
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium-catalyzed annulative approach to N–N axially chiral biaryls via C–H activation and dynamic kinetic transformation†
Xiaohan
Zhu
a,
Hongli
Wu
b,
Yishou
Wang
c,
Genping
Huang
 *b,
Fen
Wang
*a and
Xingwei
Li
ac
*b,
Fen
Wang
*a and
Xingwei
Li
ac
aSchool of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710062, China. E-mail: fenwang@snnu.edu.cn
bDepartment of Chemistry, School of Science and Tianjin Key Laboratory of Molecular Optoelectronic Sciences, Tianjin University, Tianjin 300072, China. E-mail: gphuang@tju.edu.cn
cInstitute of Molecular Science and Engineering, Institute of Frontier and Interdisciplinary Sciences, Shandong University, Qingdao 266237, China
First published on 15th July 2023
Abstract
N–N axially chiral biaryls represent a rarely explored class of atropisomeric compounds. We hereby report rhodium-catalyzed enantioselective [4 + 2] oxidative annulation of internal alkynes with benzamides bearing two classes of N–N directing groups. The coupling occurs under mild conditions via NH and CH annulation through the dynamic kinetic transformation of the directing group and is highly enantioselective with good functional tolerance. Computational studies of a coupling system at the DFT level has been conducted, and the alkyne insertion was identified as the enantio-determining as well as the turnover-limiting step.
Introduction
Axially chiral compounds have received considerable attention in the past decades owing to their wide presence in natural products and pharmaceuticals and extensive applications as chiral ligands or oganocatalysts.1 To date, the vast majority of the studies have been limited to those containing a C–C or C–N axis, and catalytic atroposelective synthesis of such molecules has been extensively investigated (Scheme 1a).2 In stark contrast, catalytic asymmetric construction of N–N atropisomers lags far behind,3 although N–N axial chirality is widely present in diverse biologically active molecules,4 natural products,5 ligands,6 and functional materials.7 For example, the schischkinii was isolated as a natural product from the seeds of Centaurea schischkinii.4b Moreover, the indoloterpenoid dixiamycins A and B have been identified as a pair of N–N atropisomers.5b,c Furthermore, 2,2′-bis(diphenylphosphanyl)-1,1′-bibenzimidazole (BIMIP) has been employed as a useful chiral bisphosphine ligand.6 In addition, DOBCz has been applied as an organic light-emitting diodes material.7 Therefore, the development of catalytic asymmetric methods to construct N–N axially chiral biaryls is under increasing demand.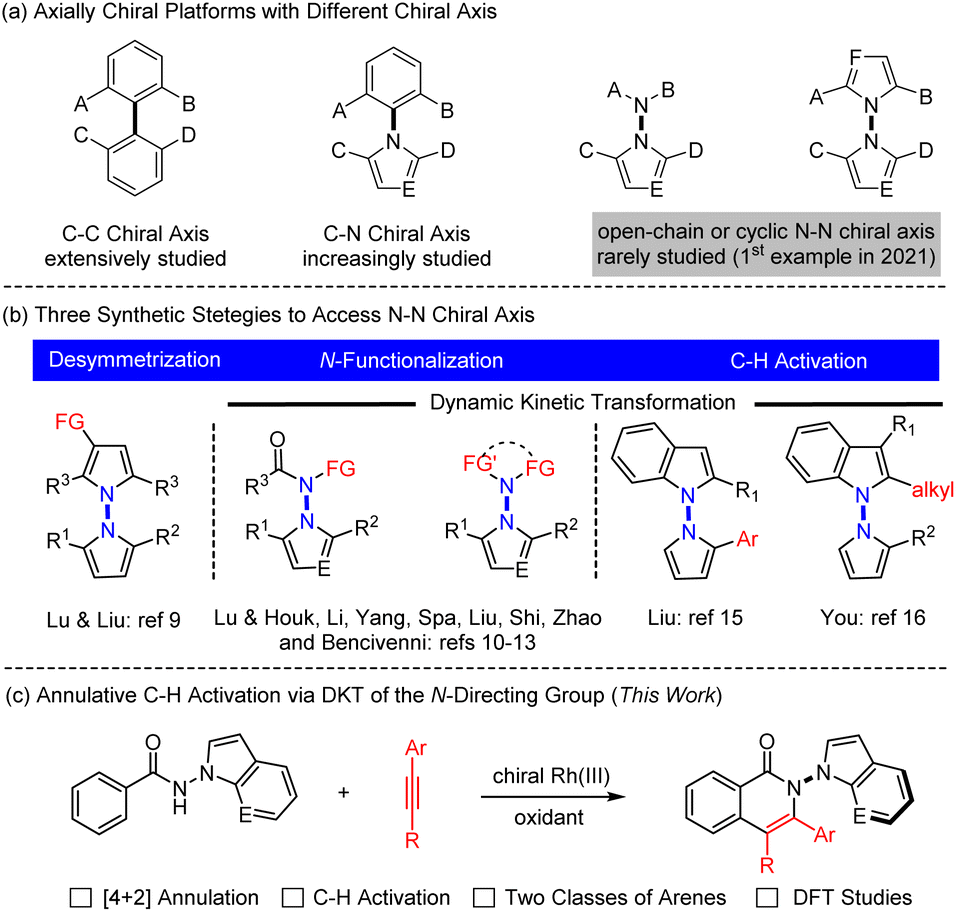 | ||
| Scheme 1 Representative atropisomers and enantioselective construction of N–N biaryl atropisomers. | ||
Three synthetic strategies have been developed toward asymmetric construction of chiral axis, namely, desymmetrization, NH-functionalization or NH2-difunctionalization, and peripheral C–H functionalization. In 2021, the group of Lu and Liu reported an enantioselective synthesis of bisindoles through Cu-catalyzed desymmetrizative Friedel–Crafts alkylation of symmetrical bisindoles (Scheme 1b left).8 Lu and Houk developed an original quinidine-catalyzed N-allylation of an amino group, accomplishing the 1st asymmetric construction of N–N axially chiral indoles (Scheme 1b middle).9 Shortly afterwards, increasing reports have documented metal- or organocatalyzed N–NH functionalization by virtue of dynamic kinetic transformations.10 Analogously, organocatalyzed [4 + 1] annulative difunctionalization of a N–NH2 group also provided an alternative avenue to access chiral indole-based biaryls in high enantioselectivity (Scheme 1b middle), as have been recently independently disclosed by Shi,11 Zhao,12 and Liu.13
Alternatively, C–H bond activation has emerged as a powerful strategy to access C–C and C–N axially chiral biaryls.14 In 2023, the Liu15 and You16 groups realized independent synthesis of N–N biaryl atropisomers through dynamic kinetic transformation (DKT) of indoles or pyrroles via palladium- or iridium-catalyzed enantioselective C–H functionalization (Scheme 1b, right). Despite the versatility of asymmetric C–H activation in construction of new chiral platforms,17 its applications toward construction of N–N axially chiral framework remain largely underdeveloped. In addition, the existing few examples (Scheme 1b, right) are also limited to simple, single-site C–H functionalization of indole rings that are intrinsically reactive.15,16 As a continuation of our ongoing interest in asymmetric synthesis of non-indole-based biaryl atropisomers via C–H functionalization,18 we now report [4 + 2] annulative coupling of readily available benzamides bearing different N-directing groups, where both proximal C–H and NH bonds are functionalized to give N–N axially chiral 5,6-biaryls in high enantioselectivity (Scheme 1c). Of note, the redox-neutral [4 + 2] annulation via N–N cleavage has been fully suppressed.
Results and discussion
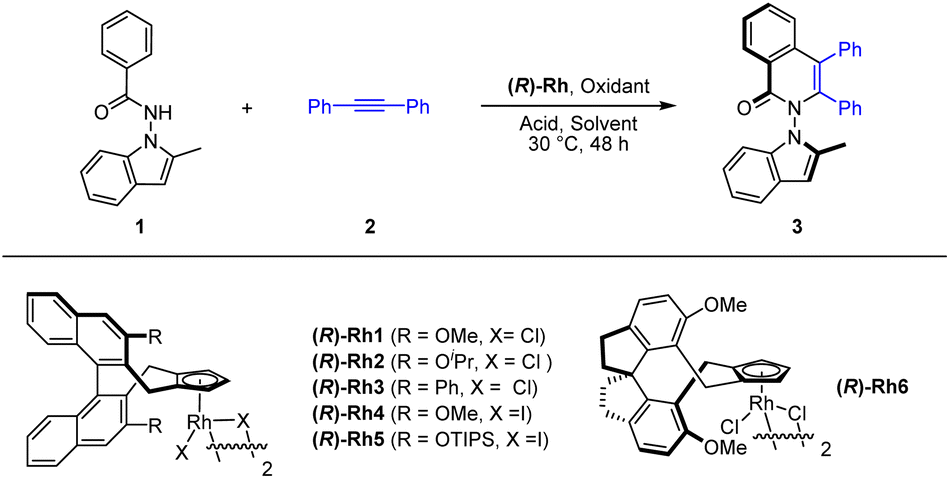
We commenced our exploration with optimization studies on the [4 + 2] annulation between benzamide 1 and diphenylacetylene (2) in the presence of a chiral CpXRh(III) catalyst (Table 1). It was found that the employment of (R)-Rh1 (3 mol%) as a catalyst in the presence of PivOH (2.0 equiv.) and Ag2O (2.0 equiv.) in MeOH at 30 °C afforded the desired product (3) in high enantioselectivity (88% ee) but with a low yield (entry 1). Further screening of solvents indicated that toluene was superior in terms of reaction efficiency, albeit with slightly lower enantioselectivity (entries 2–3). A series of chiral catalysts were then explored, and the (R)-Rh6 outperformed the rest in terms of both activity and stereoselectivity (entries 7–11), under which conditions good high and excellent enantioselectivity (−96% ee) have been secured (entry 11). In this case, the oppositely configured product 3 was obtained due to opposite spatial orientation of the chiral ligand in (R)-Rh6 despite the same nomenclature. The amount of the oxidant and additive were examined. However, reducing the amount of Ag2O and MesCO2H essentially had marginal effect on the efficiency and enantioselectivity (entry 13, Conditions A). Finally, switching the oxidant to other silver(I) oxidants only give lower efficiency or enantioselectivity (entries 14–16).|
|
||||||
|---|---|---|---|---|---|---|
| a Reaction conditions: 1 (0.1 mmol), 2 (0.15 mmol), (R)-Rh catalyst (3 mol%), oxidant (2.0 equiv.), and an acid (2.0 equiv.) in a solvent (2 mL), 30 °C, 48 h. b 60 °C. Isolated yield. The ee was determined by HPLC analysis using a chiral stationary phase. | ||||||
| Entry | Rh cat. | Oxidant (equiv.) | Acid (equiv.) | Solvent | ee (%) | Yield (%) |
| 1 | (R)-Rh1 | Ag2O (2.0) | HOPiv (2.0) | MeOH | 88 | 32 |
| 2 | (R)-Rh1 | Ag2O (2.0) | HOPiv (2.0) | DCE | 72 | 34 |
| 3 | (R)-Rh1 | Ag2O (2.0) | HOPiv (2.0) | PhMe | 84 | 49 |
| 4 | (R)-Rh1 | Ag2O (2.0) | MesCO2H (2.0) | PhMe | 88 | 55 |
| 5 | (R)-Rh1 | Ag2CO3 (2.0) | MesCO2H (2.0) | PhMe | 98 | 39 |
| 6b | (R)-Rh1 | Ag2O (1.0) | MesCO2H (2.0) | PhMe | 94 | 70 |
| Ag2CO3 (0.5) | ||||||
| 7 | (R)-Rh2 | Ag2O (2.0) | MesCO2H (2.0) | PhMe | 94 | 45 |
| 8 | (R)-Rh3 | Ag2O (2.0) | MesCO2H (2.0) | PhMe | — | <5 |
| 9 | (R)-Rh4 | Ag2O (2.0) | MesCO2H (2.0) | PhMe | 91 | 50 |
| 10 | (R)-Rh5 | Ag2O (2.0) | MesCO2H (2.0) | PhMe | — | <5 |
| 11 | (R)-Rh6 | Ag2O (2.0) | MesCO2H (2.0) | PhMe | −96 | 67 |
| 12 | (R)-Rh6 | Ag2O (1.5) | MesCO2H (2.0) | PhMe | −95 | 67 |
| 13 | (R)-Rh6 | Ag 2 O (1.5) | MesCO 2 H (1.2) | PhMe | −96 | 68 |
| 14 | (R)-Rh6 | Ag2CO3 (1.5) | MesCO2H (1.2) | PhMe | −97 | 40 |
| 15 | (R)-Rh6 | AgF (1.5) | MesCO2H (1.2) | PhMe | −84 | 43 |
| 16 | (R)-Rh6 | AgOAc (1.5) | MesCO2H (1.2) | PhMe | — | <5 |
With the optimal conditions in hand, the scope of the N-indolyl benzamide substrates was next examined (Scheme 2). The presence of alkyl and halogen groups at the 3- or 6-position in indole ring was tolerated, affording the desired products in good yield and excellent enantioselectivity (3–6, −94 to −96% ee) under the standard reaction conditions. Of note, the (R)-Rh1 catalyst could also catalyze this reaction in the presence of a mixed silver salt oxidants (entry 6, in Table 1, Conditions B) in high yield and excellent enantioselectivity ((ent)-3, 94% ee), whose absolute configuration has been determined by X-ray crystallography (CCDC 2213499). However, the Conditions B suffered from compatibility issues with various substrates (see comparisons of these two conditions for synthesis of 12 and 14). Therefore, the standard Conditions A in Table 1 (entry 13) were employed in most of our studies. Under these conditions, the presence of diverse meta and para substituents in the benzene ring were also well tolerated (7–9). An ortho substituent in the benzamide did not compromise the enantioselectivity but influenced the reaction efficiency and caused a decrease in yield (10). Symmetric diarylacetylenes bearing a range of electron-donating, electron-withdrawing, and halogen groups at para position were fully compatible, affording products 11–15 in excellent enantioselectivity. In addition to symmetric alkynes, some unsymmetrical alkynes could also react to give the desired products (16–18) with excellent regioselectivity under modified conditions. 1,3-Enynes with an alkyl or phenyl terminal group underwent [4 + 2] annulations with the benzamide, furnishing the desired products 19 and 20 in moderate yields and high enantioselectivities. Extension of the N-group to N-dihydroquinolinone also gave product 21 with a 6,6-heterocyle linkage in excellent enantioselectivity and low yield. In contrast, poor reactivity (<5% yield) was observed for the coupling of 3-hexyne under either conditions A or B (Scheme 2).
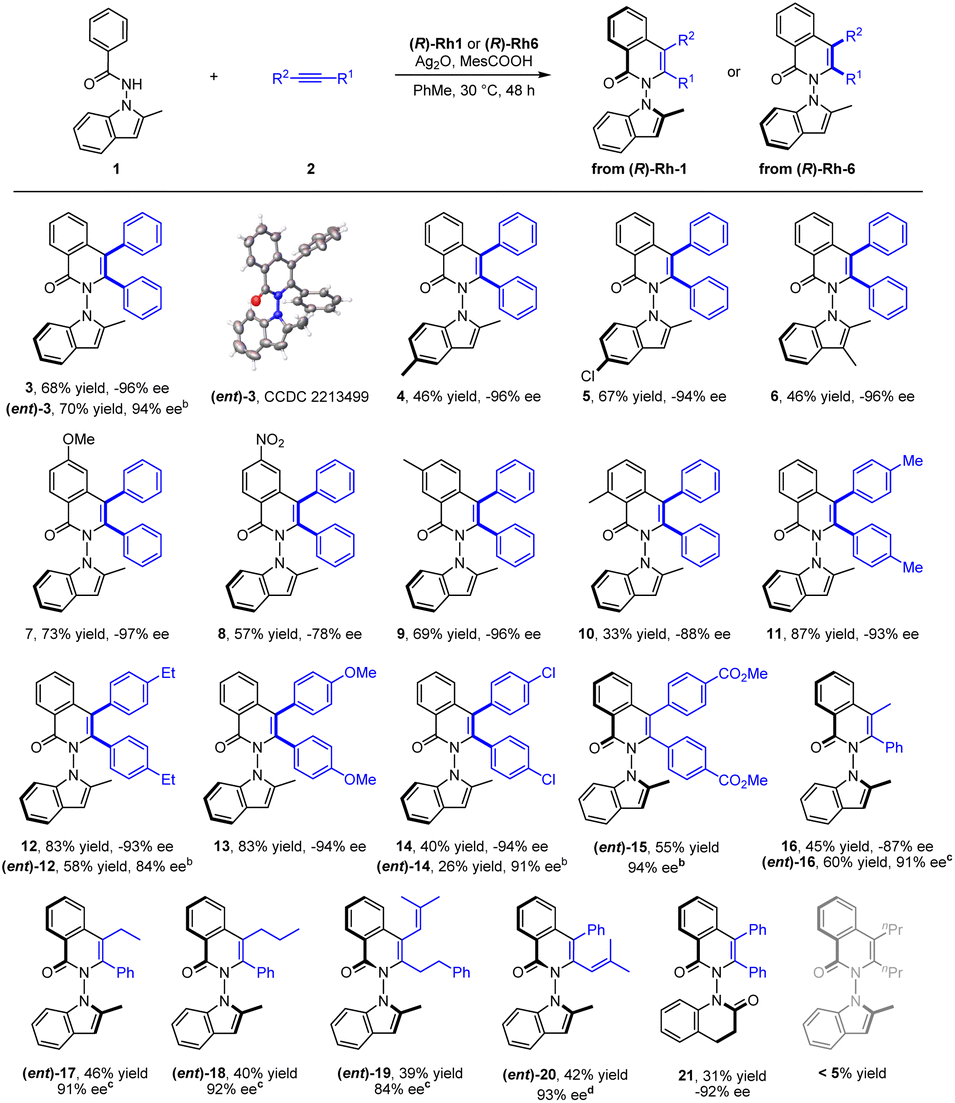 | ||
| Scheme 2 Scope of N-indolyl benzamides 1 as substrates for the synthesis of N–N atropisomers. (a) Reaction conditions A: 1 (0.1 mmol), 2 (0.15 mmol), (R)-Rh6 (3 mol%), Ag2O (1.5 equiv.), and MesCOOH (1.2 equiv.) in toluene (2 mL), 30 °C, 48 h. Isolated yield. The ee was determined by HPLC analysis using a chiral stationary phase. (b) Reaction conditions B: 1 (0.1 mmol), 2 (0.15 mmol), (R)-Rh1 (3 mol%), Ag2O (1.0 equiv.), Ag2CO3 (0.5 equiv.) and MesCOOH (2.0 equiv.) in toluene (2 mL), 60 °C, 48 h. (c) Reaction conditions C: 1 (0.1 mmol), 2 (0.15 mmol), (R)-Rh1 (3 mol%), Ag2CO3 (1.0 equiv.), Ag2O (0.5 equiv.), and HOPiv (2.0 equiv.) in toluene (2 mL), 30 °C, 48 h. (d) Reaction conditions D: 1 (0.1 mmol), 2 (0.15 mmol), (R)-Rh1 (3 mol%), Ag2O (2.0 equiv.), and HOPiv (2.0 equiv.) in toluene (2 mL), 30 °C, 48 h. | ||
To better explore the applicability of arene substrates, we next moved to benzamides bearing a N-azaindolyl group (22, Scheme 3). It was found that the modified catalytic conditions of (R)-Rh-1 (3 mol%)/AgSbF6 (12 mol%), AgOAc (2.0 equiv.) and AcOH (1.0 equiv.) allowed the formation of the desired product 23 in a high isolated yield (87%) and excellent enantioselectivity (94% ee) with DCE being a solvent. The absolute configuration of product 23 has been determined by ECD spectroscopy and confirmed by DFT studies. Thus, benzamides with a large array of methyl, methoxy, trifluoromethyl, and halogen substituents at different positions of the benzene ring were all compatible (23–29, 94–98% ee), with no significant electronic effect being observed on the enantioselectivity. In particular, a 3-thiophenecarboxamide also reacted smoothly in this reaction system (30, 92% ee). The scope of internal alkynes 2 was next examined. Symmetrical diarylalkynes bearing electron-donating (methyl and tert-butyl) and -withdrawing (trifluoromethyl and bromide) groups at the para position were well compatible, and the corresponding products 32–35 were delivered in high yields (87–93%) with excellent enantioselectivities (90–92% ee). meta-(Di)substituted alkynes also reacted smoothly to afford the corresponding product 36 and 37 in excellent stereoselectivity. Moreover, when a hereroaromatic alkyne was used, the product 38 was obtained in moderate yield and excellent enantioselectivity (92% ee). In addition, for a nonsymmetric alkyne, the corresponding product 39 was obtained only with moderate yield and enantioselectivity (75% ee). To understand the conformational stability of such N–N axial atropisomers, racemization studies have been conducted for a representative product 23 at 120 °C in mesitylene (see ESI†). The rotational barrier of 23 was estimated to be 32.8 kcal mol−1, indicating the high atropoisomeric stability of this N–N axis. Additionally, scale-up synthesis of 23 (1 mmol) was successful, with essentially no decay of enantiopurity.
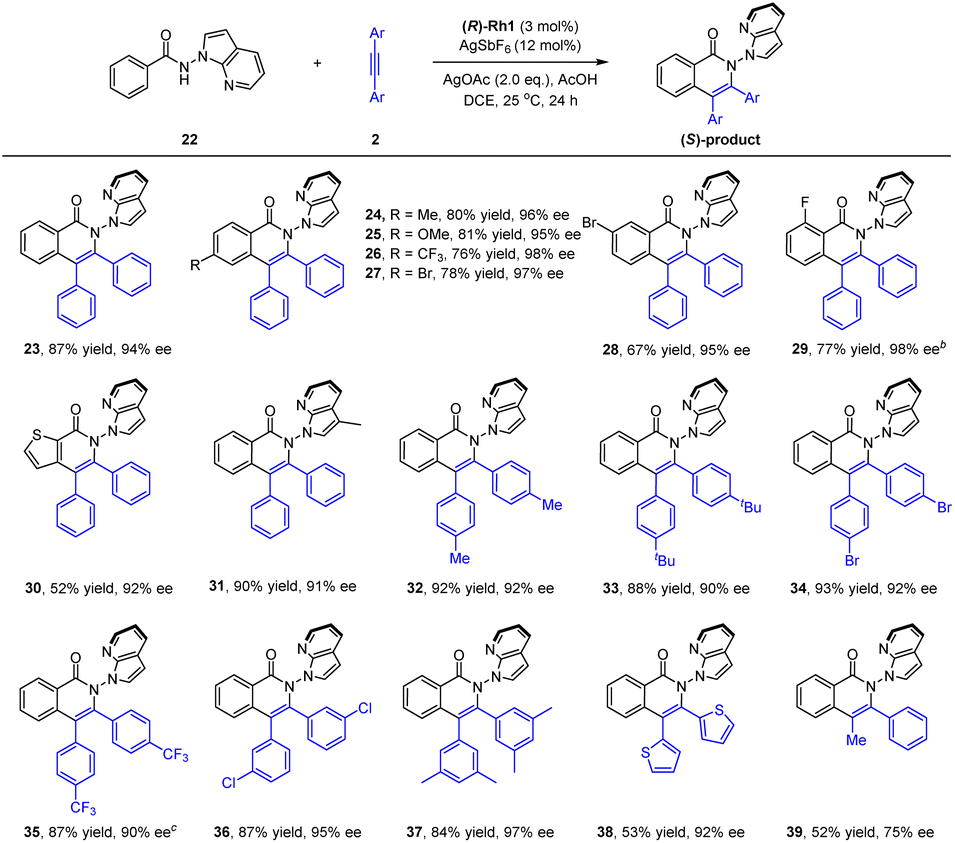 | ||
| Scheme 3 Scope of N-azaindolyl benzamides as substrates for the synthesis of N–N atropisomers. (a) Reaction conditions: 1 (0.1 mmol), 2 (0.1 mmol), (R)-Rh1 (3 mol%), AgSbF6 (12 mol%), AgOAc (2.0 equiv.), AcOH (1.0 equiv.) in DCE (1 mL), 24 h, isolated yield. The ee was determined by HPLC using a chiral stationary phase. (b) 50 °C. (c) 25 °C, 60 h. | ||
Representative products have been studied toward further synthetic or catalytic transformations as given in Scheme 4. Treatment of product (ent)-3 with NBS afforded the 3-brominated product in excellent yield (40). Ru(II)-catalyzed oxidative olefination of product (ent)-3 with styrene gave product 41 in essentially the same optical purity. Treatment of 23 with I2O5 led to selective iodination at the N-azaindole ring (42). The pyridine ring in a related axially chiral product 23 offers synthetic handles. m-CPBA oxidation of 23 afforded the N-oxide (43) in high yield. Further reaction with para-bromotoluene under palladium catalysis delivered the arylated product 44. We next applied N-oxide 43 as a chiral ligand in Pd-catalyzed insertion of an N-protected indole into a donor–acceptor carbene reagent, affording the alkylated product 45 in promising enantioselectivity (59% ee).
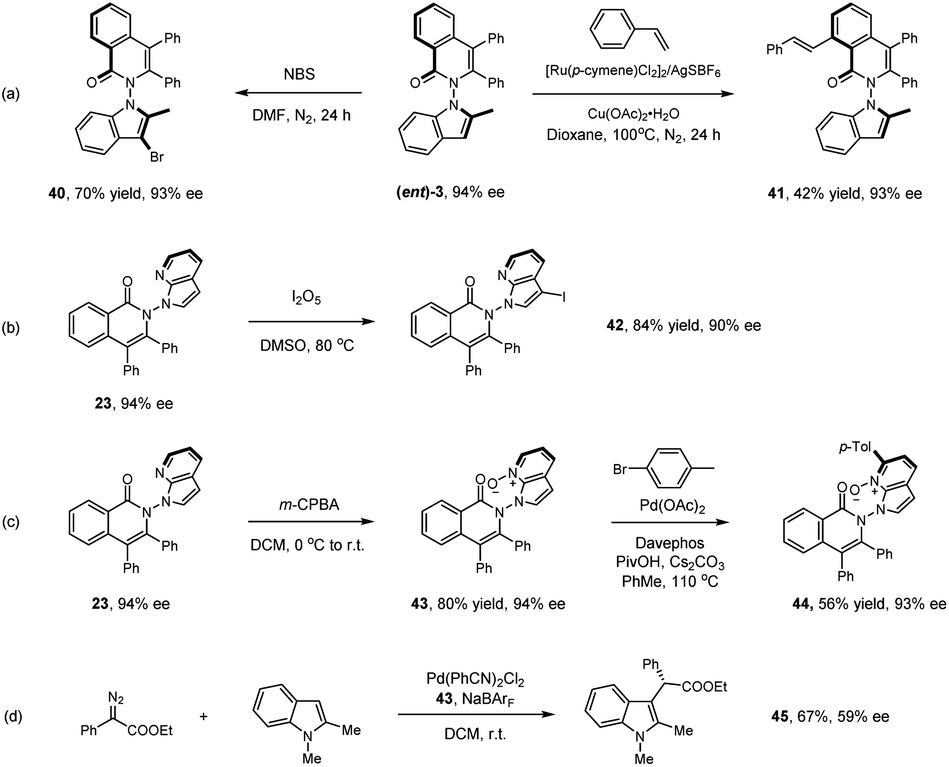 | ||
| Scheme 4 Versatile synthetic transformation of representative N–N axially chiral products. | ||
To gain a better understanding of the detailed reaction mechanism and factors that contribute to enantioselectivity, DFT calculations were performed at the M06(SMD)/def2-TZVPP//B3LYP-D3(BJ)/def2-SVP level of theory (see the ESI† for the computational details). The experimentally used benzamide 1 and diphenylacetylene 2 were selected as the model substrates in the computations (Fig. 1). The calculated energy profile of the most favorable pathway leading to [4 + 2] oxidative annulation product (SNN)-3 is given in Fig. 1. Starting from active catalyst species CAT derived from the (R)-Rh-1 catalyst, the N–H deprotonation gives the intermediate (SNN)-IM1, which was calculated to be endergonic by 5.9 kcal mol−1. The ensuing C–H bond cleavage occurs via the well-established concerted metallation deprotonation (CMD) mechanism19 through transition state (SNN)-TS1, with an energy barrier of 22.2 kcal mol−1 relative to CAT. The resulted five-membered rhodacycle intermediate (SNN)-IM2 was found to be more stable than (SNN)-IM1 by 6.1 kcal mol−1. The computations indicate that the alkyne insertion into the Rh–C bond directly from (SNN)-IM2 is kinetically unfavorable (see the ESI† for details). Instead, an isomerization step involving the rotation of Rh–Cp bond20 must occur first to give intermediate (SNN)-IM3. Subsequently, the alkyne insertion into the Rh–C bond proceeds via transition state (SNN)-TS2, leading to seven-membered rhodacycle species (SNN)-IM4. This intermediate then undergoes the C–N reductive elimination via transition state (SNN)-TS3 to forge product-coordinated intermediate (SNN)-IM4, from which product (SNN)-3 can be obtained by the re-oxidation of Rh center to regenerate active catalyst species CAT with the assistance of oxidant AgOAc.
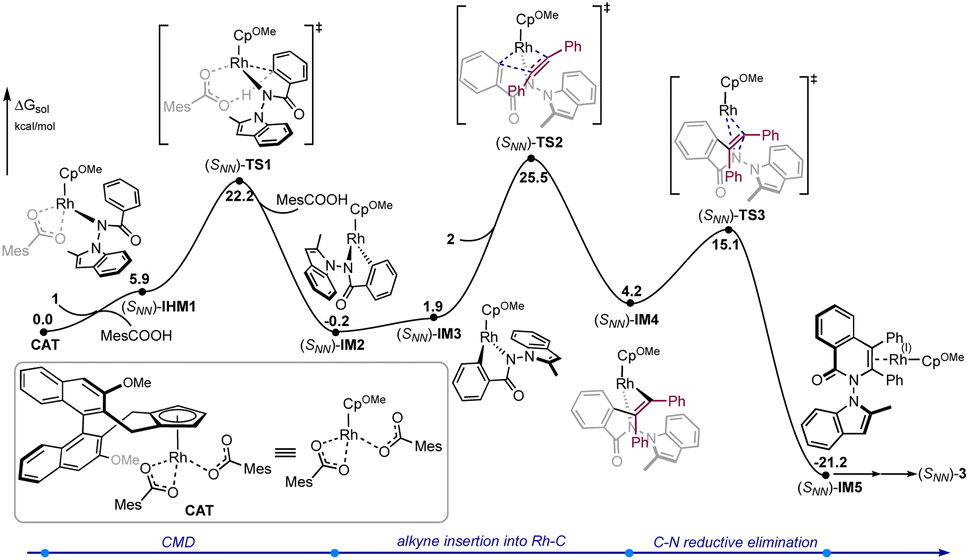 | ||
| Fig. 1 Calculated energy profiles of rhodium-catalyzed enantioselective [4 + 2] oxidative annulation of diphenylacetylene 2 with benzamide 1. | ||
The computations show that the alkyne insertion into the Rh–C bond constitutes the rate- and enantioselectivity determining step of the overall reaction.21 This is also consistent with the poor chiral control during the C–H activation step (cyclometallation). To pinpoint the origin of the enantioselectivity, the pathway leading to product (RNN)-3 was also considered (see the ESI† for the whole calculated energy profile). The calculated energy difference of 2.1 kcal mol−1 between enantiomeric insertion transition states (SNN)-TS2 and (RNN)-TS2 corresponds to a predicted enantioselectivity of 92% ee at the reaction temperature (25.5 versus 27.6 kcal mol−1, Fig. 2), which agrees very well with experimentally-observed 94% ee. The optimized geometries imply that the relative orientation of the alkyne is quite different in (SNN)-TS2 and (RNN)-TS2 (Fig. 2). Specifically, the alkyne is positioned to the right side of the benzamide moiety in (SNN)-TS2 so that the five-membered rhodacycle intermediate can readily undergo the insertion process without notable geometric change. In contrast, in (RNN)-TS2 the alkyne was located to the left side of the benzamide moiety. As a result, to proceed the insertion process, the five-membered rhodacycle intermediate must undergo significant distortion to create a vacant side for the incoming alkyne, resulting in (RNN)-TS2 higher in energy than (SNN)-TS2. It should be mentioned that the insertion transition state (RNN)-TS2′, wherein the relative orientation of the alkyne is the same as in (SNN)-TS2, was also evaluated, but it was calculated to be 3.3 kcal mol−1 higher in energy than (SNN)-TS2 (i.e., 1.2 kcal mol−1 higher in energy than (RNN)-TS2). This is likely due to that the steric repulsion between the benzamide moiety and the chiral cyclopentadienyl ligand in (RNN)-TS2′ is greater than that in (SNN)-TS2 (see the ESI† for details). Therefore, the steric repulsion results in the change of the mode of the alkyne insertion, enabling the experimentally-observed enantioselectivity. In addition, our computational free energy profile is also in good agreement with our experimentally observed kinetic isotope effect (KIE = 2.3, see ESI†). The C–H activation event (22.2 kcal mol−1) contributes a considerable portion to the overall kinetic barrier (25.5 kcal mol−1).
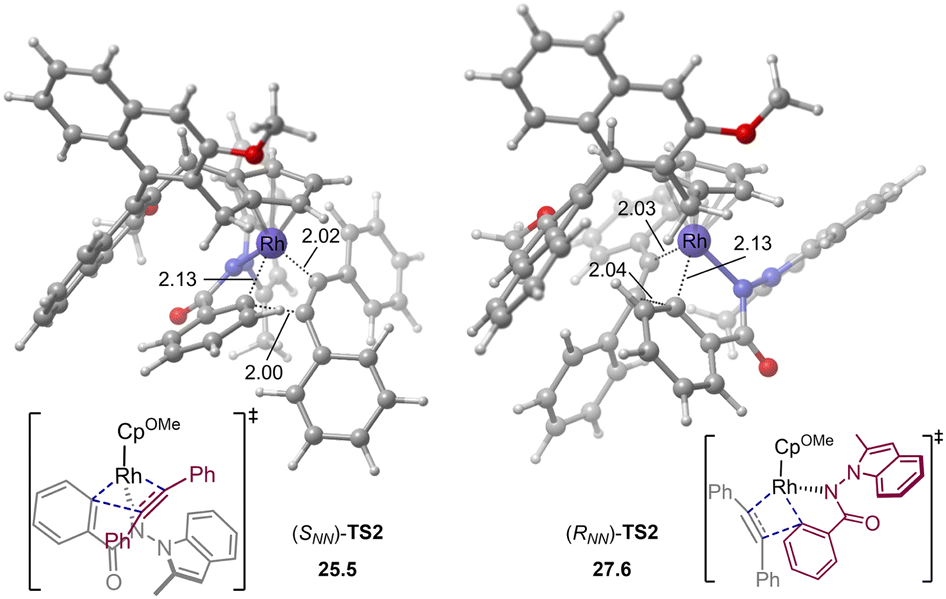 | ||
| Fig. 2 Optimized geometries of alkyne insertion transition states. Energies and bond distances are given in kcal mol−1 and Å, respectively. | ||
Conclusions
In conclusion, we have developed two classes of asymmetric [4 + 2] annulative functionalization reactions of alkynes with benzamides bearing different types of N–N directing groups. The reaction proceeds via dynamic kinetic transformation (DKT) of the directing group through RhIII-catalyzed C–H activation, which afforded a series of N–N axially chiral biaryls in high yields and excellent atroposelectivities. The reaction of benzamides bearing N-indolyl group and alkynes afforded N-quinolinoneindoles. By using benzamides with an N-azaindole ring as arene substrates, we precisely demonstrated assembly of N–N atropisomers. DFT calculations provided insight into the reaction mechanism and the enantioselective control. The strategy of de novo construction of a N–N chiral axis is currently explored toward precise synthesis of related axially chiral biaryls in our laboratory.Data availability
Further details of the experimental procedure, 1H, and 13C NMR, HPLC spectra, and X-ray crystallographic data for (ent)-3 and DFT calculations are available in the ESI.†Author contributions
X. Z. and Y. W. performed experiments. G.-P. H. performed DFT calculations. F. W., G.-P. H. and X. L. conceived and directed the project and wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We acknowledge the financial support for this work from the NSFC (22101167 and 22073066) and the Shaanxi Normal University.Notes and references
- For a review: (a) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS; (b) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed; (c) E. Kumarasamy, R. Raghunathan, M. P. Sibi and J. Sivaguru, Chem. Rev., 2015, 115, 11239–11300 CrossRef CAS PubMed; (d) J. E. Smyth, N. M. Butler and P. A. Keller, Nat. Prod. Rep., 2015, 32, 1562–1583 RSC; (e) S. T. Toenjes and J. L. Gustafson, Future Med. Chem., 2018, 10, 409–422 CrossRef CAS; (f) S. Perreault, J. Chandrasekhar and L. Patel, Acc. Chem. Res., 2022, 55, 2581–2593 CrossRef CAS PubMed; (g) M. Basilaia, M. H. Chen, J. Secka and J. L. Gustafson, Acc. Chem. Res., 2022, 55, 2904–2919 CrossRef CAS PubMed.
- For selected reviews, see: (a) G. Bringmann, A. J. P. Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS PubMed; (b) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC; (c) Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS PubMed; (d) B. Zilate, A. Castrogiovanni and C. Sparr, ACS Catal., 2018, 8, 2981–2988 CrossRef CAS; (e) H. Yang, J. Chen and L. Zhou, Chem.–Asian J., 2020, 15, 2939–2951 CrossRef CAS; (f) V. Corti and G. Bertuzzi, Synthesis, 2020, 52, 2450–2468 CrossRef CAS; (g) D.-J. Cheng and Y.-D. Shao, Adv. Synth. Catal., 2020, 362, 3081–3099 CrossRef CAS; (h) Y.-C. Zhang, F. Jiang and F. Shi, Acc. Chem. Res., 2020, 53, 425–446 CrossRef CAS; (i) J. A. Carmona, C. Rodrĺguez-Franco, R. Fernández, V. Hornillos and J. M. Lassaletta, Chem. Soc. Rev., 2021, 50, 2968–2983 RSC; (j) O. Kitagawa, Acc. Chem. Res., 2021, 54, 719–730 CrossRef CAS; (k) B.-M. Yang, X. Q. Ng and Yu Zhao, Chem Catalysis, 2022, 2, 3048–3076 CrossRef CAS.
- G. Centonze, C. Portolani, P. Righi and G. Bencivenni, Angew. Chem., Int. Ed., 2023, e3138202303966 Search PubMed.
- (a) J. T. Klein, L. Davis, G. E. Olsen, G. S. Wong, F. P. Huger, C. P. Smith, W. W. Petko, M. Cornfeldt, J. C. Wilker, R. D. Blitzer, E. Landau, V. Haroutunian, L. L. Martin and R. C. Effland, J. Med. Chem., 1996, 39, 570–581 CrossRef CAS; (b) K. Suzuki, I. Nomura, M. Ninomiya, K. Tanaka and M. Koketsu, Bioorg. Med. Chem. Lett., 2018, 28, 2976–2978 CrossRef CAS PubMed.
- (a) M. Shoeb, S. Celik, M. Jaspars, Y. Kumarasamy, S. M. MacManus, L. Nahar, P. K. Thoo-Lin and S. D. Sarker, Tetrahedron, 2005, 61, 9001–9006 CrossRef CAS; (b) Q. Zhang, A. Mándi, S. Li, Y. Chen, W. Zhang, X. Tian, H. Zhang, H. Li, W. Zhang, S. Zhang, J. Ju, T. Kurtán and C. Zhang, Eur. J. Org. Chem., 2012, 5256–5262 CrossRef CAS; (c) Z. Xu, M. Baunach, L. Ding and C. Hertweck, Angew. Chem., Int. Ed., 2012, 51, 10293–10297 CrossRef CAS.
- T. Benincori, E. Brenna, F. Sannicolò, L. Trimarco, P. Antognazza, E. Cesarotti, F. Demartin, T. Pilati and G. Zotti, J. Organomet. Chem., 1997, 529, 445–453 CrossRef CAS.
- X.-Y. Liu, Y.-L. Zhang, X. Fei, L.-S. Liao and J. Fan, Chem.–Eur. J., 2019, 25, 4501–4508 CrossRef CAS PubMed.
- X.-M. Wang, P. Zhang, Q. Xu, C.-Q. Guo, D.-B. Zhang, C.-J. Lu and R.-R. Liu, J. Am. Chem. Soc., 2021, 143, 15005–15010 CrossRef CAS PubMed.
- (a) G.-J. Mei, J. J. Wong, W. Zheng, A. A. Nangia, K. N. Houk and Y. Lu, Chem, 2021, 7, 2743–2757 CrossRef CAS; (b) Q. Xu, H. Zhang, F.-B. Ge, X.-M. Wang, P. Zhang, C.-J. Lu and R.-R. Liu, Org. Lett., 2022, 24, 3138–3143 CrossRef CAS PubMed.
- (a) M. Pan, Y.-B. Shao, Q. Zhao and X. Li, Org. Lett., 2022, 24, 374–378 CrossRef CAS PubMed; (b) W. Lin, Q. Zhao, Y. Li, M. Pan, C. Yang, G.-H. Yang and X. Li, Chem. Sci., 2022, 13, 141–148 RSC; (c) C. Portolani, G. Centonze, S. Luciani, A. Pellegrini, P. Righi, A. Mazzanti, A. Ciogli, A. Sorato and G. Bencivenni, Angew. Chem., Int. Ed., 2022, 61, e202209895 CrossRef CAS PubMed; (d) V. Hutskalova and C. Sparr, Synthesis, 2023, 55, 1770–1782 CrossRef CAS; (e) L.-Y. Wang, J. Miao, Y. Zhao and B.-M. Yang, Org. Lett., 2023, 25, 1553–1557 CrossRef CAS PubMed.
- K.-W. Chen, Z.-H. Chen, S. Yang, S.-F. Wu, Y.-C. Zhang and F. Shi, Angew., Chem. Int. Ed., 2022, 61, e202116829 CAS.
- Y. Gao, L.-Y. Wang, T. Zhang, B.-M. Yang and Y. Zhao, Angew. Chem. Int., Ed., 2022, 61, e202200371 CAS.
- P. Zhang, Q. Xu, X.-M. Wang, J. Feng, C.-J. Lu, Y. Li and R.-R. Liu, Angew. Chem., Int. Ed., 2022, 61, e202212101 CAS.
- (a) G. Liao, T. Zhou, Q.-J. Yao and B.-F. Shi, Chem. Commun., 2019, 55, 8514–8523 RSC; (b) C.-X. Liu, W.-W. Zhang, S.-Y. Yin, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2021, 143, 14025–14040 CrossRef CAS PubMed.
- Y. Wang, C.-J. Lu, L.-W. Zhan, Y. Wu, J. Feng and R.-R. Liu, Angew. Chem., Int. Ed., 2023, 62, e202218871 CrossRef PubMed.
- S.-Y. Yin, Q. Zhou, C.-X. Liu, Q. Gu and S.-L. You, Angew. Chem., Int. Ed., 2023, e202305067 Search PubMed.
- For selected reviews, see: (a) C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS PubMed; (b) D.-W. Gao, Q. Gu, C. Zheng and S.-L. You, Acc. Chem. Res., 2017, 50, 351–365 CrossRef CAS PubMed; (c) A. Link and C. Sparr, Chem. Soc. Rev., 2018, 47, 3804–3815 RSC; (d) T. K. Achar, S. Maiti, S. Jana and D. Maiti, ACS Catal., 2020, 10, 13748–13793 CrossRef CAS; (e) J. Wencel-Delord and F. Colobert, SynOpen, 2020, 4, 107–115 CrossRef CAS; (f) Q. Zhang, L.-S. Wu and B.-F. Shi, Chem, 2022, 8, 384–413 CrossRef CAS; (g) M. I. Lapuh, S. Mazeh and T. Besset, ACS Catal., 2020, 10, 12898–12919 CrossRef CAS; (h) B. Su and J. F. Hartwig, Angew. Chem., Int. Ed., 2022, 61, e202113343 CAS.
- (a) B. Shen, B. Wan and X. Li, Angew. Chem., Int. Ed., 2018, 57, 15534–15538 CrossRef CAS PubMed; (b) R. Mi, G. Zheng, Z. Qi and X. Li, Angew. Chem., Int. Ed., 2019, 58, 17666–17670 CrossRef CAS PubMed; (c) M. Tian, D. Bai, G. Zheng, J. Chang and X. Li, J. Am. Chem. Soc., 2019, 141, 9527–9532 CrossRef CAS; (d) F. Wang, Z. Qi, Y. Zhao, S. Zhai, G. Zheng, R. Mi, Z. Huang, X. Zhu, X. He and X. Li, Angew. Chem., Int. Ed., 2020, 59, 13288–13294 CrossRef CAS; (e) R. Mi, H. Chen, X. Zhou, N. Li, D. Ji, F. Wang, Y. Lan and X. Li, Angew. Chem., Int. Ed., 2022, 61, e202111860 CrossRef CAS PubMed; (f) D. Ji, J. Jing, Y. Wang, Z. Qi, F. Wang, X. Zhang, Y. Wang and X. Li, Chem, 2022, 8, 3346–3362 CrossRef CAS; (g) P. Wang, H. Wu, X.-P. Zhang, G. Huang, R. H. Crabtree and X. Li, J. Am. Chem. Soc., 2023, 145, 8417–8429 CAS.
- (a) D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754–13755 CrossRef CAS PubMed; (b) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496–16497 CrossRef CAS PubMed.
- C. Zheng and S.-L. You, ACS Catal., 2016, 6, 262–271 CrossRef CAS.
- Q. Wang, Y.-H. Nie, C.-X. Liu, W.-W. Zhang, Z.-J. Wu, Q. Gu, C. Zheng and S.-L. You, ACS Catal., 2022, 12, 3083–3093 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2213499. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02800c |
| This journal is © The Royal Society of Chemistry 2023 |