
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMerging the Norrish type I reaction and transition metal catalysis: photo- and Rh-promoted borylation of C–C σ-bonds of aryl ketones†
Yuki
Fujimaki
,
Nobuharu
Iwasawa
and
Jun
Takaya
 *
*
Department of Chemistry, Tokyo Institute of Technology, O-okayama, Meguro-ku, Tokyo 152-8551, Japan. E-mail: takayajun@chem.titech.ac.jp
First published on 26th January 2023
Abstract
Synthesis of arylboronates via borylation of C–C σ-bonds of aryl ketones was achieved by the combined use of photoenergy and a Rh catalyst. The cooperative system enables α-cleavage of photoexcited ketones to generate aroyl radicals via the Norrish type I reaction, which are successively decarbonylated and borylated with the rhodium catalyst. This work establishes a new catalytic cycle merging the Norrish type I reaction and Rh catalysis and demonstrates the new synthetic utility of aryl ketones as aryl sources for intermolecular arylation reactions.
Introduction
Transition metal-catalyzed arylation reactions have been attracting much attention as highly useful methods for the synthesis of aromatic compounds from both academic and practical viewpoints. Recent advancements enable the utilization of various aromatic compounds containing C–X bonds (X = OR, NR2, SR, CN, COOR etc.) as aryl sources instead of aryl halides in various cross-coupling reactions and functionalization reactions.1 On the other hand, the use of aryl ketones as substrates has been a great challenge because cleavage of strong C–C σ-bonds is involved to generate arylmetal species. Although catalytic C–C bond activation reactions of ketones have been developed extensively since the discovery of the Rh-catalyzed hydrogenolysis of the C–C σ-bond α to a carbonyl group in 1994,2 substrates are mostly limited to small-membered cyclic ketones and well-designed ketones bearing directing groups to facilitate oxidative addition of C–C bonds.3,4 The use of simple and versatile aryl ketones as aryl sources for arylation reactions has rarely been realized. There are several reports on Rh- or Ni-promoted intramolecular decarbonylative coupling reactions of simple aryl ketones (ArCOR, R = aryl, alkynyl, silyl) although both an aryl moiety and a coupling partner have to be furnished in the substrate (Fig. 1-a).5 Dong developed the transient directing group strategy for C–C bond activation of ketones via N-pyridylketimine intermediates, realizing insertion reactions of alkenes and alkynes into sp2C–sp2C σ-bonds of cyclic ketones (acylarylation) such as indanone and isatin derivatives (Fig. 1-b).6 Recently, Dai developed a 2-step protocol for the utilization of simple alkyl aryl ketones as aryl sources in various arylation reactions, where ketones were pre-transformed into oxime esters in advance as activated forms for Pd-mediated C–C bond cleavage (Fig. 1-c).7 However, the need for a pre-activation step reduces its atom and step economy. Therefore, the development of new and straightforward strategies that enable direct utilization of simple aryl ketones for intermolecular arylation reactions is highly desired.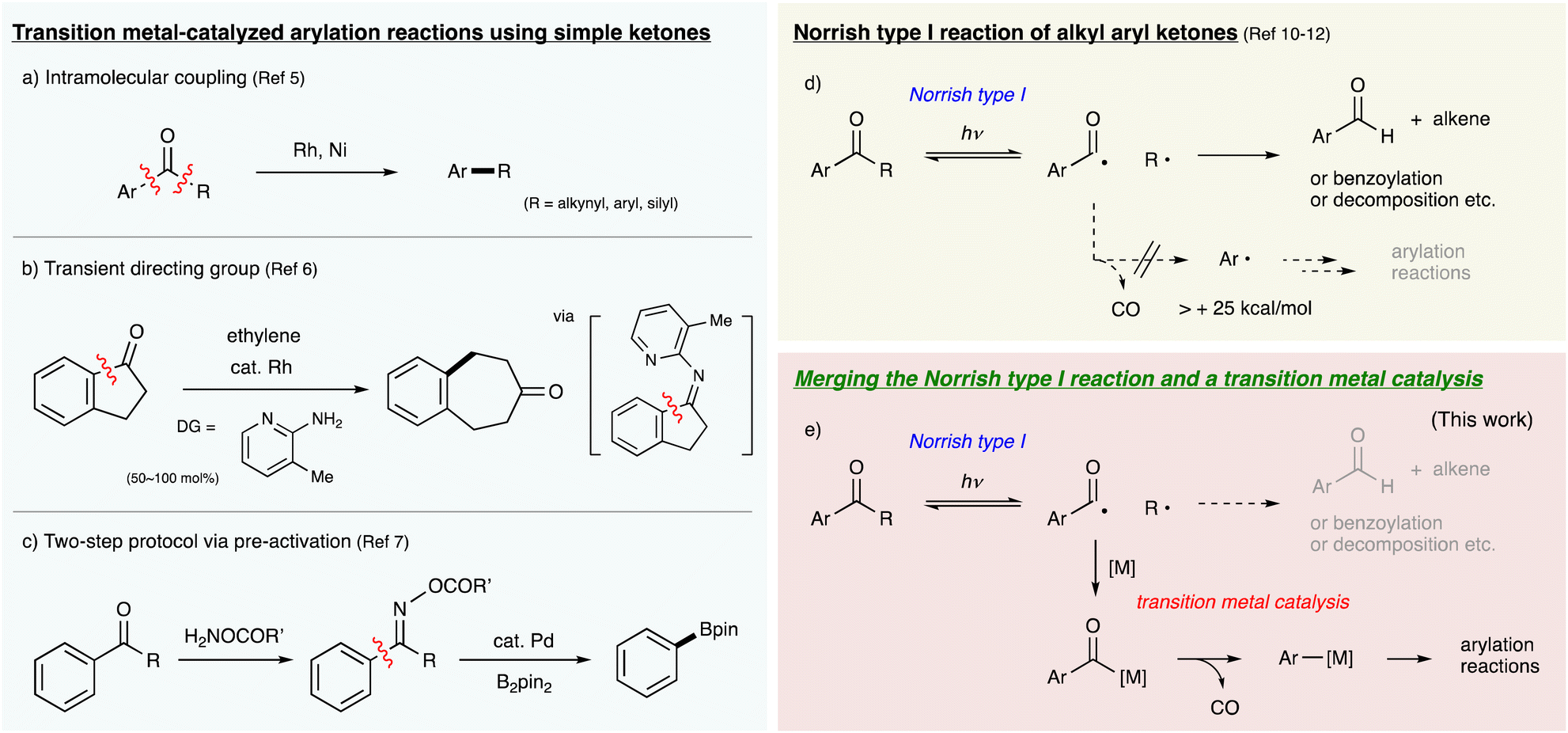 | ||
| Fig. 1 Arylation reactions using simple aryl ketones as aryl sources. | ||
Meanwhile, photoexcited organic molecules undergo various reactions involving cleavage of unreactive bonds such as C–H and C–C σ-bonds. The Norrish type I reaction is one of the representative photoreactions of ketones to cleave C–C σ-bonds α to the carbonyl moiety (α-cleavage reaction) upon ultraviolet irradiation.8 Although its synthetic application has been largely devoted to intramolecular reactions of cyclic dialkyl ketones,9 it is also reported that alkyl aryl ketones (ArCOR) generate a pair of an alkyl radical (R˙) and an aroyl radical (ArCO˙) reversibly, which undergo hydrogen abstraction to give an aldehyde along with an alkene.10 This could be a promising approach toward intermolecular arylation reactions using ketones as aryl sources if the generated aroyl radical decarbonylates efficiently to form an aryl radical (Ar˙), which could be utilized for arylation of other substrates (Fig. 1-d). However, such arylation reactions via the Norrish type I reaction have not been reported to date due to the following problems: (1) various side-reactions are easily caused by the highly reactive excited states and radical species in intermolecular reactions.11 (2) The decarbonylation of aroyl radicals is thermodynamically unfavourable and hard to generate aryl radicals because of their instability.12 This is in sharp contrast to the case of acyl radicals (alkylCO˙), which easily provide alkyl radicals. Therefore, there are several examples of benzoylation reactions using alkyl aryl ketones via the Norrish type I reaction, but none of arylation reactions.13,14
To solve these problems aiming to realize intermolecular arylation reactions via the Norrish type I reaction, we designed a new catalytic cycle merging the Norrish type I reaction and transition metal catalysis (Fig. 1-e). We envisioned that aroyl radicals generated by the α-cleavage of photoexcited alkyl aryl ketones could be captured by transition metal complexes to form aroylmetal intermediates, which could undergo decarbonylation to form arylmetal species. The decarbonylation could be reversible, but its kinetics and thermodynamics are controllable with the suitable choice of metals and supporting ligands. Diverse reactivities of the generated arylmetal species would enable a variety of intermolecular arylation reactions. Such a cooperative system between the Norrish type I reaction and transition metal catalysis has not been reported, and it is necessary to develop the appropriate metal complex that efficiently facilitates the sequence of each step and is tolerant to the photoirradiation conditions.
Herein, we demonstrate a proof of concept for this strategy that enables an intermolecular arylation reaction using simple ketones directly as aryl sources by the combined use of photoenergy and a rhodium catalyst. Synthesis of arylboronates from pivalophenone derivatives (tert-butyl aryl ketones) and other alkyl aryl ketones was successfully achieved via cleavage of C–C σ-bonds. Mechanistic studies clarified that the Norrish type I reaction and rhodium catalysis cooperate to realize the unprecedented reaction.
Results and discussion
The feasibility of the aforementioned strategy was examined by employing pivalophenone 1a as an aryl source because the photoreactivity of pivalophenone derivatives has been studied in detail, and 1a was reported to undergo α-cleavage upon photoirradiation at 313–366 nm.10 We envisioned intermolecular borylation of an arylmetal species generated from 1a using pinacolborane (HBpin) as a borylating reagent. After an extensive screening of transition metal catalysts and reaction conditions, it was found that the combination of [RhCl(cod)]2 and 1,2-bis(diethylphosphino)ethane (depe) acts as an efficient catalyst for the desired reaction under photoirradiation at 365 nm in DME at 80 °C,15 affording phenylboronic acid pinacol ester 2a in 68% yield (Table 1, Entry 1). The reaction with bis(pinacolato)diboron (B2pin2) instead of HBpin proceeded slightly to give 2a in 14% yield (Entry 2). Depe, which is a bidentate phosphine bearing compact alkyl substituents on phosphorus atoms, was the best ligand among the tested (Entries 3–5 and Table S1†). The reaction proceeded even at 25 °C, but the yield of 2a decreased to 19% (Entry 6). It should be noted that the reaction under 1 atm CO proceeded smoothly to give 2a in a comparable yield to that under an Ar atmosphere (Entry 7). This result may support that decarbonylation is not involved in the turnover-limiting step. Furthermore, the reaction without [RhCl(cod)]2 gave a complex mixture including a trace amount of 2a (Entry 8), and no reaction occurred without photoirradiation at 365 nm (Entries 9 and 10). These results clearly demonstrate that both photoenergy and Rh catalysis are essential to promote the unprecedented borylation reaction of the C–C σ-bond of pivalophenone.|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yield of 2aa/% |
| a Determined by 1H NMR using 1,1,2,2-tetrachloroethane as an internal standard. b Isolated yield. c dppe = 1,2-bis(diphenylphosphino)ethane. | ||
| 1 | — | 68 (58)b |
| 2 | B2pin2 instead of HBpin | 14 |
| 3 | Without depe | Trace |
| 4 | dppec instead of depe | 44 |
| 5 | 10 mol% PEt3 instead of depe | 3 |
| 6 | 25 °C instead of 80 °C | 19 |
| 7 | 1 atm CO | 64 |
| 8 | Without [RhCl(cod)]2 | Trace |
| 9 | Without hν | No reaction |
| 10 | 395 nm instead of 365 nm | No reaction |
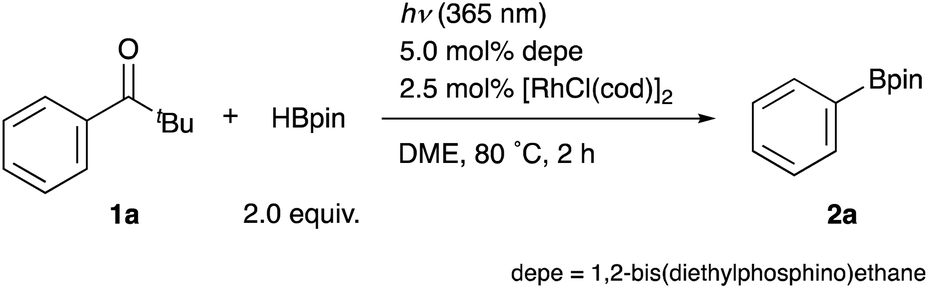
With the optimal catalyst and reaction conditions in hand, the substrate scope of this reaction was investigated (Table 2). Pivalophenone derivatives bearing electron-donating and -withdrawing substituents on the arene at the para-position were borylated smoothly to afford corresponding arylboronates 2b–d in good yields.16 Halogens such as F- and Cl- were tolerant to the reaction conditions to give 4-fluorophenyl- and 4-chlorophenylboronates 2e and 2f in good yields although a 4-Br substituent was not acceptable due to cleavage of the C–Br bond. In contrast, 4-Ph substituted pivalophenone 1g did not react at all surprisingly, providing a useful insight into the reaction mechanism (vide infra). It is noteworthy that the reaction was applicable to substrates having methyl ester, amide, nitrile, phenolic ester, and ketal moieties, giving corresponding functionalized arylboronates 2h–l in moderate to good yields without loss of these functional groups. 4-Hydroxypivalophenone was also borylated using 3 equiv. of HBpin to give 2m in high yield.17p-Trimethylsilyl and p-boryl substituents were also tolerant under the reaction conditions, affording bis(metalloid)-arenes 2n and 2o. Furthermore, meta-substituted arylboronic esters 2p–s were also synthesized in moderate to good yields from the corresponding pivalophenone derivatives. It should be noted that 1,3-bis(pivaloyl)benzene underwent double borylation at both C–C σ-bonds to give 1,3-benzenediboronate 2t. F-, Cl-, and MeO-substituents at ortho-positions were also employable without any problems to afford 2u–w in high yields, demonstrating compatibility with steric hindrance.
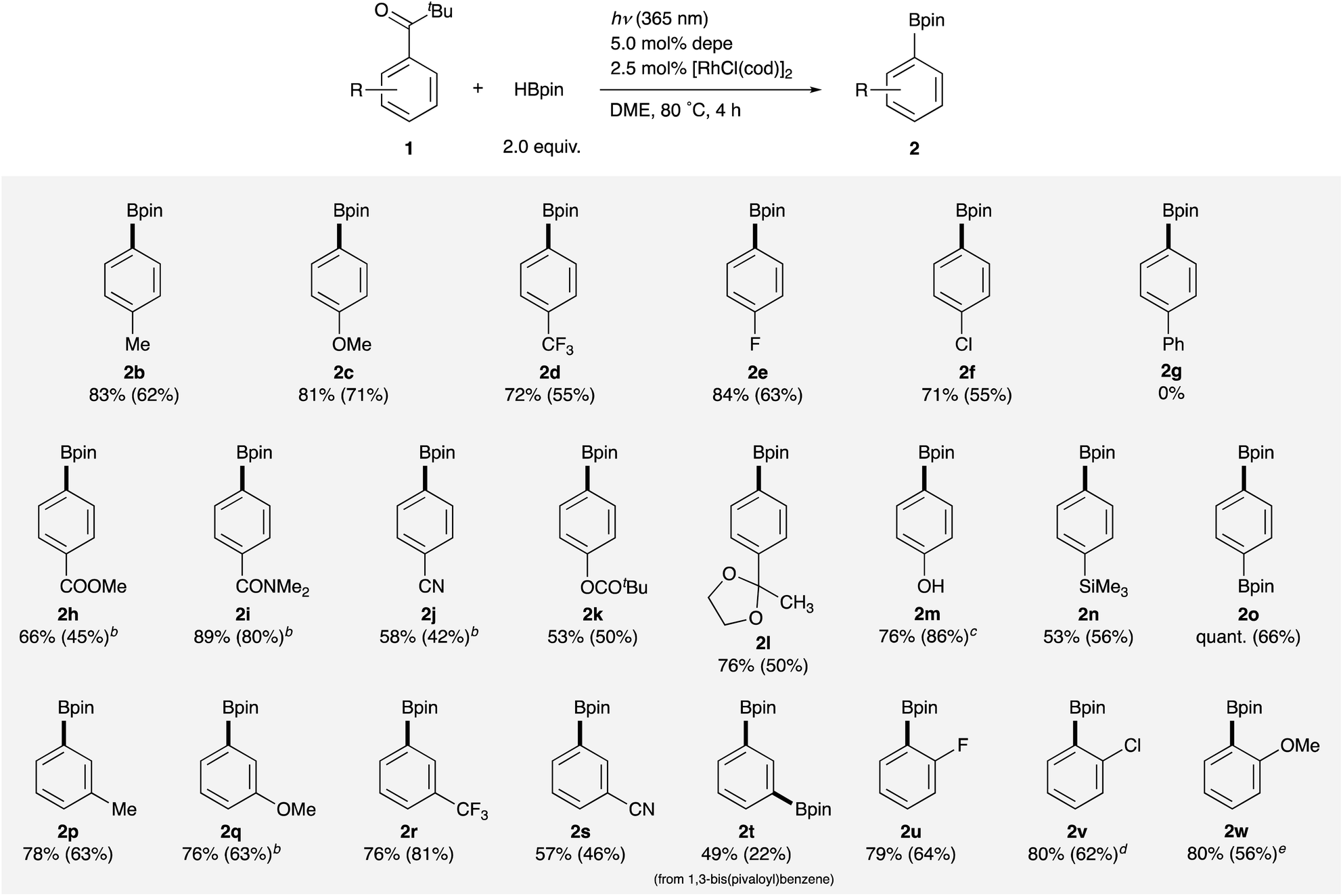
Next, the variation of alkyl groups on the carbonyl carbon instead of the tert-butyl was investigated. Although cyclohexyl phenyl ketone 3a failed to react, it was found that primary, secondary, and tertiary alkyl groups having a phenyl or an –OR group were employable to promote the C–C σ-bond borylation (Table 3). Benzyl, methylbenzyl, and dimethylbenzyl phenyl ketones 3b–d were successfully borylated to give phenylboronate 2a in moderate to good yields. The yield increased as the number of substituents at the α-position increased possibly reflecting the stability of the generated alkyl radical species. The reaction of α-hydroxyketones 3f–h also proceeded, giving the borylation product 2a in rather low yields. On the other hand, α-methoxy derivative 3e displayed better reactivity than 3f to afford 2a in moderate yield, exhibiting good applicability to various alkyl aryl ketones. These results successfully demonstrate the novelty and synthetic utility of this arylation reaction employing simple ketones as aryl sources. This reaction also provides a new method to remove and transform a pivaloyl group and other aroyl groups on arenes on demand, which can be utilized as ortho-directing groups in transition metal-catalyzed C–H activation reactions18 and as meta-directors in electrophilic aromatic substitution reactions.
| a Yields were determined by 1H NMR using 1,1,2,2-tetrachloroethane as an internal standard. Isolated yields after silica gel column chromatography were depicted in parentheses. b 100 °C, 8 h. c 3.0 equiv. HBpin. d The borylation product was isolated as PhBF3K instead of 2a by the treatment of the crude mixture with KHF2. |
|---|
|
To shed light on the reaction mechanism, several control experiments were carried out in the presence of a radical scavenger, 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) (Table 4). The reaction of 1b, HBpin, and TEMPO under the standard conditions afforded the borylation product 2b in 19% yield along with 2,2,6,6-tetramethylpiperidine-1-yl 4-methylbenzoate 4b in 52% yield, clearly supporting the generation of a 4-methylbenzoyl radical intermediate in the reaction (Entry 1). The radical-TEMPO adduct 4b was formed quantitatively in the absence of the Rh catalyst (Entry 2), and no reaction occurred without photoirradiation (Entry 3). These results clarified that only photoenergy contributes to the generation of an aroyl radical via α-cleavage of 1b, which is then captured, decarbonylated, and borylated by the Rh catalyst. In addition, it turned out that the trapping of the aroyl radical with the Rh catalyst occurs fast enough to compete with TEMPO.
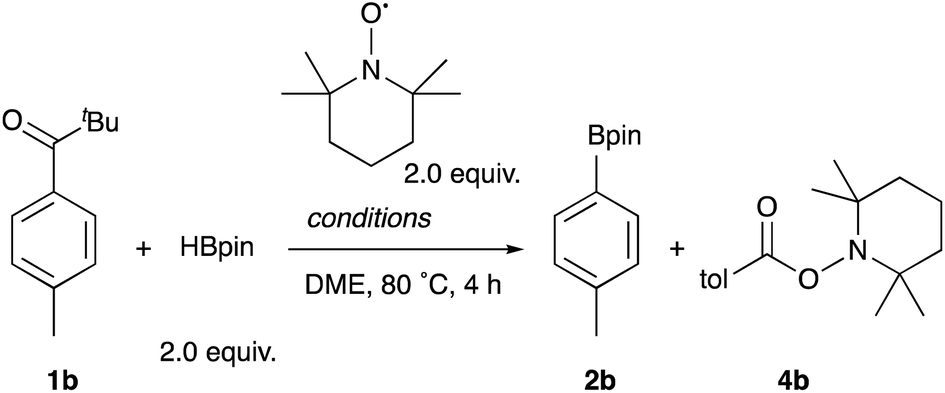
When the reaction of 1b was conducted in benzene-d6 using a sealed NMR tube, the 1H NMR spectrum of the crude mixture exhibited the formation of isobutane (57%) and 2-methylpropene (8%) as side-products (Scheme 1). These could have originated from a tert-butyl radical generated via α-cleavage of 1b along with an aroyl radical. No formation of p-tolualdehyde was observed. We also confirmed that 2b was not formed using p-tolualdehyde as a substrate instead of 1b (Scheme 2),19 thus indicating that a stepwise reaction is not plausible, in which the aroyl radical is converted to arylaldehyde via α-cleavage followed by hydrogen atom abstraction, and then the Rh catalyst cleaves the aldehydic C–H bonds.20 Furthermore, a deuterium labeling experiment using DBpin instead of HBpin revealed that the deuterium was transferred to the methine carbon of isobutane (Scheme 3).21 Therefore, it is highly indicative that the generated tert-butyl radical abstracts a hydrogen atom from HBpin to form isobutane.
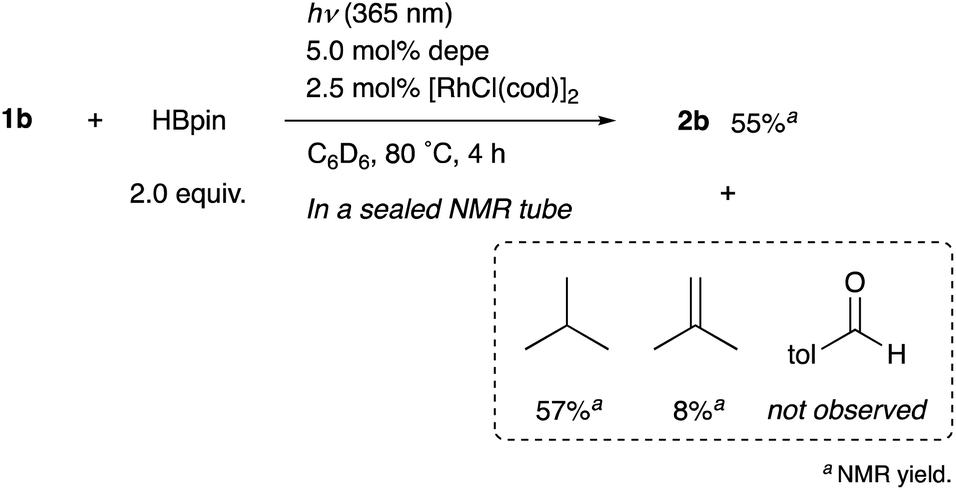 | ||
| Scheme 1 Side-products of the reaction. | ||
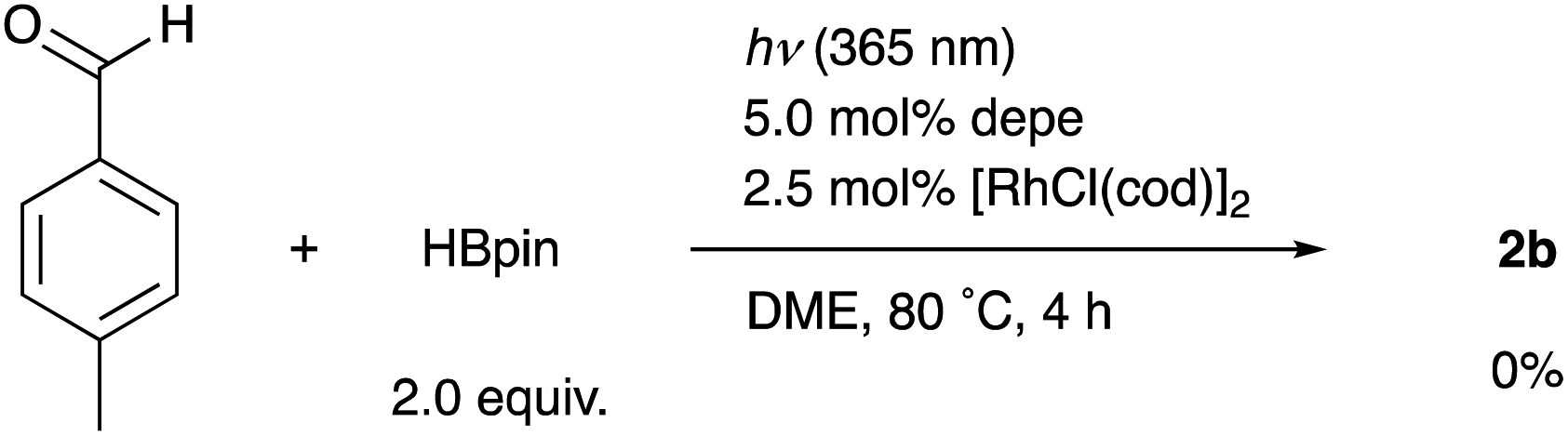 | ||
| Scheme 2 Reaction of p-tolualdehyde instead of 1b. | ||
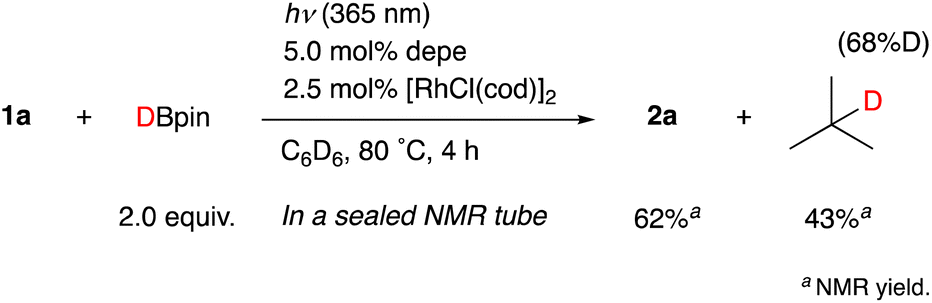 | ||
| Scheme 3 A deuterium labeling experiment using DBpin. | ||
A series of mechanistic studies prove (1) the generation of an aroyl radical upon photoirradiation, (2) no intermediacy of arylaldehyde, and (3) hydrogen atom transfer from HBpin to a tert-butyl radical, thus supporting our initial proposal of the cooperative catalytic cycle between the Norrish type I reaction and the transition metal catalysis. One of the plausible reaction mechanisms is depicted in Fig. 2 although more detailed studies are necessary for full clarification (Fig. 2). Photoirradiation at 365 nm induces α-cleavage (Norrish type I reaction) of a pivalophenone derivative to generate an aroyl radical A and a tert-butyl radical B reversibly. A rhodium complex E captures the aroyl radical A and a boryl radical generated by hydrogen atom abstraction with tert-butyl radical B, affording an aroyl(boryl)rhodium complex C. Decarbonylation of C forms an aryl(boryl)rhodium complex D, which undergoes C–B bond-forming reductive elimination to give the borylation product with regeneration of the rhodium complex E.22 This mechanism also explains the total lack of reactivity of 1g (Table 2). It is widely recognized that the Norrish type I reaction proceeds via the n-π* triplet state, not via the energetically lower π–π* triplet state, which is the case of the π-extended pivalophenone derivative such as 4-phenylpivalophenone.23
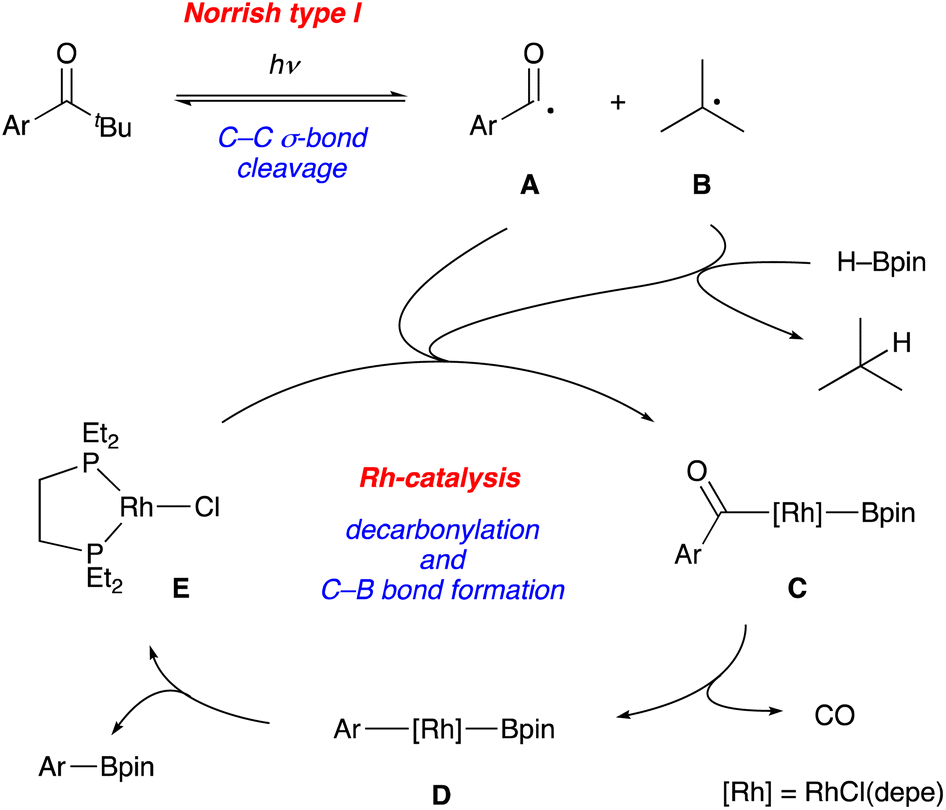 | ||
| Fig. 2 A proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed a borylation reaction of C–C σ-bonds of alkyl aryl ketones enabled by the combined use of photoenergy and Rh catalysis. Wide generality and functional group compatibility demonstrate the new synthetic utility of aryl ketones as aryl sources in intermolecular arylation reactions. This work establishes a new design of catalytic cycles, in which the Norrish type I reaction and Rh catalysis cooperate in transforming C–C σ-bonds of aryl ketones. Detailed mechanistic studies and further application of this strategy to other functionalization reactions are ongoing in our group.Data availability
Details of experimental procedures and spectral data can be found in the ESI.†Author contributions
Y. F. and J. T. conceived the idea of the research. Y. F. performed all experiments and collected experimental data with the help of J. T. and N. I. J. T. supervised the project and wrote the manuscript. All authors contributed to editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by JSPS KAKENHI Grant Numbers 20H02732 (Gran-in-Aid for Scientific Research (B)) and 22H05348 (Digi-TOS).Notes and references
- (a) R. Takise, K. Muto and J. Yamaguchi, Chem. Soc. Rev., 2017, 46, 5864–5888 RSC; (b) M. C. Willis, Chem. Rev., 2010, 110, 725–748 CrossRef CAS PubMed; (c) M. Kashihara and Y. Nakao, Acc. Chem. Res., 2021, 54, 2928–2935 CrossRef CAS PubMed; (d) N. Lalloo, C. E. Brigham and M. S. Sanford, Acc. Chem. Res., 2022, 55, 3430–3444 CrossRef CAS PubMed; (e) M. Tobisu, T. Kodama and H. Fujimoto, in Comprehensive Organometallic Chemistry IV, ed. I. A. Tonks, Elsevier, 4th edn, 2022, vol. 12, pp. 347–420 Search PubMed.
- M. Murakami, H. Amii and Y. Ito, Nature, 1994, 370, 540–541 CrossRef CAS.
- Selected reviews. (a) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610–618 RSC; (b) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410–9464 CrossRef CAS PubMed; (c) F. Song, T. Gou, B.-Q. Wang and Z.-J. Shi, Chem. Soc. Rev., 2018, 47, 7078–7115 RSC; (d) L. Deng and G. Dong, Trends Chem., 2020, 2, 183–198 CrossRef CAS; (e) H. Lu, T.-Y. Yu, P.-F. Xu and H. Wei, Chem. Rev., 2021, 121, 365–411 CrossRef CAS PubMed.
- Selected examples of arylation reactions using aryl ketones having directing groups as aryl sources. (a) A. M. Dreis and C. J. Douglas, J. Am. Chem. Soc., 2009, 131, 412–413 CrossRef CAS PubMed; (b) T.-T. Zhao, W.-H. Xu, Z.-J. Zheng, P.-F. Xu and H. Wei, J. Am. Chem. Soc., 2018, 140, 586–589 CrossRef CAS PubMed; (c) Z.-Q. Lei, F. Pan, H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang, Y.-X. Li, J. Sun and Z.-J. Shi, J. Am. Chem. Soc., 2015, 137, 5012–5020 CrossRef CAS PubMed; (d) C. Jiang, Z.-J. Zheng, T.-Y. Yu and H. Wei, Org. Biomol. Chem., 2018, 16, 7174–7177 RSC; (e) C. Jiang, W. Wu, H. Lu, T. Yu, W. Xu and H. Wei, Asian J. Org. Chem., 2019, 8, 1358–1362 CrossRef CAS.
- (a) O. Daugulis and M. Brookhart, Organometallics, 2004, 23, 527–534 CrossRef CAS; (b) A. Dermenci, R. E. Whittaker, Y. Gao, F. A. Cruz, Z.-X. Yu and G. Dong, Chem. Sci., 2015, 6, 3201–3210 RSC; (c) R. E. Whittaker and G. Dong, Org. Lett., 2015, 17, 5504–5507 CrossRef CAS PubMed; (d) T. Morioka, A. Nishizawa, T. Furukawa, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2017, 139, 1416–1419 CrossRef CAS PubMed; (e) S. Nakatani, Y. Ito, S. Sakurai, T. Kodama and M. Tobisu, J. Org. Chem., 2020, 85, 7588–7594 CrossRef CAS PubMed.
- (a) Y. Xia, S. Ochi and G. Dong, J. Am. Chem. Soc., 2019, 141, 13038–13042 CrossRef CAS PubMed; (b) R. Zhang, Y. Xia and G. Dong, Angew. Chem., Int. Ed., 2021, 60, 20476–20482 CrossRef CAS PubMed; (c) R. Zeng and G. Dong, J. Am. Chem. Soc., 2015, 137, 1408–1411 CrossRef CAS PubMed; (d) R. Zeng, P. Chen and G. Dong, ACS Catal., 2016, 6, 969–973 CrossRef CAS PubMed.
- (a) H. Li, B. Ma, Q. Liu, M. Wang, Z. Wang, H. Xu, L. Li, X. Wang and H. Dai, Angew. Chem., Int. Ed., 2020, 59, 14388–14393 CrossRef CAS PubMed; (b) M.-L. Wang, H. Xu, H.-Y. Li, B. Ma, Z.-Y. Wang, X. Wang and H.-X. Dai, Org. Lett., 2021, 23, 2147–2152 CrossRef CAS PubMed; (c) H. Xu, B. Ma, Z. Fu, H.-Y. Li, X. Wang, Z.-Y. Wang, L.-J. Li, T.-J. Cheng, M. Zheng and H.-X. Dai, ACS Catal., 2021, 11, 1758–1764 CrossRef CAS; (d) Z.-Q. Guo, H. Xu, X. Wang, Z.-Y. Wang, B. Ma and H.-X. Dai, Chem. Commun., 2021, 57, 9716–9719 RSC; (e) Z.-Y. Wang, B. Ma, H. Xu, X. Wang, X. Zhang and H.-X. Dai, Org. Lett., 2021, 23, 8291–8295 CrossRef CAS PubMed; (f) H. Li, M.-L. Wang, Y.-W. Liu, L.-J. Li, H. Xu and H.-X. Dai, ACS Catal., 2022, 12, 82–88 CrossRef CAS; (g) X. Wang, L.-J. Li, Z.-Y. Wang, H. Xu and H.-X. Dai, iScience, 2022, 25, 104505 CrossRef CAS PubMed.
- R. G. W. Norrish and C. H. Bamford, Nature, 1936, 138, 1016 CrossRef CAS.
- (a) P. Sivaguru, Z. Wang, G. Zanoni and X. Bi, Chem. Soc. Rev., 2019, 48, 2615–2656 RSC; (b) S. Majhi, Photochem. Photobiol. Sci., 2021, 20, 1357–1378 CrossRef CAS PubMed.
- (a) F. D. Lewis and J. G. Magyar, J. Org. Chem., 1972, 37, 2102–2107 CrossRef CAS; (b) M. V. Encina, E. A. Lissi, E. Lemp, A. Zanocco and J. C. Scaiano, J. Am. Chem. Soc., 1983, 105, 1856–1860 CrossRef CAS; (c) T. Kang and J. R. Scheffer, Org. Lett., 2001, 3, 3361–3364 CrossRef CAS PubMed.
- (a) S. Singh, G. Usha, C. H. Tung, N. J. Turro and V. Ramamurthy, J. Org. Chem., 1986, 51, 941–944 CrossRef CAS; (b) V. Ramamurthy, D. R. Corbin and D. F. Eaton, J. Org. Chem., 1990, 55, 5269–5278 CrossRef CAS.
- R. K. Solly and S. W. Benson, J. Am. Chem. Soc., 1971, 93, 2127–2131 CrossRef CAS.
- Aroylation reactions via the Norrish type I reaction are reported. See: (a) G. Goti, B. Bieszczad, A. Vega-Peñaloza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 1213–1217 CrossRef CAS PubMed; (b) B. Bieszczad, L. A. Perego and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 16878–16883 CrossRef CAS PubMed; (c) Y. Tu, D. Zhang, P. Shi, C. Wang, D. Ma and C. Bolm, Org. Biomol. Chem., 2021, 19, 8096–8101 RSC; (d) S. Hirashima, T. Nobuta, N. Tada and A. Itoh, Synlett, 2009, 2009, 2017–2019 CrossRef.
- Preparation of metal nanoparticles utilizing alkyl radical species generated by α-cleavage of benzoin compounds via the Norrish type I reaction is reported. See: J. C. Scaiano, K. G. Stamplecoskie and G. L. Hallett-Tapley, Chem. Commun., 2012, 48, 4798–4808 RSC.
- The photoirradiation was carried out using LEDs (365 nm) at 80 °C in a thermostatic water bath.
- Isolated yields of arylboronates generally decreased due to significant loss during silica gel column chromatography. N. Oka, T. Yamada, H. Sajiki, A. Akai and T. Ikawa, Org. Lett., 2022, 24, 3510–3514 CrossRef CAS PubMed.
- The –OH moiety was spontaneously borylated when adding HBpin before the photoirradiation, and the resulting –OBpin was hydrolyzed after the reaction by the standard work-up to give 3m.
- (a) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS; (b) W. Xu and N. Yoshikai, Chem. Sci., 2017, 8, 5299–5304 RSC; (c) N. Kimura, T. Kochi and F. Kakiuchi, J. Am. Chem. Soc., 2017, 139, 14849–14852 CrossRef CAS PubMed.
- A hydroboration product of p-tolualdehyde was formed in 71% yield.
- Bisphosphine–rhodium complexes often act as good catalysts for decarbonylation reactions of aldehydes. (a) D. H. Doughty and L. H. Pignolet, J. Am. Chem. Soc., 1978, 100, 7083–7085 CrossRef CAS; (b) M. Kreis, A. Palmelund, L. Bunch and R. Madsen, Adv. Synth. Catal., 2006, 348, 2148–2154 CrossRef CAS; (c) T. Morimoto, M. Fujioka, K. Fuji, K. Tsutsumi and K. Kakiuchi, Pure Appl. Chem., 2008, 80, 1079–1087 CrossRef CAS.
- The reaction in C6H6 also afforded the isobutane with the same deuterium incorporation (68%D), thus supporting that the deuterium is derived from DBpin.
- A β-carbon elimination pathway via a rhodium alkoxide intermediate is not plausible considering the no catalytic activity of the Rh catalyst for hydroboration of 1a without 365 nm irradiation (Table 1, Entries 9 and 10).
- It was reported that 4-phenylpivalophenone did not undergo α-cleavage at all under photoirradiation at 313 nm. See ref. 10a.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures and spectral data. See DOI: https://doi.org/10.1039/d2sc06801j |
| This journal is © The Royal Society of Chemistry 2023 |