping as a function of Trp–Trp contacts and sum of d1 + d2 distance (see ESI†). (B) Energy profile between (i) and (ii) minima from (A). (C) Structural snapshots of interconverting minima (i) and (ii). Trp and Leu residues are coloured in orange and green, respectively. (D) Structures of CP10 and their analogues, 3L and LW, with highlighted Trp and Leu residues. Epifluorescence micrographs of ThT-stained samples of 3L (left) and LW (right) showing no evidence of 2D nanosheets. Arrows point at bundled 1D structures. Scale bars = 50 μm. | ||
In this region, two minima appeared with different energy profiles: a dual Trp–Trp and Leu–Leu interface (ΔG° = 2.69 kcal mol−1; Fig. 4Ci) and a less thermodynamically favoured direct Trp–Trp contact (ΔG° = 12.54 kcal mol−1; Fig. 4Cii); both configurations spontaneously exchanged during the simulation. Due to CP10's symmetric and complementary structure, with oppositely charged His and Glu residues on either side of the hydrophobic triad, both directions of rotation are equivalent to generate dual Trp–Trp and Leu–Leu contacts (Fig. 1D). An adaptable ‘tryptophan hinge’ triad was found to be an optimal supramolecular motif to provide the flexibility required within the hydrophobic interface. To assess the importance of this tryptophan hinge (i.e. Leu–Trp–Leu) for the solvophobic stabilisation of the tubular monolayers, two CP10 variants with modified hydrophobic domains were synthesised (Fig. 4D): 3L, cyclo-(L-Leu-D-Leu-L-Glu-D-His-L-Leu-D-Leu-L-Leu-D-Glu-L-His-D-Leu), bearing a Leu triad with no steric mismatch between residues, and LW, cyclo-(L-Trp-D-Leu-L-Glu-D-His-L-Glu-D-Trp-L-Leu-D-His-L-Glu-D-His), only possessing the Leu–Trp contact point needed for the most thermodynamically favoured packing (Fig. 4Ci). The two leucines removed from the structure of LW were replaced for histidine and glutamic acid in confronted positions to maintain alternating charge neutrality with polar residues. By design, neither 3L nor LW should be capable of establishing pivotal Trp hinges (Fig. 1D) due the lack of Trp or additional Leu contact, respectively. Microscopic analysis of 3L and LW samples did not show any 2D nanosheets, only high-aspect ratio assemblies that may result from bundles associated through less ordered hydrophobic packing (Fig. 4D, see arrows). These results suggest that the structural elasticity of the Trp hinge allows tubular interfaces to adjust their orientation and compensate for local misalignments during the lateral elongation of the monolayers (Fig. 1C and D).
Circular dichroism (CD) analysis of CP10 showed a positive Cotton effect in the tryptophan absorbance region (ca. 280 nm, Fig. S6†), suggesting a close spatial disposition of laterally interacting tryptophan moieties.
To further interrogate the assembly of multiple CP10-NTs, a coarse-grained (CG) model of CP10 was constructed to simulate the system beyond the calculation limits of atomistic MD simulations. Here, the atom-bead mapping follows the Martini CG approach,47 while the bond interactions between the beads are optimised with Swarm-CG software48 to reproduce the equilibrium arrangement of one single CP10 within a NT (Fig. S7†). The virtual sites of dipoles, depicted in blue and red, are included in the CG model to improve the directional orientation of CP–CP packing in a nanotube (Fig. S8A†). It is worth noting that, while the Martini CG force field (the basis of these models), is very well suited to treat the hydrophobic interactions between the NTs during the self-assembly (e.g., 2D-growth due to lateral Trp–Trp interactions), the treatment of directional hydrogen bonding is not trivial in CG models. As recently performed for other supramolecular polymers where hydrogen-bonding plays a major role,34,49,50 the addition of rigid virtual site dipoles in this CG model allows to approximate well (via dipole–dipole interactions) the effect of H-bonds in keeping these NTs as stacked and rigid as in, for example, the AA models. The relative orientation of NTs during this simulation was analysed as cos(θ) versus time; θ being the average angle created by the longitudinal axes of all possible NT pairs, leading to cos(θ) values of 0 for all perpendicular and 1 for all parallel NT axes (Fig. S8B†). According to these simulations, NT alignment and hence 2D self-assembly is significantly influenced by the oligomerisation degree, showing a minimum requirement of 4mer as nucleus to induce an all-parallel NT cluster (4CP10-NT cos(θ) = 1, Fig. S8B†). In contrast, 3mers were unable to self-organise into the parallel configuration required for the 2D propagation of nanosheets, remaining randomly oriented during the whole simulation (3CP10-NT cos(θ) ∼ 0.5, Fig. S8B†). This result is supported by the negligible ΔG values found for 3mers and shorter oligomers in lateral contact (Fig. 2D). CG modelling did not show a significant improvement in 2D association dynamics for a longer 6C10P-NT oligomer, suggesting 4CP10-NT is suitably balanced in hydrophobic strength and diffusion/rotation freedom for 2D propagation (Fig. S8B and C†).
CG-MD simulations were then performed with variations in the hydrophobic strength (Δhpho) of Trp to study the contribution of this central amino acid to the 2D-driving Trp hinge motif. As model, the self-assembly of ten 4CP10-NTs was assessed by their ability to aggregate into one single cluster of aligned nanotubes (i.e. cos(θ) = 1) as a function of Δhpho (Fig. 5). In these experiments, a Δhpho value of 0 corresponds to the native hydrophobicity of Trp—used in all previous calculations—and that showed the stable assembly of tubular monolayers by CG-MD simulations (Fig. S8†). The system was probed with an initial 50% reduction of Δhpho, showing dispersed and randomly aligned NT clusters unable to elongate in 2D (Fig. 5A–C). A milder 30% reduction of Δhpho led to a single NT cluster with aligned 1D axes, hence successfully assembling in 2D despite taking longer to reach 2D state than the native Trp (Fig. 5B and C). Regarding the hierarchical 1D-to-2D assembly pathway, comparison between 2D cluster size and cos(θ) shows that NTs first come together into a single cluster, which then reorganises cooperatively to align all 1D axes (Fig. 5D). It is also clear that the reduction of Trp's Δhpho compromises NT clustering and axial alignment. Together with the energy mapping of Trp–Trp contacts (Fig. 4), these experiments confirmed the central role of the Trp residue in driving the second hierarchy of self-assembly in 2D based on the geometrical flexibility of this interface. Importantly, Trp Δhpho reductions could not be compensated by adjacent Leu residues – unmodified across all simulations–, reinforcing the need for a hinging Trp–Trp interface for lateral elongation.
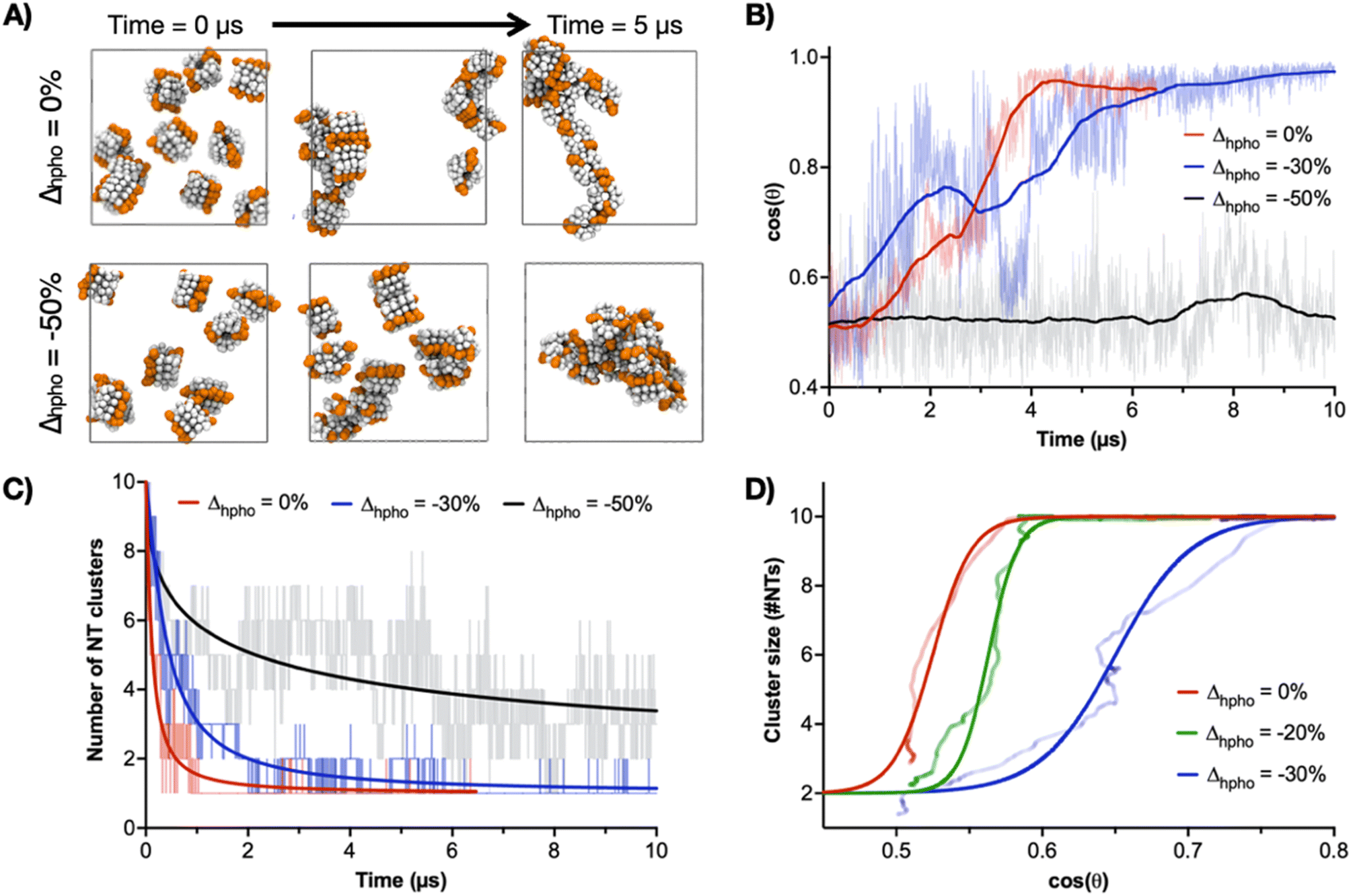 | ||
| Fig. 5 CG-MD simulations of ten 4CP10-NTs in aqueous solution with variations in the hydrophobic strength of Trp residues (Δhpho; see Methods). (A) Snapshots of the native 4CP10-NT model (Δhpho = 0) and a customised analogue with Trp beads displaying a 50% reduction of their original hydrophobic strength (Δhpho = −50%). Orange beads indicate tryptophan residues. (B) Time evolution of the average cos(θ) between the longitudinal axes of all possible NT couples. (C) Time evolution of the number of 4CP10-NT clusters versus Δhpho. (D) Self-assembly pathway as function of Δhpho comparing solvophobic 4CP10-NT clustering versus relative orientation based on Trp's hydrophobic character. | ||
Conclusions
This work studies the hierarchical supramolecular polymerisation of a new tubular monolayer architecture, designed from cyclic peptide monomers, by atomistic and coarse-grained MD models. First, we engineered a hierarchically self-assembled architecture by incorporating two hydrophobic domains into a CP monomer with increased diameter (CP10, Fig. 1C). Nanotubes from CP10 self-sorted their amphiphilic domains and generated confronted hydrophobic faces. As a result, two-dimensional elongation was induced in a 180° angle, forming a monolayered structure from peptide nanotubes in lateral association (Fig. 1). Computational simulations demonstrated that CP10 monomers preferentially undergo one dimensional elongation at short oligomerisation states (i.e. 3mer and 4mer), while longer oligomers prefer 2D propagation as their hydrophobic character is amplified along larger tubular assemblies. This is evidenced by both PMF calculations (Fig. 2D) and CG-MD modelling (Fig. 5). The supramolecular polymerisation is shown to be a stepwise process consisting of the hydrophobic clustering of nanotubes, initially in random orientation, to be then realigned in parallel for lateral extension (Fig. 5). We demonstrate that cyclic peptide monomers allow the rational disposition of their hydrophobic domains for the assembly of new supramolecular tubular monolayers. Design-wise, the key positioning of two opposite hydrophobic ‘tryptophan hinges' in CP10's sequence provides directional control for lateral assembly of a single-peptide-thick, hollow 2D structure. Control over the directionality of supramolecular propagation relies on the rigid structure of CP10's backbone and the radial disposition of its amino acid side chains in defined angles. CP10 was synthesised and assembled in tubular monolayers by heating–cooling cycles in aqueous buffered solutions. The resulting self-assembled structures were characterised by microscopy and diffraction techniques, showing lateral dimensions over 100 μm and the expected single cyclic peptide thickness. Control experiments with synthesised peptides with selected mutations confirmed the importance of the Leu–Trp–Leu triad to allow the flexible contact between nanotubes with freedom to establish exchanging Leu zippers in either direction (Fig. 1D and 4). This ‘tryptophan hinge’ provides a hydrophobic drive to the lateral association of nanotubes for subsequent 2D elongation, proving a particularly well-suited hydrophobic motif to create this dynamic non-polar interface in water. Here, we demonstrated the supramolecular versatility of cyclic peptides of alternating chirality for the rational design of hierarchical self-assembled systems across different dimensions of space. The new mechanistic concepts introduced in this work pave the way for the future assembly of complex architectures from custom peptide monomers, with direct translation to protein and peptide engineering from rational design.Data availability
Complete computational material and data pertaining to the study conducted herein are available at: https://doi.org/10.5281/zenodo.10057290. Details on the molecular models and on the MD simulations are also provided in the ESI.†Author contributions
All authors contributed to the final version of this manuscript, with shared input into experimental design and data interpretation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Spanish AEI (SAF2017-89890-R, PCI2019-103400, PID2020-117143RB-I00), the Xunta de Galicia (ED431G 2019/03, ED431C 2017/25, 2016-AD031, the Oportunius Program (Gain)), the ERC-Stg (DYNAP-677786), the ERC-PoC (TraffikGene, 838002), the HFSP (RGY0066/2017) and the ERDF. I. I. and J. B. thank AEI for their respective Ramón y Cajal fellowships (RYC2021-031367-I and RYC2021-034705-I). I. I. also thanks the European Commission for a MSCA-IF (2018-843332). S. D. thanks Xunta de Galicia for a predoctoral fellowship (ED481A-2020/137). G. M. P. acknowledges the support received by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant Agreement No. 818776 – DYNAPOL) and by the Swiss National Science Foundation (SNSF Grant IZLIZ2_183336) The authors also acknowledge the computational resources provided by the Swiss National Supercomputing Center(CSCS) and by CINECA.References
- P. K. Hashim, J. Bergueiro, E. W. Meijer and T. Aida, Prog. Polym. Sci., 2020, 105, 101250 CrossRef CAS
.
- I. Insua and J. Montenegro, Chem, 2020, 6, 1652–1682 CAS
- J. Xie, P. Yu, Z. Wang and J. Li, Biomacromolecules, 2022, 23, 641–660 CrossRef CAS PubMed
- S. Panja and D. J. Adams, Chem. Soc. Rev., 2021, 50, 5165–5200 RSC
- S. Panja, B. Dietrich, A. J. Smith, A. Seddon and D. J. Adams, ChemSystemsChem, 2022, 4, e202200008 CrossRef CAS
- N. Singh, A. Lopez-Acosta, G. J. M. Formon and T. M. Hermans, J. Am. Chem. Soc., 2022, 144, 410–415 CrossRef CAS PubMed
- S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC
- I. Insua, J. Bergueiro, A. Méndez-Ardoy, I. Lostalé-Seijo and J. Montenegro, Chem. Sci., 2022, 13, 3057–3068 RSC
- Y. Fang, Y. Yang, R. Xu, M. Liang, Q. Mou, S. Chen, J. Kim, L. Y. Jin, M. Lee and Z. Huang, Nat. Commun., 2023, 14, 2503 CrossRef CAS PubMed
- Y. Lin, M. R. Thomas, A. Gelmi, V. Leonardo, E. T. Pashuck, S. A. Maynard, Y. Wang and M. M. Stevens, J. Am. Chem. Soc., 2017, 139, 13592–13595 CrossRef CAS PubMed
- G. Zhang, X. Li, G. Chen, Y. Zhang, M. Wei, X. Chen, B. Li, Y. Wu and L. Wu, Nat. Commun., 2023, 14, 975 CrossRef CAS PubMed
- D. Lee, L. Dong, Y. R. Kim, J. Kim, M. Lee and Y. Kim, Adv. Healthcare Mater., 2023, 12, e2203136 CrossRef PubMed
- A. D. Merg, G. Touponse, E. van Genderen, T. B. Blum, X. Zuo, A. Bazrafshan, H. M. H. Siaw, A. McCanna, R. B. Dyer, K. Salaita, J. P. Abrahams and V. P. Conticello, J. Am. Chem. Soc., 2020, 142, 199956–199968 CrossRef PubMed
- S. K. Albert, S. Lee, P. Durai, X. Hu, B. Jeong, K. Park and S. Park, Small, 2021, 17, 2006110 CrossRef CAS PubMed
- J. H. Kim, S. C. Kim, M. A. Kline, E. M. Grzincic, B. W. Tresca, J. Cardiel, M. Karbaschi, D. C. Dehigaspitiya, Y. Chen, V. Udumula, T. Jian, D. J. Murray, L. Yun, M. D. Connolly, J. Liu, G. Ren, C.-L. Chen, K. Kirshenbaum, A. R. Abate and R. N. Zuckermann, ACS Nano, 2020, 14, 185–195 CrossRef CAS PubMed
- T. Kim, J. Hong, J. Kim, J. Cho and Y. Kim, J. Am. Chem. Soc., 2023, 145, 1793–1802 CrossRef CAS PubMed
- M. G. Rafique, J. M. Remington, F. Clark, H. Bai, V. Toader, D. F. Perepichka, J. Li and H. F. Sleiman, Angew. Chem., Int.
Ed., 2023, 62, e202217814 CrossRef PubMed
- J. S. Valera, H. Arima, C. Naranjo, T. Saito, N. Suda, R. Gómez, S. Yagai and L. Sánchez, Angew. Chem., Int. Ed., 2022, 61, e202114290 CrossRef CAS PubMed
- N. Sasaki, M. F. J. Mabesoone, J. Kikkawa, T. Fukui, N. Shioya, T. Shimoaka, T. Hasegawa, H. Takagi, R. Haruki, N. Shimizu, S. Adachi, E. W. Meijer, M. Takeuchi and K. Sugiyasu, Nat. Commun., 2020, 11, 3578 CrossRef PubMed
- X. Wang, H. Jun and M. Bathe, J. Am. Chem. Soc., 2022, 144, 4403–4409 CrossRef CAS PubMed
- P. J. Hurst, A. M. Rakowski and J. P. Patterson, Nat. Commun., 2020, 11, 4690 CrossRef CAS PubMed
- S. Lei, J. Tian, Y. Kang, Y. Zhang and I. Manners, J. Am. Chem. Soc., 2022, 144, 17630–17641 CrossRef CAS PubMed
- M. Zhao, K. J. Lachowski, S. Zhang, S. Alamdari, J. Sampath, P. Mu, C. J. Mundy, J. Pfaendtner, J. J. D. Yoreo, C.-L. Chen, L. D. Pozzo and A. L. Ferguson, Biomacromolecules, 2022, 23, 992–1008 CrossRef CAS PubMed
- I. J. Duti, J. R. Florian, A. R. Kittel, C. D. Amelung, V. P. Gray, K. J. Lampe and R. A. Letteri, J. Am. Chem. Soc., 2023, 145, 18468–18476 CrossRef CAS PubMed
- I. Insua and J. Montenegro, J. Am. Chem. Soc., 2020, 142, 300–307 CrossRef CAS PubMed
- B. Dai, D. Li, W. Xi, F. Luo, X. Zhang, M. Zou, M. Cao, J. Hu, W. Wang, G. Wei, Y. Zhang and C. Liu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2996–3001 CrossRef CAS PubMed
- J. Zhou, L. Venturelli, L. Keiser, S. K. Sekatskii, F. Gallaire, S. Kasas, G. Longo, T. P. J. Knowles, F. S. Ruggeri and G. Dietler, ACS Nano, 2021, 15, 944–953 CrossRef CAS PubMed
- B. Sarkhel, A. Chatterjee and D. Das, J. Am. Chem. Soc., 2020, 142, 4098–4103 CrossRef CAS PubMed
- T. O. Omosun, M.-C. Hsieh, W. S. Childers, D. Das, A. K. Mehta, N. R. Anthony, T. Pan, M. A. Grover, K. M. Berland and D. G. Lynn, Nat. Chem., 2017, 9, 805–809 CrossRef CAS PubMed
- F. Thomas, W. M. Dawson, E. J. M. Lang, A. J. Burton, G. J. Bartlett, G. G. Rhys, A. J. Mulholland and D. N. Woolfson, ACS Synth. Biol., 2018, 7, 1808–1816 CrossRef CAS PubMed
- R. Booth, I. Insua, S. Ahmed, A. Rioboo and J. Montenegro, Nat. Commun., 2021, 12, 6421 CrossRef CAS PubMed
- A. Mendez-Ardoy, A. Bayón-Fernández, Z. Yu, C. Abell, J. R. Granja and J. Montenegro, Angew. Chem., Int. Ed., 2020, 132, 6969–6975 CrossRef
- L. Zhang, N. Wang and Y. Li, Chem. Sci., 2023, 14, 5266–5290 RSC
- D. Bochicchio, M. Salvalaglio and G. M. Pavan, Nat. Commun., 2017, 8, 147 CrossRef PubMed
- A. L. de Marco, D. Bochicchio, A. Gardin, G. Doni and G. M. Pavan, ACS Nano, 2021, 15, 14229–14241 CrossRef CAS PubMed
- D. Bochicchio, S. Kwangmettatam, T. Kudernac and G. M. Pavan, ACS Nano, 2019, 13, 4322–4334 CrossRef CAS PubMed
- Y. Lin, M. Penna, M. R. Thomas, J. P. Wojciechowski, V. Leonardo, Y. Wang, E. T. Pashuck, I. Yarovsky and M. M. Stevens, ACS Nano, 2019, 13, 1900–1909 CAS
- Y. Wei, J. Tian, Z. Zhang, C. Zhu, J. Sun and Z. Li, Macromolecules, 2019, 52, 1546–1556 CrossRef CAS
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS PubMed
- M. Panciera, E. González-Freire, M. Calvelo, M. Amorín and J. R. Granja, Pept. Sci., 2020, 112, e24132 CrossRef CAS
- X. Zhang, Y. Liu, B. Zheng, J. Zang, C. Lv, T. Zhang, H. Wang and G. Zhao, Nat. Commun., 2021, 12, 4849 CrossRef CAS PubMed
- B. Bhavani, M. Mrinalini, J. V. S. Krishna, P. Basak, L. Giribabu and S. Prasanthkumar, ACS Appl. Electron. Mater., 2021, 3, 176–183 CrossRef CAS
- S. Díaz, I. Insua, G. Bhak and J. Montenegro, Chem.–Eur. J., 2020, 26, 14765–14770 CrossRef PubMed
-
A. Méndez-Ardoy, I. Insua, J. R. Granja and J. Montenegro, Cyclization and Self-Assembly of Cyclic Peptides in Peptide Macrocycles, Methods in Molecular Biology, 2022, vol. 2371, pp. 449–466 Search PubMed
- M. R. Silk, J. Newman, J. C. Ratcliffe, J. F. White, T. Caradoc-Davies, J. R. Price, S. Perrier, P. E. Thompson and D. K. Chalmers, Chem. Commun., 2017, 53, 6613–6616 RSC
- A. Barducci, G. Bussi and M. Parrinello, Phys. Rev. Lett., 2007, 100, 020603 CrossRef PubMed
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. de Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed
- C. Empereur-Mot, L. Pesce, G. Doni, D. Bochicchio, R. Capelli, C. Perego and G. M. Pavan, ACS Omega, 2020, 5, 32823–32843 CrossRef CAS PubMed
- D. Bochicchio and G. M. Pavan, ACS Nano, 2017, 11, 1000–1011 CrossRef CAS PubMed
- D. Bochicchio and G. M. Pavan, J. Phys. Chem. Lett., 2017, 8, 3813–3819 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03930g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |