
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDefect engineering of two-dimensional Nb-based oxynitrides for visible-light-driven water splitting to produce H2 and O2†
Chang
Xu
a,
Yan
Wang
*a,
Quansheng
Guo
b and
Xin
Wang
 *a
*a
aShenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, China. E-mail: xin.wang2@siat.ac.cn
bSchool of Materials Science and Engineering, Hubei University, Wuhan 430062, China
First published on 6th April 2023
Abstract
Two-dimensional (2D) Nb-based oxynitrides are promising visible-light-responsive photocatalysts for the water splitting reaction, but their photocatalytic activity is degraded by the formation of reduced Nb5+ species and O2− vacancies. To understand the influence of nitridation on the formation of crystal defects, this study synthesized a series of Nb-based oxynitrides through the nitridation of LaKNaNb1−xTaxO5 (x = 0, 0.2, 0.4, 0.6, 0.8, 1.0). During nitridation, K and Na species volatilized, which helped transform the exterior of LaKNaNb1−xTaxO5 into a lattice-matched oxynitride shell. Ta inhibited defect formation, yielding Nb-based oxynitrides with a tunable bandgap between 1.77 and 2.12 eV, straddling the H2 and O2 evolution potentials. After loading with Rh and CoOx cocatalysts, these oxynitrides exhibited good photocatalytic activity for H2 and O2 evolution in visible light (650–750 nm). The nitrided LaKNaTaO5 and LaKNaNb0.8Ta0.2O5 delivered the maximum H2 (19.37 μmol h−1) and O2 (22.81 μmol h−1) evolution rates, respectively. This work provides a strategy for preparing oxynitrides with low defect densities and demonstrates the promising performance of Nb-based oxynitrides for water splitting.
Introduction
Photocatalytic water splitting into hydrogen (H2) and oxygen (O2) is regarded as one of the most promising strategies for solving the global energy crisis and mitigating environmental problems.1–3 In the solar spectrum, UV light accounts for less than 5% of the energy in sunlight, whereas visible light constitutes approximately 54%.4–6 In order to achieve high photocatalytic efficiency, semiconductors with narrow band gap energies (Eg) that are capable of absorbing the wide wavelengths of visible light are very attractive.7,8 Recently, metal oxynitrides are being reported continuously, whose Eg is significantly narrower than that of conventional metal oxides because N 2p orbitals provide a new valence band (VB) that is more negative than that provided by O 2p orbitals.9–11Among these oxynitrides, Nb-based oxynitrides exhibit a broad light absorption band in the visible light region, with λmax values up to approximately 750 nm, which is much higher than their Ta-based counterparts.12–15 To date, various Nb-based oxynitrides, such as CaNbO2N (λmax = 600 nm), SrNbO2N (λmax = 690 nm), BaNbO2N (λmax = 740 nm) and LaNbON2 (λmax = 750 nm) have been synthesized by nitriding oxide precursors under an NH3 flow and applied for photocatalysis. For instance, CaNbO2N was found to be active for O2 evolution from a AgNO3 aqueous solution.12 The optimal nitridation temperature in that study was determined to be 1023 K. SrNbO2N prepared by nitriding Sr2Nb2O7 or SrNbO3 exhibited enhanced activity for photoelectrochemical water splitting, with a current density of 0.77 mA cm−2 at 1.2 V versus the reversible hydrogen electrode (RHE).13 Meanwhile, Hisatomi et al. improved the crystallinity and uniformity of BaNbO2N by adding BaCO3 into the Ba5Nb4O15 precursor during the synthesis procedure, and the resulting BaNbO2N exhibited enhanced O2 evolution activity by utilizing photons with wavelengths of up to 740 nm.14 Recently, Wang et al. prepared LaNbON2 with exposed metastable {010} facets by nitriding a plate-like LaKNaNbO5 precursor. In combination with a CoOx co-catalyst, LaNbON2 exhibited enhanced photocatalytic activity for O2 evolution, with an apparent quantum yield of 0.82%, whereas conventional LaNbON2 was almost inactive.15 However, nitridation is a harsh process. N doping always causes a charge imbalance as a result of aliovalent O2−/N3− exchange, yielding numerous crystal defects that are detrimental to the photocatalytic performance of oxynitrides.16,17 Like Nb-based oxynitrides, it still exhibits negligible photocatalytic activity, especially for photocatalyzing H2 evolution, because of the formation of large amounts of reduced Nb5+ species and O2− vacancies during the nitridation process. These vacancies can act as recombination and trapping centers for photoexcited charges, resulting in a decrease in charge separation efficiency.18–20 Moreover, a low concentration of the N dopant inevitably can also decrease the absorption coefficient of N-doped oxides in the visible light region.21 Therefore, undoped oxynitrides containing a stoichiometric amount of N in their crystal structure are highly desirable, which can guarantee strong visible light absorption through band-to-band electron transitions and the development of Nb-based oxynitrides with low defect densities remains a challenge.
Herein, we synthesized a series of Nb-based oxynitrides through the nitridation of LaKNaNb1−xTaxO5 (x = 0, 0.2, 0.4, 0.6, 0.8, 1.0). By introducing Ta, the formation of defects in their crystal structure is effectively inhibited yielding a tunable bandgap. In the meantime, volatilization of K and Na species during nitridation promoted LaKNaNb1−xTaxO5 crystal surfaces to be transformed into lattice-matching oxynitrides, forming a core–shell structure. Considering the above mentioned enhanced properties, these oxynitrides exhibited good photocatalytic activity for H2 and O2 evolution in visible light (650–750 nm).
Results and discussion
Fig. 1 illustrates the crystal structure of LaKNaNb1−xTaxO5, which is similar to that of LaKNaNbO5 (Fig. S1†) and has LaNaNbO5− layers along the [001] direction with K+ ions occupying the spaces between the layers. Each LaNaNbO5 layer is arranged in a “checkerboard” pattern, where NaO5 and NbO5 square pyramids are arranged in alternating rows.23,24 In LaKNaNb1−xTaxO5, a fraction of the Nb atoms are substituted with Ta atoms. Accordingly, LaNbON2 has an orthorhombic perovskite structure with the space group Pnma (Fig. 1). The unit cell parameters of LaNbON2 are a = 5.7221 Å, b = 8.0684 Å, and c = 5.7428 Å.25 As the amount of Ta5+ increases, NbxTa1−xO5 square pyramids in LaNb1−xTaxON2 become distorted because Ta–(O, N) bonds (2.056 Å) are shorter than Nb–(O, N) bonds (2.064 Å). As a result, pure LaTaON2 demonstrated a monoclinic perovskite structure, indicating the successful substitution of Nb by Ta in the oxynitride lattice with angles from 90° to 135° between the a- and c-axes (Fig. S2†).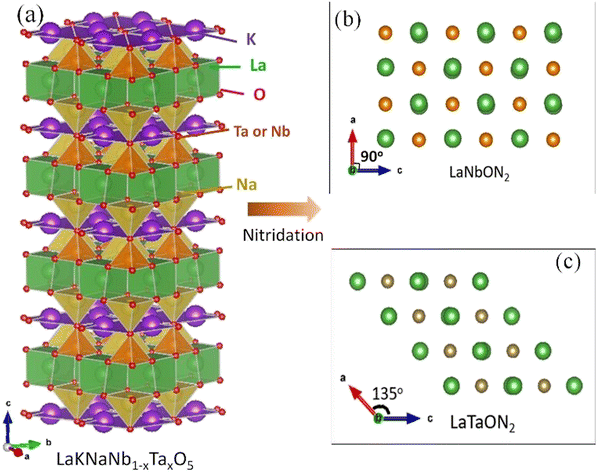 | ||
| Fig. 1 (a) Crystal structure of layered LaKNaNb1−xTaxO5. (b and c) Views of the (010) facets of (b) LaNbON2, and (c) LaTaON2. The crystal structures were visualized using the VESTA suite of programs.22 | ||
Plate-like LaKNaNb1−xTaxO5 was prepared by a NaOH/KOH flux method as previously reported.26,27 Specifically, 5 mmol of La2O3 and 5 mmol in total of Ta2O5 and Nb2O5 at various molar ratios (0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, 1:4, 2:3, 3:2, 4:1, and 5:0) were combined and finely ground in a mortar, after which the mixture was transferred to an alumina crucible containing an excess of KOH (10 g) and NaOH (5 g). Subsequently, the mixture was calcined at 873 K for 3 h and aged at 773 K for 15 h. In this synthesis, KOH and NaOH were in excess during synthesis to serve as major flux components for the crystal growth of LaKNaNb1−xTaxO5 oxides, resulting in large, highly crystalline products. A scheme of the synthetic procedure is shown in Fig. S3.†
:5, 1:4, 2:3, 3:2, 4:1, and 5:0) were combined and finely ground in a mortar, after which the mixture was transferred to an alumina crucible containing an excess of KOH (10 g) and NaOH (5 g). Subsequently, the mixture was calcined at 873 K for 3 h and aged at 773 K for 15 h. In this synthesis, KOH and NaOH were in excess during synthesis to serve as major flux components for the crystal growth of LaKNaNb1−xTaxO5 oxides, resulting in large, highly crystalline products. A scheme of the synthetic procedure is shown in Fig. S3.†
The XRD patterns of the LaKNaNb1−xTaxO5 precursor (x = 0, 0.2, 0.4, 0.6, 0.8, and 1) were nearly identical to those of the tetragonal LaKNaNbO5 phase with a space group of P4/nmm, indicating the phase purity of the flux-grown oxide crystals (Fig. 2a). Notably, the intensity of the diffraction peak at 10° for LaKNaNbO5 was enhanced, indicating the exposure of the {001} facets on the surface. Interestingly, with the increasing Ta content, the position of the diffraction peak of LaKNaNb1−xTaxO5 shifted to a slightly higher angle than that of the LaKNaNbO5 crystals, indicating that the substitution of Nb with Ta caused a decrease in the crystal size and an overall contraction of the lattice parameters in the LaKNaNb1−xTaxO5 samples.28,29 After nitridation at 1173 K for 4 h, peaks attributable to the LaNbON2 phase were observed together with intense LaKNaNbO5 peaks, which could be ascribed to the transformation of the surface of the LaKNaNbO5 precursor into LaNbON2 during nitridation, while the crystal interior remained an oxide (Fig. 2b). Interestingly, the intensity of the diffraction peaks attributed to the LaNb1−xTaxON2 phase became weaker with the increasing Ta content. This finding indicates that introducing Ta species can slow down the transformation from the LaKNaNb1−xTaxO5 precursor into its lattice-matched LaNb1−xTaxON2 owing to the lower electronegativity of the Ta species. Consequently, this transformation process can be finely tuned by varying the degree of substitution.
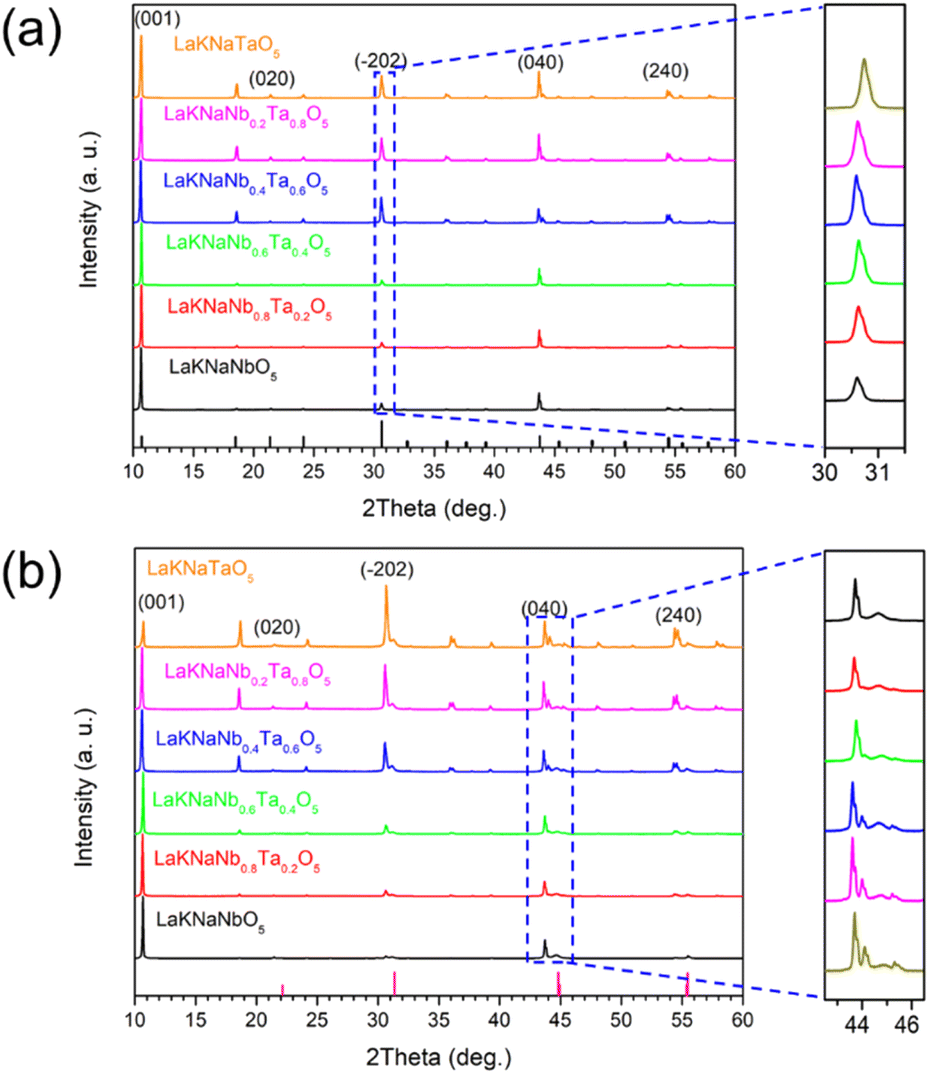 | ||
| Fig. 2 XRD patterns of the (a) layered LaKNaNb1−xTaxO5 oxide precursors and (b) nitrided LaKNaNb1−xTaxO5 (x = 0, 0.2, 0.4, 0.6, 0.8, 1.0). | ||
SEM was used to investigate the size and morphology of the resulting products. As shown in Fig. 3a and b, the oxide precursors that were not thermally aged at 773 K for 15 h exhibited an incomplete plate morphology with a rough surface. In contrast, the aged LaKNaNb1−xTaxO5 showed a plate-like shape with a smooth surface, indicating that the long-term flux process facilitates the synthesis of oxide precursors with controllable morphology and high crystallinity (Fig. S4†). After nitridation, the prepared sample maintained its plate-like shape but became porous, as illustrated in Fig. 3c and S5.† The corresponding cross-sectional SEM image in Fig. 3d shows the formation of a core–shell structure based on a porous oxynitride shell on a dense oxide core. The core–shell structure has the advantage of generating as many oxynitrides on the oxide surface as possible during the mild nitridation process while minimizing the number of crystal defects of oxynitrides as much as possible.30
 | ||
| Fig. 3 SEM images of layered LaKNaNb1−xTaxO5 (a) calcined at 873 K for 3 h and (b) then aged at 773 K for 15 h (c) surface and (d) cross-sectional SEM images of LaKNaNb1−xTaxO5 powder nitrided at 1123 K for 4 h, revealing the porous surface. | ||
The exposed facets of oxynitrides after nitridation were confirmed using TEM and high-resolution TEM (HRTEM) images (Fig. 4a and b, respectively), where the interplanar distances associated with the (200) and (002) facets of LaNb1−xTaxON2 were 0.228 and 0.202 nm, respectively, with an angle of approximately 105°. This finding indicates that the La and Nb atoms are located in the expected positions in the orthorhombic structure viewed in the [010] direction, which energetically favors the {010} facet on the surface. The energy dispersive spectroscopy (EDS) mapping images shown in Fig. 4c show that the resulting product is composed of La, K, Na, Nb, Ta, O, and N. Comparing the atomic ratios obtained from the EDS spectrum after nitridation with the known composition before nitridation reveals that the amounts of K and Na decreased dramatically during the nitridation process (Fig. S6†).
 | ||
| Fig. 4 (a) TEM and (b) HRTEM images and (c) EDS elemental maps of plate-like LaKNaNb0.8Ta0.2O5 nitrided at 1123 K for 4 h. The atomic ratios were approximately La/Nb ≈ 1.3, K/Nb < 0.05, Na/Nb < 0.05, O/Nb ≈ 1.3, and N/Nb ≈ 2.5. | ||
In the photographs of the nitrided LaKNaNb1−xTaxO5 powder samples (Fig. 5a), the color of the samples clearly changes from dark brown to orange with the increasing amount of Ta5+. Meanwhile, in the UV-vis diffraction reflectance spectra (DRS) of the nitrided LaKNaNb1−xTaxO5 samples presented in Fig. 5b, the absorption edges of the nitrided LaKNaNb1−xTaxO5 samples gradually blue-shifted from 720 to 600 nm with the increasing Ta content, indicating that the bandgap energies of these materials were greater than those of pure LaNbON2 (λ ≈ 740 nm) during mild nitridation.15 The corresponding Tauc plots of the nitrided LaKNaNb1−xTaxO5 samples (Fig. 5c) based on the UV-vis DRS spectra reveal that the bandgap energy increased monotonically from 1.77 to 2.12 eV with increasing substitution with Ta5+. Moreover, their bandgap positions were estimated based on the VB XPS data (Fig. S7 and Table S1†) along with the Tauc plots. As shown in Fig. 5d, the VB of the nitrided LaKNaNb1−xTaxO5 samples remained almost unchanged with the increasing amount of Ta5+, and the enlarged bandgaps of these samples mainly resulted from the upshift of the conduction band (CB), demonstrating that the Ta CB orbitals mainly contributed to the Ta 5d orbitals.
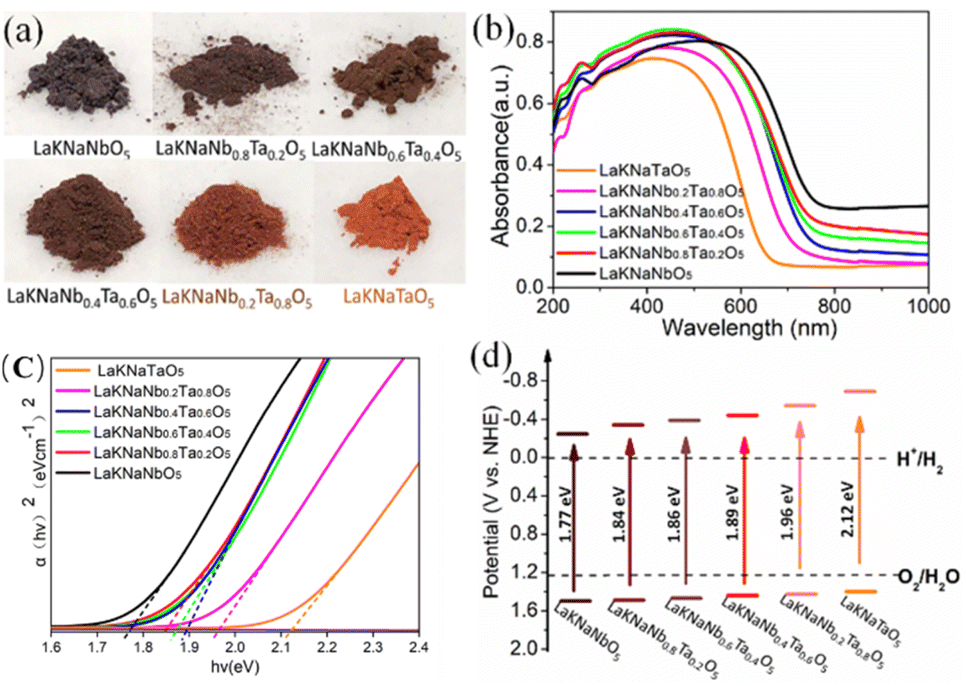 | ||
| Fig. 5 (a) Photographs, (b) UV-vis DRS, (c) corresponding Tauc plots, and (d) band structure diagrams of the LaKNaNb1−xTaxO5 samples (x = 0, 0.2, 0.4, 0.6, 0.8, 1.0) nitrided at 1123 K for 4 h. | ||
Notably, the nitrided LaKNaNbO5 samples exhibited obvious background absorption at wavelengths >650 nm, which could be ascribed to the formation of a large amount of reduced Nb5+ species as well as O2− vacancies.18,19 Reduced metallic species are known not to exhibit photocatalytic activity owing to bandgap excitation, and such defect states usually generated by prolonged exposure to NH3 during nitridation at high temperatures can act as recombination and trapping centers for photoexcited charges. In particular, the background absorption became much weaker as the amount of Ta5+ increased, indicating that fewer crystal defects formed during the nitridation process. This is because Ta is less electronegative than Nb, which slowed down the nitridation process, thus inhibiting the typical formation of crystal defects in the nitrided LaKNaNb1−xTaxO5 samples.
The chemical compositions of the nitrided LaKNaNb1−xTaxO5 samples were analyzed using high-resolution XPS. The survey spectra are shown in Fig. S8a.† They confirm the presence of Ta, Nb, N and O in the as-prepared nitrided LaKNaNb1−xTaxO5 samples. By comparing with the unnitrided sample (Fig. S8b†), the presence of N on the surface of nitrided LaKNaNb1−xTaxO5 can be confirmed. As shown in Fig. 6a, the Nb valence species at the surface of the nitrided LaKNaNb1−xTaxO5 sample were evaluated to investigate the effect of adding Ta5+ on the reduction of Nb5+ during nitridation. The Nb 3d XPS spectra were deconvoluted into peaks for three different Nb valence species,31 Nb5+, Nb4+, and Nb3+, based on the tailing of the peaks toward lower binding energies, demonstrating that more than one chemical state exists in the samples. Here, the presence of Nb4+ and Nb3+ indicates the reduction of the Nb5+ species derived from NH3 nitridation. These reduced Nb4+ and Nb3+ species can act as charge trapping and recombination centers for the photoexcited electrons and holes. Table S2† summarizes the proportions of surface Nb species in the nitrided LaKNaNb1−xTaxO5 sample with different amounts of Ta5+. With increasing Ta5+, the nitrided LaKNaNb1−xTaxO5 samples exhibited the decreased presence of reduced Nb species (both Nb3+ and Nb4+) from 82.06% for nitrided LaKNaNbO5 without Ta to 48.61% for nitrided LaKNaNb0.2Ta0.8O5. These results indicate that substitution with Ta5+ species can effectively inhibit the reduction of Nb.
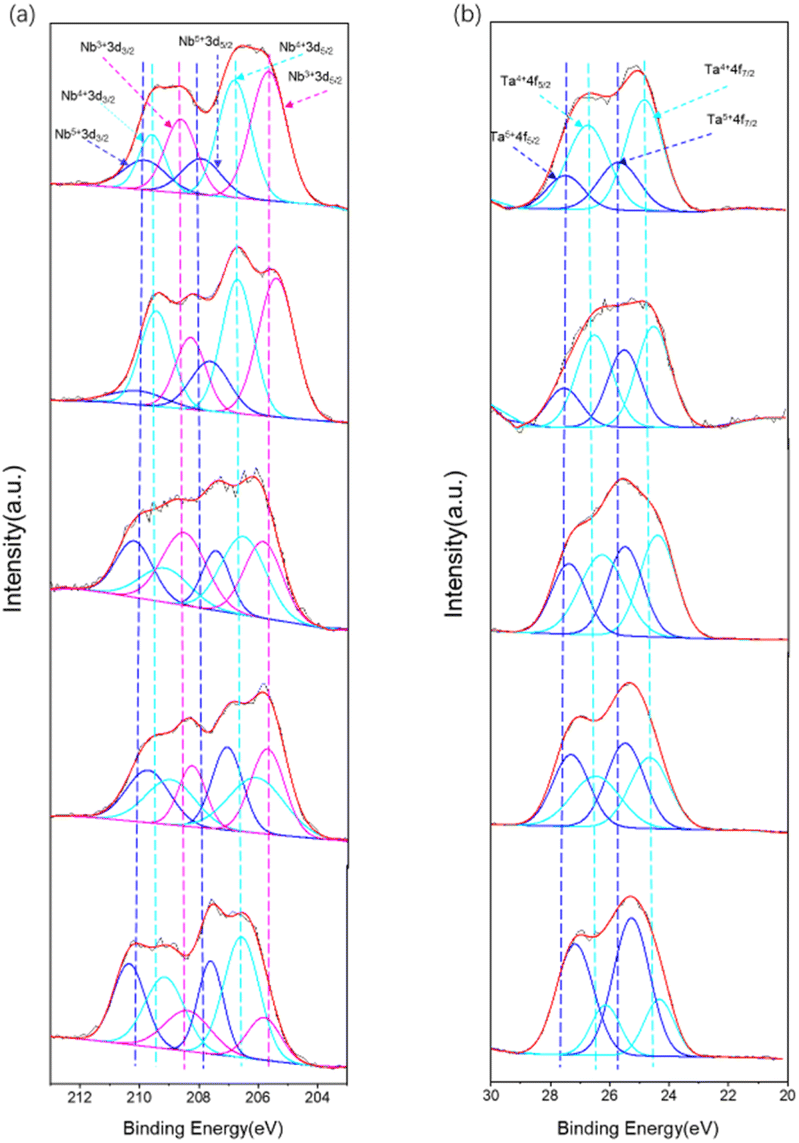 | ||
| Fig. 6 High-resolution XPS spectra of the LaKNaNb1−xTaxO5 samples nitrided at 1123 K for 4 h: (a) Nb 3d spectra for x = 0, 0.2, 0.4, 0.6 and 0.8 from up to down; (b) Ta 4f spectra for x = 0.2, 0.4, 0.6, 0.8 and 1.0 (no Nb) from up to down. | ||
A similar phenomenon was also observed for the Ta5+ species in the deconvoluted Ta 4f XPS spectra of the nitrided LaKNaNb1−xTaxO5 samples, which can be deconvoluted into two different reduced Ta species,32,33 Ta5+ and Ta4+, based on the binding energies (Fig. 6b).30 The increasing Ta5+ content was found to inhibit the reduction of Ta5+ species, from 70.72% for the nitrided LaKNaTa0.2Nb0.8O5 sample to 25.84% for the nitrided LaKNaTaO5 sample (Table S3†). Therefore, according to the band diagrams and XPS data, the replacement of Nb5+ with Ta5+ evidently hindered the reduction of both Nb and Ta during nitridation, thus tuning the band structure of the nitrided LaKNaNb1−xTaxO5 sample while maintaining the visible-light-driven photocatalytic activity.
Moreover, the O 1s state of the oxide precursor could be decomposed into two overlapping peaks centered at 529.6 and 532.5 eV (Fig. S9†). These two peaks could be attributed to lattice oxygen in the oxide (OL) and hydroxyl groups (O–H) that adsorbed on the sample surfaces during sample handling, respectively.34,35 As illustrated in Fig. S10,† a new peak centered at 531.4 eV appeared after nitridation, the intensity of which is partly related to the concentration of O2− vacancies. Notably, the intensity of the Ov peak decreased from 43.27% for LaKNaNbO5 (no Ta5+) to 20.75% for LaKNaTaO5 in which Nb5+ was completely replaced with Ta5+, indicating that fewer O2− vacancies formed in the nitrided samples (Table S4†).
The photocatalytic performance of the nitrided LaKNaNb1−xTaxO5 samples toward the evolution of H2 and O2 under visible light was examined in an aqueous methanol solution loaded with a Rh co-catalyst and an aqueous AgNO3 solution loaded with the CoOx co-catalyst. Fig. 7a shows the H2 evolution as a function of time. Evidently, all the nitrided LaKNaNb1−xTaxO5 samples showed photocatalytic activity for H2 evolution. However, the nitrided sample without Ta5+ showed the lowest activity (0.2635 μmol h−1, Table S5†), which could be ascribed to the formation of large amounts of reduced Nb5+ species and O2− vacancies in the LaNbON2 shells, as discussed in the analyses of the DRS and XPS data (Fig. 5b and 6). In contrast, the activity of the nitrided LaKNaNbO5 sample was enhanced by the increased substitution of Nb with Ta5+. Thus, introducing Ta5+ species effectively inhibited the reduction of Nb species, thereby decreasing the possibility of photogenerated charges recombining at defects and enhancing the photocatalytic activity toward H2 evolution.15,18,30 After complete Ta5+ substitution, the nitrided LaKNaTaO5 sample showed the highest photocatalytic H2 evolution activity (19.37 μmol h−1) because of the presence of a low-defect-density LaTaON2 shell on the lattice-matched LaKNaTaO5 core.
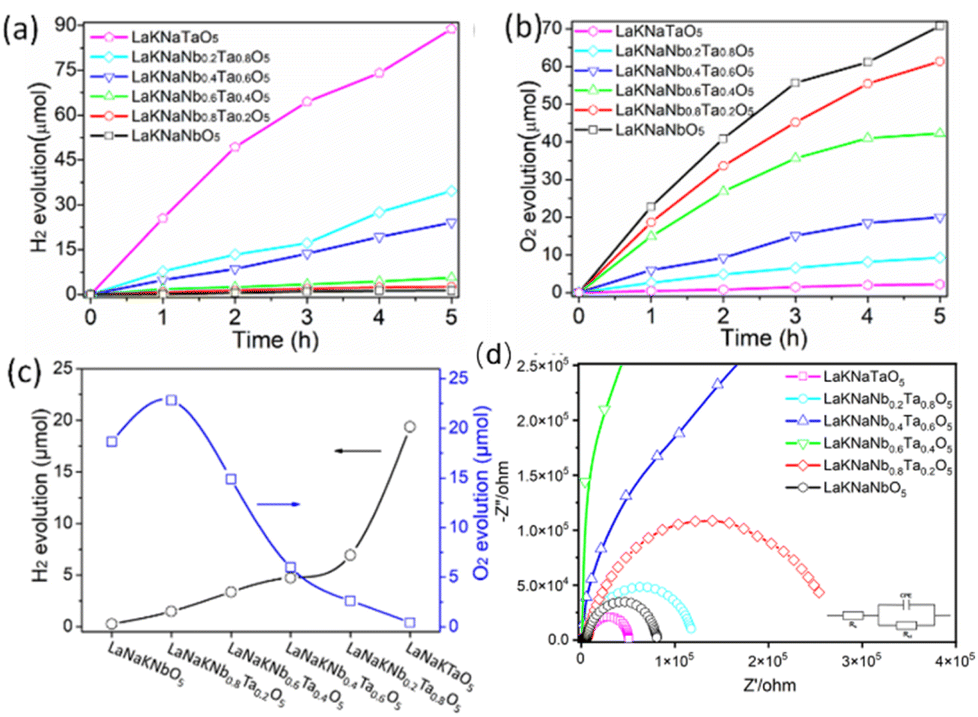 | ||
| Fig. 7 Photocatalytic performance of LaKNaNb1−xTaxO5 nitrided at 1123 K for 4 h (x = 0, 0.2, 0.4, 0.6, 0.8, 1.0): (a) H2 and (b) O2 evolution as a function of time, (c) H2 and O2 evolution rates, and (d) EIS spectra recorded under visible light illumination. Reaction conditions for H2 evolution: 150 mL of aqueous methanol (20 vol%), photocatalyst: 300 mg, co-catalyst: 0.5 wt% Rh, loading method: impregnation with H2 reduction, light source: a 300 W xenon lamp (λ ≥ 420 nm), reaction cell: a top-irradiation cell with a Pyrex window. Reaction conditions for O2 evolution: 150 mL of an aqueous 50 mM AgNO3 solution containing 0.20 g of La2O3, photocatalyst: 300 mg, co-catalyst: 0.5 wt% CoOx, light source: a 300 W xenon lamp (λ ≥ 420 nm), reaction cell: a top-irradiation cell with a Pyrex window. | ||
Moreover, the photocatalytic activity of nitrided LaKNaNb1−xTaxO5 for O2 evolution was evaluated by modifying the CoOx co-catalyst (Fig. 7b). The treatment temperature of the CoOx co-catalyst modified on the sample surface was first optimized (Fig. S11†). A low treatment temperature resulted in low photocatalytic activity for O2 evolution owing to weak interfacial bonding between the CoOx co-catalyst and LaNb1−xTaxON2. On the other hand, a high treatment temperature above 873 K dramatically decreased the photocatalytic activity for O2 evolution, which could be ascribed to the severe aggregation of the CoOx co-catalyst at high temperature. Therefore, the sample that was treated at ∼773 K showed the highest photocatalytic activity for O2 evolution owing to its well-integrated interface between the CoOx co-catalyst and LaNb1−xTaxON2.36 Although the nitrided LaKNaNbO5 sample without Ta possessed a large number of crystal defects, it showed high photocatalytic activity for O2 evolution (18.65 mol h−1), which could be ascribed to the exposure of metastable {010} facets on the surface of LaNbON2 crystals. These facets enabled photogenerated electrons to transfer from the interior of the oxynitride to its surface along the [010] direction, thus improving the charge separation efficiency.15 The photocatalytic O2 evolution activity of nitrided LaKNaNb0.8Ta0.2O5 (i.e., substituted with 20% Ta5+) was the highest (22.81 μmol h−1). However, higher Ta5+ contents decreased the photocatalytic O2 evolution activity of the photocatalysts, even though their defect densities were lower in the formed oxynitride. This could be attributed to the fact that a greater Ta content decreased the transformation of LaKNaNb1−xTaxO5 into LaNb1−xTaxON2. This phenomenon was confirmed using the XRD results shown in Fig. 2b.
Overall, the nitrided LaKNaNb1−xTaxO5 samples showed photocatalytic activity for both H2 and O2 evolution. This activity can also be finely tuned by varying the Ta5+ substitution content, which indicates that these oxynitrides have a bandgap straddling the H2 and O2 evolution potentials and thus show promise for use in the water splitting reactions (Fig. 7c). Furthermore, electrochemical impedance spectra (EIS) were recorded to further confirm the charge separation ability of these nitrided LaKNaNb1−xTaxO5 samples. As illustrated in Fig. 7d, nitrided LaKNaTaO5 exhibited a smaller arc radius than nitrided LaKNaNbO5, demonstrating a smaller resistance to charge transfer, which could be related to the lower amount of crystal defects in the former.37,38 Interestingly, pure nitrided LaKNaNbO5 and LaKNaTaO5 both exhibited a lower resistance to charge transfer than the nitrided LaKNaNb1−xTaxO5 samples, suggesting that the Ta substitution distorted the lattice in the LaKNaNb1−xTaxO5 precursor. Moreover, regarding the stability performance of the nitrided LaKNaNb1−xTaxO5 samples, we carried out XPS analysis on nitrided LaKNaNb0.8Ta0.2O5 before and after the photocatalytic H2 production reaction, which shows no obvious difference between fresh and used catalysts (Fig. S12 and S13†). The detailed peak contents are provided in Table S6.† The ratio of N 1s/Nb 3d decreased slightly from 0.33 to 0.28, and the ratio of N 1s/Ta 4f decreased from 0.86 to 0.80. This result indicates a small decrease in the amount of N on the surface of the sample, which suggests that the nitrided LaKNaNb1−xTaxO5 samples remained stable in this photocatalytic system. These findings provide the basis for further studies on optimizing the nitridation temperature and heat treatment time of oxide precursors to improve the photocatalytic activity of oxynitrides for H2 and O2 evolution.
Conclusion
We investigated the nitridation of layered LaKNaNb1−xTaxO5 oxides and its effect on both crystal defects and the bandgap energy. During nitridation, the volatilization of K and Na species promoted the transformation of the surface of LaKNaNb1−xTaxO5 crystals into lattice-matched oxynitrides, thus forming a core–shell structure. Moreover, introducing Ta species decreased the defect densities, yielding Nb-based oxynitrides with a tunable bandgap from 1.77 to 2.12 eV, which straddled the H2 and O2 evolution potentials. As a result, these oxynitrides exhibited good photocatalytic activities for H2 (19.37 μmol h−1) and O2 (22.81 μmol h−1) evolution, which utilized visible light wavelengths between 650 and 750 nm. This work provides a novel strategy for preparing oxynitrides with a tunable bandgap and low defect densities, thus uncovering the prospects of Nb-based oxynitrides for the overall water splitting reaction.Experimental section
Materials
La2O3, Ta2O5, and Nb2O5 precursors and NaOH and KOH fluxing agents were all obtained from Macklin. Co(NO3)2·6H2O and RhCl3·3H2O co-catalyst precursors were purchased from Sigma-Aldrich.:5, 1:4, 2:3, 3:2, 4:1, and 5:0) were mixed with La2O3 (5 mmol) and finely ground in a mortar. Subsequently, the mixture was added to an alumina crucible containing excess KOH (10 g) and NaOH (5 g). The crucible was then placed in a furnace heated to 873 K at a rate of 10 K min−1. This temperature was maintained for 3 h and then held at 773 K for 15 h. The furnace was subsequently allowed to cool naturally to room temperature. Afterward, the product was washed with ultrapure water at 343 K for 2 h and centrifuged twice to remove any residual KOH and NaOH. The powdered product was completely dried by heating at 313 K under vacuum overnight.
Characterization
XRD patterns of the prepared samples were recorded using a MiniFlex 300, Rigaku. using Cu- Kα radiation at 40 kV and 15 mA over a 2θ range of 10° to 60° with a step size of 0.017° and counting time of 80 s per step. SEM was performed with a JEOL JSM-7600F microscope and SEM-EDX mappings were acquired by employing a FESEM TESCAN S9000G. TEM and HRTEM images were obtained using an FEI Tecnai G2 F30 Spirit microscope operated at an accelerating voltage of 300 kV. UV-vis DRS (V-670, JASCO) was conducted over the range of 200–1000 nm using BaSO4 as the reflectance standard. XPS was conducted using a Thermo Scientific ESCALAB 250Xi spectrometer and the decumulation of the peaks was carried out using the software of Avantage. EIS measurements were performed on an electrochemical analyser (CHI660C instruments) in a standard three-electrode system utilizing the synthesized samples as the working electrodes, Ag/AgCl (saturated KCl) as a reference electrode, and a Pt wire as the counter electrode. 0.2 M Na2S and 0.04 M Na2SO3 mixed aqueous solution was used as the electrolyte.Photocatalytic reactions
The photocatalytic reactions were performed in a top-irradiation-type reaction vessel with a Pyrex window using a Perfect 6A reaction system. For both H2 and O2 evolution, 0.3 g of the photocatalyst was dispersed in 150 mL of a 20 vol% aqueous methanol solution. Before photoirradiation, the reaction system was evacuated to remove all air and then irradiated from the top using a 300 W xenon lamp equipped with a dichroic mirror and a cut-off filter (λ ≥ 420 nm). The reactant solution was maintained at 288 K using a cooling-water system during the reaction. The reaction was carried out for 5 h, and the gas production was analyzed every 30 min using an online analysis system with a gas chromatograph (GC-9790II) equipped with a thermal conductivity detector with N2 as the carrier gas.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the National Natural Science Foundation of China (22072094) and the Science and Technology Innovation Commission of Shenzhen (JCYJ20190808150815084 and JCYJ20220530154408019).Notes and references
- Y. Qi, J. Zhang, Y. Kong, Y. Zhao, S. Chen, D. Li, W. Liu, Y. Chen, T. Xie, J. Cui, K. Domen and F. Zhang, Nat. Commun., 2022, 13, 484 CrossRef CAS PubMed.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- Y. H. Hong, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2022, 144(2), 695–700 CrossRef CAS PubMed.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- Y. Tachibana, l. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- Q. Wang, M. Nakabayashi, T. Hisatomi, S. Sun, S. Akiyama, Z. Wang, Z. Pan, X. Xiao, T. Watanabe, T. Yamada, N. Shibata, T. Takata and K. Domen, Nat. Mater., 2019, 18, 827–832 CrossRef CAS PubMed.
- C. Pan, T. Takata, M. Nakabayashi, T. Matsumoto, N. Shibata, Y. Ikuhara and K. Domen, Angew. Chem., Int. Ed., 2015, 54, 2955–2959 CrossRef CAS PubMed.
- B. Tian, B. Tian, B. Smith, M. C. Scott, R. Hua, Q. Lei and Y. Tian, Nat. Commun., 2018, 9, 1397 CrossRef PubMed.
- K. Maeda, Y. Tokunaga, K. Hibino, K. Fujii, H. Nakaki, T. Uchiyama, M. Eguchi, D. Lu, S. Ida, Y. Uchimoto and M. Yashima, ACS Appl. Energy Mater., 2018, 1, 1734–1741 CrossRef CAS.
- Y. Tang, K. Kato, T. Oshima, H. Mogi, A. Miyoshi, K. Fujii, K. Yanagisawa, K. Kimoto, A. Yamakata, M. Yashima and K. Maeda, Inorg. Chem., 2020, 59, 11122–11128 CrossRef CAS PubMed.
- B. Siritanaratkul, K. Maeda, T. Hisatomi and K. Domen, ChemSusChem, 2011, 4, 74–78 CrossRef CAS PubMed.
- B. Siritanaratkul, K. Maeda, T. Hisatomi and K. Domen, ChemSusChem, 2011, 4, 74–78 CrossRef CAS PubMed.
- M. Kodera, Y. Moriya, M. Katayama, T. Hisatomi, T. Minegishi and K. Domen, Sci. Rep., 2018, 8, 15849 CrossRef PubMed.
- T. Hisatomi, C. Katayama, Y. Moriya, T. Minegishi, M. Katayama, H. Nishiyama, T. Yamada and K. Domen, Energy Environ. Sci., 2013, 6, 3595–3599 RSC.
- X. Wang, T. Hisatomi, J. Liang, Z. Wang, Y. Xiang, Y. Zhao, X. Dai, T. Takata and K. Domen, J. Mater. Chem. A, 2020, 8, 11743–11751 RSC.
- S. Ida, Y. Okamoto, M. Matsuka, H. Hagiwara and T. Ishihara, J. Am. Chem. Soc., 2012, 134, 15773–15782 CrossRef CAS PubMed.
- K. Maeda, Y. Tokunaga, K. Hibino, K. Fujii, H. Nakaki, T. Uchiyama, M. Eguchi, D. Lu, S. Ida, Y. Uchimoto and M. Yashima, ACS Appl. Energy Mater., 2018, 1, 1734–1741 CrossRef CAS.
- J. Seo, T. Hisatomi, M. Nakabayashi, N. Shibata, T. Minegishi, M. Katayama and K. Domen, Adv. Energy Mater., 2018, 8, 1800094 CrossRef.
- T. Hryniewicz, K. Rokosz and H. R. Z. Sandim, Appl. Surf. Sci., 2012, 263, 357–361 CrossRef CAS.
- Y.-I. Kim, P. M. Woodward, K. Z. Baba-Kishi and C. W. Tai, Chem. Mater., 2004, 16, 1267–1276 CrossRef CAS.
- Y. Tang, K. Kato, T. Oshima, H. Mogi, A. Miyoshi, K. Fujii, K. Yanagisawa, K. Kimoto, A. Yamakata, M. Yashima and K. Maeda, Inorg. Chem., 2020, 59, 11122–11128 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
- I. P. Roof, T.-C. Jagau, W. G. Zeier, M. D. Smith and H.-C. Loye, Chem. Mater., 2009, 21, 1955–1961 CrossRef CAS.
- J.-H. Liao and M.-C. Tsai, Cryst. Growth Des., 2002, 2, 83–85 CrossRef CAS.
- L. Wan, F.-Q. Xiong, B. Zhang, R. Che, Y. Li and M. Yang, J. Energy Chem., 2018, 27, 367–371 CrossRef.
- Y. Du, L. Yuan, K. Huang and S. Feng, J. Lumin., 2013, 135, 196–200 CrossRef CAS.
- K. Korzeniowski and M. Sobczyk, Solid State Commun., 2018, 273, 30–33 CrossRef CAS.
- V. R. Reddy, D. W. Hwang and J. S. Lee, Catal. Lett., 2003, 90, 39–43 CrossRef CAS.
- M. Hojamberdiev, E. Zahedi, E. Nurlaela, K. Kawashima, K. Yubuta, M. Nakayama, H. Wagata, T. Minegishi, K. Domen and K. Teshima, J. Mater. Chem. A, 2016, 4, 12807–12817 RSC.
- X. Wang, T. Hisatomi, Z. Wang, J. Song, J. Qu, T. Takata and K. Domen, Angew. Chem., Int. Ed., 2019, 58, 10666–10670 CrossRef CAS PubMed.
- J. Seo, S. Jeong and S. Kim, ACS Appl. Energy Mater., 2021, 4, 3141–3150 CrossRef CAS.
- V. Khanal, N. O. Balayeva, C. Günnemann, Z. Mamiyev, R. Dillert, D. W. Bahnemann and V. (Ravi) Subramanian, Appl. Catal., B, 2021, 291, 119974 CrossRef CAS.
- Y. Okamoto, S. Ida, J. Hyodo, H. Hagiwara and T. Ishihara, J. Am. Chem. Soc., 2011, 133, 18034 CrossRef CAS PubMed.
- H. Zhang, S. Wei and X. Xu, J. Catal., 2020, 383, 135–143 CrossRef CAS.
- G. R. Dillip, A. N. Banerjee, V. C. Anitha, B. D. P. Raju, S. W. Joo and B. K. Min, ACS Appl. Mater. Interfaces, 2016, 8, 5025–5039 CrossRef CAS PubMed.
- S. Chen, S. Shen, G. Liu, Y. Qi, F. Zhang and C. Li, Angew. Chem., Int. Ed., 2015, 54, 3047–3051 CrossRef CAS PubMed.
- B. Dong, J. Cui, T. Liu, Y. Gao, Y. Qi, D. Li, F. Xiong, F. Zhang and C. Li, Adv. Energy Mater., 2018, 8, 1801660 CrossRef.
- C. Peng, T. Zhou, P. Wei, H. Ai, B. Zhou, H. Pan, W. Xu, J. Jia, K. Zhang, H. Wang and H. Yu, Chem. Eng. J., 2022, 439, 135685 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00915c |
| This journal is © The Royal Society of Chemistry 2023 |