
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocyclization by a triplet–triplet annihilation upconversion pair in water – avoiding UV-light and oxygen removal†
R.
Jeyaseelan‡
,
M.
Utikal‡
,
C. G.
Daniliuc
and
L.
Næsborg
 *
*
Westfälische Wilhelms-Universität Münster, Organisch-Chemisches Institut, Corrensstraße 40, 48149 Münster, Germany. E-mail: lnaesbor@uni-muenster.de
First published on 31st August 2023
Abstract
We present a formal [2 + 2]-cycloaddition of unsaturated ketones enabled by a green-to-ultraviolet triplet–triplet annihilation upconversion (TTA-UC) pair, using commercially available Ru(bpy)32+ and pyrene as sensitizer and annihilator, respectively. In the developed protocol, visible light irradiation at λmax = 520 nm allows for the reaction to proceed without the need for UV-light and the aqueous medium eliminates the need for oxygen removing protocols. Through this study, the application of the readily available upconversion pair is broadened to include cyclization reactions. We showcase the utility of the system by generating bicyclo[2.1.1]hexanes that are valuable bioisosteres of ortho-substituted benzenes, a promising motif for pharmaceuticals.
Introduction
In the past decades visible light photochemistry has received increasing attention, especially following the commercialization of blue LEDs in the early 90s.1 Despite the progress of visible light photochemistry, some photochemical transformations require UV-light for sufficient reactivity. Compared to visible light sources, UV-light sources are energy inefficient and environmentally unfriendly, and present harsh reaction conditions.2,3 In this context, upconversion processes based on for example triplet–triplet annihilation have the potential to be a part of the solution by enabling milder reaction conditions. They exploit the energy of two photons of a lower energy wavelength to access higher energy excited states whereby visible light can provide “UV-reactivity”. The bimolecular process of TTA-UC requires a sensitizer that absorbs the light, enters the triplet state, and undergoes energy transfer to an annihilator. From two triplet state annihilators, a ground state annihilator and a higher energy singlet state annihilator can be formed potentially fluorescing in the UV region.4–7TTA-UC has gained increasing attention from the photochemistry community and many applications such as solar energy applications,8,9 bioimaging9–11 and phototherapeutics9,10 are being explored. Although efforts towards synthetic applications have been made, the opportunity to use visible light to access energetically higher excited states corresponding to UV-reactivity holds an unfulfilled promise. The underdevelopment of TTA-UC mediated synthetic protocols may be due to the complex photocatalytic system that has to work in an interplay of mechanisms that are challenging to optimize and predict. In addition, upconversion strategies rely on multiple subsequent steps that all must function well to obtain satisfactory productivity. A single nonfunctioning step could lead to false negatives in screenings for reactivity. Recently, protocols for protodehalogenation5,6,12–16 and reductive C–C couplings of aryl halides with heteroaromatics or arenes5,6,12,14,17via TTA-UC have been disclosed, whereas the application of TTA-UC for cyclizations is limited.16,18–20 Cyclizations in which TTA-UC protocols have been utilized for red-to-blue and red-to-orange systems have been previously disclosed.16,18 Cyclizations using TTA-UC systems that access UV light are rare, and have been explored to perform a [4 + 4]-cycloaddition of the annihilator itself (Scheme 1a).19 Very recently, during the preparation of this manuscript, Wenger et al. demonstrated an elegant example of visible-to-UV TTA-UC for a Paterno–Büchi reaction (Scheme 1b).20
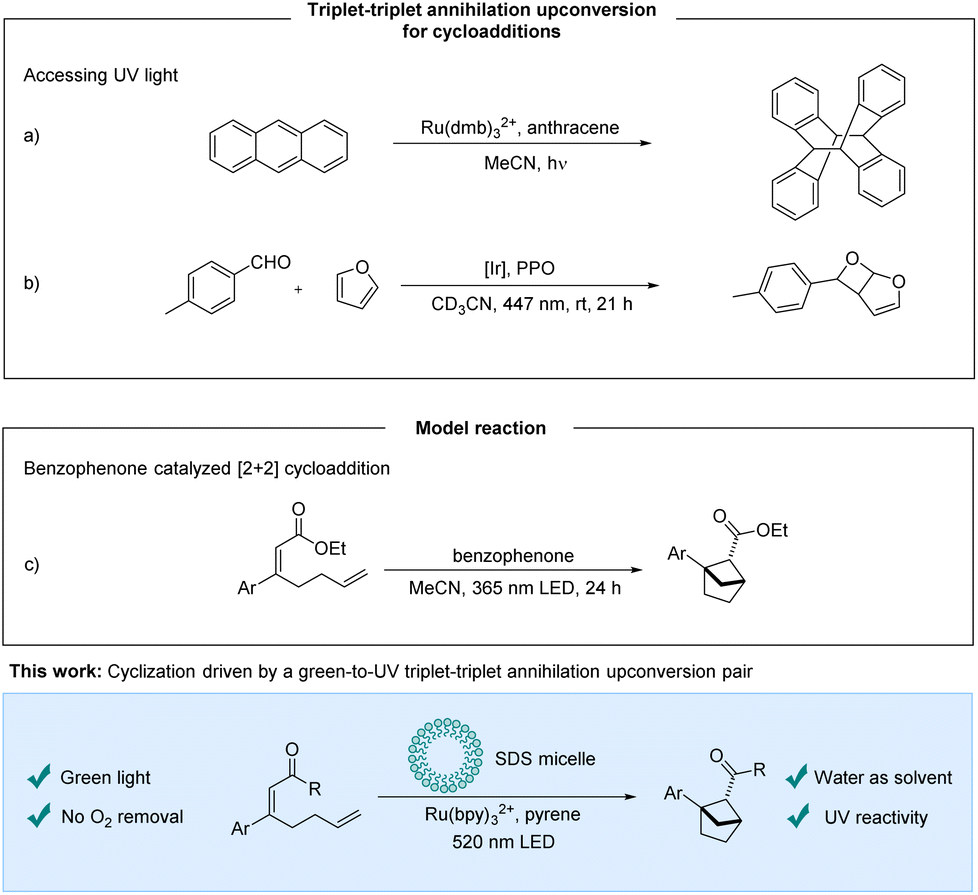 | ||
| Scheme 1 Previous work and this work. PPO = 2,5-diphenyloxazole. | ||
We sought to identify a TTA-UC system that can generate UV-light to promote cyclization reactions. Various sensitizer/annihilator pairs have been reported, providing a range of available upconverted emission wavelengths.3,4,21 One such pair, which has been reported,22,23 is [Ru]-based photosensitizers and pyrene derivatives providing upconverted emission at around 390 nm.3 We set out to expand the application of the upconversion system to include a cyclization. As a model reaction for our investigations, the intramolecular [2 + 2]-cycloaddition of α,β-unsaturated esters forming bicyclo[2.1.1]hexanes was selected (Scheme 1c). These target compounds are bioisosteres of ortho-substituted benzenes and are a valuable motif for potential pharmaceutical applications. Mykhailiuk et al. activated the esters with benzophenone as the photosensitizer using UV-light irradiation.24 We hypothesized that the singlet excited state of pyrene accessed via triplet–triplet annihilation could excite the benzophenone photosensitizer by an emission–absorption process. Preliminary experiments showed that benzophenone could not promote the desired photocycloaddition under our reaction conditions. As an alternative strategy, we designed a ketone substrate that could also be subject to an electron transfer pathway. We tested the α,β-unsaturated ketone substrate 1a, and were delighted to obtain the desired bioisostere 2a.
Results and discussion
With Ru(bpy)3(PF6)2 and pyrene as a suitable sensitizer/annihilator pair we continued our investigations into the cyclization of α,β-unsaturated ketone 1a to form the desired bicyclo[2.1.1]hexanes 2a. Using an aqueous sodium dodecyl sulfate (SDS) solution under green-light irradiation led to the desired ketone 2a in 62% NMR yield (Table 1, entry 1). Previous work from our group25 showed that an oxygen sensitive photocycloaddition promoted by [Ru(bpy)3]2+, which has been used as an oxygen sensor,26 could be performed in micellar media without freeze–pump–thaw protocols. We hoped to observe similar effects in our protocol for a highly oxygen sensitive TTA-UC process,27 and did not make any efforts to remove oxygen. This combination of micellar photocatalysis and upconversion allowed for a simple protocol that is accessible to synthetic chemists outside the field of photochemistry.|
|
||||
|---|---|---|---|---|
| Entrya | Sensitizer | Annihilator | Solvent | NMR yield |
| a For the determination of the NMR yield, toluene (5.3 μL, 0.05 mmol) was used as internal standard. b 3 W 520 nm LED. c 10 W 520 nm LED. d Yields of isolated product. e No light. SDS = sodium dodecyl sulfate, CTAC = cetyltrimethylammonium chloride. | ||||
| 1b | [Ru]2+ | Pyrene | SDS (4 wt%) | 62 |
| 2b | [Ru]2+ | Pyrene | Triton-X-100 (4 wt%) | 48 |
| 3b | [Ru]2+ | Pyrene | CTAC (4 wt%) | 12 |
| 4c | [Ru]2+ | Pyrene | SDS (4 wt%) | 76 (80d) |
| 5c | [Ru]2+ | Pyrene | Acetonitrile, no O2 | 52 |
| 6c | [Ru]2+ | Pyrene | DCM, no O2 | 45 |
| 7c | [Ru]2+ | Pyrene | MeOH, no O2 | 84 (79d) |
| 8c | [Ru]2+ | Pyrene | MeOH | 64 |
| 9c | [Ru]2+ | Pyrene | SDS (4 wt%), no O2 | 79 |
| 10c | [Ru]2+ | Pyrene | H2O, no O2 | 22 |
| 11c | [Ru]2+ | — | SDS (4 wt%) | 28 |
| 12c | — | Pyrene | SDS (4 wt%) | 0 |
| 13c | — | — | SDS (4 wt%) | 0 |
| 14e | [Ru]2+ | Pyrene | SDS (4 wt%) | 0 |
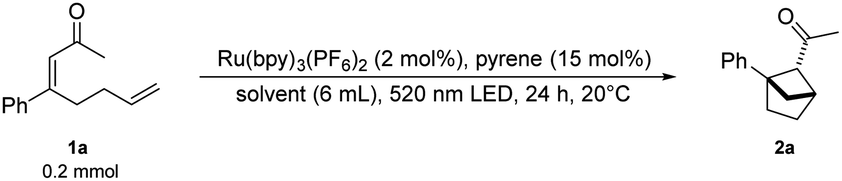
As head-group charges are expected to influence the reaction, a neutral and a positively charged amphiphile were tested. Using the neutral amphiphile Triton-X-100 resulted in a lower NMR yield of 48% (Table 1, entry 2) and the positively charged amphiphile cetyltrimethylammonium chloride (CTAC) decreased the NMR yield to a mere 12% (Table 1, entry 3). The drastic decrease of reactivity using a positively charged amphiphile may be caused by repulsion of the positively charged photosensitizer. Changing from 3 W to 10 W green LEDs increased the product formation and the bicyclo[2.1.1]hexane 2a was obtained in 78% isolated yield (Table 1, entry 4). To verify the benefit of using micellar solutions, organic solvents were tested. Acetonitrile and dichloromethane degassed by a freeze–pump–thaw protocol provided the product 2a in moderate yields (52% and 45% NMR yield respectively, Table 1, entries 5 and 6). Degassed methanol provided the desired product in 79% isolated yield (Table 1, entry 7) whereas leaving out a freeze–pump–thaw procedure decreased the NMR yield by 20% (Table 1, entry 8). We were delighted to see that the aqueous sodium dodecyl sulfate (SDS) solution provides the desired product in similar NMR yields with or without oxygen removal (Table 1, entries 4 and 9). Control experiments demonstrated that light, sensitizer, annihilator and surfactant are all necessary for an efficient formation of the desired product (Table 1, entries 10–14). To our surprise, 28% NMR yield could be observed without pyrene although the triplet energy of the substrate is expected to be too high for energy transfer to take place. This observation could be explained by the micelles enabling energy transfer from the sensitizer to the substrate. In this context we have previously proposed micellar substrate activation for energy transfer catalysis.21 Owing to the simpler reaction setup we continued our investigations using SDS-micelles as the reaction medium.
With the optimized conditions in hand, various α,β-unsaturated ketones were examined (Scheme 2A). The synthesis of starting materials led to the formation of both possible diastereomers of the substrate, both of which were applied in the reaction protocol separately when possible. Seemingly, the alkene geometry of the starting material has no influence on the diastereomeric outcome of the reaction. Different electron-withdrawing substituents in the para position such as halogens (1b–1d) a cyano- (1k) and a trifluoromethyl group (1e) are tolerated leading to the desired bicyclo[2.1.1]hexanes-derivatives in good to very good yields (2b–2e; 2k 64–89%). Introduction of an electron donating methyl group in the meta and para positions respectively resulted in the product being formed in good yields (2f, 2g 65–77%). Notably, a β-napthyl- substituted ketone underwent the targeted transformation in an excellent yield (2h 92–94%). The relative configuration and structure were verified by X-ray analysis of derivative 6a. A cyclohexyl- and a phenyl-ketone were tested (1i, 1j), each leading to very high yields of the desired bicyclic product (2i, 2j 86–87%).
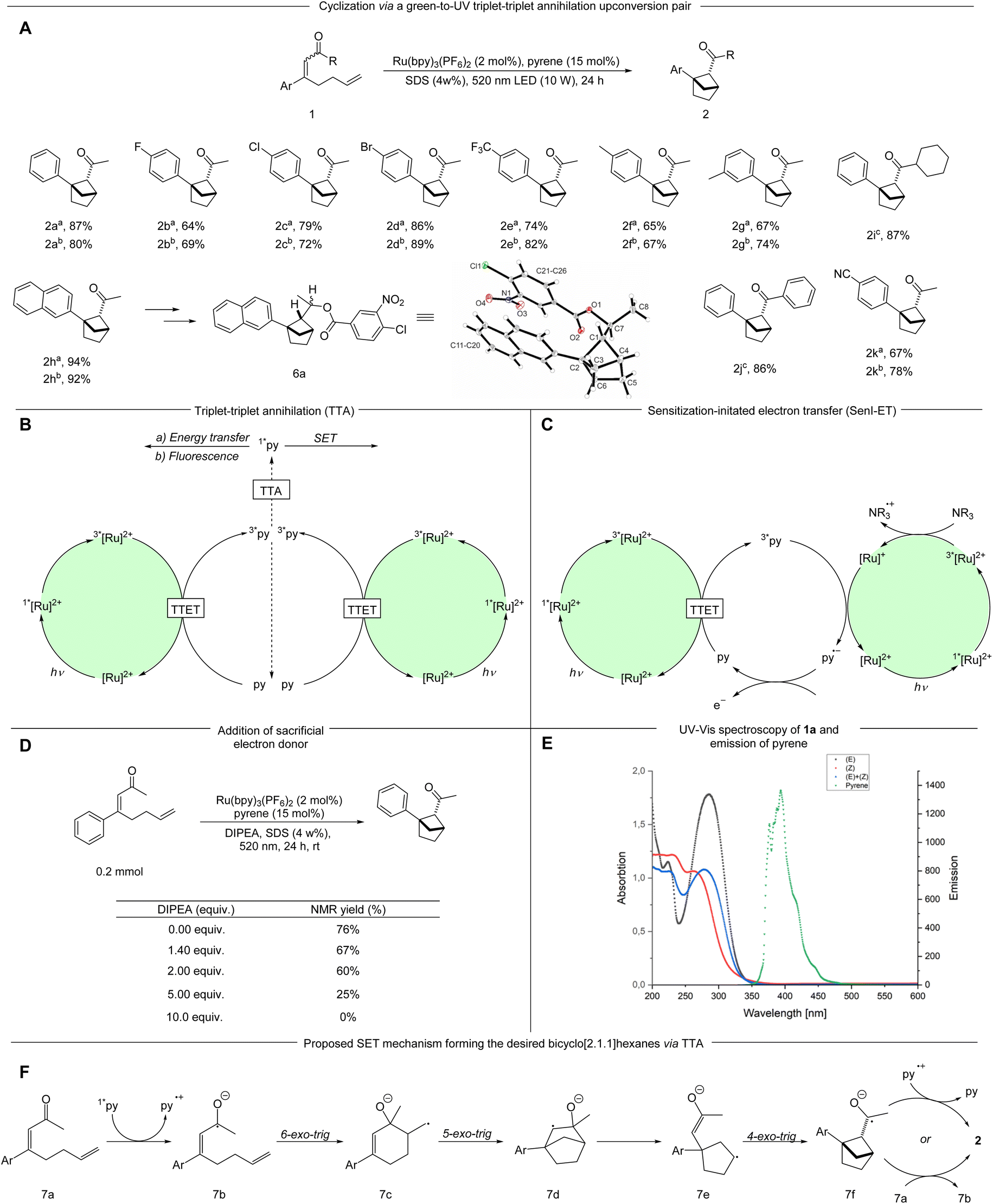 | ||
| Scheme 2 An array of substrates undergoing the cyclization mediated by a TTA-UC pair and mechanistic proposal of the formation of 2. (A) a(Z)-Isomer of the substrate 1; b(E)-isomer of the substrate 1. cMixture of (E)- and (Z) isomer of the substrate. | ||
As the reaction takes place in a complex reaction medium with a multicomponent system the determination of the underlying mechanism is not simple. Moore et al. demonstrated that pyrene can undergo TTA to form the annihilator singlet state (1*py) in the presence of [Ru(bpy)3](PF6)2 as the sensitizer (Scheme 2B).23 When an additional sacrificial electron donor i.e. N,N-diisopropylethylamine (DIPEA) is present the excited 3*[Ru(bpy)3]2+ is likely reduced via SET (single electron transfer) to [Ru(bpy)3]+, which in turn can reduce the 3*py to the pyrene radical anion, thereby severely diminishing the TTA pathway (Scheme 2C).23 The formed radical anion has a high reduction potential (2.1 V vs. SCE), which has been applied for reductive C–C couplings between aryl halides and heteroaromatics or arene derivatives.28 In this context, we investigated the effect of adding varying amounts of DIPEA to the reaction. Upon addition of 1.4 eq. of DIPEA, which is previously reported to be important for a sensitization-initiated electron transfer (SenI-ET) pathway,28 the NMR yield decreased to 67%. Increasing amounts of the electron donor led to further reductions in the NMR yields (Scheme 2D). These experiments support formation of the 1*py via TTA as a likely pathway in our reaction as the presence of sacrificial electron donor is known to form the pyrene radical anion via SenI-ET.23,28 Furthermore, we applied another upconversion system with fac-[Ir(ppy)3] and pyrene for this transformation. In comparison to Ru(bpy)32+, fac-[Ir(ppy)3] is more difficult to reduce by a sacrificial electron donor and thereby favours the TTA over the SenI-ET pathway.15 Using this system we obtained 64% NMR yield of the desired product. Control experiments without pyrene resulted in 28% NMR yield (see ESI†). Additionally, the alternate system only shows a minor decrease in NMR yield with DIPEA present, further strengthening TTA as a likely activation mode (see ESI†).
From the 1*py accessed by TTA we consider three possibilities: (1) the relaxation of 1*py with emission of upconverted, more energetic photons that can be absorbed by the substrate, (2) Foerster resonance energy transfer (FRET) or (3) direct SET reduction of the substrate by 1*py. The first two pathways require spectral overlap, and were investigated via spectroscopic analyses: UV-vis absorption spectra of both diastereomers of 4-phenylocta-3,7-dien-2-one (1a) were measured in SDS solution and compared to the fluorescence emission of pyrene (in SDS solution). No productive overlap could be identified, making these pathways unlikely (Scheme 2E and Fig. S9 and S10†).
The third pathway is enabled by direct SET from 1*py which could lead to the ketyl radical 7b. The ketyl radical 7b can undergo a 6-exo-trig cyclization forming the primary radical 7c, which, after a subsequent cyclization and fragmentation-cyclization sequence could lead to the product through SET oxidation, possibly by a pyrene radical cation or another substrate molecule (Scheme 2F). To examine the feasibility of the SET reduction, the SET reduction potential of both diastereomers of the substrate 1 were measured in acetonitrile. Given the estimated reduction potential of singlet state pyrene (−2.1 V vs. SCE)29 and the measured reduction potential of the substrates (−1.87 V vs. SCE for the (E)-isomer and −1.82 V vs. SCE for the (Z)-isomer, see ESI†), the SET process should be viable. To further support our mechanistic hypothesis, we set out to perform pyrene fluorescence quenching experiments with substrate 1a as the quencher. As the substrate and pyrene absorb in the same region, we produced pyrene fluorescence using an SDS-micelle solution of Ru(bpy)32+ and pyrene, irradiating at 450 nm (see ESI† for further details). A weak pyrene fluorescence signal could be produced (Fig. 1, black) and addition of substrate 1a in varying amounts could quench the signal (Fig. 1, red and blue). These results support a mechanism where pyrene is quenched by the substrate as outlined in our proposed mechanism (Scheme 2F).
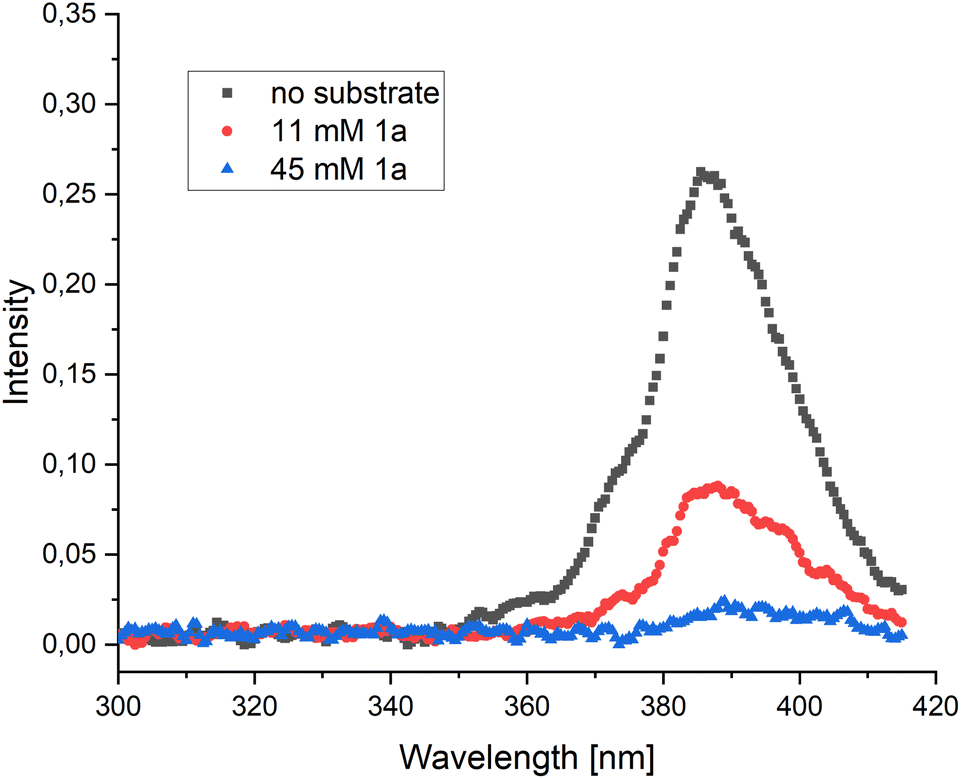 | ||
| Fig. 1 Pyrene fluorescence, produced via Ru(bpy)32+ and pyrene in aqueous SDS solution, without the presence of substrate (black) and with varying amounts of substrate 1a added as a quencher (red, blue). | ||
Conclusion
In conclusion, we have expanded the current synthetic possibilities using green light for “UV-reactivity” in water without the need for oxygen removing protocols. A commercially available green-to-UV TTA-UC pair was used in a formal [2 + 2]-cycloaddition to generate bioisosteres. We hope that by broadening the application of triplet–triplet annihilation upconversion, we can start to perform “UV-light photochemistry” under milder and more benign conditions.Data availability
The data supporting the findings in this article are presented in the manuscript or available in the ESI.†Author contributions
Experiments were performed by R. J. and M. U. The project was guided by L. N. All authors contributed to writing the manuscript. C. G. D. performed the crystallographic analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Westfälische Wilhelms-Universität and the Fonds der Chemischen Industrie for funding (Liebig fellowship for R. J. and L. N.). We thank Matthew James Milner and Elena Sophia Horst for measuring cyclic voltammetry and Lorenz Borsdorf for measuring the UV spectra. The authors also thank Dr Dirk Leifert, Dr Ruairi Oliver Mc Court, Dr Fabio Rizzo and Julian Carl Gregor Kürschner for scientific discussion and for proof-reading our manuscript. We thank our reviewers for constructive feedback that ultimately allowed us to improve the work presented in this article.Notes and references
- M. Sender and D. Ziegenbalg, Chem. Ing. Tech., 2017, 89, 1159 CrossRef CAS.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed.
- M. Uji, T. J. B. Zähringer, C. Kerzig and N. Yanai, Angew. Chem., Int. Ed., 2023, e202301506 CAS.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560 CrossRef CAS.
- R. Pérez-Ruiz, Top. Curr. Chem., 2022, 380, 23 CrossRef.
- F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem., Int. Ed., 2020, 59, 10266 CrossRef CAS PubMed.
- J. Zhao, S. Ji and H. Guo, RSC Adv., 2011, 1, 937 RSC.
- (a) L. Naimovičius, P. Bharmoria and K. Moth-Poulsen, Mater. Chem. Front., 2023, 7, 2297–2315 RSC; (b) T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103 RSC; (c) T. Dilbeck and K. Hanson, J. Phys. Chem. Lett., 2018, 9, 5810 CrossRef CAS PubMed; (d) A. J. Carrod, V. Gray and K. Börjesson, Energy Environ. Sci., 2022, 15, 4982 RSC; (e) B. S. Richards, D. Hudry, D. Busko, A. Turshatov and I. A. Howard, Chem. Rev., 2021, 121, 9165 CrossRef CAS PubMed.
- S. E. Seo, H.-S. Choe, H. Cho, H.-I. Kim, J.-H. Kim and O. S. Kwon, J. Mater. Chem. C, 2022, 10, 4483 RSC.
- T. Schloemer, P. Narayanan, Q. Zhou, E. Belliveau, M. Seitz and D. N. Congreve, ACS Nano, 2023, 17, 3259 CrossRef CAS PubMed.
- (a) Q. Dou, L. Jiang, D. Kai, C. Owh and X. J. Loh, Drug Discovery Today, 2017, 22, 1400 CrossRef CAS PubMed; (b) J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395 CrossRef CAS PubMed.
- F. Glaser and O. S. Wenger, JACS Au, 2022, 2, 1488 CrossRef CAS PubMed.
- (a) M. Häring, R. Pérez-Ruiz, A. Jacobi von Wangelin and D. D. Díaz, Chem. Commun., 2015, 51, 16848 RSC; (b) C. Kerzig and O. S. Wenger, Chem. Sci., 2018, 9, 6670 RSC; (c) M. Majek, U. Faltermeier, B. Dick, R. Pérez-Ruiz and A. Jacobi von Wangelin, Chem. –Eur. J., 2015, 21, 15496 CrossRef CAS PubMed; (d) B. Pfund, D. M. Steffen, M. R. Schreier, M.-S. Bertrams, C. Ye, K. Börjesson, O. S. Wenger and C. Kerzig, J. Am. Chem. Soc., 2020, 142, 10468 CrossRef CAS PubMed.
- W. Liang, C. Nie, J. Du, Y. Han, G. Zhao, F. Yang, G. Liang and K. Wu, Nat. Photon., 2023, 17, 346 CrossRef CAS.
- F. Glaser, C. Kerzig and O. S. Wenger, Chem. Sci., 2021, 12, 9922 RSC.
- B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343 CrossRef CAS PubMed.
- (a) Y. Du, X. Ai, Z. Li, T. Sun, Y. Huang, X. Zeng, X. Chen, F. Rao and F. Wang, Adv. Photonics Res., 2021, 2, 2000213 CrossRef CAS; (b) B. Yao, H. Sun, Y. He, S. Wang and X. Liu, Int. J. Mol. Sci., 2022, 23 Search PubMed; (c) C. G. López-Calixto, M. Liras, V. A. de La Peña O'Shea and R. Pérez-Ruiz, Appl. Catal., B, 2018, 237, 18 CrossRef; (d) L. Zeng, L. Huang, W. Lin, L.-H. Jiang and G. Han, Nat. Commun., 2023, 14, 1102 CrossRef CAS.
- F. Glaser and O. S. Wenger, Chem. Sci., 2022, 14, 149 RSC.
- R. R. Islangulov and F. N. Castellano, Angew. Chem., Int. Ed., 2006, 45, 5957 CrossRef CAS PubMed.
- H. Li, C. Wang, F. Glaser, N. Sinha and O. S. Wenger, J. Am. Chem. Soc., 2023, 145, 11402 CrossRef CAS PubMed.
- P. Bharmoria, H. Bildirir and K. Moth-Poulsen, Chem. Soc. Rev., 2020, 49, 6529 RSC.
- (a) K. A. El Roz and F. N. Castellano, Chem. Commun., 2017, 53, 11705 RSC; (b) S. Mutsamwira, E. W. Ainscough, A. C. Partridge, P. J. Derrick and V. V. Filichev, Chem. –Eur. J., 2016, 22, 10376 CrossRef CAS PubMed.
- M. S. Coles, G. Quach, J. E. Beves and E. G. Moore, Angew. Chem., Int. Ed., 2020, 59, 9522 CrossRef CAS PubMed.
- A. Denisenko, P. Garbuz, S. V. Shishkina, N. M. Voloshchuk and P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2020, 59, 20515 CrossRef CAS PubMed.
- J. C. G. Kürschner, L. Brüss and L. Naesborg, Chem. –Eur. J., 2023, e202300627 CrossRef.
- (a) S. Oguzlar, Opt. Mater., 2020, 101, 109772 CrossRef CAS; (b) P. Zhang, J. Guo, Y. Wang and W. Pang, Mater. Lett., 2002, 53, 400 CrossRef CAS.
- M. A. Filatov, S. Baluschev and K. Landfester, Chem. Soc. Rev., 2016, 45, 4668 RSC.
- I. Ghosh, R. S. Shaikh and B. König, Angew. Chem., Int. Ed., 2017, 56, 8544 CrossRef CAS PubMed.
- (a) I. Ghosh, J. I. Bardagi and B. König, Angew. Chem., Int. Ed., 2017, 56, 12822 CrossRef CAS PubMed; (b) M. Montalti, A. Credi, L. Prodi and M. T. M. T. Gandolfi, Handbook of Photochemistry, CRC, Taylor & Francis, Boca Raton, 3rd edn, 2006 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2264151. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03242f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |