 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural modifications to platinum(II) pincer complexes resulting in changes in their vapochromic and solvatochromic properties†
Mathew J.
Bryant
a,
Sara
Fuertes
 b,
Lauren E.
Hatcher
c,
Lynne H.
Thomas
a and
Paul R.
Raithby
*a
b,
Lauren E.
Hatcher
c,
Lynne H.
Thomas
a and
Paul R.
Raithby
*a
aDepartment of Chemistry, University of Bath, Bath BA2 7AY, UK. E-mail: p.r.raithby@bath.ac.uk
bDepartamento de Quimica Inorgánica, Universidad de Zaragoza, Zaragoza 50009, Spain. E-mail: sfuertes@unizar.es
cSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK. E-mail: HatcherL1@cardiff.ac.uk
First published on 9th February 2023
Abstract
There is a need to develop rapidly responsive chemical sensors for the detection of low concentrations of volatile organic solvents (VOCs). Platinum pincer complexes have shown promise as sensors because of their colours and vapochromic and solvatochromic properties, that may be related to the non-covalent interactions between the pincer complexes and the guest VOCs. Here we report an investigation into a series of Pt(II) complexes based on the 1,3-di(pyridine)benzene tridentate (N⁁C⁁N) skeleton with the formula [Pt(N⁁C(R)⁁N)(CN)] (R = C(O)Me 2, C(O)OEt 3, C(O)OPh 4) with the fourth coordination site occupied by a cyanide ligand. Solid-state samples of the complexes have been tested with a range of volatiles including methanol, ethanol, acetone, dichloromethane and water, and while 2 displays thermochromism, 3 and 4 display rapidly reversible vapochromism and solvatochromism. These results are correlated with X-ray powder and single crystal X-ray structural data including an assessment of the crystal packing and the void space in the crystalline space. The cyanide ligand and the R substituents are involved in hydrogen bonding that creates the voids within the structures and interact with the solvent molecules that influence the Pt⋯Pt separation in the crystalline state.
Introduction
Square planar platinum(II) complexes are among the classes of small molecules that have shown promise as chemical sensors.1,2 There are many examples of these complexes that show vapochromic or solvatochromic responses to simple analytes in the solid state.3–9 These colour changes and changes in luminescent properties have been attributed to alterations in intermolecular Pt⋯Pt contacts and changes in intermolecular non-covalent interactions upon the sorption of volatile organic compounds (VOCs).3Within the general class of square planar platinum(II) complexes the platinum terpyridine pincer complexes [Pt(tpy)X][Y] (where tpy = 2,2′;6′,2′′-terpyridine; X = a monodentate anionic ligand; Y = a monoanionic counterion) (Fig. 1(a)) have been extensively studied for their luminescent properties.10–13 The luminescence observed in these systems, when adjacent molecules pack with Pt⋯Pt separations of 3.5 Å or less, originates from a low mixed-metal-to-ligand charge-transfer (MMLCT) state, involving an unoccupied π* state on the terpyridine ligand and a filled dσ* orbital on the Pt centre, primarily derived from the interaction of the dz2 orbitals of adjacent Pt centres.
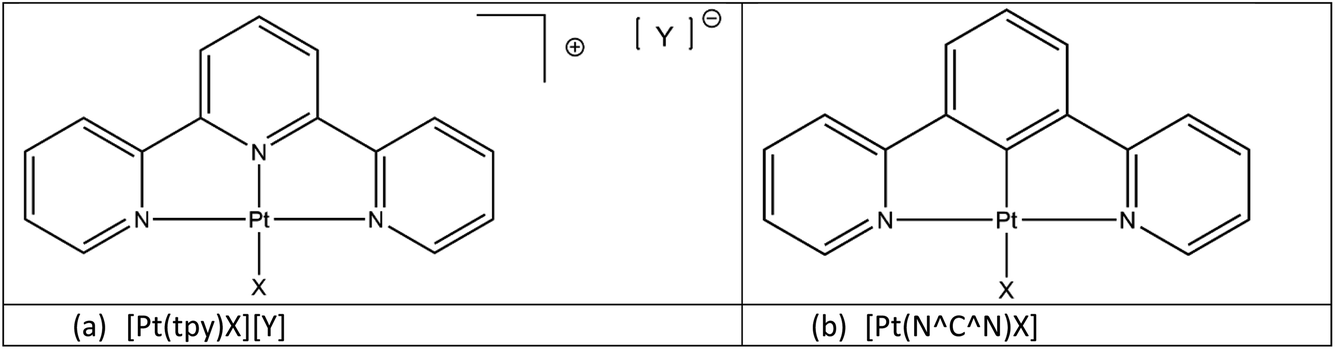 | ||
| Fig. 1 Classes of (a) terpyridyl (N⁁N⁁N) and (b) cyclometallated (N⁁C⁁N) platinum(II) pincer complexes. | ||
Several of these luminescent platinum terpyridine pincer complexes or their derivatives display vapochromic or solvatochromic properties. For example, [Pt(Me2bzimpy)Cl][PF6] (Me2bzimpy = 2,6-bis(N-methylbenzimidazol-2-yl)pyridine), which crystallises as an orange dimethylformamide (DMF) solvate, turns purple within a matter of seconds when exposed to acetonitrile vapour.3,4 The related complex [Pt(Me2bzimpy)Cl]Cl interacts with methanol, ethanol, chloroform and acetonitrile, showing a colour change from orange to red.3 The solid red complex [Pt(Nttpy)Cl]Cl (Nttpy = 4′-(p-nicotinamide-N-methylphenyl)-2,2′:6′,2′′-terpyridine) changes to an orange colour at room temperature when exposed to methanol.13
Also related to the terpyridine systems are the planar cyclometallated pincer complexes, [Pt(N⁁C⁁N)X], based on the 1,3-di(2-pyridyl)benzene tridentate (N⁁C⁁N) platinum(II) unit (Fig. 1(b)). These complexes are neutral if the fourth ligand position is occupied by a monoanionic ligand (X), and the crystal packing in the solid-state is not moderated by the need for the presence of a counterion, unlike the tpy analogues. Their steric and electronic properties can also easily be modified by the inclusion of substituents on the N⁁C⁁N rings. Since the synthesis of the first polyaromatic N⁁C⁁N pincer complex [Pt(N⁁C⁁N)Cl] in 199914 the range of complexes has expanded enormously through the substitution in the para-position on the central arene ring,15–18 or on the two pyridyl rings.19–22 With these variations it is possible to tune these brightly coloured complexes to exhibit absorptions and emissions spanning the visible spectrum and beyond.11,23–25 Several of these complexes show colour changes when exposed to solvent vapours or liquids2,26,27 prompting our investigations into the solvatochromic and vapochromic properties of these materials and their potential use as chemical sensors.
Crystallographic studies of platinum pincer complexes that display either vapochromic or solvatochromic properties indicate that the structures, in the solid-state, contain channels that permit the diffusion of small gaseous or solvent molecules through the crystal lattice. The presence of these molecules and their non-covalent intermolecular interactions with the pincer molecules in the lattice changes the Pt⋯Pt interactions, particularly the extent of the dz2 overlap, and causes the colour change.28,29
With these factors in mind we embarked on the synthesis of platinum(II) cyclometallated pincer complexes with a series of different substituents on the para-position on the central arene ring and using the anionic cyanide ligand in the fourth coordination site on the Pt(II) centre. The substituents on the central arene ring of the pincer ligand act as spacer groups that alter the separation between adjacent molecules within the crystalline lattice, and thus, potentially, alter the size of the channels within the crystal structure, or at least alter the overall porosity of the crystalline material. The cyanide ligand is an extremely strong-field ligand that is likely to promote luminescence in the complex by displacing the antibonding dx2–y2 metal orbital, that is known to quench emissive states, to inaccessibly high energies.23,24 Additionally, in the solid-state the cyanide ligand can act as an effective hydrogen-bond acceptor in interactions with gas or solvent molecules (see Fig. 2).
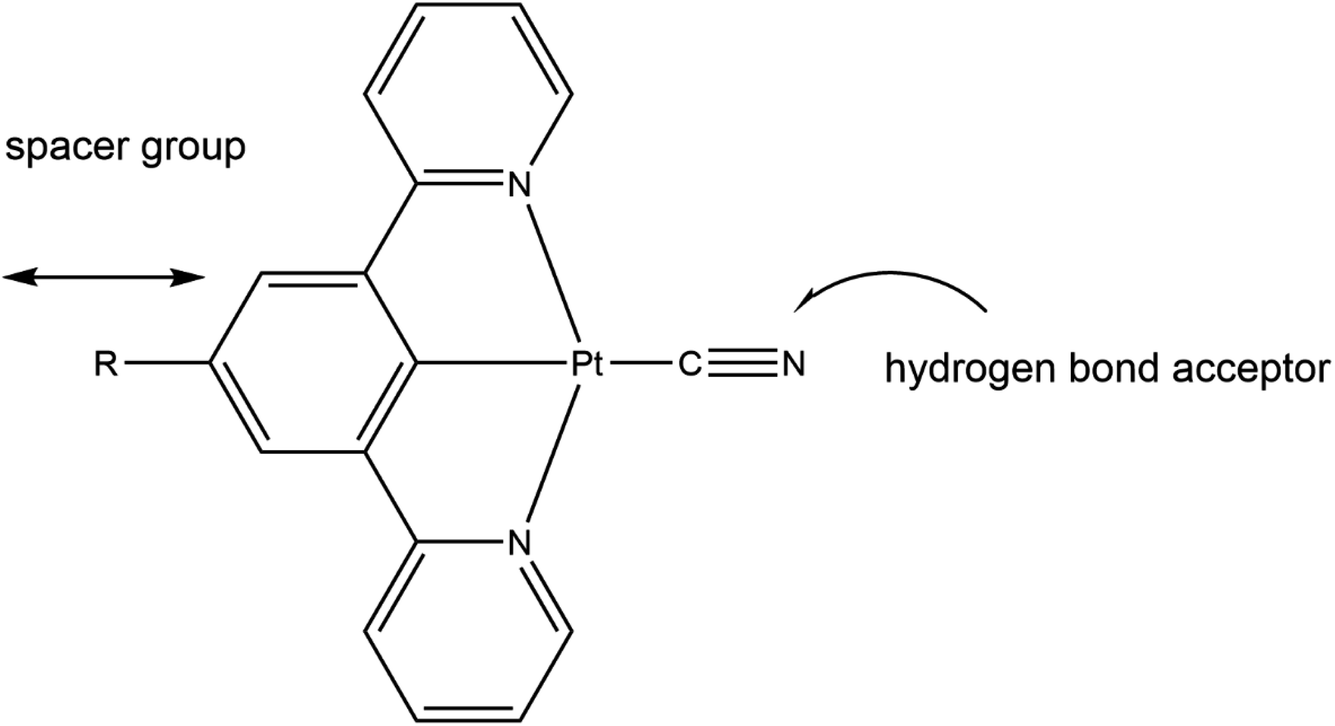 | ||
| Fig. 2 The role of the substituent R and the cyanide ligand in the [Pt(N⁁C(R)⁁N)(CN)] pincer complexes. | ||
In our preliminary results we found that the complex [Pt(N⁁C(R)⁁N)(CN)] (1, R = C(O)OMe) displayed rapidly-reversible absorptive and emissive vapochromic properties with water and methanol.30 The complex exhibits a yellow anhydrous form that converts reversibly on a subsecond timescale to a red hydrate in the presence of parts-per-thousand quantities of atmospheric water vapour. Similarly, exposure of the anhydrous form to methanol results in a rapid and reversible colour change to a blue methanol solvate. The colour changes are a result of the water and methanol molecules forming intermolecular hydrogen-bonds with the cyanide ligand of the complex perturbing the Pt⋯Pt interaction distance in the molecular stacks within the crystal. The presence of the methoxy substituent reduces the crystal packing density and aids the dispersion of the water and methanol molecules through the crystalline solid. The complex can be incorporated as microcrystals into polymer membranes where the vapochromic behaviour remains the same, and the complex has been used to measure the concentration of a gaseous throughflow at the solid–fluid interface.31
We now report an extension of this work to investigate the influence of systematically changing the nature of the R group in the [Pt(N⁁C(R)⁁N)(CN)] complexes (Fig. 3) on the crystal structure and packing and how these changes alter the vapochromic and solvatochromic response to interactions with a range of volatile solvents and vapours. Through these measurements we hope to identify specific selectivities within the series and establish the rapidity of the colour changes involved to assess whether these complexes might be suitable chemical sensors detecting the presence of volatile solvents.
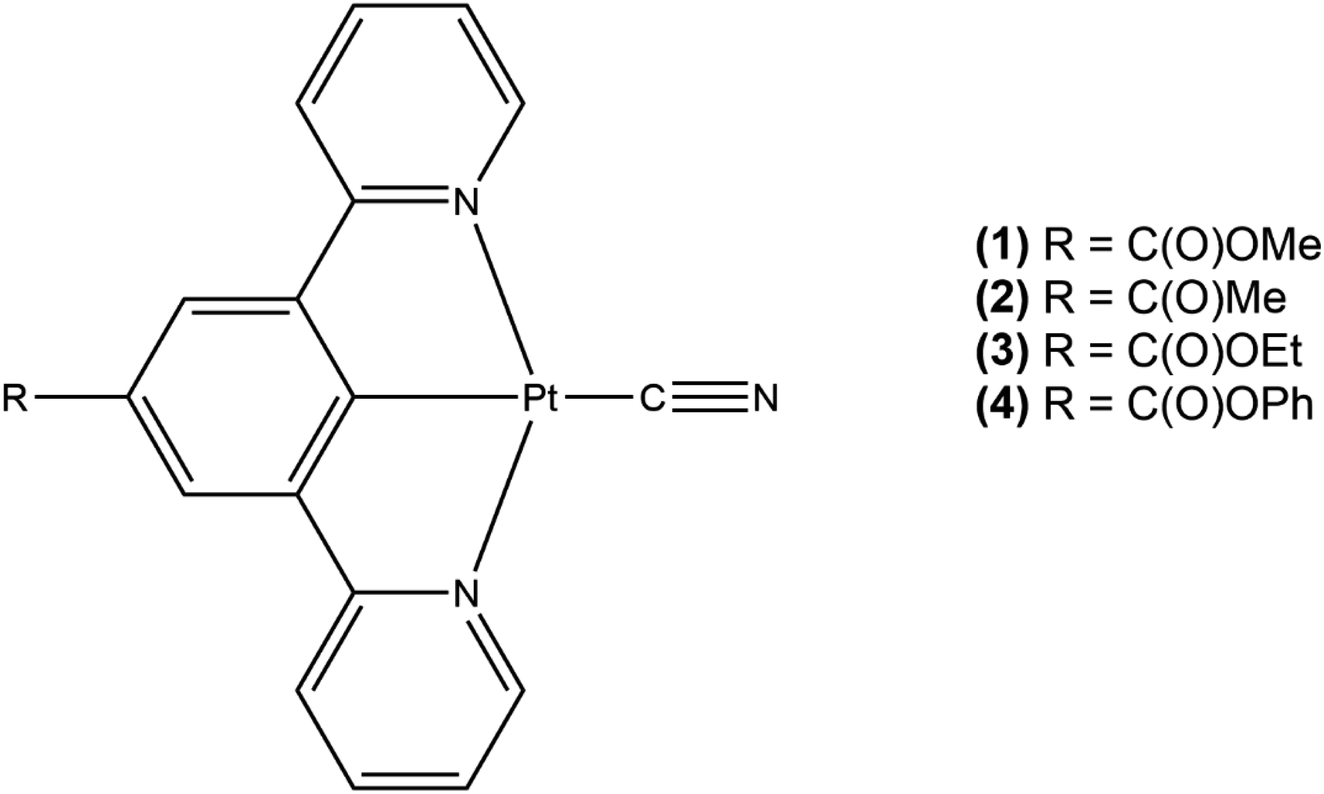 | ||
| Fig. 3 The series of the platinum(II) pincer complexes to be studied and compared. Complex 1 has already been reported.30 | ||
Experimental
All reactions were carried out under an atmosphere of dry nitrogen using standard Schlenk line techniques. All solvent were dried prior to use using either an automated solvent purification system or through distillation over an appropriate drying agent and stored over molecular sieves. All starting materials were purchased from commercial sources and used without further purification unless otherwise stated. The precursors chloro[acetyl 3,5-di(2-pyridyl)phenyl]platinum, chloro[ethyl 3,5-di(2-pyridyl)benzoato]platinum and chloro[phenyl 3,5-di(2-pyridyl)benzoate]platinum were prepared by modified literature procedures15 and their formulation was confirmed by 1H NMR spectroscopy and mass spectroscopic data (see ESI†).IR spectra were recorded on a PerkinElmer Spectrum One spectrometer using neat solids. 1H NMR and 13C NMR spectra were recorded on either a Bruker Avance 400 MHz or 500 MHz instrument. Chemical shifts (expressed in parts per million) were referenced to residual solvent peaks. Mass spectra were acquired using a micro TOF electrospray time-of-flight (ESI-TOF) mass spectrometer (Bruker Daltonik GmbH). UV-visible spectra were recorded in the solution state using a PerkinElmer Lambda 650 UV/vis Spectrometer. Thermogravimetric analyses (TGA) were recorded on a PerkinElmer TGA4000 thermogravimetric analyser. Photoluminescence spectroscopy was performed using a PerkinElmer LS55 luminescence spectrometer.
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), chloro[acetyl 3,5-di(2-pyridyl)phenyl]platinum (100 mg, 0.199 mmol) was added, and heated under reflux for 24 h at 85 °C under a nitrogen atmosphere. The resulting suspension was dried under reduced pressure, and then stirred with dichloromethane (30 ml) until the residues were dissolved. The pale-yellow mixture was then filtered, and solvents removed in vacuo. The resulting green solid, was washed with an excess of diethyl ether resulting in pure product. Yield: 30 mg, 30.6%.1H NMR (400 MHz, CDCl3) δH: 8.90 (d, 3JH–H = 5.0 Hz, 3JH–Pt = 46.3 Hz, 2H, ortho-Py), 7.95 (t, 3JH–H = 8.0 Hz, 2H, para-Py), 7.88 (s, 2H, pincer-Ph), 7.71 (d, 3JH–H = 7.7 Hz, 2H, meta-Py), 7.19 (t, 3JH–H = 6.0 Hz, 2H, meta-Py), 2.56 (s, 3H, COCH3). 13C24,30 NMR (101 MHz, CDCl3) δC: 201.5, 171.0, 159.0, 146.9, 143.4, 137.4, 128.4, 127.5, 123.9, 30.1. Low solubility did not allow detection of all quaternary carbons. IR (cm−1): ν(C
:1), chloro[acetyl 3,5-di(2-pyridyl)phenyl]platinum (100 mg, 0.199 mmol) was added, and heated under reflux for 24 h at 85 °C under a nitrogen atmosphere. The resulting suspension was dried under reduced pressure, and then stirred with dichloromethane (30 ml) until the residues were dissolved. The pale-yellow mixture was then filtered, and solvents removed in vacuo. The resulting green solid, was washed with an excess of diethyl ether resulting in pure product. Yield: 30 mg, 30.6%.1H NMR (400 MHz, CDCl3) δH: 8.90 (d, 3JH–H = 5.0 Hz, 3JH–Pt = 46.3 Hz, 2H, ortho-Py), 7.95 (t, 3JH–H = 8.0 Hz, 2H, para-Py), 7.88 (s, 2H, pincer-Ph), 7.71 (d, 3JH–H = 7.7 Hz, 2H, meta-Py), 7.19 (t, 3JH–H = 6.0 Hz, 2H, meta-Py), 2.56 (s, 3H, COCH3). 13C24,30 NMR (101 MHz, CDCl3) δC: 201.5, 171.0, 159.0, 146.9, 143.4, 137.4, 128.4, 127.5, 123.9, 30.1. Low solubility did not allow detection of all quaternary carbons. IR (cm−1): ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) 2114, ν(C
N) 2114, ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) 1677. Mass spectrometry (positive loop injection): measured (MeOH) m/z – 517.0678, calculated for [C19O1N3H13Pt][Na]+ – 517.0678 – correct isotope pattern.
:1), chloro[ethyl 3,5-di(2-pyridyl)benzoato]platinum (106 mg, 0.199 mmol) was added, and heated under reflux for 24 h at 85 °C under a nitrogen atmosphere. The resulting suspension was dried under reduced pressure, and then stirred with dichloromethane (30 ml) until the residues were dissolved. The pale-yellow mixture was then filtered, and solvents removed in vacuo. The resulting yellow/orange solid, was washed with an excess of diethyl ether resulting in pure product. Yield: 30 mg, 30.6%. 1H NMR (500 MHz, CDCl3) δH: 9.28 (d, 3JH–H = 5.8 Hz, 3JH–Pt = 47.3 Hz, 2H, ortho-Py), 8.20 (s, 2H, pincer-Ph), 8.05 (t, 3JH–H = 7.8 Hz, 2H, para-Py), 7.84 (d, 3JH–H = 7.8 Hz, 2H, meta-Py), 4.46 (q, 3JH–H = 7.1 Hz, 2H, CO2CH2), 1.48 (t, 3JH–H = 7.1 Hz, 3H, CO2CH2CH3). 13C30 NMR (101 MHz, CDCl3) δC: 179.9, 167.7, 166.6, 155.7, 153.5, 143.2, 139.6, 136.9, 126.7, 125.0, 124.6, 120.3, 61.3, 14.5. IR (cm−1): ν(CN) 2111, ν(CO) 1702. Mass spectrometry (positive loop injection): measured (MeOH) m/z – 547.0688, calculated for [C20O2N3H15Pt1][Na]+ – 547.070599. Correct isotope pattern.
:1), chloro[phenyl 3,5-di(2-pyridyl)benzoato]platinum (115 mg, 0.199 mmol) was added, and heated under reflux for 24 h at 85 °C under a nitrogen atmosphere. The resulting suspension was dried under reduced pressure, and then stirred with dichloromethane (30 ml) until the residues were dissolved. The pale-yellow mixture was then filtered, and solvents removed in vacuo. The resulting purple solid, was washed with an excess of diethyl ether resulting in pure product. Yield: 30 mg, 30.6%. 1H NMR (400 MHz, CDCl3) δH: 9.32 (d, 3JH–H = 5.3 Hz, 3JH–Pt = 46.1 Hz, 2H, ortho-Py), 8.36 (s, 2H, pincer-Ph), 8.10 (td, 3JH–H = 7.7 Hz, 4JH–H = 1.5 Hz, 2H, para-Py), 7.90 (d, 3JH–H = 7.7 Hz, 2H, meta-Py), 7.53 (t, 3JH–H = 7.6 Hz, 2H, meta-ester-Ph), 7.37 (t, 3JH–H = 7.3 Hz, 4H, meta-Py + para-ester-Ph), (ortho-ester-Ph obscured by CDCl3). 13C {1H} NMR (125 MHz, CDCl3) δC: 180.4, 167.4, 165.4, 155.6, 150.8, 147.4, 143.5, 139.8, 136.1, 129.6, 126.1, 125.5, 124.9, 121.6, 120.4. Low solubility did not allow detection of all quaternary carbons. IR (cm−1): ν(CN) 2116, ν(CO) 1725. Mass spectrometry (positive loop injection): measured (MeOH) m/z – 595.0787, calculated for [C24O2N3H15Pt1][Na]+ – 595.070634 – correct isotope pattern.
O) 1677. Mass spectrometry (positive loop injection): measured (MeOH) m/z – 517.0678, calculated for [C19O1N3H13Pt][Na]+ – 517.0678 – correct isotope pattern.
:1), chloro[ethyl 3,5-di(2-pyridyl)benzoato]platinum (106 mg, 0.199 mmol) was added, and heated under reflux for 24 h at 85 °C under a nitrogen atmosphere. The resulting suspension was dried under reduced pressure, and then stirred with dichloromethane (30 ml) until the residues were dissolved. The pale-yellow mixture was then filtered, and solvents removed in vacuo. The resulting yellow/orange solid, was washed with an excess of diethyl ether resulting in pure product. Yield: 30 mg, 30.6%. 1H NMR (500 MHz, CDCl3) δH: 9.28 (d, 3JH–H = 5.8 Hz, 3JH–Pt = 47.3 Hz, 2H, ortho-Py), 8.20 (s, 2H, pincer-Ph), 8.05 (t, 3JH–H = 7.8 Hz, 2H, para-Py), 7.84 (d, 3JH–H = 7.8 Hz, 2H, meta-Py), 4.46 (q, 3JH–H = 7.1 Hz, 2H, CO2CH2), 1.48 (t, 3JH–H = 7.1 Hz, 3H, CO2CH2CH3). 13C30 NMR (101 MHz, CDCl3) δC: 179.9, 167.7, 166.6, 155.7, 153.5, 143.2, 139.6, 136.9, 126.7, 125.0, 124.6, 120.3, 61.3, 14.5. IR (cm−1): ν(CN) 2111, ν(CO) 1702. Mass spectrometry (positive loop injection): measured (MeOH) m/z – 547.0688, calculated for [C20O2N3H15Pt1][Na]+ – 547.070599. Correct isotope pattern.
:1), chloro[phenyl 3,5-di(2-pyridyl)benzoato]platinum (115 mg, 0.199 mmol) was added, and heated under reflux for 24 h at 85 °C under a nitrogen atmosphere. The resulting suspension was dried under reduced pressure, and then stirred with dichloromethane (30 ml) until the residues were dissolved. The pale-yellow mixture was then filtered, and solvents removed in vacuo. The resulting purple solid, was washed with an excess of diethyl ether resulting in pure product. Yield: 30 mg, 30.6%. 1H NMR (400 MHz, CDCl3) δH: 9.32 (d, 3JH–H = 5.3 Hz, 3JH–Pt = 46.1 Hz, 2H, ortho-Py), 8.36 (s, 2H, pincer-Ph), 8.10 (td, 3JH–H = 7.7 Hz, 4JH–H = 1.5 Hz, 2H, para-Py), 7.90 (d, 3JH–H = 7.7 Hz, 2H, meta-Py), 7.53 (t, 3JH–H = 7.6 Hz, 2H, meta-ester-Ph), 7.37 (t, 3JH–H = 7.3 Hz, 4H, meta-Py + para-ester-Ph), (ortho-ester-Ph obscured by CDCl3). 13C {1H} NMR (125 MHz, CDCl3) δC: 180.4, 167.4, 165.4, 155.6, 150.8, 147.4, 143.5, 139.8, 136.1, 129.6, 126.1, 125.5, 124.9, 121.6, 120.4. Low solubility did not allow detection of all quaternary carbons. IR (cm−1): ν(CN) 2116, ν(CO) 1725. Mass spectrometry (positive loop injection): measured (MeOH) m/z – 595.0787, calculated for [C24O2N3H15Pt1][Na]+ – 595.070634 – correct isotope pattern.
X-ray crystallography
32 and refined by full-matrix least-squares based on F2 using SHELXL-201433 in the OLEX2 suite.34 All non-hydrogen atoms were refined with anisotropic displacement parameters and hydrogen atoms were placed in idealised positions and allowed to ride on the relevant carbon atoms. Isotropic displacement parameters for the hydrogen atoms were set at 1.2 Ueq of the aromatic carbon atoms and 1.5 Ueq for the methyl hydrogen atoms; the methyl groups were refined as rigid bodies. The hydrogen atoms on the solvent water molecules were not located and were not included in the refinement cycles. In structure 2′ an attempt was made to model the disordered solvent in the channels (probably water and acetone) by assigning the largest peaks as water molecules and assigning them isotropic displacement parameters. Refinements were continued until convergence was reached and the residual electron density maps showed no chemically sensible residual features.
Results and discussion
The platinum(II) pincer complexes 2, 3 and 4 were prepared following the same general synthetic methodology as previously reported for complex 1 (Fig. 3).30 The complexes were characterised by IR and 1H and 13C NMR spectroscopies and by mass spectrometry. In the IR spectra of the three complexes each showed stretching frequencies characteristic of the cyanide and carbonyl groups around 2114 cm−1 and 1700 cm−1, respectively. The TOF electrospray time-of-flight (ESI-TOF) mass spectra showed ions corresponding to the [Na]+ salt and the correct isotope patterns for the chemical formula.The study of complex 1 had shown that the vapochromic and solvatochromic behaviour in the solid state was associated with the intermolecular interactions between the pincer complex and the water or methanol molecules and also the Pt⋯Pt stacking interactions. It was appropriate to determine the crystal structures of the new complexes in the presence or absence of solvent molecules and establish whether Pt⋯Pt stacking occurred in the anhydrous and solvated forms, if possible, correlating the structural data with the vapochromic or solvatochromic behaviour. In 1 in the red form, [Pt(N⁁C(C(O)OMe)⁁N)(CN)]·H2O, there is an extended hydrogen-bonding network that bridges between the cyanide ligands on adjacent pincer complexes, and brings the Pt-centres close enough to interact at a distance of 3.452(3) Å. This degree of metal⋯metal interaction facilitates triplet metal–metal-to-ligand charge-transfer (3MMLCT), also observed in related platinum complexes.36,37 In the yellow anhydrous form of 1, [Pt(N⁁C(C(O)OMe)⁁N)(CN)], there is no solvent mediated hydrogen-bonding network, and in the crystal structure there is a staggered arrangement of pincer molecules with an increase in Pt⋯Pt separation to 3.663(1) Å. While in the blue methanol solvated form of 1, [Pt(N⁁C(C(O)OMe)⁁N)(CN)]·MeOH, the methanol molecules participate in a hydrogen-bonding, with each methanol molecule hydrogen bonding to one cyanide group on a nearby pincer molecule. This facilitates a shorter Pt⋯Pt stacking interaction with the shortest Pt⋯Pt distance of the three at 3.388(1) Å. These small changes in the Pt⋯Pt interaction distance and the metal⋯metal orbital overlap influence the 3MMLCT transitions in each of the three forms and thus provide an explanation for the vapochromic response. This combination of vapochromic and structural data provides a benchmark for the studies described for complexes 2, 3 and 4, described here.
Cyano[acetyl 3,5-di(2-pyridyl)phenyl]platinum 2
 | ||
| Fig. 4 Hot-stage microscope images of 2 displaying the colour transformation. | ||
In order to confirm whether or not the colour change involves the loss of water, IR spectra were run on the solid-state before and after the transformation. These could be recorded straightforwardly because of the irreversibility of the transition. The spectra are largely identical (Fig. S1†), however upon transformation to form-II, there is the complete loss of a broad signal at 3405 cm−1. This broad signal is highly characteristic of water as part of a hydrogen bonding network.38 The only other differences between the two spectra can be seen as small shifts in the peaks assigned to the νCN and νCO: to 2108 cm−1 and 1670 cm−1, respectively. These small changes are consistent with changes in the hydrogen bonding associated with the loss of water. The loss of water from the green form of 2 was further confirmed by a thermogravimetric analysis. The result indicates that there is a two-step loss of 3.45% which corresponds to 17.67 g mol−1 or one equivalent of water (Fig. S2†).
Powder X-ray diffraction has been used to assess whether the green to yellow transition was a result of a structural transition. First, a powder pattern was recorded for a sample of the green powder in an open-ended capillary. This was then heated until the sample had visibly changed completely to yellow, and the diffraction pattern remeasured. The patterns produced are of low resolution (measured using an in-house diffractometer), but they serve well as a fingerprinting tool, and it is at first immediately clear that both the forms are crystalline, giving defined diffraction patterns. It is also apparent that the crystal structure has transformed as a result of heating, producing a markedly different pattern (Fig. S3†), indicating that a very definite structural transition coincides with the colour change, as observed for 1.
A further powder diffraction experiment was performed to track the structural changes between the forms, a second capillary of the green powder was prepared (with one open end for water to leave), and the temperature scanned between 300–390 K measuring the diffraction pattern at 10 K intervals. The experiment (Fig. S4†) resulted in a phasing-out of the green form-I pattern with simultaneous phasing-in of the yellow form-II pattern, as the temperature increases. This suggests a definite and rapid crystalline switch, with no detectable intermediates.
During the crystallisation experiments, it was possible to obtain alternative green crystals from the solvent mixture that do not display the same diffraction pattern with that of the bulk powder but may be a different solvate of the thermochromic green material with a different solvent mixture. The structure of these crystals was determined, admittedly to a low quality, partly attributed to the quantity of disordered solvent present in the crystal. While this structure does not accurately represent the structure of the bulk thermochromic powder, it does offer us some insights into the possible structure of the powder. The similar colouration for example suggests there may be a comparable level of intermolecular interaction between the platinum centres.
The crystal of the green form of 2 (designated 2′ to differentiate it from the structure of the bulk form) was obtained by vapour diffusion of n-hexane into a water/acetonitrile solution. 2′ crystallised in the orthorhombic space group Ibam, with the asymmetric unit consisting of half a neutral molecule of 2′, one defined molecule of water, and a large quantity of disordered solvent (Fig. 5). The complex lies on a crystallographic mirror plane so that the whole structure is planar with the C(O)Me substituent required to be in the same plane as the three six-membered rings, the Pt atom and the cyanide group. The complex adopts the expected square planar coordination geometry at the Pt(II) centre, with the cyanide group being situated trans to the coordinated aryl ring. The angles at the Pt(II) display minor distortions from the idealized geometry (N1–Pt1–N2 159.4 (10)°) because of the pincer bite angle, with bond parameters in the expected ranges (Table 1).39–41
 | ||
| Fig. 5 The asymmetric unit of 2′ (green form) as determined by single crystal X-ray diffraction. The crystal was not of sufficient quality to resolve the water hydrogen atoms. Disordered solvent molecules have been omitted for clarity. Displacement ellipsoids are set at 50% probability. | ||
| Bond | Length (Å) |
|---|---|
| Pt1–N1 | 2.024(18) |
| Pt1–N2 | 2.083(22) |
| Pt1–C6 | 1.946(28) |
| Pt1–C19 | 2.033(31) |
| C19–N3 | 1.182(38) |
The extended structure of the crystal consists of layers of planar units of 2′ arranged into columns, with significant levels of platinum–platinum interaction (Pt–Pt–Pt angle of 177.20 (6)°). The molecules also alternate between two planar orientations, separated by a torsion of 47.93° (taken as angle between planes [C19, Pt1, C19#, Pt1#] and [C19$, Pt1$, C19&, Pt1&] where # = (x, y, 1 + z), $ = (x, ½ − y, ½ + z), & = (x, ½ −y, −½ + z)) (Fig. 6). The Pt⋯Pt interaction distance along these stacks is 3.319(1) Å which is slightly shorter than the Pt⋯Pt distances of 3.452(3) Å and 3.388(1) Å in the red and blue forms of 1, respectively. Each molecule of 2′ is hydrogen bonded to the structurally ordered water molecule via the cyanide ligand, which are then arranged into a network of water that extends along the c-axis (Table 2).
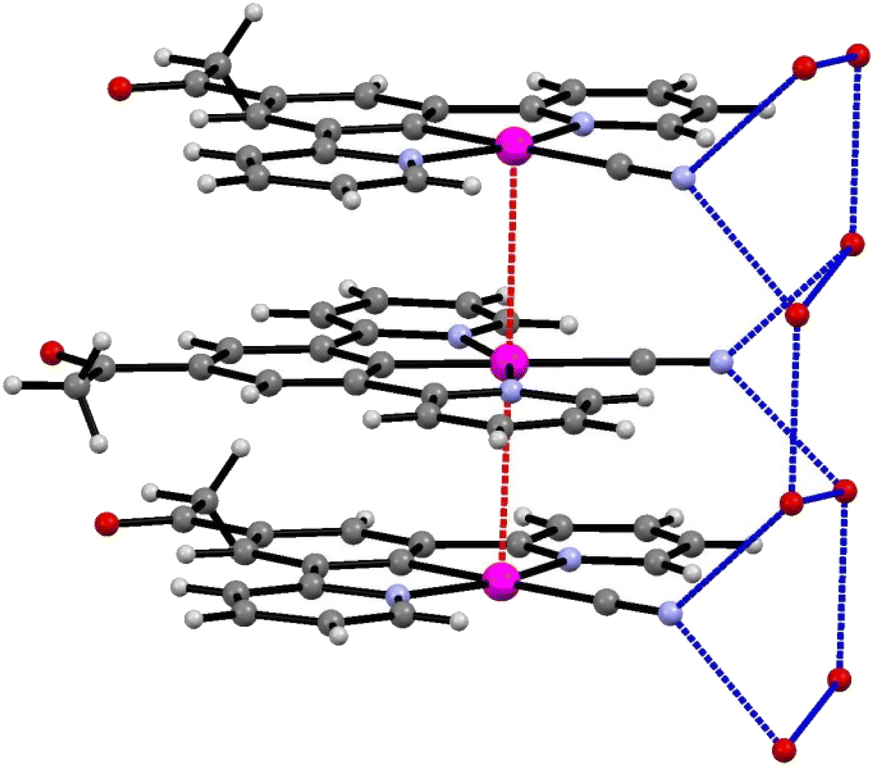 | ||
| Fig. 6 The stacks of 2′, highlighting the platinum–platinum interactions (red) and hydrogen bonding of the water to the cyanide groups (blue). | ||
The crystal structure is porous, with a large void that runs along the c-axis. By omitting the occupying solvent molecules, the channels take up 35.6% of the total crystal volume. The channel contains disordered solvent that it was not possible to resolve crystallographically, although an attempt was made to model some of the largest residual peaks by assigning them as water molecules. It is that the channels contain a mixture of water and acetonitrile (Fig. 7). The formation of such a large pore is unusual in this class of complexes. As can be seen in Fig. 6 the stacks of molecules along the c-axis are similar to those observed in 1 with planar molecules, in two alternating orientations and Pt⋯Pt interactions.30 It is interesting to understand how a minor change in structure between 1 and 2′, replacing an ester group by a smaller acetyl substituent, has such a major influence on the crystal packing, generating large pores through the structure, resulting in different solvatochromic properties. In the crystal structure of 2′ the weak C–H donor hydrogen bonding interactions play a different role to those in 1 forming rings between the pincer molecules in the same layer of the crystal (Fig. 8). Unlike in 1 there are no interactions between the cyanide and the substituent group (acetyl) in 2′ and the orientation is completely different such that that interaction to this group bridged by solvent is impossible. It appears that the replacement of the ester with an acetyl group has prevented this type of inter-stack interaction. In the absence of this interaction, there are in-plane weak hydrogen bonding interactions from the pincer to the cyanide and carbonyl groups, which in turn leads to the formation of a large ring of molecules. These rings then stack via Pt⋯Pt interactions to create the pores of the structure. Details of these hydrogen bonding interactions are listed in Table 3.
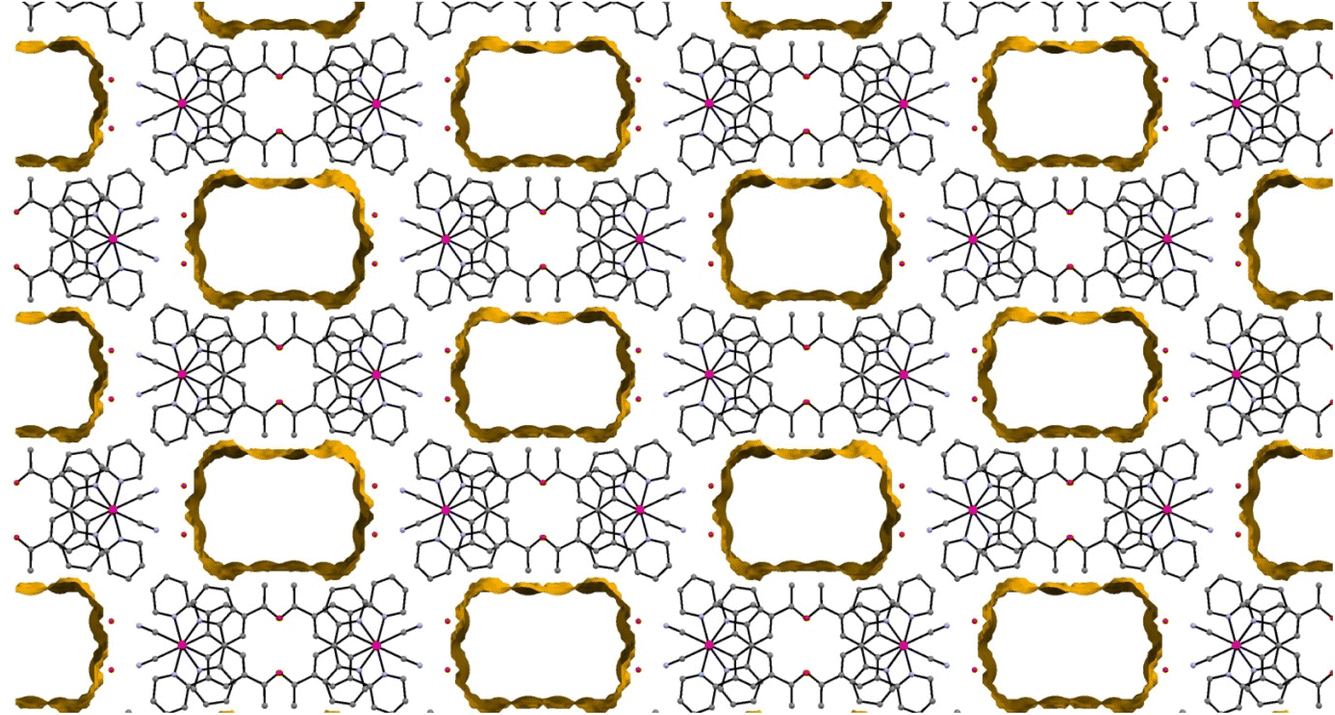 | ||
| Fig. 7 The view along the c-axis of 2′ with the large pores highlighted. The disordered solvent within these pores is not shown. | ||
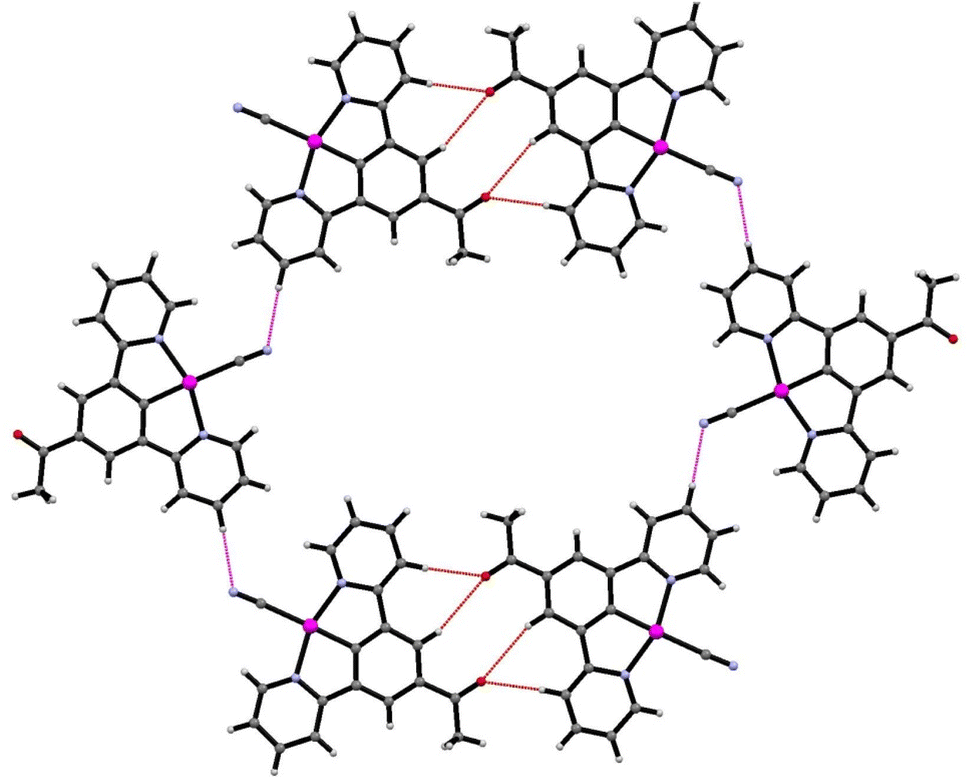 | ||
| Fig. 8 Details of the weak hydrogen bonding interactions in the crystal structure of 2′. | ||
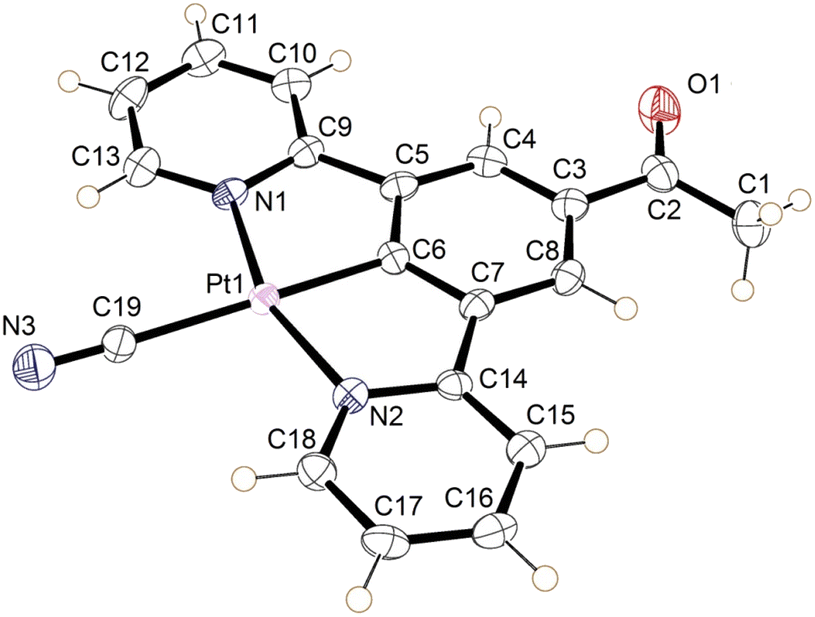 | ||
| Fig. 9 The molecular structure of the yellow form of 2 (form-II). Displacement ellipsoids are set at 50% probability. | ||
| Bond | Length (Å) |
|---|---|
| Pt1–N1 | 2.034(3) |
| Pt1–N2 | 2.043(3) |
| Pt1–C6 | 1.934(3) |
| Pt1–C19 | 2.058(3) |
| C19–N3 | 1.148(5) |
The crystal packing in the structure of the yellow complex 2 (Fig. 10 and 11) consists of stacks of end-to end pairs of pincer molecules, themselves arranged into a herringbone orientation (two orientations of stacks) so that there are essentially no longer planar layers in the crystal with Pt⋯Pt stacks perpendicular to them. Platinum–platinum separations within the pairs are 4.813 (1) Å and 5.341 (2) Å which indicates that there are no direct metal⋯metal interactions in the structure as is the case in the yellow form of 1. Weak C–H donor hydrogen bonding interactions are present both within each pair, between pairs in the stacks, and between the stacks, which involve either the cyanide or carboxylate groups. Details of these interactions can be found in Table 5.
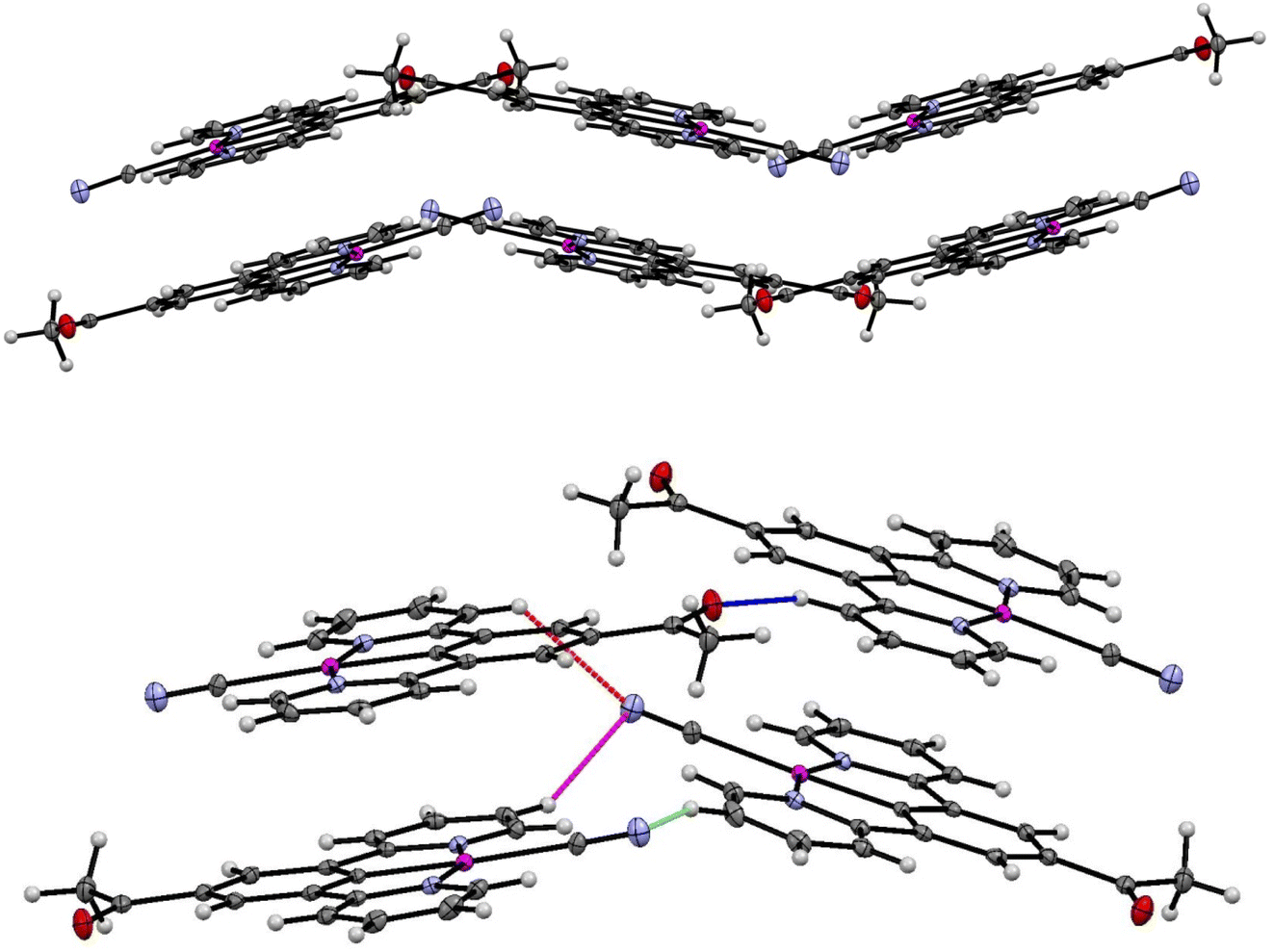 | ||
| Fig. 10 (Top) Herringbone arrangement in the yellow form of 2. (Bottom) Weak hydrogen bonding interactions between the two orientations of the herringbone structure. | ||
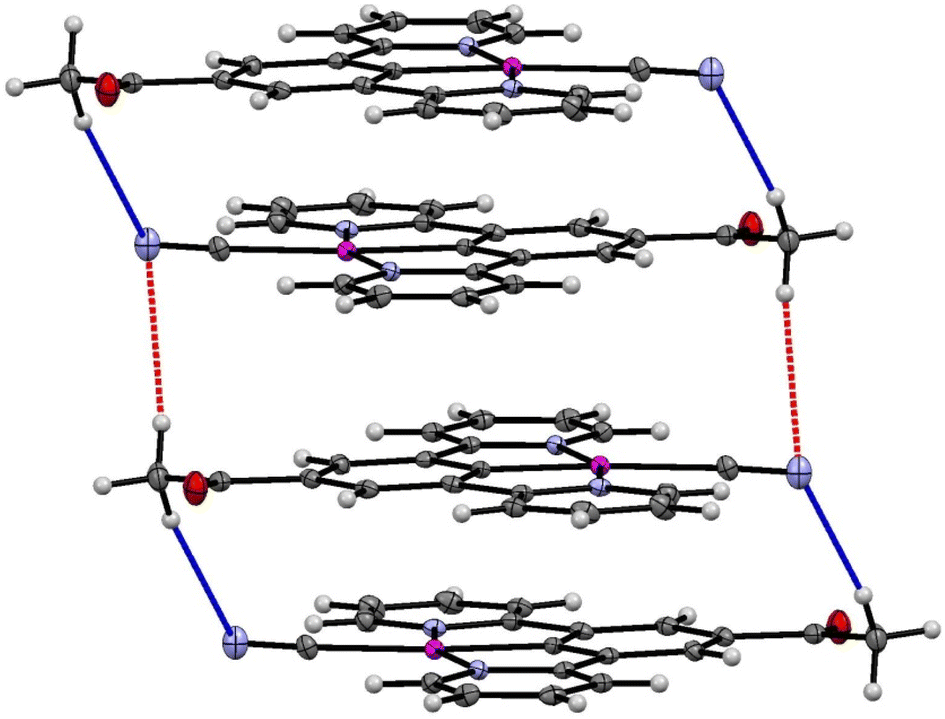 | ||
| Fig. 11 Weak hydrogen bonding interactions within and between pairs of molecules of the yellow form of 2 (form-II). | ||
| Bonda | Distance (D⋯A) (Å) | Distance (H⋯A) (Å) | Angle (D⋯H⋯A) (°) |
|---|---|---|---|
| a 1 = (−x, −y, 1 − z), 2 = (1 − x, −y, 1 − z), 3 = (x, ½ − y, ½ + z), 4 = (x, −½ − y, −½ + z) 5 = (x, ½ − y, −½ + z), 6 = (1 − x, −½ + y, 1.5 − z). | |||
| C1–H1C–N31 | 3.736 (6) | 2.891 | 147.42 |
| C1–H1B–N32 | 3.742 (6) | 2.792 | 170.35 |
| C12–H12–N33 | 3.315 (5) | 2.526 | 142.80 |
| C15–H15–O14 | 3.148 (5) | 2.447 | 132.20 |
| C17–H17–N35 | 3.721 (5) | 2.867 | 153.29 |
| C10–H10–N36 | 3.460 (5) | 2.788 | 130.08 |
It is clear from the extended structure (Fig. 10) that there are significant differences in arrangement from those observed in the crystal packing of 1, and from the packing in the green form of 2′ (form-I). As with the green crystal, there is no interaction between the acetyl group and adjacent cyanides, but it is also non-planar. The absence of this interaction between the cyanide and the substituted group of the pincer could be the important difference that prevents reversible transitions between hydrates/solvates and the anhydrous form for this complex, when compared to 1. It may be the case looking at 1 that the interaction between the ester and adjacent cyanides stabilises the solvent-free structure in a planar configuration that allows for re-insertion of water or other solvents, leading to the observed vapochromic and solvatochromic properties.
Although for 2 the structure of the thermochromic green form has not been determined, however, based on other experimental data and the nature of the alternative green form produced 2′, it may be hypothesised that they share similarities. It is likely that the green form-I is a planar, platinum stacked structure, as evidenced by the green coloration. It has also been shown that water is present in the structure.
Based on this, it may be proposed that due to a lack of the weak intermolecular interactions to adjacent cyanides, provided by the ester group, planar structures facilitating platinum–platinum interactions for complex 2 are only stable in the presence of water. The transition to a buckled herringbone yellow form, unable to re-accept water occurs once water is evacuated, and it may follow that the dissimilarity of this structure to any seen for this class of complexes so far (i.e. lack of planarity) may be the cause for this lack of reversibility.
Cyano[ethyl 3,5-di(2-pyridyl)benzoato]platinum (3)
An ethyl analogue of the pincer ester was synthesised by esterifying 1,3-dibromobenzoic acid, and then performing Stille-coupling,42 then performing cyclometallation15 and salt metathesis steps as with 1 to obtain 3.
Complex 3 was fully characterised by NMR and IR spectroscopy, mass spectrometry and X-ray crystallography. The ethyl signal was clearly visible in the 1H NMR spectrum. The IR spectrum shows a strong, sharp absorption at 2111 cm−1 assignable to the νCN of a cyanide group. This value is consistent with those recorded for a series of related compounds.43,44 Also seen is a single strong absorption at 1702 cm−1 assignable to the νCO of the ester group, which is a typical value for an ethyl benzoate ester.
 | ||
| Fig. 12 Photographs showing the solvent interactions of 3. (Left) Application of dichloromethane vapour to a drop cast sample on a glass slide. (Right) Application of water to the same sample to return it to the orange form. | ||
The reversibility of this transition is also different to that observed for 1. For 3, some thin film samples, the transition to green became slower/less complete with repeated transitions, and unless re-precipitated the sample eventually became permanently orange. Investigation into why this may be, led to the realisation that the substance has a high affinity for crystallisation with dichloromethane, and it is proposed that this non-vapochromic solvate may result in a more stable structural configuration, resulting in eventual transformation to this form. In response to this, the solid was dissolved in acetone and repeatedly dried rapidly under heat and vacuum to try and remove residual crystallised dichloromethane from the reaction conditions. The solid was then dissolved in hot isopropanol and precipitated as green solid. This solid transitions between green and orange forms which were then fully reversible.
The nature of these transitions was investigated in the same manner as for 1, with the additional advantage of having a detailed understanding of the process in 1. It would, for instance, be a reasonable assumption that the rapid switching between coloured forms is again due to a structural rearrangement facilitating varying degrees of inter-metallic overlap. It may also be hypothesised that due to the orange form being produced upon exposure to water, that a form structurally similar to that of the red form of 1 could be produced, with the water hydrogen bonding to the cyanide ligands.
The green form however is not as easily explainable. The compound does not initially appear to uptake any solvent into the structure, given that multiple solvents result in the same visual response, including the non-hydrogen bonding solvent dichloromethane (although residual water may be present). It was therefore proposed that the green form is perhaps analogous to the solvent-free, yellow form of 1, and the solvent vapour acts to displace the bound water molecules. However, transition to the green form was not producible with heat, vacuum, or dry gas flow. An alternative explanation could be that both forms, orange and green, are water adducts and the difference between them is the number of water molecules absorbed. The orange form could contain more water molecules that prevents stacking between the Pt centres while the green form with fewer water molecules, using only solvent vapour, results in better Pt⋯Pt overlap in the stacks.
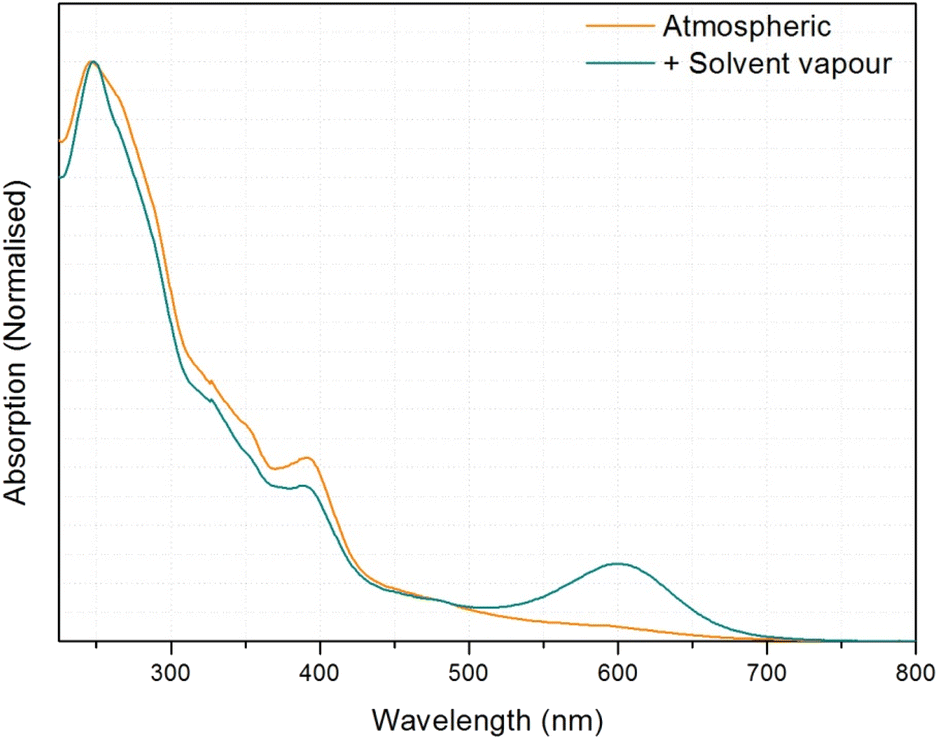 | ||
| Fig. 13 UV-visible spectrum of 3 before and after solvent vapour application. | ||
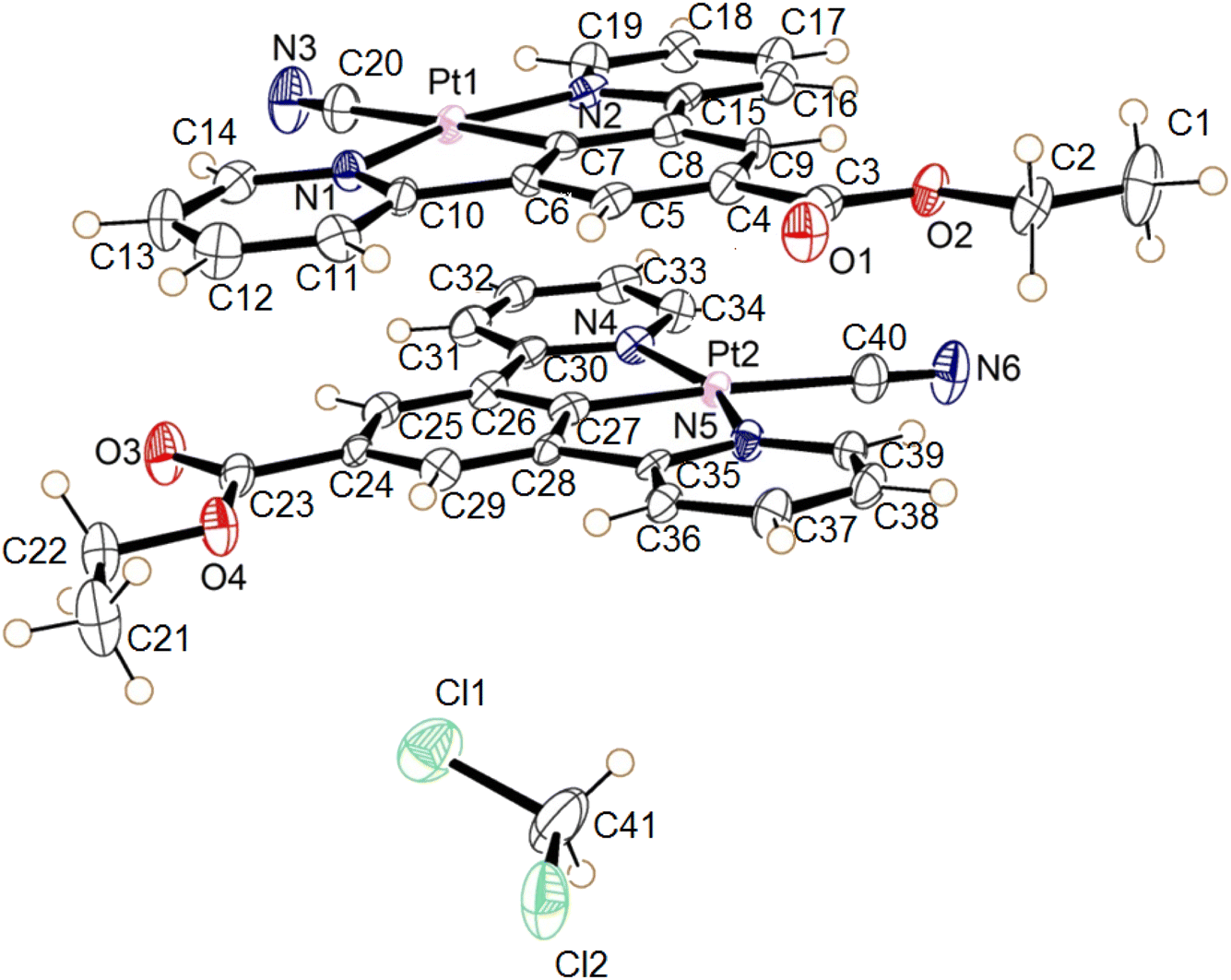 | ||
| Fig. 14 The molecular structures of the two independent molecules of the dichloromethane solvate of 3, the displacement ellipsoids are shown with 50% probability. | ||
The complex 3 crystallised in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with two independent neutral molecules of 3, and one of dichloromethane, in the asymmetric unit of the unit cell. The molecular structure of the independent molecules is similar to that of 1, with the ester groups lying close to the plane of the pincer ligand. The platinum centres adopt the expected square planar geometry with minor distortion (N1–Pt1–N2 angle 160.3° (2) and N4–Pt2–N5 angle 159.7 (2)°) due to the pincer bite angle. Bond parameters are within expected range (Table 6).39–41 The extended structure has the molecules lying in planes, however Pt⋯Pt separation between the layers is 4.6139(5) Å suggesting no interaction. Weak C–H donor hydrogen-bonding to the dichloromethane, cyanide and carboxylate groups is also present in the structure. Details of these interactions are listed in Table 7.
, with two independent neutral molecules of 3, and one of dichloromethane, in the asymmetric unit of the unit cell. The molecular structure of the independent molecules is similar to that of 1, with the ester groups lying close to the plane of the pincer ligand. The platinum centres adopt the expected square planar geometry with minor distortion (N1–Pt1–N2 angle 160.3° (2) and N4–Pt2–N5 angle 159.7 (2)°) due to the pincer bite angle. Bond parameters are within expected range (Table 6).39–41 The extended structure has the molecules lying in planes, however Pt⋯Pt separation between the layers is 4.6139(5) Å suggesting no interaction. Weak C–H donor hydrogen-bonding to the dichloromethane, cyanide and carboxylate groups is also present in the structure. Details of these interactions are listed in Table 7.
| Bond | Length (Å) |
|---|---|
| Pt1–N1 | 2.023 (5) |
| Pt1–N2 | 2.050 (7) |
| Pt1–C7 | 1.943 (6) |
| Pt1–C20 | 2.089 (7) |
| C20–N3 | 1.120 (10) |
| Bond | Length (Å) |
|---|---|
| Pt2–N4 | 2.051 (6) |
| Pt2–N5 | 2.054 (6) |
| Pt2–C27 | 1.938 (7) |
| Pt2–C40 | 2.078 (7) |
| C40–N6 | 1.129 (9) |
| Bonda | Distance (D⋯A) (Å) | Distance (H⋯A) (Å) | Angle (D⋯H⋯A) (°) |
|---|---|---|---|
| a 1 = (x, 1 + y, z), 2 = (1 − x, 1 − y, −z), 3 = (1 − x, 1 − y, 1 − z), 4 = (1 − x, 2 − y, −z), 5 = (1 − x, 2 − y, 1 − z), 6 = (1 + x, 1 + y, z). | |||
| C31–H23–Cl21 | 3.759 (8) | 2.840 | 170.16 |
| C13–H10–Cl22 | 3.469 (9) | 2.820 | 127.87 |
| C12–H9–Cl22 | 3.532 (8) | 2.939 | 122.93 |
| C38–H29–N63 | 3.546 (10) | 2.777 | 140.80 |
| C21–H17–N34 | 3.486 (11) | 2.730 | 136.15 |
| C18–H14–N65 | 3.450 (12) | 2.590 | 153.93 |
| C13–H10–O34 | 3.2215 (09) | 2.471 | 137.90 |
| C33–H25–N65 | 3.557 (10) | 2.867 | 131.84 |
| C32–H24–O16 | 3.574 (9) | 2.651 | 172.03 |
Cyano[phenyl 3,5-di(2-pyridyl)benzoato]platinum (4)
The pincer complex 4 was fully characterised by NMR and IR spectroscopy, and by mass spectrometry. The presence of the phenyl group was clearly indicated by 1H NMR spectroscopy. The IR spectrum shows a strong, sharp absorption at 2116 cm−1, assignable to the νCN of a cyanide group. This value is consistent with those recorded for a series of related compounds.43,44 Also seen is a single strong absorption at 1725 cm−1 assignable to the νCO of the ester group, which is a typical value for a benzoate ester.
 | ||
| Fig. 15 Observation of the vapochromic properties of a drop-cast-thin film (from methanol/dcm solution) (ca. 2 cm in diameter) of 4 and its response to a gentle flow of methanol vapour and after washing with methanol and drying. (a) Purple film on precipitation, (b) turning red upon a gentle flow of solvent vapour, (c) red colouration after solvent vapour treatment, (d) after washing with methanol and drying. | ||
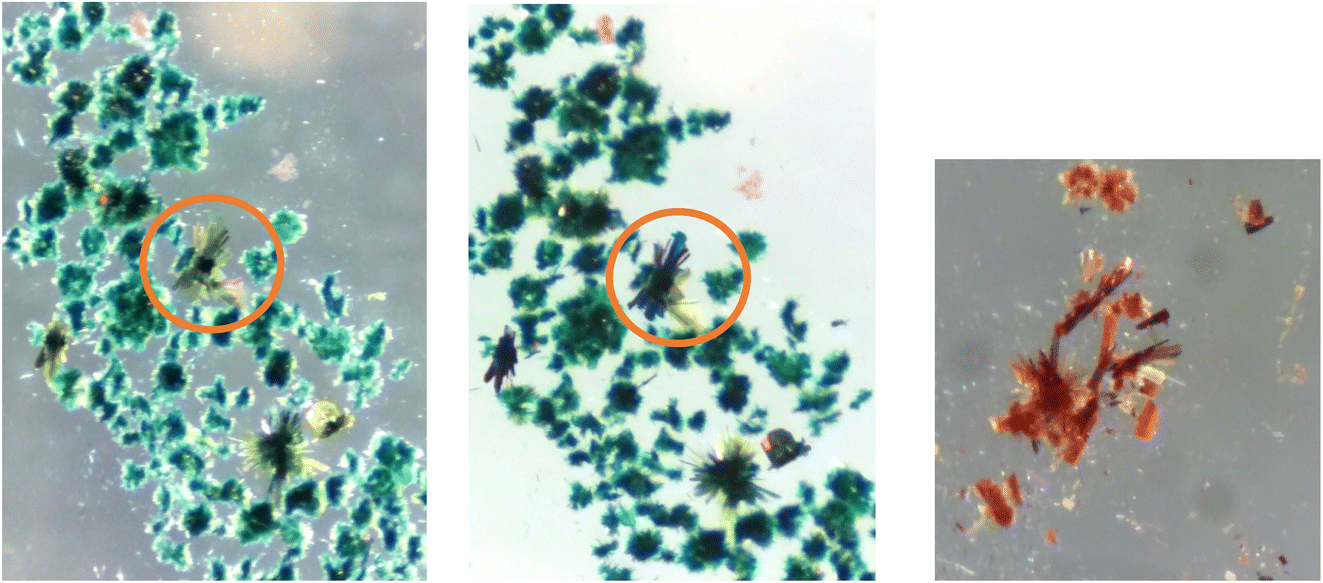 | ||
| Fig. 16 (Left/middle) Dichroism seen in crystals of 4 under two orientations of polarising filter. Within the circle in both photographs is the same cluster of crystals. (Right) The same crystals after exposure to air. | ||
Although the structure could not be established it is likely by comparison with the other members of the series and because of its observed vapochromic behaviour is likely that the crystal is porous, and hydrogen bonds will be formed with hydrogen bond donor solvents. By changing the solvent that the crystalline material is exposed to, through exchange of solvents in the cavities within the crystal a variety of crystalline forms will result, exhibiting different metal⋯metal interactions and different colours.
Conclusions
This study shows that the incorporation of an acyl or ester substituent (R) on the central ring in a (N⁁C⁁N) pincer ligand coupled with the coordination of a small, strong field cyanide ligand to the central Pt atom, with the general formula [Pt(N⁁C(R)⁁N)(CN)], results in the formation of complexes that undergo thermochromic or vapochromic/solvatochromic behaviour. The presence of the smaller acyl substituent results in a thermochromic solid, as solvent water is lost from the complex (2), while with the larger ethyl (3) and phenyl (4) esters, along with the previously reported methyl ester analogue (1)30 the solids display a range of rapidly reversible vapochromic or solvatochromic behaviour with a range of solvents and show some selectivity.An analysis of the factors that can be correlated with the behaviour suggests that the cyanide ligand plays an important role, since simple [Pt(N⁁C⁁N)Cl] complexes have been extensively studied and their luminescent properties investigated3,11 but have not been reported to show vapochromic behaviour, to our knowledge, although substituted pincer ligand systems have. The cyanide ligand may have certain advantages over the chloride. In the case of complexes 1–4 the cyanide ligand serves not only as a hydrogen bonding site for solvent uptake, but as a facilitator for the colour changing interactions by allowing the stabilisation of the HOMOs with which it shares π-interactions, allowing for a prominent non-bonding dz2 orbital relative to the frontier orbitals.
The size and hydrogen bonding capabilities of the substituents on the central ring of the pincer ligand also play a significant role in determining the vapochromic properties. For example, in our hands, the simple [Pt(N⁁C⁁N)(CN)] complex, with no substituent on the central ring, does not show vapochromic responses to common volatile organic solvents.45 The bulkier the substituent the less efficiently do the pincer molecules pack in the solid state, thus creating a greater void space for the solvents or vapours to diffuse through the crystal. Similar effects have been seen for pincer complexes with substituents on the two pyridyl rings of the N⁁C⁁N pincer ligand.21,22 Additionally, the acetyl and ester groups form hydrogen bonded networks running through the crystals, generating channels through which the solvents and vapours can pass, allowing the rapid dynamic colour changes to occur.
In some cases, the pincer complexes show particular selectivity towards particular solvents and vapours. For complex 1 the solid is red in the presence of water and blue in the presence of methanol,30 while for complexes 3 and 4, the colour change is less dependent on the specific solvent used. For example, 3 shows the same colour change for methanol, ethanol or dichloromethane (although residual water may be present in the solvents), and for 4 the colour changes are even more complex. This selectivity can be related to the hydrogen bonding in the material and the influence that this has on the stacking of the layers and the intermetallic dz2–dz2 interactions between the adjacent Pt centres, which is at least partially responsible for the colour of the complexes.
While this study does not rationalise the vapochromic behaviour of platinum pincer complexes fully, it does highlight some of the factors that favour rapidly reversible vapochromic behaviour, and benchmark some of the crystal engineering protocols that would be useful for the design of such materials. There would be many industrial applications of such a protocol with platinum vapochromic compounds as practical chemosensor devices being considered a promising prospect.46 Several attempts have been made at immobilising a vapochromic material into a polymer substrate in order to create “smart coatings” for chemical sensing applications.31,47
Author contributions
M. J. B. and S. F. are responsible for the synthesis reported in the paper, and M. J. B. performed the spectroscopic and vapochromic and solvatochromic studies. M. J. B., L. E. H. and L. H. T. undertook the structural studies and interpretation of the data. M. J. B. and L. H. T. are responsible for the first draft of the paper. P. R. R. is responsible for the conceptualisation of the project, for reviewing and editing the manuscript, and for project administration and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Engineering and Physical Sciences Research Council (EPSRC) for a Programme Grant (grant no. EP/K004956/1). The project was also supported by EPSRC grant EP/101974X. SF is grateful for the support of a fellowship from the European Commission, under the Marie Curie Intra European Fellowship Scheme (PIEF-GA-2009-252883; SFL-PRR).References
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC.
- M. Kato, Bull. Chem. Soc. Jpn., 2007, 80, 287–294 CrossRef CAS.
- L. J. Grove, J. M. Rennekamp, H. Jude and W. B. Connick, J. Am. Chem. Soc., 2004, 126, 1594–1595 CrossRef CAS PubMed.
- L. J. Grove, A. G. Oliver, J. A. Krause and W. B. Connick, Inorg. Chem., 2008, 47, 1408–1410 CrossRef CAS PubMed.
- J. Fornies, S. Fuertes, J. A. Lopez, A. Martin and V. Sicilia, Inorg. Chem., 2008, 47, 7166–7176 CrossRef CAS PubMed.
- Y. Shigeta, A. Kobayashi, T. Ohba, M. Yoshida, T. Matsumoto, H. C. Chang and M. Kato, Chem.–Eur. J., 2016, 22, 2682–2690 CrossRef CAS PubMed.
- S. D. Taylor, W. Howard, N. Kaval, R. Hart, J. A. Krause and W. B. Connick, Chem. Commun., 2010, 46, 1070–1072 RSC.
- S. Chatterjee, A. E. Norton, M. K. Edwards, J. M. Peterson, S. D. Taylor, S. A. Bryan, A. Andersen, N. Govind, T. E. Albrecht-Schmitt, W. B. Connick and T. G. Levitskaia, Inorg. Chem., 2015, 54, 9914–9923 CrossRef CAS PubMed.
- V. M. Shingade, L. J. Grove and W. B. Connick, Dalton Trans., 2020, 49, 9651–9661 RSC.
- M. L. Muro, C. A. Daws and F. N. Castellano, Chem. Commun., 2008, 6134–6136 RSC.
- V. W. W. Yam, K. H. Y. Chan, K. M. C. Wong and N. Y. Zhu, Chem.–Eur. J., 2005, 11, 4535–4543 CrossRef PubMed.
- P. W. Du, J. Schneider, P. Jarosz, J. Zhang, W. W. Brennessel and R. Eisenberg, J. Phys. Chem. B, 2007, 111, 6887–6894 CrossRef CAS PubMed.
- T. J. Wadas, Q. M. Wang, Y. J. Kim, C. Flaschenreim, T. N. Blanton and R. Eisenberg, J. Am. Chem. Soc., 2004, 126, 16841–16849 CrossRef CAS PubMed.
- D. J. Cardenas, A. M. Echavarren and M. C. R. de Arellano, Organometallics, 1999, 18, 3337–3341 CrossRef CAS.
- J. A. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609–8611 CrossRef CAS PubMed.
- J. A. G. Williams, Chem. Soc. Rev., 2009, 38, 1783–1801 RSC.
- S. Fuertes, S. K. Brayshaw, P. R. Raithby, S. Schiffers and M. R. Warren, Organometallics, 2012, 31, 105–119 CrossRef CAS.
- S. Fuertes, C. H. Woodall, P. R. Raithby and V. Sicilia, Organometallics, 2012, 31, 4228–4240 CrossRef CAS.
- Y. Nishiuchi, A. Takayama, T. Suzuki and K. Shinozaki, Eur. J. Inorg. Chem., 2011, 1815–1823 CrossRef CAS.
- C. Y. Lien, Y. F. Hsu, Y. H. Liu, S. M. Peng, T. Shinmyozu and J. S. Yang, Inorg. Chem., 2020, 59, 11584–11594 CrossRef CAS PubMed.
- P. Pander, A. Sil, R. J. Salthouse, C. W. Harris, M. T. Walden, D. S. Yufit, J. A. G. Williams and F. B. Dias, J. Mater. Chem. C, 2022, 10, 15084–15095 RSC.
- S. Hattori, T. Nakano, N. Kobayashi, Y. Konno, E. Nishibori, T. Galica and K. Shinozaki, Dalton Trans., 2022, 51, 15830–15841 RSC.
- S. J. Farley, D. L. Rochester, A. L. Thompson, J. A. K. Howard and J. A. G. Williams, Inorg. Chem., 2005, 44, 9690–9703 CrossRef CAS PubMed.
- J. A. G. Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef.
- W. A. Tarran, G. R. Freeman, L. Murphy, A. M. Benham, R. Kataky and J. A. G. Williams, Inorg. Chem., 2014, 53, 5738–5749 CrossRef CAS PubMed.
- A. Kobayashi, T. Yonemura and M. Kato, Eur. J. Inorg. Chem., 2010, 2465–2470 CrossRef CAS.
- Y. Kunugi, K. R. Mann, L. L. Miller and C. L. Exstrom, J. Am. Chem. Soc., 1998, 120, 589–590 CrossRef CAS.
- K. M. C. Wong and V. W. W. Yam, Acc. Chem. Res., 2011, 44, 424–434 CrossRef CAS PubMed.
- S. C. F. Kui, S. S. Y. Chui, C. M. Che and N. Y. Zhu, J. Am. Chem. Soc., 2006, 128, 8297–8309 CrossRef CAS PubMed.
- M. J. Bryant, J. M. Skelton, L. E. Hatcher, C. Stubbs, E. Madrid, A. R. Pallipurath, L. H. Thomas, C. H. Woodall, J. Christensen, S. Fuertes, T. P. Robinson, C. M. Beavers, S. J. Teat, M. R. Warren, F. Pradaux-Caggiano, A. Walsh, F. Marken, D. R. Carbery, S. C. Parker, N. B. McKeown, R. Malpass-Evans, M. Carta and P. R. Raithby, Nat. Commun., 2017, 8, 1800 CrossRef CAS PubMed.
- C. M. Sangan, O. J. Pountney, J. A. Scobie, B. Cochrane, C. Stubbs and P. R. Raithby, Int. J. Heat Mass Transfer, 2018, 127, 437–446 CrossRef CAS.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori, D. Siliqi and R. Spagna, J. Appl. Crystallogr., 2007, 40, 609–613 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Nolze and W. Kraus, Powdercell 2.3, Federal Institute for Materials Research and Testing, Berlin, 2000 Search PubMed.
- W. Lu, B.-X. Mi, M. C. W. Chan, Z. Hui, C.-M. Che, N. Zhu and S.-T. Lee, J. Am. Chem. Soc., 2004, 126, 4958–4971 CrossRef CAS PubMed.
- S. J. Choi, J. Kuwabara, Y. Nishimura, T. Arai and T. Kanbara, Chem. Lett., 2012, 41, 65–67 CrossRef CAS.
- K. Mizuse, Spectroscopic Investigations of Hydrogen Bond Network Structures in Water Clusters, Springer Science & Business Media, 2013 Search PubMed.
- M. S. Khan, M. K. Al-Suti, M. R. A. Al-Mandhary, B. Ahrens, J. K. Bjernemose, M. F. Mahon, L. Male, P. R. Raithby, R. H. Friend, A. Kohler and J. S. Wilson, Dalton Trans., 2002, 65–73 Search PubMed.
- J. R. Berenguer, E. Lalinde and J. Torroba, Inorg. Chem., 2007, 46, 9919–9930 CrossRef CAS PubMed.
- Y. Chen, K. Li, W. Lu, S. S.-Y. Chui, C.-W. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2009, 48, 9909–9913 CrossRef CAS PubMed.
- J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef.
- A. A. Lemus-Santana, J. Rodríguez-Hernández, L. F. delCastillo, M. Basterrechea and E. Reguera, J. Solid State Chem., 2009, 182, 757–766 CrossRef CAS.
- T. Kayano, S. Takayasu, K. Sato and K. Shinozaki, Chem.–Eur. J., 2014, 20, 16583–16589 CrossRef CAS PubMed.
- M. J. Bryant, Platinum Pincer Complexes: in Pursuit of Switchable Materials, PhD Thesis, University of Bath, 2015.
- A. Kobayashi and M. Kato, Eur. J. Inorg. Chem., 2014, 2014, 4469–4483 CrossRef CAS.
- J. R. Kumpfer, S. D. Taylor, W. B. Connick and S. J. Rowan, J. Mater. Chem., 2012, 22, 14196–14204 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2238847–2238849. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3fd00025g |
| This journal is © The Royal Society of Chemistry 2023 |