
Net anionic poly(β-amino ester)s: synthesis, pH-dependent behavior, and complexation with cationic cargo†
Mara K.
Kuenen
 ,
Alexa M.
Cuomo
,
Vincent P.
Gray
and
Rachel A.
Letteri
*
,
Alexa M.
Cuomo
,
Vincent P.
Gray
and
Rachel A.
Letteri
*
Department of Chemical Engineering, University of Virginia, Charlottesville, VA 22903, USA. E-mail: rl2qm@virginia.edu
First published on 21st December 2022
Abstract
As hydrolytically-labile, traditionally-cationic polymers, poly(β-amino ester)s (PBAEs) adeptly complex anionic compounds such as nucleic acids, and release their cargo as the polymer degrades. To engineer fully-degradable polyelectrolyte complexes and delivery vehicles for cationic therapeutics, we sought to invert PBAE net charge to generate net anionic PBAEs. Since PBAEs can carry up to a net charge of +1 per tertiary amine, we synthesized a series of alkyne-functionalized PBAEs that allowed installation of 2 anionic thiol-containing molecules per tertiary amine via a radical thiol–yne reaction. Finding dialysis in aqueous solution to lead to PBAE degradation, we developed a preparative size exclusion chromatography method to remove unreacted thiol from the net anionic PBAEs without triggering hydrolysis. The net anionic PBAEs display non-monotonic solution behavior as a function of pH, being more soluble at pH 4 and 10 than in intermediate pH ranges. Like cationic PBAEs, these net anionic PBAEs degrade in aqueous environments with hydrophobic content-dependent hydrolysis, as determined by 1H NMR spectroscopy. Further, these net anionic PBAEs form complexes with the cationic peptide (glycine-arginine)10, which disintegrate over time as the polymer hydrolyzes. Together, these studies outline a synthesis and purification route to make previously inaccessible net anionic PBAEs with tunable solution and degradation behavior, allowing for user-determined complexation and release rates and providing opportunities for degradable polyelectrolyte complexes and cationic therapeutic delivery.
Introduction
As it becomes critical to engineer polymers with controlled lifetimes to address challenges in plastic waste pollution, drug delivery, templating porous materials, and more, poly(β-amino ester)s (PBAEs) are an attractive polymer class due to their facile synthesis, rapid degradation timescales, and compositional diversity. PBAEs feature a protonatable amine in each repeating unit, imparting pH-responsive cationic character and allowing them to capably complex anionic species, such as DNA.1–8 Indeed, PBAEs have been used extensively for gene delivery2,5–7,9–11 and as degradable components in polyelectrolyte multilayer assemblies.2,4,12,13Formed simply from amines and acrylates, PBAEs can incorporate a variety of functional groups, as evidenced by several published combinatorial libraries with hundreds of monomer permutations.2,6,14–16 Notably absent from these libraries, however, are monomers containing multiple anionic groups, such as carboxylic acids or sulfonates, which reduce organic solubility (limiting synthesis and processing conditions) and/or are not commercially available. This inability to incorporate net anionic charge prevents PBAEs from carrying cationic therapeutics (e.g., antimicrobial peptides which have a short half-life in the bloodstream, limiting clinical implementation17–23) and complexing cationic polymers.4,24,25
As PBAEs contain one or more protonatable amine in each repeating unit, accessing net anionic PBAEs requires the addition of two anionic groups per tertiary amine.1 We reasoned that generating alkyne-functionalized PBAEs and using thiol–yne chemistry to add two anionic thiols to each alkyne would provide a suitable synthetic route to net anionic PBAEs.26–29 Moreover, alkyne-functionalized amines (e.g., propargylamine which has previously been used as a PBAE monomer30) and anionic thiol-containing molecules (e.g., sodium 3-mercapto-1-propanesulfonate, MPS) are commercially available, contributing to the synthetic accessibility of this approach. As these net anionic PBAEs have pH-dependent opposite charges, we investigate solution behavior as a function of pH. To demonstrate the potential of these materials to act as degradable components of polyelectrolyte complexes and drug delivery vehicles, we complex them with the cationic peptide (GR)10. Together, these studies provide a template for generating net anionic PBAEs as well as PBAEs with other, previously inaccessible, functionalities.
Experimental section
Materials
Poly(ethylene glycol) diacrylate (PEGDA, Mn = 250 g mol−1, Đ = 1.46 with 100 ppm monomethyl ether hydroquinone [MEHQ] as a radical inhibitor), 1,6-hexanediol diacrylate (HDDA, with 100 ppm MEHQ, ≥80%), propargylamine (≥98%), dimethylsulfoxide-d6 (DMSO-d6, 99.9 atom% D), potassium dihydrogen phosphate (≥99%), dipotassium hydrogen phosphate (≥98%), 37 wt% ACS reagent grade hydrochloric acid (HCl), sodium acetate (≥99%), sodium carbonate (≥99.5%), sodium bicarbonate (≥99.7%), 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (photoinitiator, 98%), 5,5′-dithiobis(2-nitrobenzoic acid) (Ellman's reagent, ≥98%), deuterium oxide (D2O, 99.9 atom% D, contains 1 wt% 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt), dimethylformamide (DMF, ≥99.8%), diethyl ether (≥99.0%, contains butylated hydroxytoluene as inhibitor), triisopropylsilane (98%), piperidine (≥99%), 2,2′-(ethylenedioxy)diethanethiol (95%), diisopropyl carbodiimde (99%), deuterium chloride (DCl, 35 wt% in D2O, ≥99 atom% D), sodium deuteroxide (NaOD, 40 wt% in D2O, 99 atom% D), and trifluoroacetic acid (TFA, 99%) were purchased from Sigma Aldrich. Sodium hydroxide (NaOH, ≥98%), glacial acetic acid (≥99.7%), sodium chloride (NaCl, ≥99.0%), sodium 3-mercapto-1-propanesulfonate (MPS, >85%), and dimethylsulfoxide (DMSO, ≥99.9%) were purchased from VWR. Formic acid (LC/MS grade, ≥99.0%) and methanol (LC/MS grade, ≥99.9%) were purchased from Fisher Scientific. Water (LC/MS grade) for mass spectrometry was purchased from Alfa Aesar. Rink resin SS (0.5 mmol g−1 loading, 100–200 mesh, 1% divinylbenzene), Fmoc-Arg(Pbf)-OH, Fmoc-Gly-OH, and Oxyma Pure were purchased from Advanced Chemtech. All reagents were used as received. Water was obtained from an in-house reverse osmosis (RO) system. Bio-Gel® P-2 media (fine, 45–90 μm [wet]) was purchased from Bio-Rad Laboratories.Characterization
1H nuclear magnetic resonance (NMR) spectroscopy was conducted on a 500 MHz Varian NMR spectrometer or an 800 MHz Bruker Avance III NMR spectrometer equipped with a 5 mm HCN cryoprobe in DMSO-d6 or D2O. Chemical shifts were referenced to the solvent residual peak (2.50 or 4.79 ppm for DMSO or D2O, respectively).13C NMR spectroscopy was conducted on an 800 MHz Bruker Avance III NMR spectrometer equipped with a 5 mm HCN cryoprobe in DMSO-d6. Chemical shifts were referenced to the residual DMSO peak at 39.52 ppm.
Fourier-transform infrared (FTIR) spectroscopy was conducted on a PerkinElmer Frontier spectrometer using 16 scans and 1 cm−1 resolution.
Size exclusion chromatography (SEC) was performed using a TOSOH EcoSec system eluting in TFE with 0.02 M sodium trifluoroacetate (NaTFAc) at 0.3 mL min−1. The system was equipped with a refractive index detector, a TSKgel SuperAWM-H (4.6 mm × 3.5 cm, 9 μm diameter beads) mixed-bed guard column, and two TSKgel SuperAWM-H (6 mm × 15 cm, 9 μm diameter beads) mixed-bed columns. Number average molecular weight (Mn) and dispersity (Đ) were determined relative to poly(methyl methacrylate) (PMMA) standards. As we previously found TFE to degrade PBAE esters,8 we ran samples immediately after dissolution. Though the polymer likely degrades somewhat in TFE, other solvents like THF do not dissolve the net anionic PBAEs. Therefore, we kept the time between dissolution and sample injection minimal (<5 min) to reduce artificial differences in molecular weight measurements between samples.
Solution pH was measured with a Mettler Toledo Benchtop pH meter calibrated with buffered standards (pH = 1.68, 4.01, 7.00, and 10.01).
Alkyne PBAE synthesis
Alkyne-functionalized PBAEs were synthesized by solvent-free Michael Addition polymerization of PEGDA, HDDA, and propargylamine according to a procedure adapted from Safranski et al.31 To vary hydrophobicity, polymerizations were conducted with 0 to 100 mol% hydrophobic HDDA per diacrylate monomer. For example, to synthesize a PBAE containing 75 mol% HDDA-containing repeat units, PEGDA (0.89 g, 3.4 mmol), HDDA (2.32 g, 10 mmol), and propargylamine (0.92 mL, 14 mmol) were added to a 20 mL scintillation vial and stirred at 90 °C for 24 h. To prevent crosslinking or branching during the subsequent radical reaction, an amine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :diacrylate molar ratio of 1.05:1 was selected to promote acrylate consumption and favor amine end-groups. However, 1H NMR spectroscopy after the polymerization revealed peaks at 5.77–5.85, 6.06–6.18, and 6.34–6.44 ppm, characteristic of acrylates (Fig. S1B†). To consume the remaining acrylates, we end-capped the polymers with propargylamine (in the case of 75% HDDA, 0.46 mL, 7.2 mmol) at room temperature for 1 h, as done by Green and coworkers.10 Disappearance of the acrylate peaks in the 1H NMR spectra confirmed acrylate consumption following the end-capping reaction (Fig. S1C†). Of note, we found both heating and/or longer end-capping reaction times led to later SEC retention times, indicative of chain scission, likely mediated by the excess nucleophilic amine (ESI section S1†). To minimize ester hydrolysis, the polymers were stored at 4 °C if not used immediately (Fig. S3†).
:diacrylate molar ratio of 1.05:1 was selected to promote acrylate consumption and favor amine end-groups. However, 1H NMR spectroscopy after the polymerization revealed peaks at 5.77–5.85, 6.06–6.18, and 6.34–6.44 ppm, characteristic of acrylates (Fig. S1B†). To consume the remaining acrylates, we end-capped the polymers with propargylamine (in the case of 75% HDDA, 0.46 mL, 7.2 mmol) at room temperature for 1 h, as done by Green and coworkers.10 Disappearance of the acrylate peaks in the 1H NMR spectra confirmed acrylate consumption following the end-capping reaction (Fig. S1C†). Of note, we found both heating and/or longer end-capping reaction times led to later SEC retention times, indicative of chain scission, likely mediated by the excess nucleophilic amine (ESI section S1†). To minimize ester hydrolysis, the polymers were stored at 4 °C if not used immediately (Fig. S3†).
Synthesis of net anionic PBAEs using thiol–yne click chemistry
Amine-terminated alkyne-functionalized PBAEs (ca. 1 g, 5 mmol alkynes, 1 mol eq.), MPS (4 mol eq.), and photoinitiator (0.7 mol eq.) were dissolved in DMSO (200 mg reactants per mL solvent) in a 50 mL round bottom flask. The mixture was deoxygenated with N2(g) for 30 min prior to exposure to 365 nm UV light (Analytik Jena UVP Crosslinker, CL-3000L) for 1 h, with stirring via a battery-operated stir plate (Bel-Art H37017-0000). To encourage alkyne conversion, we dosed the mixture with additional photoinitiator (0.35 molar equivalents relative to alkynes) under N2(g) prior to exposure to 365 nm UV light for another 1 h with stirring.32 This dosing procedure was repeated one additional time for a total photoinitiator loading of 1.4 equivalents per alkyne and 3 h of UV exposure in the thiol–yne functionalization (see ESI section S2† for more information).To aid in polymer 1H NMR spectra peak assignments, we performed a model thiol–yne reaction under the same conditions as the polymer functionalization with the small molecules propargylamine and MPS (ESI section S2†).
Purification
Unreacted MPS was removed from the functionalized polymer following UV exposure using preparative SEC according to the following procedure. Samples were passed through a 4′′ × 1′′ P-2 Bio-gel size exclusion resin bed packed into a column with a coarse frit (see ESI section S4† for column packing details). Prior to loading onto the column, DMSO was removed from the reaction mixture (the resin is not compatible with organic solvents) by precipitation into cold acetone (5 mL reaction mixture per ca. 40 mL acetone) and centrifugation for 5 min at 4816g. After decanting the solution, the pellets were dried overnight in the fume hood. Dry pellets from two centrifuge tubes were combined and dissolved in preparative SEC running solvent (5 mL, 20 mM NaCl adjusted to pH 4 or pH 9 with HCl or NaOH, respectively, vacuum degassed for ca. 30 min with house vacuum line based on manufacturer's instructions). After the pellets were fully dissolved, the pH was readjusted to 4 or 9 and the mixture briefly degassed (ca. 5 min) under 400 mmHg vacuum. A serological pipette was then used to transfer the solution dropwise to the column bed before adding 100 mL running solvent to gravimetrically elute the polymer. Sample collection was started shortly before the colored polymer reached the bottom of the column (after ca. 20 mL had eluted from the column) and was stopped before 30 mL had eluted (the elution volume of MPS, see ESI section S4† for elution volume determination details). The collected polymer-containing fraction (ca. 10 mL) was immediately frozen in liquid nitrogen and lyophilized.Ellman's reagent assay
Unreacted thiol remaining after SEC purification was quantified with an Ellman's reagent assay according to the following procedure, adapted from the manufacturer's instructions. A sample of lyophilized, purified, polymer was dissolved in 100 mM pH 8 potassium phosphate buffer at 1 mg mL−1. This solution (180 μL) was added to each of 6 wells of a 96 well plate (Costar clear flat bottom black polystyrene 96 well plates). Ellman's reagent solution (20 μL, 4 mg mL−1 in 100 mM pH 8 potassium phosphate buffer) was added to each of 3 of these wells while buffer (20 μL, 100 mM pH 8) was added to each of the remaining 3 wells as a blank. Absorbance at 405 nm was read using a Biotek Synergy 4 plate reader. The triplicate measurements were averaged and the difference in absorbance between the Ellman's reagent-containing and buffer samples compared to a calibration curve of known concentrations of MPS with Ellman's reagent (Fig. S5†) to calculate the mass fraction of thiol remaining in the purified polymer samples. Samples containing less than 4 wt% unreacted thiol were used for further experiments.Absorbance measurements
Solution properties of the anionic PBAEs were evaluated as a function of pH and time by measuring absorbance at 550 nm on a Biotek Synergy 4 plate reader in different buffers (1 mg mL−1 polymer in 50 mM pH 4, 6, 7, 8, or 10 buffer) in a 96 well plate (Costar clear flat bottom black polystyrene 96 well plates). Solutions (200 μL) were added to each of 3 wells. Absorbance was then measured at 550 nm at specified time points (1 h apart) and reported as an average of the triplicate measurements.pH titrations
To study the pH-responsive behavior after functionalization with sulfonates and as a function of hydrophobicity, polymers were titrated with HCl (0.5 M) using a Mettler Toledo EasyPlus Titrator Easy pH instrument. Before titrating, samples (ca. 20 mg) were dissolved in RO water (15 mL) with NaOH (5 mL, 0.5 M). Samples were automatically titrated using the Easy pH interface with control set to “normal” and samples were stirred continuously during titration. See ESI section S15† for additional experimental and analysis details.NMR degradation studies
Net anionic PBAE degradation was monitored over 100 h in basic aqueous conditions using 1H NMR (800 MHz) spectroscopy according to the following procedure. Samples (20 mg mL−1) degraded in pD 10 carbonate buffer (500 mM) in D2O, prepared by combining 2.3 mmol sodium bicarbonate and 2.7 mmol sodium carbonate with 10 mL of D2O. Solution pD was estimated from the measured pH by:33,34| pD ≈ measured pH + 0.4. |
The estimated pD was adjusted to 10, as needed, immediately after samples were dissolved. The apparent solution pH of the most hydrophilic sample was measured with a pH meter (pH electrode InLab NMR, Mettler Toledo) prior to each time point; we found negligible change in apparent pH occurred over the timescale of the degradation experiments (ca. 100 h). 1H NMR spectra were then acquired at specified time points on an 800 MHz Bruker Avance III NMR spectrometer equipped with a 5 mm HCN cryoprobe. Chemical shifts were referenced to the residual HOD peak at 4.79 ppm. The disappearance of protons near the hydrolysable esters was tracked as done similarly by Rydholm, Anseth, and Bowman.35 Ester hydrolysis was monitored by the decrease in relative integration of the peak corresponding to protons alpha to the ester carbonyl (4.44–3.98 ppm). We tracked the percentage of esters remaining intact at a given time by normalizing peak integration to the initial peak integration by
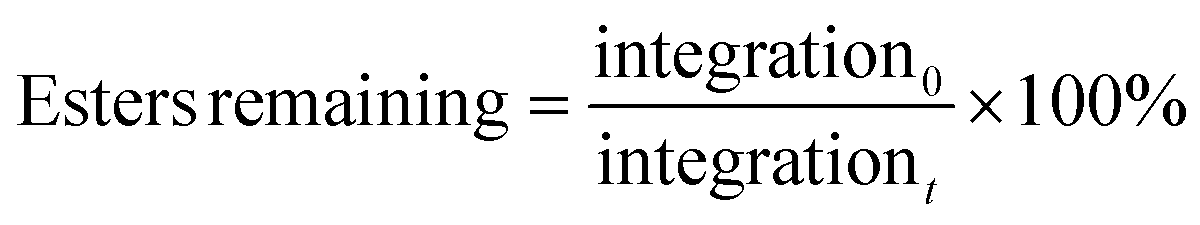
Cationic peptide synthesis
To demonstrate the ability of net anionic PBAEs to complex cationic cargo, we prepared an alternating glycine-arginine peptide, (GR)10. The peptide was synthesized via standard solid phase Fmoc methods on a Rink amide resin (0.5 mmol g−1 loading) using a CEM Liberty Blue microwave-assisted peptide synthesizer. The peptide was deprotected and cleaved from the resin using a solution of 92.5% trifluoroacetic acid, 2.5% 2,2′-(ethylenedioxy) diethanethiol, 2.5% triisopropylsilane, and 2.5% RO water (v/v). The peptide was precipitated in cold diethyl ether, dried under vacuum, and lyophilized, then stored at −20 °C until ready for use. 1H NMR spectroscopy (800 MHz, D2O): δ = 4.48–4.20 (m, 10H, Arg-α-H), 3.83–4.03 (m, 20H, Gly-α-H), 3.16–3.25 (m, 20H, Arg-δ-H), 1.75–1.95 (m, 20H, Arg-β-H), 1.59–1.74 (m, 20H, Arg-γ-H). 1H NMR spectra were referenced to the D2O residual at 4.79 ppm. Calculated m/z for (GR)10 [M + H]+: 2149.2, observed: 2149.3. See ESI section S12 and Fig. S17–S19† for additional experimental details and 1H NMR, mass, and circular dichroism spectra of (GR)10.Complexation with cationic peptide
Solutions of polymer and peptide (both 1 mg mL−1) were prepared separately in 50 mM pH 10 carbonate buffer. Solutions (200 μL) of polymer, peptide, or their mixtures (1:1 by volume, corresponding to a 1:1 charge ratio) were added to each of 3 wells in a 96 well plate and absorbance was measured and reported in the same manner as for the solution properties studies described above.
Additional experimental details, including buffer preparation, peptide synthesis details, and spectroscopic characterization, are provided in the ESI.†
Results and discussion
Net anionic PBAE synthesis
To generate net anionic PBAEs via thiol–yne chemistry, we first synthesized a suite of alkyne-functionalized PBAEs by polymerizing diacrylate monomers with propargylamine which were later reacted with an anionic thiol-containing molecule via radical thiol–yne chemistry (Scheme 1). As a tuning parameter for solution and degradation behavior, we generated these PBAEs with varied hydrophobicity by adjusting the ratio of hydrophilic PEGDA and hydrophobic HDDA. Since thiol–yne chemistry proceeds by a radical mechanism, the precursor alkyne PBAEs need amine end-groups (as opposed to acrylates) to avoid crosslinking or branching during the thiol–yne reaction. The 13C NMR spectra of these polymers show peaks at 74.9 and 79.4 ppm (Fig. 1a and Fig. S11–S13†), characteristic of alkynes, while number-average molecular weight (Mn) measured via SEC eluting in TFE relative to PMMA standards are similar across the suite of polymers (Table 1 and Fig. S15†). We attribute the higher dispersity of the alkyne PBAE with 75% HDDA repeating units to aggregation in SEC solvent (ESI section S11†). With the alkyne-functionalized PBAEs in hand, we proceeded to our thiol–yne step with an anionic sulfonate-thiol, MPS.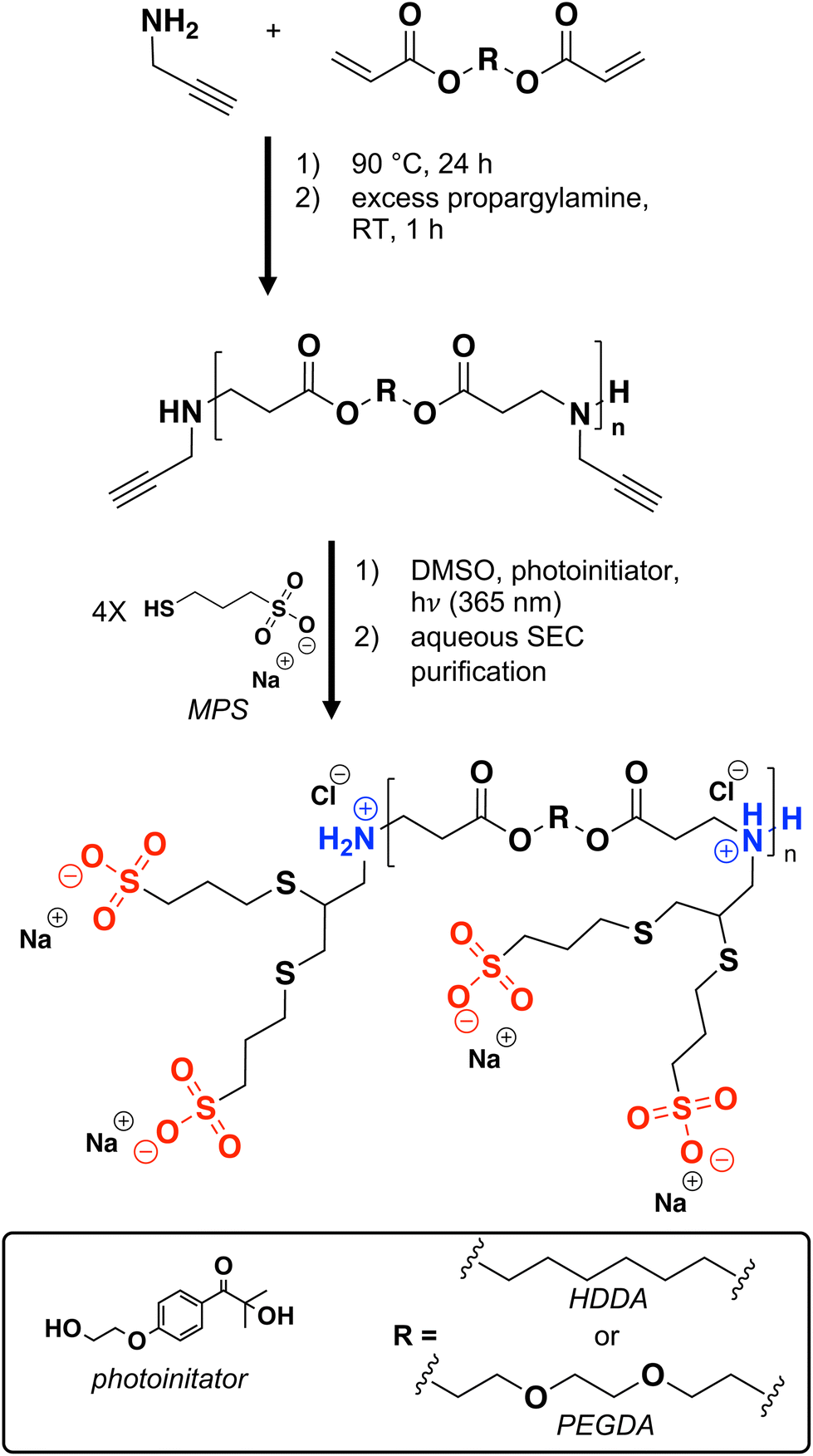 | ||
| Scheme 1 Net anionic PBAE synthesis. | ||
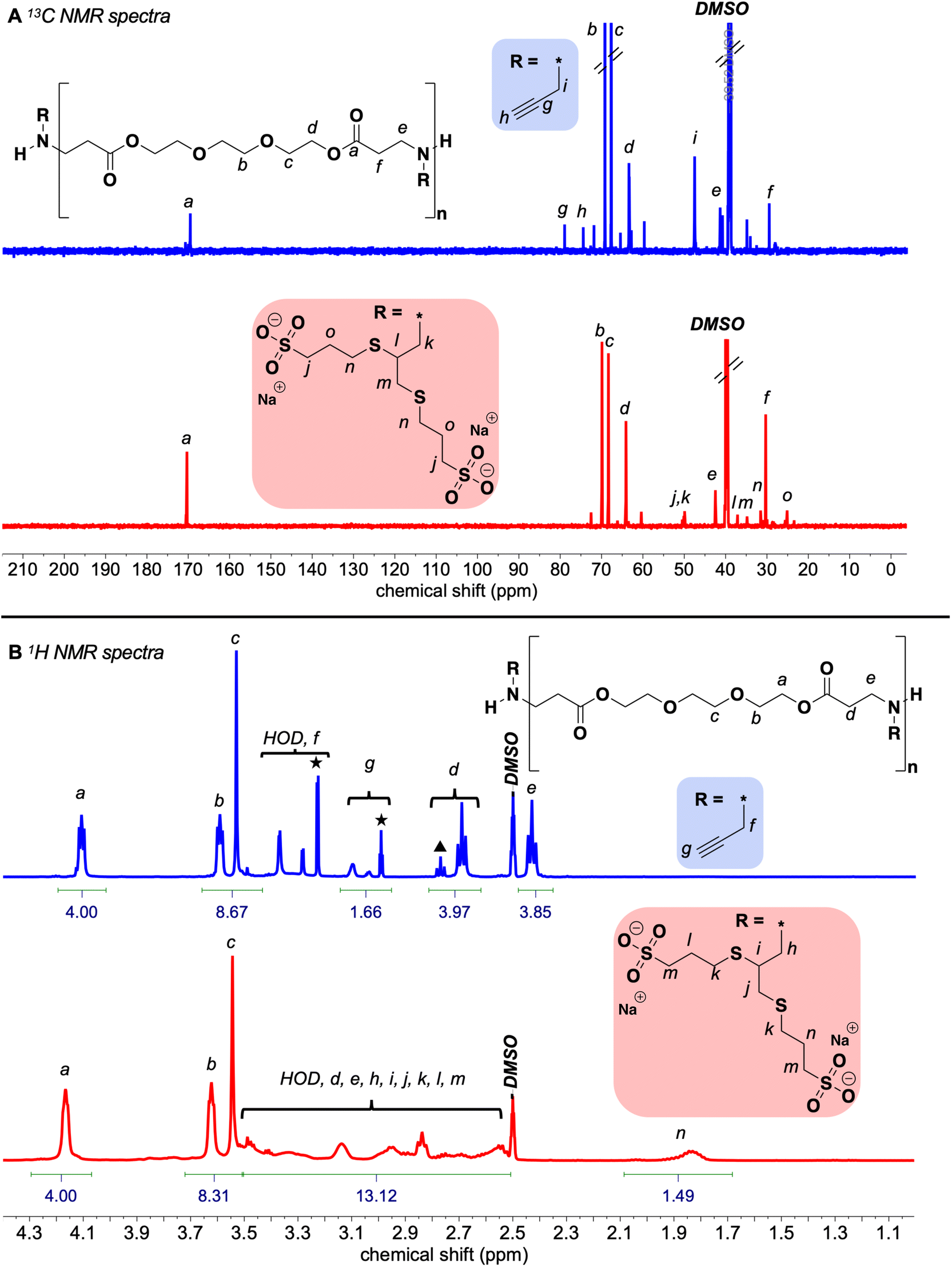 | ||
| Fig. 1 NMR spectroscopy characterization of alkyne-functionalized and net anionic PBAEs: (A) 13C NMR (200 MHz) spectra of (top) the alkyne functionalized PBAE (0% HDDA) and (bottom) the corresponding net anionic PBAE; and (B) 1H NMR (500 MHz) spectra of (top) the crude alkyne functionalized PBAE (0% HDDA) end-capped with excess amine (unreacted amine peaks are marked with a star; we attribute the d peak labeled with a triangle to d protons near the chain ends) and (bottom) the corresponding net anionic PBAE. Disappearance of alkyne resonances and appearance of resonances from MPS indicate successful modification of the alkyne PBAEs with MPS. | ||
| Target HDDA-containing repeat units [mol%] | Alkyne-functionalized Mna [g mol−1] |
Alkyne-functionalized Đa | Purified anionic PBAE Mna [g mol−1] |
Purified anionic Đa | HDDA-containing repeat units in purified anionic PBAEb [mol%] |
|---|---|---|---|---|---|
| a Estimated by SEC in TFE with 0.02 M NaTFAc relative to PMMA standards. b Determined by 1H NMR spectroscopy from the relative integrations of the peaks at 3.9–4.4 ppm in the PEGDA-containing repeating units and at 1.2–1.7 ppm in the HDDA-containing repeating units. c The higher dispersity is attributed to aggregation in SEC eluent as the high molecular weight shoulder is absent when run in tetrahydrofuran, discussed further in ESI section S11.† | |||||
| 0 | 5600 | 3.1 | 5300 | 2.7 | 0 |
| 50 | 4400 | 2.7 | 4300 | 2.3 | 48 |
| 75 | 4400 | 6.0c | 3600 | 2.1 | 71 |
| 100 | 3700 | 3.3 | 4500 | 4.8 | 100 |
To install anionic groups on our polymers, we used thiol–yne chemistry to attach two anionic thiols (MPS) to the alkyne in each repeating unit. While step-growth polymerization of PBAEs occurs without solvent, we added solvent in the post-polymerization thiol–yne step to dissolve the sulfonate thiol MPS and, as previously noted,26 to improve conversion by increasing polymer mobility. Our choice of solvent was severely limited by MPS solubility, as MPS is usually only soluble in water or water-containing solvent mixtures, which we avoided to prevent PBAE hydrolysis. We therefore narrowed down our solvent selection to DMSO, in which MPS is soluble at 0.1 g mL−1 at room temperature, though we note that dissolution often took >1 h with stirring/vortexing. We used excess MPS (2×) to further encourage alkyne consumption which, along with the polymers, remained soluble in DMSO throughout the thiol–yne reaction.
Net anionic PBAE purification
Following the thiol–yne reaction, we needed to remove excess unreacted thiol. Purification following similar post-polymerization reactions commonly involves dialysis for extended times in aqueous solution. However, even under acidic conditions where PBAEs are generally stable,8 analytical SEC elution time increased after just 5.5 h of dialysis (Fig. S5†), indicating degradation had occurred. We then turned to another size-based separation method common in protein engineering, preparative SEC, which allows faster purification than dialysis (minutes as opposed to days). In a further effort to reduce hydrolysis during purification, we ran our columns in acidic water (with 20 mM NaCl, adjusted to pH 4). To check for degradation during purification, we used analytical SEC and found similar retention times between even the most hydrophilic alkyne-functionalized PBAE (0% HDDA) and the corresponding purified net anionic PBAE in SEC, indicating minimal hydrolysis during purification (Fig. S10†). To quantify the efficacy of our purification, we used an Ellman's reagent assay to demonstrate ≥90% removal of the excess thiol from the polymer. We do, however, note relatively low yields from this purification method (ca. 40%) which likely could be improved by optimizing column size, flow rate, and sample concentration, though potentially at the cost of purity. For the structure–property relationships we describe next, the importance of purity outweighed the cost of low yields, so we proceeded with this purification method for subsequent polymers.With a purification strategy established for the most hydrophilic (0% HDDA) net anionic PBAEs, we moved to purifying our more hydrophobic derivatives. We purified net anionic PBAEs with 50% HDDA-containing repeating units similarly to our 0% HDDA net anionic PBAEs and also found no change in SEC elution time between the alkyne and sulfonate functionalized PBAEs (Fig. S10†). Unlike the 0 and 50% HDDA polymers, we found a loss of the high molecular weight shoulder after purifying our net anionic PBAE with 75% HDDA. However, upon closer inspection, we suspect this is due to aggregation of the alkyne-functionalized polymer in the trifluoroethanol SEC eluent, discussed further in ESI section S11,† and is not indicative of degradation during purification, further supported by the absence of multiple carbonyl peaks in the 13C NMR spectrum (Fig. S12†). For net anionic PBAEs with both 50 and 75% HDDA, we saw large reductions in thiol, similarly to the most hydrophilic net anionic PBAE. When we attempted to purify the 100% HDDA anionic PBAE, however, we noticed the presence of precipitates under acidic conditions. These precipitates were unable to pass through the column and only eluted after passing copious amounts (ca. 1 L) of 0.1 M NaOH as a cleaning solution through the column. Since these hydrophobic polymers remain very soluble in basic aqueous solution (pH > 9), we attempted a similar purification but eluting in 20 mM NaCl adjusted to pH 9 instead of pH 4, anticipating that some degradation might occur under these alkaline conditions. The analytical SEC chromatogram, however, showed no change in elution time (Fig. 2B) while Ellman's assay revealed large reductions in thiol content, indicating that our use of acidified water to slow degradation was likely unnecessary and base can be used to improve solubility without triggering degradation on these short timescales.
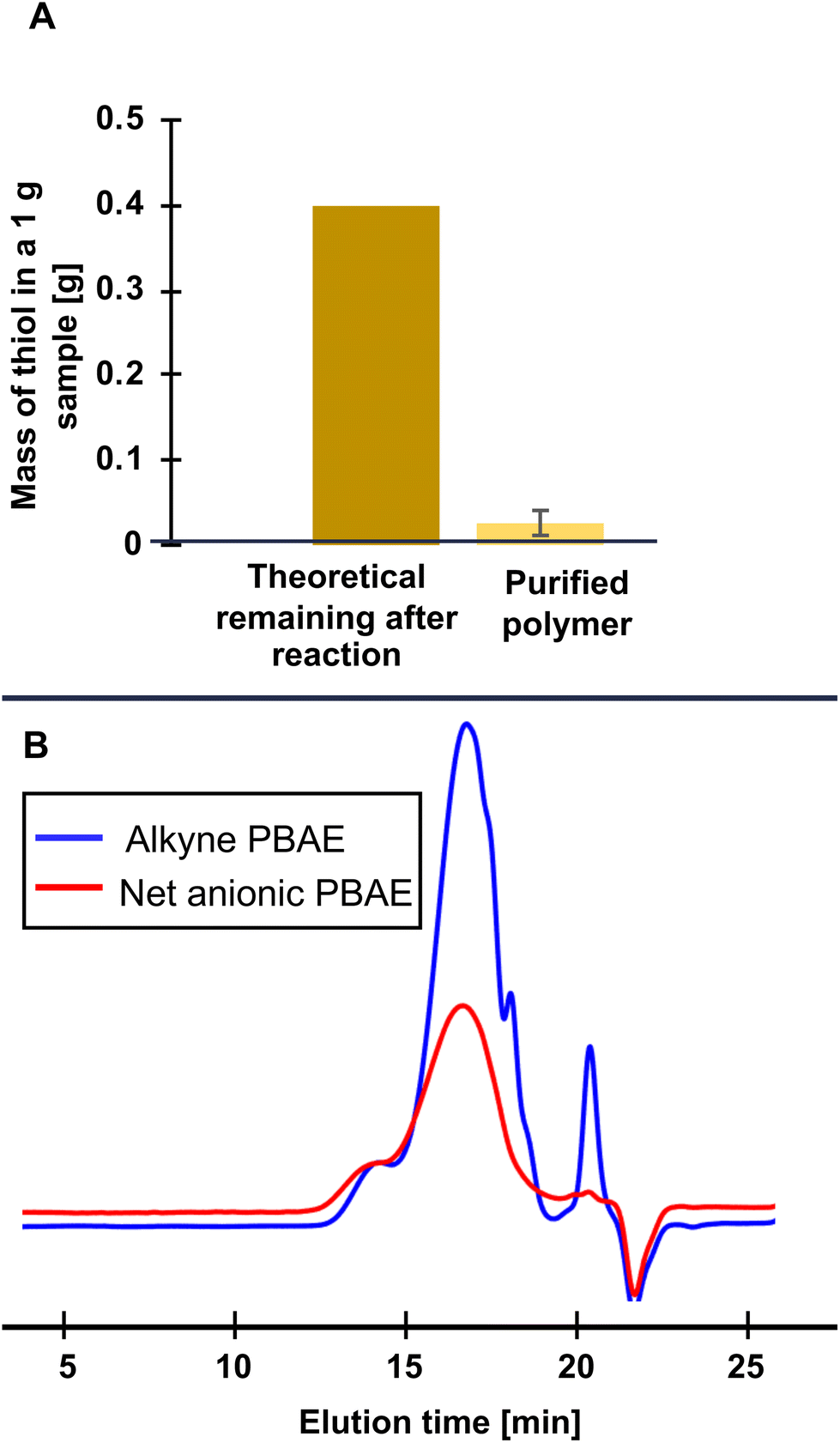 | ||
| Fig. 2 Purification of net anionic PBAE by preparative SEC: (A) the reduction of thiol content averaged across all samples, measured by Ellman's assay, after preparative SEC purification, the error bar represents standard deviation (n = 7); and (B) differential refractive index analytical SEC chromatogram of the 100% HDDA alkyne functionalized PBAE and the same polymer after thiol–yne functionalization and preparative SEC under basic (pH = 9) conditions. | ||
Net anionic PBAE spectroscopic characterization
After purifying our suite of net anionic PBAEs, we used NMR and IR spectroscopy to confirm the consumption of alkynes and installation of anionic groups. The disappearance of the characteristic alkyne peaks at 74.9 and 79.4 ppm in the 13C NMR spectra (Fig. 1A) and 3280 cm−1 in the IR spectra (Fig. S9†) after the thiol–yne reaction and purification indicate quantitative alkyne consumption. Since Ellman's assay showed minimal remaining thiol after purification, the appearance of MPS associated peaks indicates successful thiol–yne functionalization. To quantify thiol attachment efficiency, we compared the relative integration of the MPS peak at 1.83 ppm in the 1H NMR spectra to the integration of the protons alpha to the ester (labeled n and a in Fig. 1B, respectively) and found 70–95% functionalization among all our reactions. Altogether, the spectroscopic evidence points to successful functionalization of our PBAEs into net anionic, sulfonated PBAEs (see Fig. S7–S14† for spectra of 50, 75, and 100% HDDA net anionic PBAEs).Solution behavior
As our net anionic PBAEs are polyampholytes with both positive and negative charge, we expected them to have different solution behavior compared to their parent cationic PBAEs. Traditional cationic PBAEs have a fairly straightforward relationship between solubility and pH; as pH decreases and more backbone amines protonate, the polymers becoming increasingly soluble in aqueous media.8,36 Similar to cationic PBAEs, our net anionic PBAEs have a pH-dependent protonatable amine but, unlike traditional PBAEs, have two anionic groups that remain charged across the pH range tested here (pH = 4–10). To learn how the installation of these anionic groups changes PBAE solution behavior, we monitored pH-, hydrophobicity-, and time-dependent absorbance (at 550 nm) of our net anionic PBAEs in addition to visual assessments (photos in Fig. S19†). Over the conditions examined here (pH 4–10), the hydrophilic 0% HDDA net anionic PBAE remained very soluble with minimal variation in absorbance across the entire pH range. The net anionic PBAEs with more hydrophobic units meanwhile, displayed pH- and hydrophobicity-dependent solution behavior. When PBAE backbone amines are largely deprotonated at pH 10, all of the net anionic PBAEs, including the most hydrophobic derivative (100% HDDA), had very low absorbance indicating they were soluble under these alkaline conditions (Fig. 3A). At pH 8, where some backbone amines begin to protonate, the solutions of net anionic PBAEs with hydrophobic HDDA units (50, 75, or 100% HDDA) were more turbid than at pH 10. When we lowered the pH further to 7, we were unable to suspend the polymers with HDDA units (a portion of the polymer remained stuck to the vial making absorbance measurements unreliable). Further acidifying the solution to pH 6, allowed the net anionic PBAEs with intermediate hydrophobicity (50 and 75% HDDA) to disperse and form a turbid suspension (Fig. 3A). Meanwhile, the 100% HDDA PBAE remained stuck to the side of the vial at pH 6. Lowering the pH to 4, where most PBAE amines are protonated, the 50 and 75% HDDA net anionic PBAEs were quite soluble with absorbances matching pH 10 conditions. Based on our purification method development, we had already suspected our most hydrophobic anionic PBAE (100% HDDA) would have low solubility in pH 4 solution and indeed, we were unable to suspend the 100% HDDA net anionic PBAE in pH 4, 6, or 7 buffers. However, when titrated with HCl, the 100% HDDA net anionic PBAE looked visually dissolved once the pH was below ca. 3 (Fig. 3D). These net anionic PBAEs with hydrophobic units display non-monotonic solution behavior where they readily dissolve at low and high pH but form turbid suspensions or cannot suspend in more neutral pH solutions (pH 6–8). Over time, the measured absorbance of all the polymers that aggregated decreased as the polymer degraded and the aggregates broke apart (Fig. 3B). | ||
| Fig. 3 Solution behavior of net anionic PBAEs: (A) absorbance at 550 nm immediately after dissolution in pH 4, 6, 7, 8, or 10 buffer. Lines between data points are drawn as guides to the eye. The standard deviation between triplicate measurements (of the same solution) is smaller than the height of the data points presented here. (B) 3D plot of absorbance measurements at 550 nm as a function of time, where the initial time points are the 2D data presented in part A; (C) schematic of net anionic PBAE charge states at various pH, which we suspect creates a regime at neutral pH that allows hydrophobic interactions to dominate; and (D) a titration of the 100% HDDA net anionic PBAE with HCl with photographic insets at pH 10, 7, and 3 to demonstrate solubility changes as a function of pH. | ||
We suspect these behaviors are explained by two opposing phenomena: like-charge repulsion that extends polymer chains and promotes solubility at high pH and increased charge density at low pH increasing solubility, together, creating a scenario at moderate pH where hydrophobic interactions dominate (Fig. 3C). When the pH is high (pH = 10), PBAE backbone amines are mostly deprotonated (based on both published titration curves10,37,38 and our own titration curves in Fig. 3D and S21†) and the PBAE is a polyanion with two anionic charges per repeating unit. To reduce the proximity of these like-charges, we suspect the chains stretch out, preferring to interact with surrounding water than other polymers, known as the polyelectrolyte effect.39–44 At lower pH (pH = 6–8) some of the backbone amines protonate and the polymers are now polyampholytes with both cationic and anionic charge. The cationic charge along the backbone reduces the repulsion of anionic units along the chain, reducing the level to which the chains extend.41,43,44 At the same time, reduced neighboring chain repulsion allow more intermolecular interactions.45 This then allows the polymers with hydrophobic portions to interact and create larger structures that result in the turbid solutions we observe.43 In acidic pH (pH = 4) most amines protonate and we suspect the increased charge density (a net charge of −1 per repeating unit) compared to the neutral pH range where only some amines are protonated makes the polymer more hydrophilic and thus, more soluble, as evidenced by the lower absorbance at pH 4 for 50% and 75% HDDA net anionic PBAEs relative to pH 6 and 8. While the hydrophobic 100% HDDA PBAE remains insoluble at pH 4, further lowering the pH to ca. 3, as in the titration (Fig. 3D), triggers dissolution and shows this polymer to display the same non-monotonic solution behavior as the less hydrophobic 50% and 75% HDDA derivatives. The tunable pH- and hydrophobicity-dependent behavior of these materials provides opportunities for designing therapeutic carriers and pH-responsive materials.
Degradation monitored by 1H NMR spectroscopy
While we anticipated the hydrolytically cleavable nature of PBAE esters would extend to our net anionic PBAEs, assessing degradation behavior of these materials by SEC presented challenges as the polymers are insoluble or degrade in most common organic SEC solvents. Therefore, we evaluated anionic PBAE degradation by monitoring changes in the 1H NMR spectra as a function of time in 500 mM pD 10 carbonate buffer. We selected basic conditions, where we expected ester hydrolysis to be faster than in more acidic conditions.8 We found several signals to increase, decrease, and/or shift in the 1H NMR spectra over the 100 h experiment, but the disappearance of the signal corresponding to protons adjacent to the ester (a in Fig. 1B) provided the most straightforward way to monitor hydrolysis, as the peak was well separated from other peaks and decreases in intensity as the polymer hydrolyzes (Fig. 4). Though degradation occurred on a similar timescale for all of our net anionic PBAEs, the hydrophilic PBAEs hydrolyzed faster than more hydrophobic derivatives, consistent with reports of hydrophobic units slowing degradation in PBAEs.31,46 For example, after 22 h in carbonate buffer, 93% of the esters were intact in the 100% HDDA-containing net anionic PBAEs while only 52% of the esters remained in the more hydrophilic 0% HDDA derivative after the same time period. While the degradation data reported here are the result of one experimental run, we repeated degradation experiments for two polymers (50 and 75% HDDA) and found very good agreement between the two runs (Fig. S20†). The ability to control hydrolysis rate by simply adjusting the feed ratio of monomers during PBAE synthesis will be critical as we start to examine these materials for applications such as releasing active drug molecules commensurate with patient healing.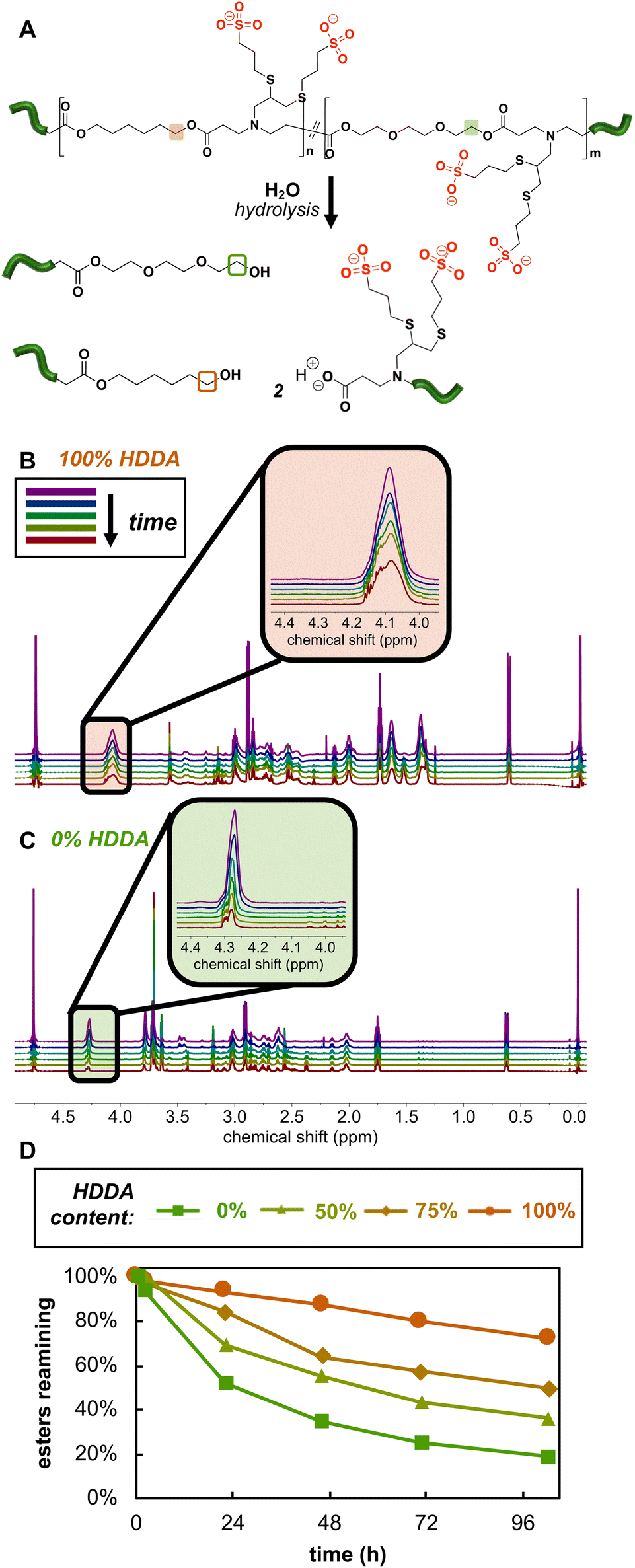 | ||
| Fig. 4 Net anionic PBAE degradation: (A) net anionic PBAE degradation scheme with the protons of interest in the HDDA- and PEGDA-containing repeating units highlighted in orange and green, respectively; 1H NMR (800 MHz) spectra of net anionic PBAEs with (B) 100% HDDA-containing repeating units and (C) 0% HDDA-containing repeating units degrading in 500 mM carbonate buffer (pD = 10); (D) integration of the peaks corresponding to protons adjacent to the esters was normalized to the initial integration and plotted as a function of time for net anionic PBAEs with 0, 50, 75, or 100% HDDA-containing repeating units. | ||
Complexation with model peptide
To gauge the ability of these net anionic PBAEs to complex and release cationic species, we mixed polymer solutions with a model cationic peptide, (GR)10, in pH 10 solution, where the net anionic PBAEs are soluble and have the highest anionic charge density, giving them the best chance at complexing (GR)10.45 Solutions of polymer and peptide alone were both visually clear at 1 mg mL−1 in 50 mM pH 10 carbonate buffer and mixtures of the most hydrophilic net anionic PBAE (0% HDDA-containing repeating units), and (GR)10 solutions at a 1:1 volume ratio (corresponding to a 1:1 charge ratio) also remained clear. However, when we mixed solutions of hydrophobic net anionic PBAE and (GR)10 they became visually turbid. We quantified turbidity by monitoring absorbance as a function of time and polymer hydrophobicity (Fig. 5). We found complexes made from 75% and 100% HDDA net anionic PBAEs had higher absorbance than hydrophilic 0% HDDA derivatives, even though the polymer solutions alone were all very clear with minimal absorbance. As time progressed, the absorbance of these hydrophobic net anionic PBAE–peptide complexes decreased, reaching a plateau after ca. 5 h at which point all mixtures showed the same absorbance as that of the starting polymer or peptide alone. These preliminary complexation experiments demonstrate the potential for PBAEs, a versatile class of polymers used for anionic therapeutic delivery, to act as carriers for cationic therapeutics and degradable polyanions, greatly expanding their use.
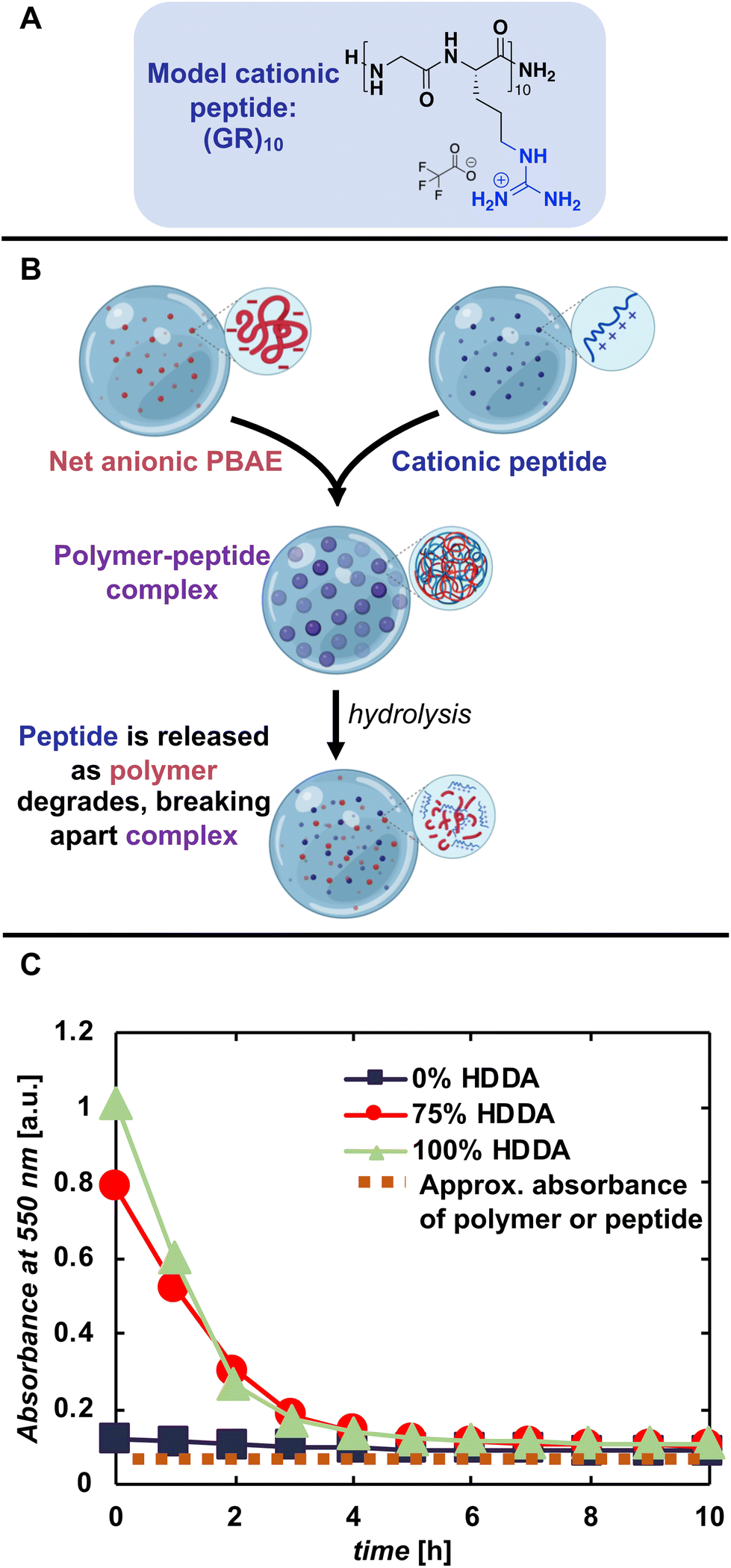 | ||
| Fig. 5 Complexation of net anionic PBAEs with cationic species: (A) structure of model cationic peptide (GR)10; (B) scheme showing proposed complexation of (GR)10 with net anionic PBAEs; (C) absorbance at 550 nm as a function of time of complexes of (GR)10 and net anionic PBAEs with 0, 75, or 100% HDDA-containing repeating units at a 1:1 charge ratio. | ||
Conclusions
Through post-polymerization modifications and chromatographic purification, we access PBAEs with previously restricted functionality: net anionic charge. While cationic PBAE solubility improves in acid, the net anionic versions are quite soluble in basic conditions and exhibit complex solution behavior that varies non-monotonically with solution pH. We suspect this solution behavior results from the balance between like-charge repulsion and charge density changes allowing hydrophobic interactions to dominate at neutral pH. The pH-responsive behavior and hydrophobicity-dependent degradation of these polymers enable user-tuned properties that evolve over time as the polymers degrade. For example, these polymers form complexes with a cationic peptide that disintegrate as the polymer hydrolyzes. Therefore, we envision these polymers as delivery vehicles for sensitive cationic therapeutics, including antimicrobial peptides, and as degradable net anionic constituents in polyelectrolyte complexes.Author contributions
M. K. K. and R. A. L. conceptualized the study. M. K. K., A. M. C., and V. P. G. collected data and drafted the manuscript. M. K. K. and R. A. L. revised the manuscript. R. A. L. acquired funding and supervised the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the National Institutes of Health (R35GM147424) and a seed grant from the Center for Engineering in Medicine at UVA. M. K. K. and V. P. G. acknowledge financial support from Dean's Scholar Fellowships from the School of Engineering and Applied Science at UVA. V. P. G. acknowledges financial support from a National Institutes of Health Biotechnology Training Program (T32GM136615). A. M. C. acknowledges financial support from the Center for Advanced Biomanufacturing Internship Program at UVA. The authors thank Dr Jeffrey Ellena for helpful NMR discussions, Dr Earl Ashcraft for ESI experiments, and the BioNMR Core at UVA. Schematics in Fig. 3C and 5B were made with the help of BioRender.com.References
- D. M. Lynn and R. Langer, J. Am. Chem. Soc., 2000, 122, 10761–10768 CrossRef CAS.
- Y. Liu, Y. Li, D. Keskin and L. Shi, Adv. Healthcare Mater., 2019, 8, 1801359 Search PubMed.
- D. Shenoy, S. Little, R. Langer and M. Amiji, Pharm. Res., 2005, 22, 2107–2114 CrossRef CAS PubMed.
- Y. Al Thaher, S. Latanza, S. Perni and P. Prokopovich, J. Colloid Interface Sci., 2018, 526, 35–42 CrossRef CAS PubMed.
- N. Segovia, P. Dosta, A. Cascante, V. Ramos and S. Borrós, Acta Biomater., 2014, 10, 2147–2158 CrossRef CAS PubMed.
- J. C. Sunshine, M. I. Akanda, D. Li, K. L. Kozielski and J. J. Green, Biomacromolecules, 2011, 12, 3592–3600 CrossRef CAS PubMed.
- X. Deng, N. Zheng, Z. Song, L. Yin and J. Cheng, Biomaterials, 2014, 35, 5006–5015 CrossRef CAS PubMed.
- M. K. Kuenen, J. A. Mullin and R. A. Letteri, J. Polym. Sci., 2021, 59, 2212–2221 CrossRef CAS.
- D. R. Wilson, M. P. Suprenant, J. H. Michel, E. B. Wang, S. Y. Tzeng and J. J. Green, Biotechnol. Bioeng., 2019, 116, 1220–1230 CrossRef CAS PubMed.
- J. C. Sunshine, D. Y. Peng and J. J. Green, Mol. Pharm., 2012, 9, 3375–3383 CrossRef CAS PubMed.
- U. Capasso Palmiero, J. C. Kaczmarek, O. S. Fenton and D. G. Anderson, Adv. Healthcare Mater., 2018, 7, 1–6 Search PubMed.
- N. J. Shah, M. N. Hyder, J. S. Moskowitz, M. A. Quadir, S. W. Morton, H. J. Seeherman, R. F. Padera, M. Spector and P. T. Hammond, Sci. Transl. Med., 2013, 5(191), 191ra83 Search PubMed.
- S. L. Bechler and D. M. Lynn, Biomacromolecules, 2012, 13, 542–552 CrossRef CAS PubMed.
- D. M. Lynn, D. G. Anderson, D. Putnam and R. Langer, J. Am. Chem. Soc., 2001, 123, 8155–8156 CrossRef CAS PubMed.
- D. G. Anderson, C. A. Tweedie, N. Hossain, S. M. Navarro, D. M. Brey, K. J. Van Vliet, R. Langer and J. A. Burdick, Adv. Mater., 2006, 18, 2614–2618 CrossRef CAS.
- R. B. Metter, J. L. Ifkovits, K. Hou, L. Vincent, B. Hsu, L. Wang, R. L. Mauck and J. A. Burdick, Acta Biomater., 2010, 6, 1219–1226 CrossRef CAS PubMed.
- H. Sun, Y. Hong, Y. Xi, Y. Zou, J. Gao and J. Du, Biomacromolecules, 2018, 19, 1701–1720 CrossRef CAS PubMed.
- W. Li, F. Separovic, N. M. O'Brien-Simpson and J. D. Wade, Chem. Soc. Rev., 2021, 50, 4932–4973 RSC.
- Y. Zhang, J. Liao, T. Wang, W. Sun and Z. Tong, Adv. Funct. Mater., 2018, 28, 1–9 CAS.
- L. S. Biswaro, M. G. d. C. Sousa, T. M. B. Rezende, S. C. Dias and O. L. Franco, Front. Microbiol., 2018, 9, 1–14 CrossRef PubMed.
- J. Lei, L. C. Sun, S. Huang, C. Zhu, P. Li, J. He, V. Mackey, D. H. Coy and Q. Y. He, Am. J. Transl. Res., 2019, 11, 3919–3931 CAS.
- C. H. Chen and T. K. Lu, Antibiotics, 2020, 9, 1–20 Search PubMed.
- H. B. Koo and J. Seo, Pept. Sci., 2019, 111, 5 Search PubMed.
- M. Tirrell, ACS Cent. Sci., 2018, 4, 532–533 CrossRef CAS PubMed.
- W. C. Blocher McTigue and S. L. Perry, Small, 2020, 16, 1–17 CrossRef PubMed.
- S. S. Naik, J. W. Chan, C. Comer, C. E. Hoyle and D. A. Savin, Polym. Chem., 2011, 2, 303–305 RSC.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745–4750 RSC.
- J. C. Worch, C. J. Stubbs, M. J. Price and A. P. Dove, Chem. Rev., 2021, 121, 6744–6776 CrossRef CAS PubMed.
- A. B. Lowe, Polymer, 2014, 55, 5517–5549 CrossRef CAS.
- K. Yao, G. Gong, Z. Fu, Y. Wang, L. Zhang, G. Li and Y. Yang, ACS Macro Lett., 2020, 9, 1391–1397 CrossRef CAS PubMed.
- D. Safranski, M. A. Lesniewski, B. S. Caspersen, V. M. Uriarte and K. Gall, Polymer, 2010, 51, 3130–3138 CrossRef CAS.
- A. E. Neitzel, Y. N. Fang, B. Yu, A. M. Rumyantsev, J. J. De Pablo and M. V. Tirrell, Macromolecules, 2021, 54, 6878–6890 CrossRef CAS PubMed.
- A. K. Covington, M. Paabo, R. A. Robinson and R. G. Bates, Anal. Chem., 1968, 40, 700–706 CrossRef CAS.
- D. Lundberg and K. Holmberg, J. Surfactants Deterg., 2004, 7, 239–246 CrossRef CAS.
- A. E. Rydholm, K. S. Anseth and C. N. Bowman, Acta Biomater., 2007, 3(4), 449–455 CrossRef CAS PubMed.
- W. Song, Z. Tang, M. Li, S. Lv, H. Yu, L. Ma, X. Zhuang, Y. Huang and X. Chen, Macromol. Biosci., 2012, 12, 1375–1383 CrossRef CAS PubMed.
- J. S. Lee, X. Deng, P. Han and J. Cheng, Macromol. Biosci., 2015, 15, 1314–1322 CrossRef CAS PubMed.
- F. Gao, Q. Wang and X. Yang, Colloid Polym. Sci., 2020, 298, 303–312 CrossRef CAS.
- Y. Takeoka, B. A. Nihat, D. Rose, E. Takashi, G. Alexander, K. Mehran, O. Taro, T. Kazunori, W. Guoqiang, Y. Xiaohong and T. Toyoichi, Phys. Rev. Lett., 1999, 82, 4863–4865 CrossRef CAS.
- S. E. Kudaibergenov and N. Nuraje, Polymer, 2018, 10, 1146 Search PubMed.
- P. G. Higgs and J. F. Joanny, J. Chem. Phys., 1991, 94, 1543–1554 CrossRef CAS.
- H. Yang, Q. Zheng and R. Cheng, Colloids Surf., A, 2012, 407, 1–8 CrossRef CAS.
- A. V. Dobrynin, R. H. Colby and M. Rubinstein, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 3513–3538 CrossRef CAS.
- J. Dinic, A. B. Marciel and M. V. Tirrell, Curr. Opin. Colloid Interface Sci., 2021, 54, 101457 CrossRef CAS.
- D. E. Hastings, J. C. Bozelli, R. M. Epand and H. D. H. Stöver, Macromolecules, 2021, 54, 11427–11438 CrossRef CAS.
- A. Muralidharan, R. R. McLeod and S. J. Bryant, Adv. Funct. Mater., 2022, 32, 1–16 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py01319c |
| This journal is © The Royal Society of Chemistry 2023 |