
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiol modifier effects of diphenyl diselenides: insight from experiment and DFT calculations†
Pablo A.
Nogara
 ab,
Cláudia S.
Oliveira
cd,
Andrea
Madabeni
e,
Marco
Bortoli
f,
João Batista T.
Rocha
a and
Laura
Orian
*e
ab,
Cláudia S.
Oliveira
cd,
Andrea
Madabeni
e,
Marco
Bortoli
f,
João Batista T.
Rocha
a and
Laura
Orian
*e
aDepartamento de Bioquímica e Biologia Molecular, Centro de Ciências Naturais e Exatas, Universidade Federal de Santa Maria, Av. Roraima 1000, 97105-900 Santa Maria, RS, Brazil
bInstituto Federal de Educação, Ciência e Tecnologia Sul-rio-grandense (IFSul), Av. Leonel de Moura Brizola, 2501, 96418-400, Bagé, RS, Brazil
cInstituto de Pesquisa Pelé Pequeno Príncipe (IPPPP), Av. Silva Jardim, 1632, 80250-060, Curitiba, PR, Brazil
dFaculdades Pequeno Príncipe, Av. Iguaçu, 333, 80230-020, Curitiba, PR, Brazil
eDipartimento di Scienze Chimiche, Università degli Studi di Padova, Via Marzolo 1, 35131, Padova, Italy. E-mail: laura.orian@unipd.it
fDepartment of Chemistry and Hylleraas Centre for Quantum Molecular Sciences, University of Oslo, P.O. Box 1033, Blindern, 0315 Oslo, Norway
First published on 13th February 2023
Abstract
A combination of spectroscopic, chromatographic and computational approaches was employed to investigate the reaction of several diselenides of formula (R-PhSe)2 (R = CH3O, CH3, H, Cl, CF3) with a thiolate nucleophile, leading to the breaking of the selenium–selenium (Se–Se) bond. This process has fundamental importance in biological environments and provides a rationale to analyze the so-called thiol modifier effect of diselenides, which may be exploited in pharmacology and toxicology. Our data suggest that withdrawing substituents favor the reaction, effectively making the reaction energy more negative, but strong electron-withdrawing groups also prompt structural modification on the starting reactant, increasing the reaction barrier. Thus, the nature (electron rich or electron poor) of the diselenides can play an essential role in the reactivity and biological activity of these molecules.
1. Introduction
Diaryl diselenides are an important class of organodiselenides characterized by the presence of a Se–Se bond bridging two aromatic moieties, whose activity can in principle be tuned by varying the chemical nature, as well as the position of different substituents on the rings. The archetypal diphenyl diselenide, (PhSe)2, was first synthesized in 1888 by Chabrie, who likely identified it as phenylselenol, as postulated six years later by Krafft and Lyons. The historical background was constructed by Bradt and Green.1 In the past two decades, several authors have thoroughly studied the applications of diphenyl diselenides in organic catalysis, as reported by Wirth.2–6 Santi and co-workers investigated their use in green chemistry7–11 and pharmaceutical applications.12–16 The chemical interest is mainly due to the catalytic properties of diphenyl diselenides, which are largely used in important classes of organic reactions, like Baeyer–Villiger oxidations of aldehydes and ketones, oxidations of alcohols and nitrogen containing compounds and alkene epoxidations.2,17,18 In fact, in the presence of hydrogen peroxide (H2O2), (PhSe)2 is readily oxidized to a selenoxide,19 in which the Se–Se bond is weakened and upon reaction with further equivalents of H2O2 an autocatalytic mechanism is established leading to benzeneperoxyseleninic acid which may act as an oxidizing agent, as described and rationalized by Ribaudo et al.20Like all organoselenides, (PhSe)2 has attracted a lot of interest also for its glutathione peroxidase (GPx) mimic potential, i.e., its antioxidant properties.21–25 In the enzymatic mechanism, the selenocysteine residue is oxidized by H2O2 and then reduced in two steps by two equivalents of reduced glutathione (GSH), leading first to a selenylsulfide (Se–S) intermediate and then to the initial selenol.26 Conversely, the GPx-like function of a diselenide first requires Se–Se bond breaking, fostered by a thiolate, leading to the formation of a selenylsulfide and a selenolate, and the latter is active in reducing peroxides (Fig. 1).27,28 Both in chemistry and in biology, the oxidation of organoselenides, including diselenides, has been largely investigated and its mechanistic details have been elucidated.19,29,30 Conversely, the reaction of diselenides with thiolates has been explored by Mugesh et al.,23,31 Rocha et al.,21,32–34 Santi et al.,35,36 and Braga et al.37–40 but only a few theoretical studies have been reported, among which are those by Bachrach et al.,41,42 Bortoli et al.43 and Yáñez et al.44
 | ||
| Fig. 1 Catalytic activity of diphenyl diselenide: GPx-like activity (A) and thiol oxidase activity (B). | ||
From a biological point of view, diphenyl diselenide, (PhSe)2, is the most studied diaryl diselenide, demonstrating pharmacological potential, such as neuroprotective, antidepressant, anxiolytic, anti-inflammatory, and anticancer properties among others.45,46 Many derivatives have been synthesized by replacing hydrogen atoms of the phenyl ring with different chemical groups. Interestingly, their biological activity is not significantly affected by these substitutions when compared to the parent compound.45 However, selenium organic derivatives, including diselenides, present toxicological effects. An example is the inhibition of the aminolevulinate dehydratase (δ-AlaD) enzyme (involved in the heme biosynthesis). In this case, diaryl diselenides, (R-PhSe)2, have comparable effects (the half-maximal inhibitory concentration (IC50) for R = H, m-CF3, p-Cl, and p-CH3O is 4.6, 6.6, 4.3, and 4.3 μM, respectively).47 In addition, the half-maximal lethal dose (LD50) of these compounds for a single acute oral administration in mice is very similar (higher than 1 mmol kg−1).48 Their pharmacological and toxicological mechanisms of action are believed to involve the reduction of the Se–Se bond by thiol/thiolate groups from endogenous molecules (proteins, enzymes, and low-molecular-mass thiols (LMM-SH) like cysteine (Cys), GSH, coenzyme A (CoA), and lipoic acid), leading to a species with a Se–S bond, which can disrupt the cellular function. Recent studies suggest that this thiol-modifier effect (thiol oxidase, Fig. 1) of organoselenium compounds can explain their biological action better than their GPx-like activity.49 In addition, diphenyl diselenide and analogs can be substrates for the mammalian thioredoxin reductase (TrxR) enzyme, where the Se–Se is reduced to two selenol species, by the action of thiol and selenol groups from cysteine and selenocysteine residues, respectively.21 According to Sausen et al.,21 there is a correlation between thiol oxidase activity and the effectiveness of diselenides to act as substrates of TrxR, suggesting that the first reaction step, i.e., the thiol attack on the Se–Se bond might play an essential role in the organoselenium activity.
An important mechanistic aspect, which remains elusive, is the experimental characterization of the selenol/selenolate (–SeH/–Se−), mainly due to its short lifetime (it is quickly oxidized to the diselenide form) and the lack of adequate analytical techniques.50–53 In this work, we investigate the formation of the Se–S intermediate and selenol/selenolate products by reacting diselenides with thiols (Fig. 2) through a combined experimental and computational approach. In addition, we analyze the effect of different substituents on the phenyl rings on the energetics and kinetics of the process.
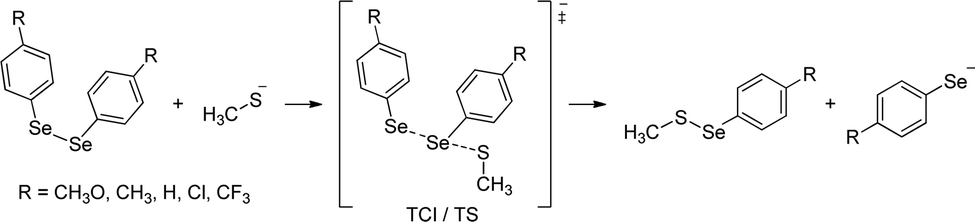 | ||
| Fig. 2 Proposed reaction of diselenides and methylthiolate. | ||
2. Experimental section
2.1. Chemicals
All chemicals used were obtained from either Sigma Aldrich or Fluka suppliers and presented analytical grade.2.2. Oxygen consumption assay
The rate of thiol oxidation was determined in a medium containing 100 mM potassium phosphate buffer, pH 7.4, dithiothreitol (DTT) 2 mM, and the diselenides at 50 μM in the presence or absence of iodoacetic acid (IAcOH, 10, 50, and 100 μM), through Oxygraph Version 1.02 (Hansatech Instruments, Norfolk, England).2.3. Thiol oxidase activity: colorimetric assay
The ability of a thiol to reduce the Se–Se bond was measured indirectly by the Ellman method.21,22 The reaction medium (5% water) containing 1 mM dithiothreitol (DTT; a thiol source), 25 or 50 μM (PhSe)2, and 50 μM methylmercury chloride (MeHg+) or iodoacetic acid IAcOH (–SeH trap molecules) was incubated at 37 °C for 24 h. Aliquots (20 μL) were collected at 0, 1, 2, 3, 4, 5, 6, and 24 h for color reaction. In the colorimetric assay, aliquots were mixed with 180 μL of 2 mM 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB; chromophore agent) and the absorbance was measured spectrophotometrically at 412 nm.2.4. Detection of the selenyl–sulfide intermediate: HPLC assay
High performance liquid chromatography with a photodiode array detector (HPLC-DAD) was performed with a Shimadzu SPD-20A UV/Vis Detector, CBM-20A communication bus module and DGU-20A5 Degasser prominence HPLC controlled by the LCSolution software system. The separation was achieved on a VerticalTM VertiSep GES C18 HPLC column (4.6 × 150 mm) packed with 5 μm diameter particles; the mobile phase contained acetonitrile and acidified water (0.5% H3PO4) (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v, 80:20) when DTT was used as a –SH source. The flow rate was 1 mL min−1 and the injection volume was 50 μL. The peaks were detected at 230 nm. The reaction medium (80% water) containing 2 mM DTT, 50 μM (PhSe)2, and/or 0–120 μM methylmercury chloride (–SeH trap molecule) was prepared and after 3 minutes was analyzed in the HPLC.
:v, 80:20) when DTT was used as a –SH source. The flow rate was 1 mL min−1 and the injection volume was 50 μL. The peaks were detected at 230 nm. The reaction medium (80% water) containing 2 mM DTT, 50 μM (PhSe)2, and/or 0–120 μM methylmercury chloride (–SeH trap molecule) was prepared and after 3 minutes was analyzed in the HPLC.
2.5. Density functional theory (DFT) calculations
Geometry optimization of the reactants, products and transition states was carried out using the Amsterdam Density Functional (ADF) engine of the Amsterdam Modeling Suite (AMS 2020.103).54,55 The OLYP density functional was used, in combination with the TZ2P basis set, according to the literature,56 with a gradient convergence of 10−5 Hartree (10−4 for R = CF3). Scalar relativistic effects were taken into account using the zeroth-order regular approximation (ZORA).57 The level of theory is denoted as ZORA-OLYP/TZ2P. Frequency calculations were used to assess the correctness of the computed minima and transition states. The calculations in the condensed phase were carried out employing the conductor-like screening model58–60 as implemented in ADF61 (level of theory: COSMO-ZORA-OLYP/TZ2P). Intrinsic reaction coordinate (IRC) profiles62,63 were computed at the same level of theory starting from the imaginary frequency of the transition state structure and proceeding downhill to reactants and products with the algorithms implemented in AMS. The electrophilicity index was calculated with ADF according to the work of Parr et al.64 which is defined as the ratio between the square of the chemical potential and twice the chemical hardness. The Hirshfeld charges65 were analysed since they yield a good overall reactivity prediction.66,67 The Gibbs free energy was calculated using a standard statistical thermodynamic relationship at a temperature of 298.15 K and a concentration of 1 M.3. Results and discussion
3.1. Oxygen consumption assay
The diselenides are catalysts of the thiol oxidation reaction (thiol oxidase activity), and the process involves the formation of the selenol intermediate, which is then oxidized by oxygen regenerating the original diselenide (Fig. 1). Thus, the consumption of oxygen (O2) during the thiol oxidase cycle may be a strategy to understand the reactions. For this purpose, (PhSe)2, 4-4′-bischlorodiphenyl diselenide – (ClPhSe)2, and 4-4′-bismethoxydiphenyl diselenide – (CH3OPhSe)2 were studied.As shown in Fig. 3, the thiol oxidation is dependent on oxygen consumption. In addition, it is possible to observe that IAcOH almost completely blocks thiol oxidation by trapping the selenol intermediate. In fact, by increasing the IAcOH concentration, less oxygen is consumed in the reaction. Thus, the assay demonstrates that the thiol oxidase activity of (PhSe)2 is similar to that of (ClPhSe)2, while (CH3OPhSe)2 presents the lowest activity, suggesting that deactivating and activating substituents may play a role in the catalytic cycle. In order to investigate the selenol trapping, spectroscopic and chromatographic assays with (PhSe)2 were performed.
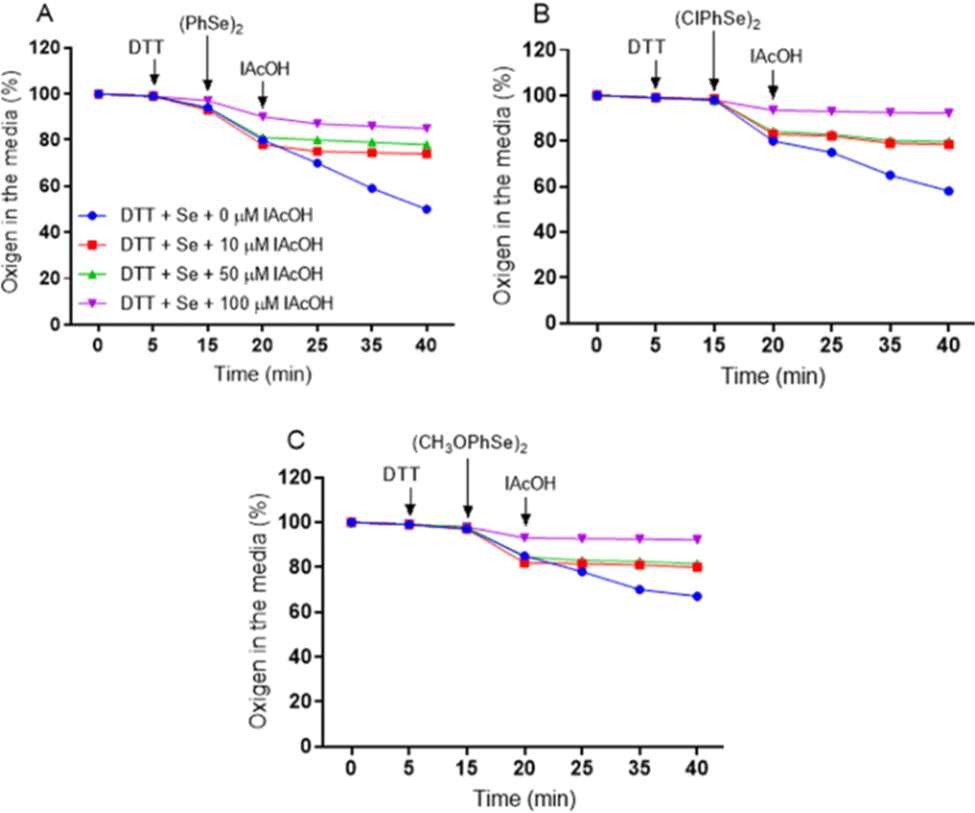 | ||
| Fig. 3 Oxygen consumption during the thiol oxidase activity of (A) diphenyl diselenide, (B) 4-4′-bischlorodiphenyl diselenide and (C) 4-4′-bismethoxydiphenyl diselenide. The oxygen of the medium was measured (Oxytherm, Hansatech) and its decrease indicated that it was consumed from the reaction media. The arrows indicate the time when a given reagent was added to the reaction media. The final concentration was 2 mM. | ||
3.2. Thiol oxidase activity
To study the thiol oxidase activity of (PhSe)2, the Ellman method was applied, using DTT as a thiol source. Fig. 4 shows the reduction of Se–Se by DTT and the trap of –SeH by MeHg+, using the colorimetric reaction with DTNB. It is possible to observe that the vehicle, (PhSe)2, and MeHg+per se do not interact with DTNB (the absence of yellow color). As expected, DTT alone reduced the DTNB forming TNB (yellow color), and the presence of MeHg+ (1 mM DTT + 50 μM MeHg+) does not interfere with the DTT + DTNB reaction and color formation. About 3 hours after the beginning of the reaction, the presence of 50 μM (PhSe)2 in the reaction medium catalyzed almost the total oxidation of 1 mM DTT. However, in the presence of 50 μM MeHg+ plus 50 μM (PhSe)2, the oxidation of DTT by (PhSe)2 was slower than that in the presence of only 50 μM (PhSe)2. Interestingly, 50 μM MeHg+ blocked completely the oxidation caused by 25 μM (PhSe)2, suggesting a stoichiometric reaction between its selenol intermediate and MeHg+. This result can be explained by the fact that the reduction of 50 μM (PhSe)2 by DTT leads to the formation of two equivalents of selenol, i.e., 100 μM of PhSeH. Thus, 50 μM MeHg+ can only trap 50 μM PhSeH, leaving the remaining of it (∼25 μM (PhSe)2) free to oxidize DTT. Hence, the result with 50 μM (PhSe)2 + 50 μM MeHg+ was very similar to the oxidation caused by 25 μM (PhSe)2 alone. The presence of 50 μM MeHg+ in the reaction medium totally prevents the oxidation of DTT by 25 μM (PhSe)2, probably because the MeHg+ traps all the PhSeH present in the medium. In the same pattern, the presence of 50 μM IAcOH in the reaction medium totally prevents the oxidation of 1 mM DTT by 25 μM (PhSe)2 (or 50 μM PhSeH). This suggests that IAcOH traps all the selenol present in the reaction medium (reaction: PhSeH + ICH2COOH → PhSe–CH2COOH + HI). Furthermore, the presence of 50 μM IAcOH slows down the oxidation of 1 mM DTT when incubated with 50 μM (PhSe)2 (Fig. 4(B)).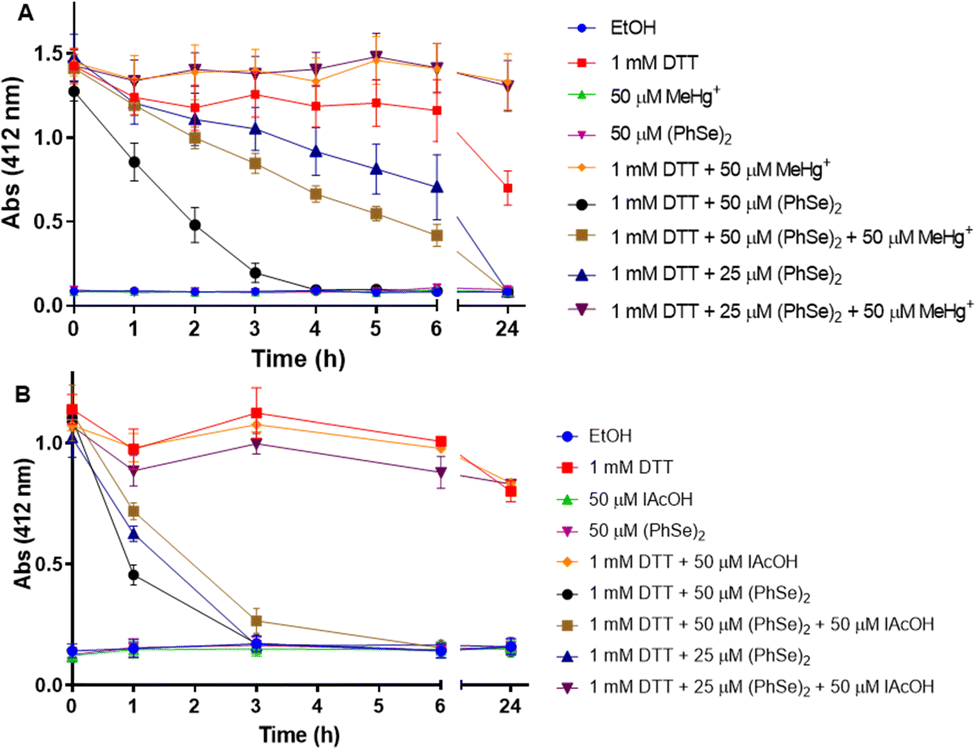 | ||
| Fig. 4 Thiol oxidase activity of (PhSe)2 and its inhibition by (A) MeHg+ and (B) IAcOH. The lines and symbols for MeHg+ (green) or (PhSe)2 (purple) are harder to be visualized because they are almost coincident with that of EtOH (dark-blue). | ||
3.3. HPLC assay
Aiming at detecting PhSeH and the Se–S intermediate, chromatographic assays were carried out (Fig. 5). When a high concentration of DTT was used (Fig. 5(A)), it was possible to detect the PhSeH and Se–S intermediate peak a few minutes after the reaction medium preparation. When MeHg+ was present in the medium at a low concentration (20 μM) (Fig. 5(B)), it was possible to observe the PhSe–HgMe and PhSeH peaks, as well as a decrease in the Se–S intermediate peak area. On the other hand, when MeHg+ at a high concentration (120 μM) was in the medium (Fig. 5(C)), it was possible to observe the absence of the PhSeH peak, as well as a decrease in the Se–S intermediate and (PhSe)2 peaks. Unfortunately, the DTT reduced (DTT-(SH)2) and oxidized (DTT-(SS)) form elute practically at the same retention time, and their peaks overlap. In this sense, this assay does not show if MeHg+ inhibits the DTT oxidation; however, the PhSe–HgMe adduct has been identified, suggesting the inhibition of the (PhSe)2 thiol oxidase activity. Thus, by combining these results with the colorimetric assay, the formation of the PhSeH and Se–S intermediate and its relevance for the diselenide thiol modifier activity can be inferred.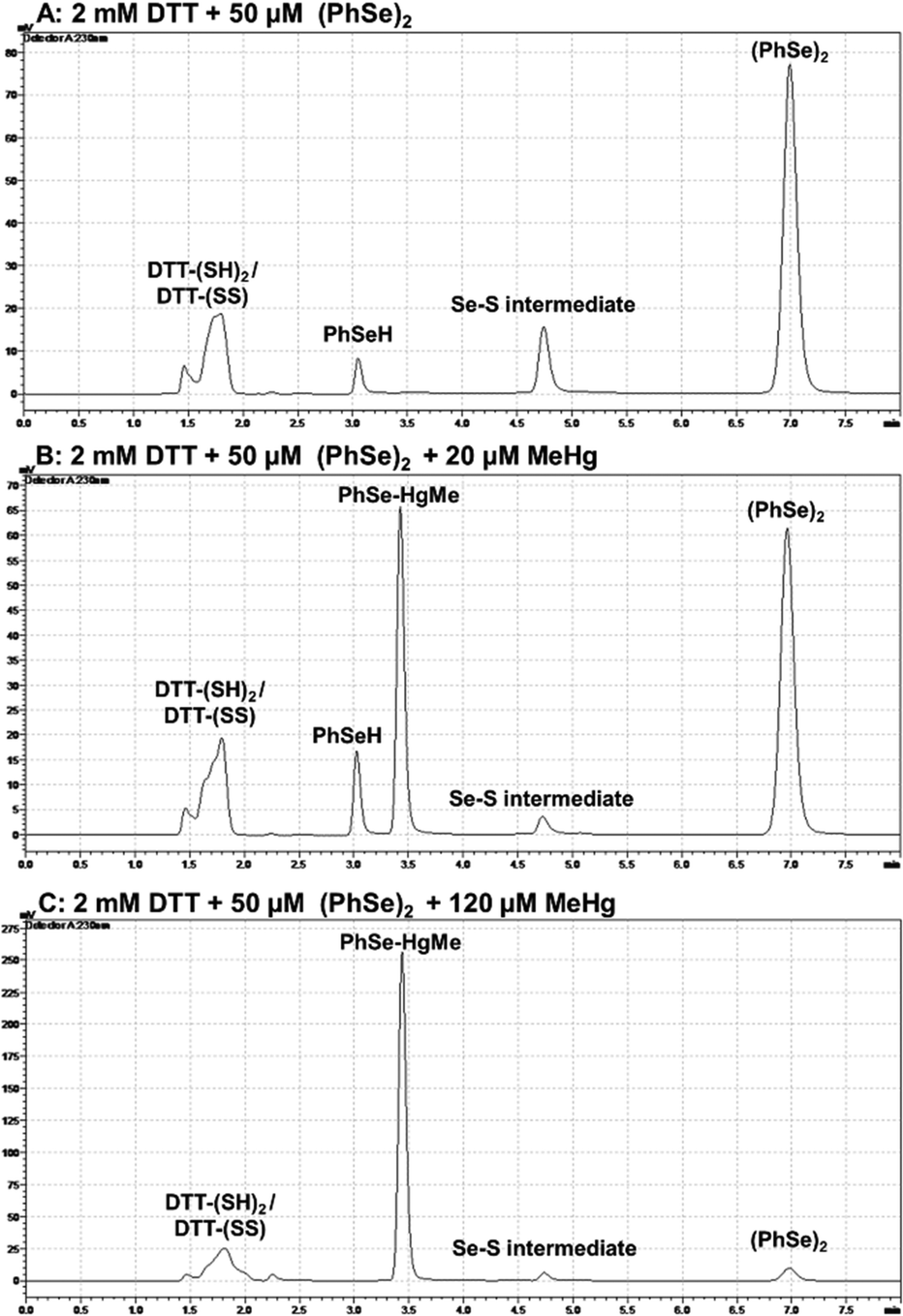 | ||
| Fig. 5 Chromatographic detection of thiol oxidase intermediates using DTT as the thiol source. The peaks of reduced (DTT-(SH)2) and oxidized DTT (DTT-(SS)) are overlapped. The data suggested that a near stoichiometric reaction occurred between MeHg+ and the PhSeH intermediate. Furthermore, there was a clear preference for the PhSeH to react first with MeHg+ or to displace the MeHg+ that could be coordinated to the –SH groups of DTT. Note that the y-axis was not identical in parts A (max. 70 mV), B (max. 80 mV) and C (max. 275 mV). | ||
3.4. DFT calculations
Aiming at better understanding the Se–Se reactivity with thiols, we studied the nucleophilic attack of methyl thiolate at different diphenyl diselenides (with activating and deactivating groups on the rings) using a validated DFT approach (see the Experimental section).(PhSe)2 and its analogues can be found in different conformations, due to the flexibility of the Se–Se bond and the possibility of the aryl rings rotating along the C–Se bonds. This aspect was studied in depth previously,19,68 and the relevant outcomes were reproduced also for the systems studied in the present work, in the gas phase as well as in water: all the diselenides adopt a skewed conformation with the C–Se1–Se2–C dihedral (φ1, Fig. 6 and Table 1) close to 90°. In addition, for all the reactants, the most stable conformation of the phenyl rings was found to be ‘open’ (φ1 = C2–C1–Se1–Se2 and φ2 = Se1–Se2–C1′–C2′, dihedrals close to 90°, Table 1) except for R = CF3 (the most electron withdrawing substituent) which prefers a ‘closed’ conformation (φ1 and φ2 dihedrals close to 0°, Table 2). Notably, in the study by Torsello et al.,69 the NO2 group, which has well known electron-withdrawing characteristics, influences the minimum energy conformation to be closed. So far, the correlation between the ring substituent and the conformation is unexplored.
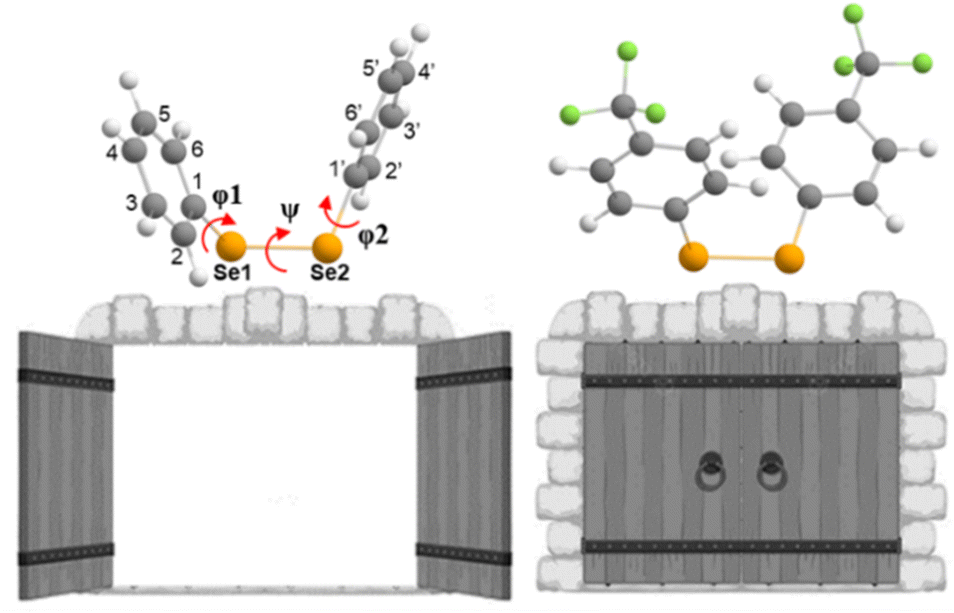 | ||
| Fig. 6 Schematic molecular structures of (PhSe)2 and (CF3PhSe)2, in their open (left) and closed (right) conformations (in the water phase), respectively. ψ, φ1 and φ2 dihedral angles are shown. Molecules are represented by the ball-and-stick model, colour code H: white, C: grey, F: green and Se: orange. Level of theory: COSMO-ZORA-OLYP/TZ2P. | ||
| Structure | R | Distances (Å) | Angles (°) | ||||
|---|---|---|---|---|---|---|---|
| Se1–Se2 | Se2–S | Se1–Se2–S | ψ | φ1 | φ2 | ||
| ψ = C1–Se1–Se2–C1′; φ1 = C2–C1–Se1–Se2; and φ2 = Se1–Se2–C1′–C2′. | |||||||
| React | CH3O | 2.37 | — | — | −87.12 | −84.14 | −80.59 |
| CH3 | 2.36 | — | — | −86.12 | −84.60 | −76.89 | |
| H | 2.36 | — | — | −84.56 | −77.15 | −78.20 | |
| Cl | 2.36 | — | — | −86.21 | −84.04 | −76.20 | |
| CF3 | 2.35 | — | — | −84.21 | −74.19 | −60.39 | |
| TCI | CH3O | 2.86 | 2.45 | 172.15 | −78.96 | −55.91 | −77.17 |
| CH3 | 2.87 | 2.44 | 170.97 | −80.67 | −06.58 | −62.81 | |
| H | 2.87 | 2.43 | 170.22 | −81.33 | 00.42 | −58.65 | |
| Cl | 2.90 | 2.42 | 169.43 | −80.56 | 03.29 | −51.75 | |
| CF3 | 3.05 | 2.36 | 160.20 | −82.24 | 11.31 | −13.75 | |
| Struct | R | Distances (Å) | Angles (°) | ||||
|---|---|---|---|---|---|---|---|
| Se1–Se2 | Se2–S | Se1–Se2–S | ψ | φ1 | φ2 | ||
| ψ = C1–Se1–Se2–C1′; φ1 = C2–C1–Se1–Se2; and φ2 = Se1–Se2–C1′–C2′. | |||||||
| React | CH3O | 2.38 | — | — | −87.97 | −84.54 | −82.12 |
| CH3 | 2.37 | — | — | −86.80 | −84.26 | −76.53 | |
| H | 2.36 | — | — | −85.42 | −77.01 | −75.33 | |
| Cl | 2.37 | — | — | −86.83 | −83.52 | −74.17 | |
| CF3 | 2.32 | — | — | −89.00 | 06.69 | 00.86 | |
| TS | CH3O | 2.44 | 3.42 | 163.79 | −84.82 | −80.34 | −81.92 |
| CH3 | 2.42 | 3.51 | 163.79 | −84.18 | −77.54 | −80.61 | |
| H | 2.41 | 3.59 | 163.62 | −84.00 | −74.69 | −80.13 | |
| Cl | 2.40 | 3.65 | 163.75 | −83.61 | −76.20 | −79.36 | |
| CF3 | 2.37 | 3.75 | 162.40 | −83.03 | −16.23 | −63.72 | |
In the gas phase, the reaction proceeds with the formation of a stable three-center intermediate (TCI), which is consistent with an addition–elimination mechanism.56 Calculations of the Gibbs free energies show that both the initial addition (ΔGadd), leading to the TCI, and the final elimination (ΔGelm), to afford the products, are thermodynamically favored for all the investigated substituents. Moreover, no activation barriers were found for both the addition and the elimination process, indicating that these processes should be diffusion limited in the gas phase. The Gibbs reaction energies of the nucleophilic attack of CH3S− at the diselenide substrates (Table 3) suggest that, in the gas phase, an increasing electron withdrawing character of the substituent groups makes both ΔGadd and ΔGelm more negative due to a more positively polarized selenium atom that has a stronger electrostatic interaction with the incoming thiolate and the increased stabilization of the produced selenolate anion which can more effectively delocalize its charge in the presence of strong electron withdrawing groups. The structural parameters support these results: in the reactants, there is only a very small effect on the Se1–Se2 bond length but in the TCIs a clear trend is established that is the Se2–S bond length decreases and the Se1–Se2 bond length becomes larger with increasingly more electron withdrawing substituents, moving towards a more product-like TCI (Table 1). The different conformation of the R = CF3 reactant, which is maintained also in the TCI, does not alter this trend. Hirshfeld charge analysis shows how the addition of the thiolate results in a strong polarization of the Se1–Se2 bond with an accumulation of negative charge on Se1 (Table 4). Moreover, this effect is accentuated by the nature of the R groups on the phenyl rings with the more electron withdrawing ones having a stronger effect.
| R | Gas phase | Water | ||
|---|---|---|---|---|
| ΔGadd | ΔGelm | ΔG‡ | ΔG | |
| CH3O | −14.16 | −17.86 | 14.61 | −4.99 |
| CH3 | −15.47 | −19.04 | 13.17 | −4.30 |
| H | −18.96 | −20.45 | 11.71 | −6.10 |
| Cl | −25.00 | −25.82 | 10.99 | −9.04 |
| CF3 | −33.88 | −36.69 | 16.23 | −9.62 |
| Struct | Partial charges (eV) | ω | |||
|---|---|---|---|---|---|
| Se1 | Se2 | S | |||
| React | MeS− | — | — | −0.738 | — |
| CH3O | 0.017 | 0.017 | — | 0.083 | |
| CH3 | 0.024 | 0.024 | — | 0.090 | |
| H | 0.030 | 0.030 | — | 0.097 | |
| Cl | 0.035 | 0.035 | — | 0.112 | |
| CF3 | 0.052 | 0.052 | — | 0.129 | |
| TCI | CH3O | −0.276 | 0.010 | −0.219 | — |
| CH3 | −0.254 | 0.021 | −0.209 | — | |
| H | −0.251 | 0.023 | −0.204 | — | |
| Cl | −0.247 | 0.031 | −0.185 | — | |
| CF3 | −0.249 | 0.062 | −0.127 | — | |
In water, the reaction mechanism shifts towards a bimolecular substitution displaying a transition state structure with a tri-coordinated Se2 atom resembling the gas phase TCI (Fig. 7), in agreement with previous results on SN2 mechanisms involving heavy central atoms.56,70 In all the reactions, the starting conformation of the reactant is maintained also in the TS, meaning that, apart from R = CF3 (which in water is found in the ‘closed’ conformation), in all the TSs φ1 and φ2 dihedrals are found to be close to 90°. To confirm this change in mechanism, intrinsic reaction profiles in water were computed for all the reactions (Fig. 8). Calculations agree with the predicted SN2 mechanism with reaction energies that become more negative with the substituent order CH3O > CH3 > H > Cl > CF3. A similar trend is seen in the electronic activation energies which decrease from activating to deactivating groups. In addition, it is important to observe that R = Cl and R = CF3 have the lowest values and very similar reaction barriers (Fig. 8).
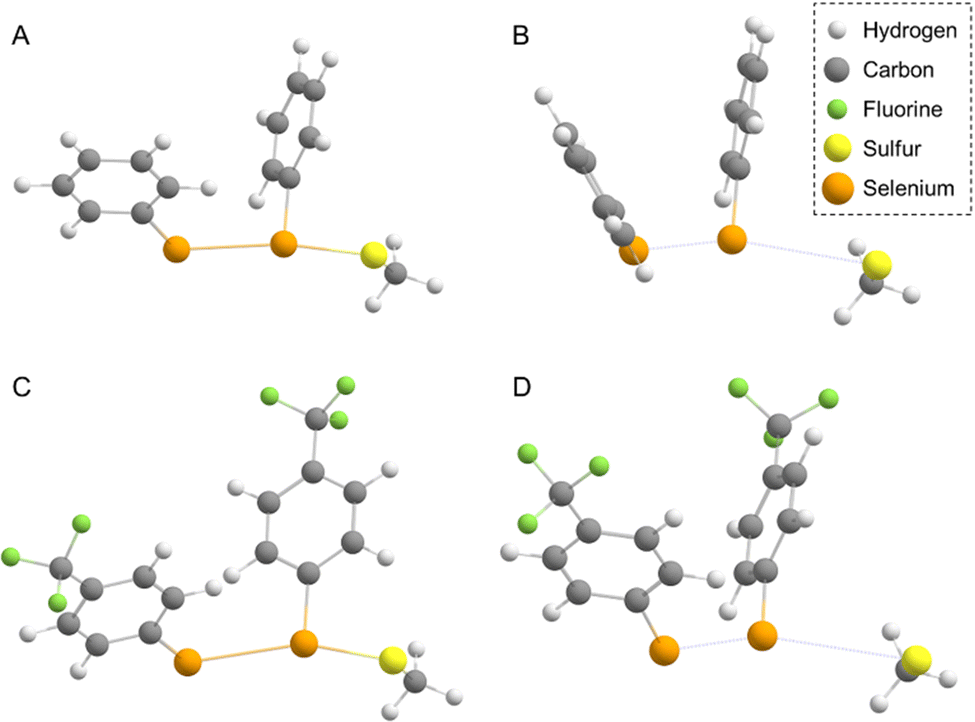 | ||
| Fig. 7 Optimized geometries of TCI (A) and (C) and TS (B) and (D) from (PhSe)2 (A), (B) and (CF3PhSe)2 (C), (D), respectively. Molecules are represented by the ball-and-stick model, colour code H: white, C: grey, F: green and Se: orange. Level of theory: (COSMO)-ZORA-OLYP/TZ2P. | ||
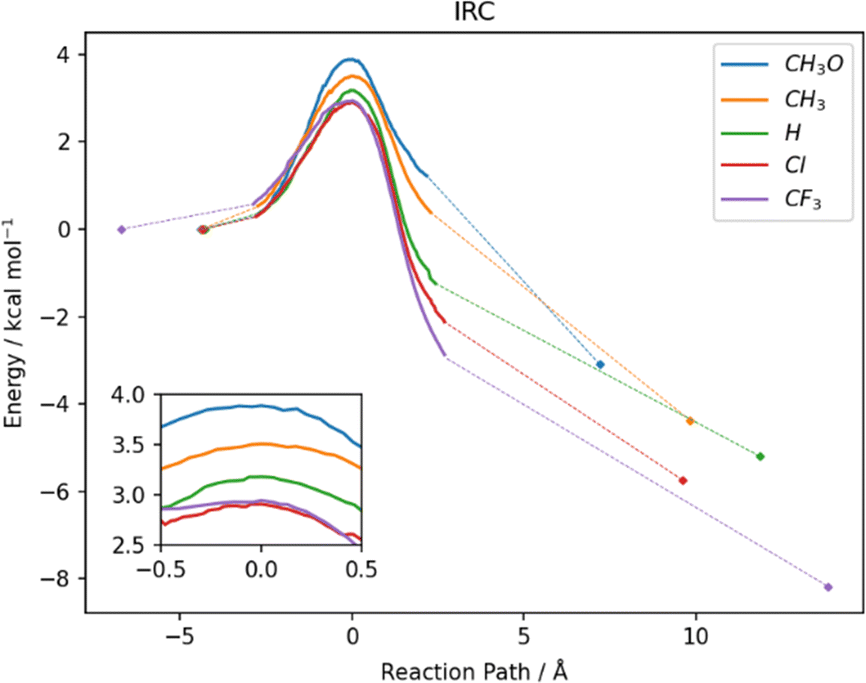 | ||
| Fig. 8 IRC profiles of the investigated reactions in water. The dashed lines represent the path between the last computed IRC point and the geometry optimized structures of reactants and products (colored diamonds). The TS region is shown in more detail in the inset. Level of theory COSMO-ZORA-OLYP/TZ2P. | ||
Taking into account the thermal contributions, the Gibbs free energies of the reaction in solution are less negative than those in the gas phase, since the presence of the solvent electrostatic field stabilizes the charged species found in the reactants and products with a stronger effect on the reacting thiolate than on the produced selenolate, thus mitigating the energy difference of the two anions.71,72 The reaction with R = CF3 presents the largest negative energy, being around twice the one computed for R = CH3O (ΔΔG = 4.62 kcal mol−1, Table 3) confirming the beneficial effect of the electron withdrawing groups in yielding more favourable thermodynamics.
From a kinetic point of view, the cleavage of the diselenide bond by thiolate is seen to display a lower free activation energy as the electron withdrawing nature of the substituents increases, in accordance with what was computed for electronic energies in the IRC profiles (Fig. 8). An exception to this trend, that is seen only in free activation energies, involves R = CF3. Most likely due to the peculiar configuration adopted by the reactants and transition state in the process (vide supra) the free activation energy computed for R = CF3 is 16.23 kcal mol−1, which is found to be the highest in the series. The change in configuration displayed by R = CF3 (the most electron withdrawing substituents among those investigated) and the subsequent increase in the free activation energy point out that there seems to be a pivot point after which increasing the electron withdrawing character of the substituent results in a decrease in the rate of the reaction due to the conformational change from ‘open’ to ‘closed’. Inspection of the TS structures shows that for all the substituents the Se2–S distance is found to be larger than the Se1–Se2 one, which is computed to be close to that of the free reactants, i.e. the TSs are more reactant-like. In addition, the effect of the different substituents is noticeable since an increase in the Se2–S distance and a parallel decrease of that of Se1–Se2 are computed upon increasing their electron withdrawing character.
By analyzing the Hirshfeld partial charges of TSs and reactants (Tables 4 and 5), we can see that there is polarization of the Se1–Se2 bond, albeit being less pronounced than in the gas phase, due to the greater Se2–S distance found in the TSs with respect to the TCIs. The Se nucleus becomes more positive when going from CH3O to CF3, which is in agreement with the electron withdrawing trend. In addition, these data fit well with the electrophilicity index (ω) obtained from ADF, where diselenides with stronger electron withdrawing groups are more electrophilic.
| Struct | Partial charges | ω | |||
|---|---|---|---|---|---|
| Se1 | Se2 | S | |||
| React | MeS− | — | — | −0.848 | — |
| CH3O | 0.000 | 0.000 | — | 0.100 | |
| CH3 | 0.012 | 0.012 | — | 0.103 | |
| H | 0.022 | 0.022 | — | 0.107 | |
| Cl | 0.023 | 0.030 | — | 0.115 | |
| CF3 | 0.101 | 0.102 | — | 0.120 | |
| TS | CH3O | −0.071 | −0.032 | −0.686 | — |
| CH3 | −0.050 | −0.022 | −0.704 | — | |
| H | −0.032 | −0.008 | −0.719 | — | |
| Cl | 10.017 | −0.002 | −0.726 | — | |
| CF3 | 0.031 | 0.050 | −0.741 | — | |
4. Conclusions
In this work, we have investigated the reaction of diphenyl diselenides with thiols by chromatographic, spectroscopic and computational methods. The experimental data show that selenol is formed by the cleavage of the Se–Se bond by thiols and successfully trapped by MeHg+ and IAcOH electrophiles. The thiol oxidase activity of diselenides proves that deactivating and activating groups have an influence on the catalytic cycle, with (ClPhSe)2 and (PhSe)2 presenting similar catalytic activity and (CH3OPhSe)2 presenting the lowest one, which is in accordance with the literature.21 In addition, the calculations show a barrierless process in the gas phase which turns to an activated SN2 reaction in the presence of the solvent. The effect of different substituents on the rings was analyzed in silico. Two distinct effects on the kinetics of these substitutions in the condensed phase can be observed: (i) more electron withdrawing substituents on the rings favor the reaction effectively reducing the activation energy and (ii) the presence of a very strong electron withdrawing moiety on the phenyl rings, such as CF3, causes the starting reactant to adopt a different conformation which could explain the increase in the reaction barrier found for this particular substituent. These data suggest that the thiol modifier effect, i.e., the reaction of diselenides with thiol/thiolate targets (for instance proteins, enzymes, and LMM-SH), is dependent on the substituents on the phenyl rings. Not only the electronic effects of these moieties affect the electron density around the Se atom, but a geometrical modification, found as we move towards strong electron withdrawing groups, affects the structural features of the reactant causing a different starting conformation.This interplay between electronic and geometric features combined with the different nature of nucleophiles and the environment (solution, cell, organism, etc.) found in biological systems can indeed play an important role in the activity of this class of molecules.
Author contributions
Conceptualization: P. A. N., J. B. T. R., and L. O.; investigation: P. A. N., C. S. O., A. M., and M. B.; supervision: J. B. T. R. and L. O.; writing – initial draft: P. A. N., C. S. O., A.M., M.B., J. B. T. R., and L. O.; writing – review and editing: P. A. N., C. S. O., A. M., M. B., J. B. T. R., and L. O.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the financial support from the Coordination for Improvement of Higher Education Personnel CAPES/PROEX (no. 23038.005848/2018-31 and no. 0737/2018). J. B. T. R. and P. A. N. were funded by CAPES (Edital 09-88887.505377/2020-00 and 88887.511828/2020-00).The authors acknowledge the C3P high performance computing facility of the Dipartimento di Scienze Chimiche at Università degli Studi di Padova (Padova, Italy) and the cloud@CNAF resources (project Insight on Nitrogen Chalcogen Interaction, INCIpit, P.I.: M. B.) granted by the CNAF center of the Italian institute of Nuclear Physics (INFN, Bologna, Italy) for the computational infrastructure. Part of the calculations has been carried out also using CINECA computational facilities, thanks to the ISCRA C project PROSIT2, P.I.: L. O. Open access funding provided by Università degli Studi di Padova within the CRUI-CARE agreement.
References
- W. E. Bradt and J. F. Green, J. Org. Chem., 1937, 1, 540–543 CrossRef CAS
.
- F. V. Singh and T. Wirth, Catal. Sci. Technol., 2019, 9, 1073–1091 RSC
- D. M. Browne, O. Niyomura and T. Wirth, Org. Lett., 2007, 9, 3169–3172 CrossRef CAS PubMed
- F. V. Singh and T. Wirth, Org. Lett., 2011, 13, 6504–6507 CrossRef CAS PubMed
- T. G. Back and Z. Moussa, J. Am. Chem. Soc., 2003, 125, 13455–13460 CrossRef CAS PubMed
- T. G. Back and M. V. Krishna, J. Org. Chem., 1988, 53, 2533–2536 CrossRef CAS
- D. M. Freudendahl, S. Santoro, S. A. Shahzad, C. Santi and T. Wirth, Angew. Chem., Int. Ed., 2009, 48, 8409–8411 CrossRef CAS PubMed
- S. Santoro, B. Battistelli, B. Gjoka, C. Si, L. Testaferri, M. Tiecco and C. Santi, Synlett, 2010, 9, 1402–1406 Search PubMed
- S. Santoro, C. Santi, M. Sabatini, L. Testaferri and M. Tiecco, Adv. Synth. Catal., 2008, 350, 2881–2884 CrossRef CAS
- A. A. Vieira, J. B. Azeredo, M. Godoi, C. Santi, E. N. da Silva Júnior and A. L. Braga, J. Org. Chem., 2015, 80, 2120–2127 CrossRef CAS PubMed
- S. Santoro, J. B. Azeredo, V. Nascimento, L. Sancineto, A. L. Braga and C. Santi, RSC Adv., 2014, 4, 31521–31535 RSC
- C. Santi, C. Tidei, C. Scalera, M. Piroddi and F. Galli, Curr. Chem. Biol., 2013, 7, 25–36 CrossRef CAS
- A. J. Pacula, F. Mangiavacchi, L. Sancineto, E. J. Lenardão, J. Ścianowski and C. Santi, Curr. Chem. Biol., 2016, 9, 97–112 CrossRef
- L. Sancineto, M. Piccioni, S. De Marco, R. Pagiotti, V. Nascimento, A. L. Braga, C. Santi and D. Pietrella, BMC Microbiol., 2016, 16, 220 CrossRef PubMed
- M. H. M. Sari, B. da C. W. Fulco, L. M. Ferreira, N. S. Pegoraro, E. da S. Brum, K. K. Casola, M. C. L. Marchiori, S. M. de Oliveira, C. W. Nogueira and L. Cruz, Eur. J. Pharm. Sci., 2020, 153, 105500 CrossRef CAS PubMed
- L. M. Ferreira, M. H. M. Sari, J. H. Azambuja, E. F. da Silveira, V. F. Cervi, M. C. L. Marchiori, S. S. Maria-Engler, M. R. Wink, J. G. Azevedo, C. W. Nogueira, E. Braganhol and L. Cruz, Invest. New Drugs, 2020, 38, 662–674 CrossRef CAS PubMed
- T. G. Back, Curr. Green Chem., 2016, 3, 76–91 CrossRef CAS
- L. Sancineto, F. Mangiavacchi, C. Tidei, L. Bagnoli, F. Marini, A. Gioiello, J. Scianowski and C. Santi, Asian J. Org. Chem., 2017, 6, 988–992 CrossRef CAS
- M. Bortoli, F. Zaccaria, M. Dalla Tiezza, M. Bruschi, C. F. Guerra, F. Matthias Bickelhaupt and L. Orian, Phys. Chem. Chem. Phys., 2018, 20, 20874–20885 RSC
- G. Ribaudo, M. Bellanda, I. Menegazzo, L. P. Wolters, M. Bortoli, G. Ferrer-Sueta, G. Zagotto and L. Orian, Chem. – Eur. J., 2017, 23, 2405–2422 CrossRef CAS PubMed
- A. Sausen de Freitas, A. De Souza Prestes, C. Wagner, J. Haigert Sudati, D. Alves, L. Oliveira Porciúncula, I. J. Kade and J. B. Teixeira Rocha, Molecules, 2010, 15, 7699–7714 CrossRef CAS PubMed
- J. H. Sudati, P. A. Nogara, R. A. Saraiva, C. Wagner, E. E. Alberto, A. L. Braga, R. Fachinetto, P. C. Piquini and J. B. T. Rocha, Org. Biomol. Chem., 2018, 16, 3777–3787 RSC
- G. Mugesh, A. Panda, H. B. Singh, N. S. Punekar and R. J. Butcher, J. Am. Chem. Soc., 2001, 123, 839–850 CrossRef CAS PubMed
- R. Morgenstern, I. A. Cotgreave and L. Engman, Chem. – Biol. Interact., 1992, 84, 77–84 CrossRef CAS PubMed
- S. R. Wilson, P. A. Zucker, R. R. C. Huang and A. Spector, J. Am. Chem. Soc., 1989, 111, 5936–5939 CrossRef CAS
- L. Flohé, S. Toppo and L. Orian, Free Radical Biol. Med., 2022, 187, 113–122 CrossRef PubMed
- M. Dalla Tiezza, G. Ribaudo and L. Orian, Curr. Org. Chem., 2019, 23, 1381–1402 CrossRef
- L. Orian and S. Toppo, Free Radical Biol. Med., 2014, 66, 65–74 CrossRef CAS PubMed
- M. Bortoli, M. Bruschi, M. Swart and L. Orian, New J. Chem., 2020, 44, 6724–6731 RSC
- L. Orian and L. Flohé, Antioxidants, 2021, 10, 1–22 CrossRef PubMed
- B. K. Sarma and G. Mugesh, J. Am. Chem. Soc., 2005, 127, 11477–11485 CrossRef CAS PubMed
- W. Hassan and J. B. Teixeira Rocha, Molecules, 2012, 17, 12287–12296 CrossRef CAS PubMed
- P. A. Nogara, L. Orian and J. B. T. Rocha, Comput. Toxicol., 2020, 15, 100127 CrossRef PubMed
- L. S. Galant, J. Rafique, A. L. Braga, F. C. Braga, S. Saba, R. Radi, J. B. T. da Rocha, C. Santi, M. Monsalve, M. Farina and A. F. de Bem, Neurochem. Res., 2021, 46, 120–130 CrossRef CAS PubMed
- C. Tidei, M. Piroddi, F. Galli and C. Santi, Tetrahedron Lett., 2012, 53, 232–234 CrossRef CAS
- V. Nascimento, P. S. Cordeiro, M. Arca, F. Marini, L. Sancineto, A. L. Braga, V. Lippolis, M. Iwaoka and C. Santi, New J. Chem., 2020, 44, 9444–9451 RSC
- G. V. Botteselle, W. C. Elias, L. Bettanin, R. F. S. Canto, D. N. O. Salin, F. A. R. Barbosa, S. Saba, H. Gallardo, G. Ciancaleoni, J. B. Domingos, J. Rafique and A. L. Braga, Molecules, 2021, 26, 4446 CrossRef CAS PubMed
- V. Nascimento, N. L. Ferreira, R. F. S. Canto, K. L. Schott, E. P. Waczuk, L. Sancineto, C. Santi, J. B. T. Rocha and A. L. Braga, Eur. J. Med. Chem., 2014, 87, 131–139 CrossRef CAS PubMed
- J. Rafique, S. Saba, R. F. S. Canto, T. E. A. Frizon, W. Hassan, E. P. Waczuk, M. Jan, D. F. Back, J. B. T. Da Rocha and A. L. Braga, Molecules, 2015, 20, 10095–10109 CrossRef CAS PubMed
- V. Nascimento, E. E. Alberto, D. W. Tondo, D. Dambrowski, M. R. Detty, F. Nome and A. L. Braga, J. Am. Chem. Soc., 2012, 134, 138–141 CrossRef CAS PubMed
- S. M. Bachrach, D. W. Demoin, M. Luk and J. v Miller, J. Phys. Chem. A, 2004, 108, 4040–4046 CrossRef CAS
- S. M. Bachrach, C. J. Walker, F. Lee and S. Royce, J. Org. Chem., 2007, 72, 5174–5182 CrossRef CAS PubMed
- M. Bortoli, L. P. Wolters, L. Orian and F. M. Bickelhaupt, J. Chem. Theory Comput., 2016, 12, 2752–2761 CrossRef CAS PubMed
- G. S. Heverly-Coulson, R. J. Boyd, O. Mó and M. Yáñez, Chem. – Eur. J., 2013, 19, 3629–3638 CrossRef CAS PubMed
-
P. A. Nogara, C. S. Oliveira and J. B. T. Rocha, in Organoselenium Chemistry, ed. B. C. Ranu and B. Banerjee, De Gruyter, Berlin, 2020, pp.305–346 Search PubMed
- C. W. Nogueira, N. v Barbosa and J. B. T. Rocha, Arch. Toxicol., 2021, 95, 1179–1226 CrossRef CAS PubMed
- J. B. T. Rocha, R. A. Saraiva, S. C. Garcia, F. S. Gravina and C. W. Nogueira, Toxicol. Res., 2012, 1, 85–102 CrossRef CAS
- C. W. Nogueira and J. B. T. Rocha, J. Braz. Chem. Soc., 2010, 21, 2055–2071 CrossRef CAS
- N. V. Barbosa, C. W. Nogueira, P. A. Nogara, A. F. de Bem, M. Aschner and J. B. T. Rocha, Metallomics, 2017, 9, 1703–1734 CrossRef CAS PubMed
- S. Flemer, Molecules, 2011, 16, 3232–3251 CrossRef CAS PubMed
-
E. J. Lenardão, L. Sancineto and C. Santi,New frontiers in organoselenium compounds, Springer, Switzerland, 2018 Search PubMed
-
V. K. Jain, Organoselenium Compounds in Biology and Medicine: Synthesis, Biological and Therapeutic Treatments, Royal Society of Chemistry, Cambridge, 2017, pp.1–33 Search PubMed
- H. J. Reich and R. J. Hondal, ACS Chem. Biol., 2016, 11, 821–841 CrossRef CAS PubMed
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS
-
AMS 2020.103, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, 2020, http://www.scm.com Search PubMed
- M. Bortoli, L. P. Wolters, L. Orian and F. M. Bickelhaupt, J. Chem. Theory Comput., 2016, 12, 2752–2761 CrossRef CAS PubMed
- E. Van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS
- A. Klamt and V. Jonas, J. Chem. Phys., 1996, 105, 9972–9981 CrossRef CAS
- C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396–408 Search PubMed
- L. Deng, T. Ziegler and L. Fan, J. Chem. Phys., 1993, 99, 3823–3835 CrossRef CAS
- L. Deng and T. Ziegler, Int. J. Quantum Chem., 1994, 52, 731–765 CrossRef CAS
- R. G. Parr, L. v Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS
- B. Wang, C. Rong, P. K. Chattaraj and S. Liu, Theor. Chem. Acc., 2019, 138, 124 Search PubMed
- S. Liu, J. Phys. Chem. A, 2015, 119, 3107–3111 CrossRef CAS PubMed
- F. Zaccaria, L. P. Wolters, C. Fonseca Guerra and L. Orian, J. Comput. Chem., 2016, 37, 1672–1680 CrossRef CAS PubMed
- M. Torsello, A. C. Pimenta, L. P. Wolters, I. S. Moreira, L. Orian and A. Polimeno, J. Phys. Chem. A, 2016, 120, 4389–4400 CrossRef CAS PubMed
- T. A. Hamlin, M. Swart and F. M. Bickelhaupt, ChemPhysChem, 2018, 19, 1315–1330 CrossRef CAS PubMed
- T. A. Hamlin, B. van Beek, L. P. Wolters and F. M. Bickelhaupt, Chem. – Eur. J., 2018, 24, 5927–5938 CrossRef CAS PubMed
- G. Serdaroğlu, Int. J. Quantum Chem., 2011, 111, 3938–3948 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj05976b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |