
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amenamevir by Ugi-4CR†
Xin
Li
 a,
Tryfon
Zarganes-Tzitzikas
b,
Katarzyna
Kurpiewska
c and
Alexander
Dömling
*d
a,
Tryfon
Zarganes-Tzitzikas
b,
Katarzyna
Kurpiewska
c and
Alexander
Dömling
*d
aUniversity of Groningen, Department of Drug Design, A. Deusinglaan 1, 9713 AV, Groningen, The Netherlands
bAlzheimer's Research UK Oxford Drug Discovery Institute, University of Oxford, Oxford OX3 7FZ, UK
cJagiellonian University, Faculty of Chemistry, Department of Crystal Chemistry and Crystal, Physics, Biocrystallography Group, Gronostajowa 2, 30-387 Krakow, Poland
dPalackȳ University, CATRIN, Department of Innovative Chemistry, Šlechtitelů 241/27, 779 00, Olomouc – Holice, Czech Republic. E-mail: alexander.domling@upol.cz
First published on 1st February 2023
Abstract
We report a concise, convenient and sustainable synthesis of the approved anti-herpes zoster drug, Amenamevir. Based on the Ugi-4CR, our approach can access Amenamevir by a simple, rapid and one-pot synthesis. Compared to other reported syntheses, ours is more sustainable, shorter, and higher yielding. The X-ray crystal structures of key intermediate 3-(4-isocyanophenyl)-1,2,4-oxadiazole 4 and Amenamevir are reported for the first time. Computational retrosynthesis of Amenamevir using four popular freeware could recapitulate the described multi-step approaches but disappointingly did not propose our one-pot synthesis.
Introduction
Amenamevir was first introduced to the Japanese market in 2017 for the treatment of herpes zoster (HZ) infection.1 Up to now, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 240000 patients with HZ have been treated in Japan.2 As a helicase-primase inhibitor (HPI), Amenamevir has a novel action mechanism from previously reported synthetic nucleoside compounds for the treatment of HZ, including Acyclovir, and Valacyclovir (Fig. 1A).3 In contrast to Acyclovir, the non-nucleoside Amenamevir inhibits helicase-primase, thereby suppressing the replication fork progression that separates double DNA strands into two single strands during DNA synthesis. Amenamevir showed superior antiviral activity compared to Acyclovir during the DNA synthesis stage when it was used to treat herpes simplex virus (HSV) and varicella-zoster virus (VZV) related diseases,4,5 Unlike Acyclovir, its anti-VZV and anti-HSV activities were not attenuated by viral DNA synthesis in the infected cells. Among four available HPIs, Amenamevir has both anti-HSV and anti-VZV activity, while T157602, Pritelivir and BILS 22 BS only have anti-HSV activity.6,7 Amenamevir is N-(2-((4-(1,2,4-oxadiazol-3-yl)phenyl)amino)-2-oxoethyl)-N-(2,6-dimethylphenyl)tetrahydro-2H-thiopyran-4-carboxamide-1,1-dioxide. It consists of α-aminoacyl-amide backbone with a C-terminal thiopyrane dioxide, an N-terminal p-1,2,4-oxadiazol-3-yl phenyl, and a 2,6-dimethylphenyl anilid sub-structure.
240000 patients with HZ have been treated in Japan.2 As a helicase-primase inhibitor (HPI), Amenamevir has a novel action mechanism from previously reported synthetic nucleoside compounds for the treatment of HZ, including Acyclovir, and Valacyclovir (Fig. 1A).3 In contrast to Acyclovir, the non-nucleoside Amenamevir inhibits helicase-primase, thereby suppressing the replication fork progression that separates double DNA strands into two single strands during DNA synthesis. Amenamevir showed superior antiviral activity compared to Acyclovir during the DNA synthesis stage when it was used to treat herpes simplex virus (HSV) and varicella-zoster virus (VZV) related diseases,4,5 Unlike Acyclovir, its anti-VZV and anti-HSV activities were not attenuated by viral DNA synthesis in the infected cells. Among four available HPIs, Amenamevir has both anti-HSV and anti-VZV activity, while T157602, Pritelivir and BILS 22 BS only have anti-HSV activity.6,7 Amenamevir is N-(2-((4-(1,2,4-oxadiazol-3-yl)phenyl)amino)-2-oxoethyl)-N-(2,6-dimethylphenyl)tetrahydro-2H-thiopyran-4-carboxamide-1,1-dioxide. It consists of α-aminoacyl-amide backbone with a C-terminal thiopyrane dioxide, an N-terminal p-1,2,4-oxadiazol-3-yl phenyl, and a 2,6-dimethylphenyl anilid sub-structure.
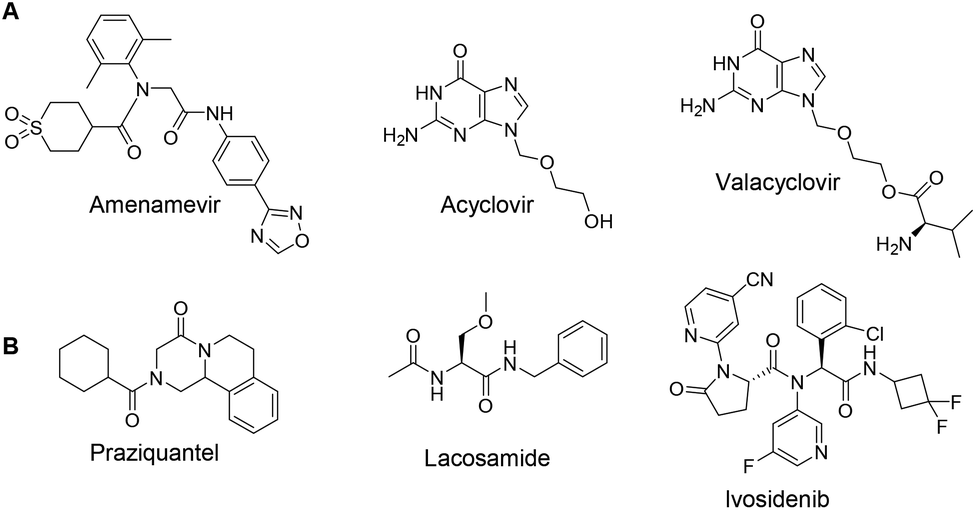 | ||
| Fig. 1 (A) Three marketed anti-herpes zoster drugs. (B) Examples of drugs advantageously synthesizable by Ugi-4CR chemistry. | ||
The Ugi four component reaction (Ugi-4CR), is one of the most widely used and versatile multicomponent reactions (MCRs) in organic synthesis and medicinal chemistry, allowing the rapid construction of the chemical compounds with α-aminoacyl amide scaffolds.8–11 A few examples of Ugi-4CR-derived drug synthesis are listed in Fig. 1, such as praziquantel,12,13 lacosamide,14 or ivosidenib (Fig. 1B).15
Results and discussion
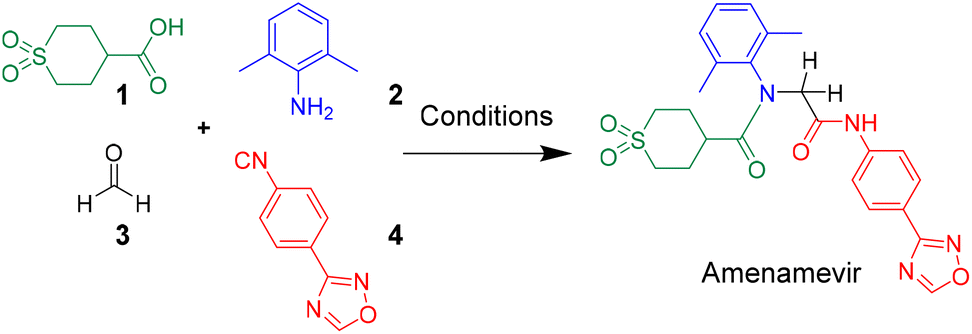
Herein we report a novel synthesis of Amenamevir exploiting the Ugi-4CR with carboxylic acid 1, 2,6-dimethylaniline 2, paraformaldehyde 3, and 4-(1,2,4-oxadiazol-3-yl)-phenyl isocyanide 4. Based on our previous patent application, here we further optimized the reaction condition (Table 1).16 Extensive optimization boosted the initial 18% yield (MeOH, rt, 3 days) to 65% (TFE, 50 °C. 12 h).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Aldehyde amount | Concentration | Solvent | Time | Temp. | Yield |
| a Reaction condition in our patent: 1 (1.2 mmol, 1.2 equiv.), 2 (1 mmol, 1 equiv.), 3 (1.2 mmol, 1.2 equiv.), 4 (1.2 mmol, 1.2 equiv.), MeOH (2 mL), isolated yields. b Reaction conditions: 1 (1.2 mmol, 1.2 equiv.), 2 (1 mmol, 1 equiv.), 3 (1.2 mmol, 1.0–2.0 equiv.), 4 (1.2 mmol, 1.2 equiv.), solvent (1–2 mL), isolated yields. c Under microwave irradiation. | ||||||
| 1 | 1.2 equiv. | 0.5 M | MeOH | 3 d | rt | 18%a |
| 2 | 1.2 equiv. | 0.5 M | TFE | 12 h | 50 °C | 65%b |
| 3 | 1.2 equiv. | 0.5 M | MeOH | 12 h | 50 °C | 47%b |
| 4 | 1.2 equiv. | 0.5 M | 2-Propanol | 12 h | 50 °C | 34%b |
| 5 | 1.5 equiv. | 0.5 M | TFE | 12 h | 50 °C | 71%b |
| 6 | 2.0 equiv. | 0.5 M | TFE | 12 h | 50 °C | 55%b |
| 7 | 1.0 equiv. | 0.5 M | TFE | 12 h | 50 °C | 60%b |
| 8 | 1.5 equiv. | 1 M | TFE | 12 h | 50 °C | 54%b |
| 9 | 1.5 equiv. | 0.5 M | TFE | 12 h | 50 °C | 62%c |
The reaction time (12 h) and temperature (50 °C) were kept still, and we changed the solvent to MeOH and 2-propanol, both providing lower yields, 47% and 34%, respectively. Next, we investigated the influence of the stoichiometry of aldehyde on the reaction, and it turned out that 1.5 equivalent of aldehyde resulted in a superior yield (71%, entry 5) than 1.0 equivalent (60%, entry 7), 1.2 equivalent (65%, entry 2) or 2.0 equivalent (55%, entry 6). Attempts to improve the yield by increasing the overall concentration failed, only providing 54% yield (entry 8).
Also microwave irradiation didn't provide any benefit to the reaction, giving a relatively low yield instead (62%, entry 9).
After identifying the optimal reaction condition, we then applied it to the gram-scale synthesis of Amenamevir (Scheme 1). We ran the Ugi-4CR in TFE with 15 mmol 1, 12.5 mmol 2, 18.75 mmol 3 and 15 mmol 4, to isolate 3.8 gram of Amenamevir at 63% yield, indicating that our method may be also applied to larger scale synthesis.
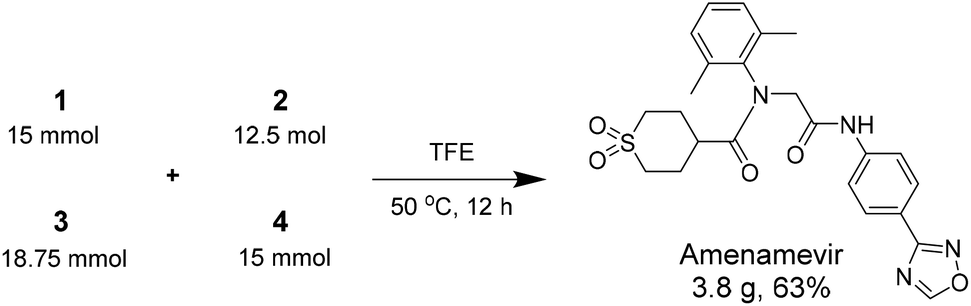 | ||
| Scheme 1 Gram-scale synthesis. | ||
The industrial synthesis of Amenamevir was first released by Kontani in their patent from 2005 (Scheme 2).17 Aniline 2 first underwent alkylation with ethyl bromoacetate 5 to intermediate 6, the amidation of 6 with synthesized acid chloride 7 in pyridine formed compound 8. 8 was then saponified and followed by condensation with aniline 10 to yield Amenamevir. The synthesis employs chlorinated solvents (DCM, CHCl3) during five steps. Based on the synthetic method of kontani, xumeng developed a superior strategy.
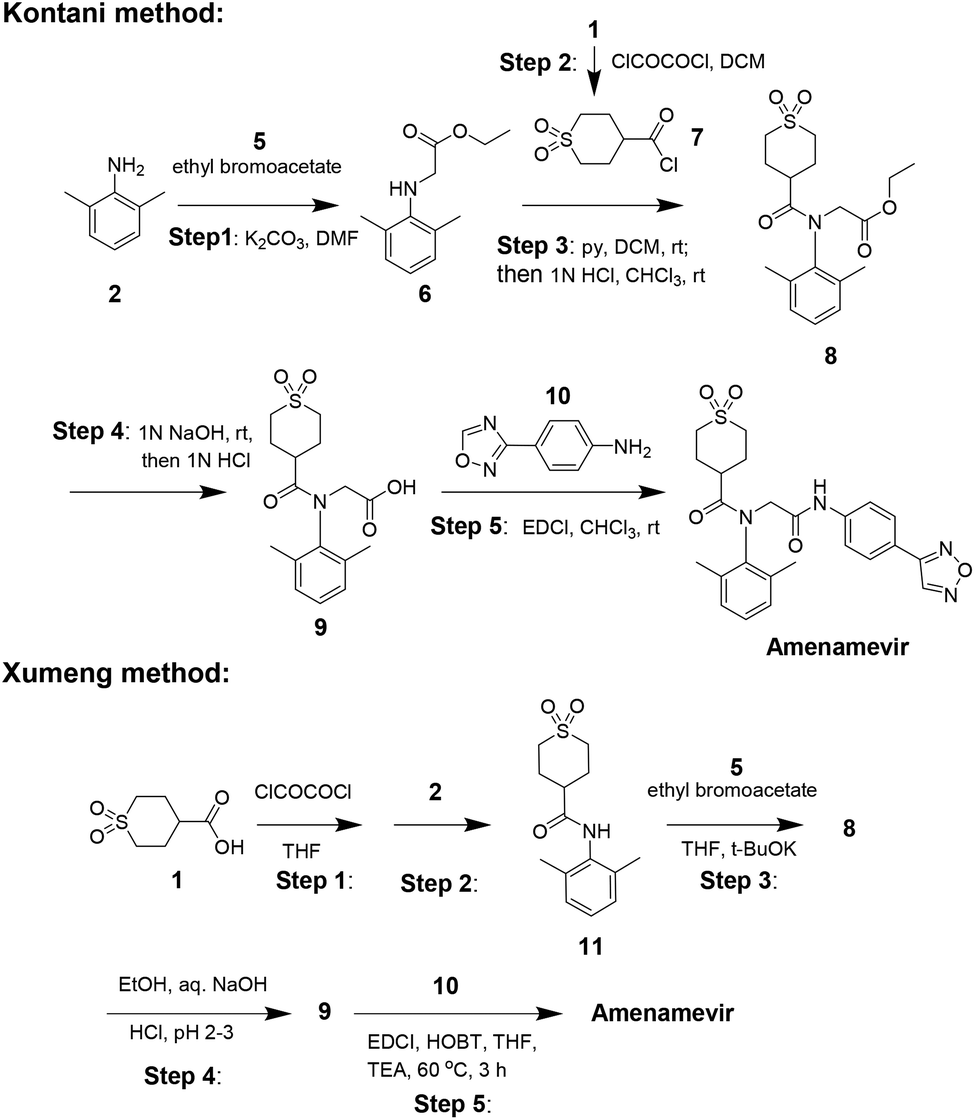 | ||
| Scheme 2 Kontani's and Xumeng's synthetic route to Amenamevir. | ||
Different from Kontani, Xumeng first reacted acid 1 with oxalyl chloride, then followed by the nucleophilic addition with aniline 2 to provide amide intermediate 11. Then 11 sequentially subjected to N-alkylation, ester hydrolysis and amidation reaction to obtain Amenamevir.
We first reported the synthesis of Amenamevir via Ugi-4CR in our patent on 2020.16 In this work, we further optimized the reaction condition and finished the gram-scale synthesis with the optimal solvent and reaction temperature. To demonstrate the green chemistry merits of our synthetic approach over the existing ones, we quantified and compared various reaction parameters such as the total yield, reaction time (h), amount of inorganic and organic solvents (mL), number of steps, process mass intensity (PMI), the E-factor and atom economy (AE) (Fig. 2 and ESI†). Compared with the Kontani and Xumeng routes,17,18 our current method not only gives a comparable yield (56%, including the isocyanide synthesis) to Xumeng route (63%, optimized from Kontani route), But outperforms both methods in other categories, such as the reaction time, amount of solvents (organic and inorganic), number of steps, the E-factor and AE, which will result in significant time and cost savings for industrial production. In addition, both Kontani and Xumeng's method require the use of HCl and chlorinated solvents (see ESI†), which does not meet the requirement of green chemistry and produces lots of inorganic waste. Last but not least, both competitor routes are more time and solvent amount consuming, hence less sustainable.
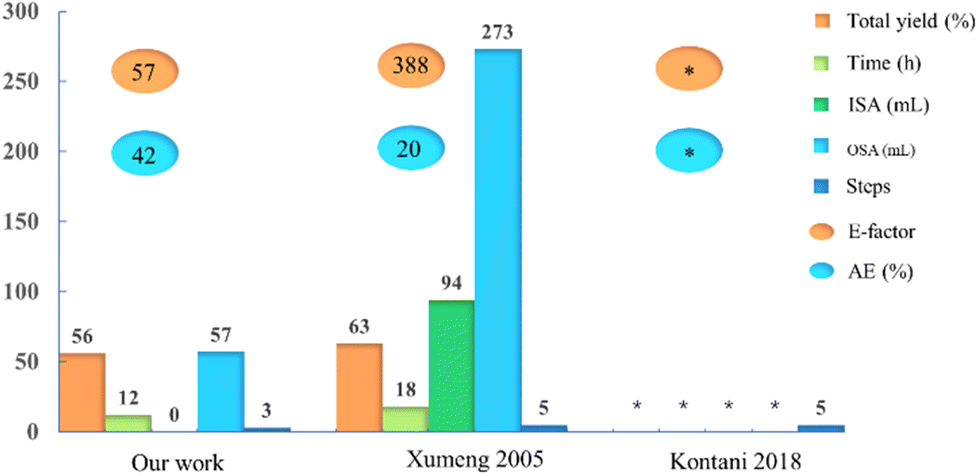 | ||
| Fig. 2 Bar chart comparison between two representative procedures and our work (including the isocyanide synthesis) for the preparation of Amenemevir, including the total yield, reaction time, inorganice and organic waste, numbers of steps, the E-factor and atom economy (AE). *Not reported. The total yield of Kontani method was not reported in the original patent, and total yield of Xumeng method was calculated from the five steps together, the total yield of our method was calculated from three steps including the isocyanide synthesis. | ||
The single-crystal X-ray structures of isocyanide 4 and Amenamevir were first reported in our work (Fig. 3). It reveals that Amenamevir tertiary phenyl amide is losing conjugation through out of plane rotation of the sterically hindered 2,6-dimethyl phenyl substitutents. Moreover, a hydrogen bonding between the secondary amide and a neighbor molecules sulfone O. The isocyanide building block 4 also shows interesting short contacts in the crystal.
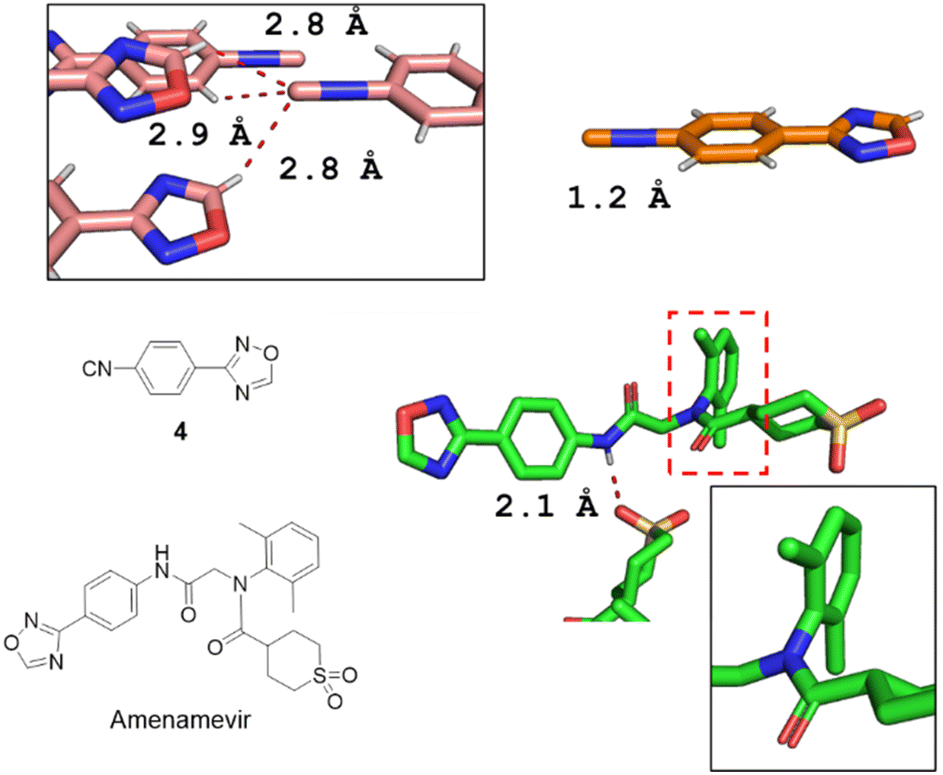 | ||
| Fig. 3 X-ray diffraction structures of 4 (CCDC 2213853†) and Amenamevir (CCDC 2213854†). The isocyanide 4 shows a short 2.2 Å triple bond between C and N. Interestingly, the isocyanide-C features a trifurcated interaction with short contacts to two adjacent oxadiazole-H's (2.8 Å) and an ortho-phenyl-H (2.9 Å) in a trigonal pyramidal geometry. Amenamevir's amide-H displays a 2.1 Å short hydrogen bond to an adjacent sulfone-O. Noteworthy, the anilid is not in conjugation but 180° twisted with the amide due to the bulky 2,6-dimethyl substitution. | ||
Automated computational retrosynthesis based on machine learning algorithms is now considered to provide efficient synthetic pathways with the claim to outperform expert-guided reaction planning.19,20 Several programs can be accessed freely. We tested popular freely available computational retrosynthesis tools for the target molecule Amenamevir (Table 2, more details see ESI†). All programs resulted in several multistep retrosynthesis pathways and could reconstitute the described Kotani and Xumeng routes. However, no program proposed a retrosynthesis of Amenamevir based on the Ugi-4CR in our patent. It is sometimes argued that for seldomly used reactions there's often not enough data to build robust models. However, the Ugi reaction is clearly a widely used reaction.
| No. | Program | Reconstitution of Kotani and Xumeng routes | Reconstitution of Ugi route |
|---|---|---|---|
| 1 | SciFinder-n | Yes | No |
| 2 | IBM RXN | No | No |
| 3 | Spaya | Yes | No |
| 4 | ASKCOS | Yes | No |
Conclusions
In conclusion, in this work, we reported a novel synthetic method to synthesize Amenamevir, a drug which is high demand by patients who suffered from herpes simplex virus and varicella-zoster virus-related diseases with a novel mode-of-action. Based on Ugi-4CR, our method can lead to Amenamevir in a one-pot, simple and green way, which made it synthetically superior to other anti-HZ drugs like Acyclovir and Valacyclovir. As compared to competitor syntheses, our approach features avoidance of chlorinated solvents, better yields, less steps, less side products, less inorganic and organic waste, very short time to product, hence outperforms hitherto described synthesis pathways in sustainability and cost of goods. Since there is no final cyclization or deprotection step, our method still has advantages over other Ugi-4CR-built drugs like praziquantel or lacosamide. Moreover, a 12.5 mmol scale reaction was performed to support the possibility of industrial application of this method. Our work also cautions to ‘blindly’ trust computational approaches to find ‘best’ routes for the synthesis of a target molecule.Author contributions
A. D. conceived the research project and raised funding. X. L. performed the syntheses and collected the analytical data. K. K. determined the single crystal X-ray structure. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank W.H.C. Huibers (University of Groningen) for their help in HRMS analysis. X. L. acknowledge the China Scholarship Council for supporting.References
- N. Shoji, K. Tanese, A. Sasaki, T. Horiuchi, Y. Utsuno, K. Fukuda, Y. Hoshino, S. Noda, H. Minami and W. Asakura, J. Dermatol., 2020, 47, 683–688 CrossRef CAS PubMed.
- K. Shiraki, S. Yasumoto, N. Toyama and H. Fukuda, Viruses, 2021, 13, 1547 CrossRef CAS.
- K. Chono, K. Katsumata, T. Kontani, M. Kobayashi, K. Sudo, T. Yokota, K. Konno, Y. Shimizu and H. Suzuki, J. Antimicrob. Chemother., 2010, 65, 1733–1741 CrossRef CAS.
- M. Yajima, H. Yamada, M. Takemoto, T. Daikoku, Y. Yoshida, T. Long, T. Okuda and K. Shiraki, Antiviral Res., 2017, 139, 95–101 CrossRef CAS PubMed.
- K. Shiraki, L. Tan, T. Daikoku, M. Takemoto, N. Sato and Y. Yoshida, Antiviral Res., 2020, 180, 104829 CrossRef CAS.
- J. J. Crute, C. A. Grygon, K. D. Hargrave, B. Simoneau, A.-M. Faucher, G. Bolger, P. Kibler, M. Liuzzi and M. G. Cordingley, Nat. Med., 2002, 8, 386–391 CrossRef CAS PubMed.
- G. Kleymann, R. Fischer, U. A. Betz, M. Hendrix, W. Bender, U. Schneider, G. Handke, P. Eckenberg, G. Hewlett and V. Pevzner, Nat. Med., 2002, 8, 392–398 CrossRef CAS PubMed.
- A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef.
- T. Zarganes-Tzitzikas, C. G. Neochoritis and A. Dömling, ACS Med. Chem. Lett., 2019, 10, 389–392 CrossRef CAS.
- T. Zarganes-Tzitzikas and A. Dömling, Org. Chem. Front., 2014, 1, 834–837 RSC.
- R. C. Cioc, E. Ruijter and R. V. Orru, Green Chem., 2014, 16, 2958–2975 RSC.
- A. Dömling and K. Khoury, ChemMedChem, 2010, 5, 1420–1434 CrossRef.
- H. Cao, H. Liu and A. Dömling, Chem. – Eur. J., 2010, 16, 12296–12298 CrossRef CAS.
- H. Wehlan, J. Oehme, A. Schäfer and K. Rossen, Org. Process Res. Dev., 2015, 19, 1980–1986 CrossRef CAS.
- J. Popovici-Muller, R. M. Lemieux, E. Artin, J. O. Saunders, F. G. Salituro, J. Travins, G. Cianchetta, Z. Cai, D. Zhou and D. Cui, ACS Med. Chem. Lett., 2018, 9, 300–305 CrossRef CAS.
- A. Dömling and T. Zarganes-Tzitzikas, WO2020038812A1, 2020.
- T. Kontani, J. Miyata, W. Hamaguchi, T. Kawano, A. Kamikawa, H. Suzuki and K. Sudo, US20050032855A1, 2005.
- X. M. Zhang, X. Zhao, C. Gao, Z. Zhang, J. Chen and Y. Zhen, CN108623577A, 2018.
- A. Bender and I. Cortés-Ciriano, Drug Discovery Today, 2021, 26, 511–524 CrossRef CAS.
- Y. Shen, J. E. Borowski, M. A. Hardy, R. Sarpong, A. G. Doyle and T. Cernak, Nat. Rev. Methods Primers, 2021, 1, 1–23 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experiment procedure, analytical data, NMR spectrums, X-ray data. CCDC 2213853 and 2213854. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2gc04869h |
| This journal is © The Royal Society of Chemistry 2023 |