
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of non-metal single atomic phosphorus active sites for the CO2 reduction reaction†
Hong
Bin Yang‡
a,
Cong-Qiao
Xu‡
 b,
Sambath
Baskaran
b,
Ying-Rui
Lu
c,
Chengding
Gu
d,
Wei
Liu
e,
Jie
Ding
a,
Jincheng
Zhang
a,
Qilun
Wang
a,
Wei
Chen
e,
Jun
Li
*bg,
Yanqiang
Huang
f,
Tao
Zhang
f and
Bin
Liu
*a
b,
Sambath
Baskaran
b,
Ying-Rui
Lu
c,
Chengding
Gu
d,
Wei
Liu
e,
Jie
Ding
a,
Jincheng
Zhang
a,
Qilun
Wang
a,
Wei
Chen
e,
Jun
Li
*bg,
Yanqiang
Huang
f,
Tao
Zhang
f and
Bin
Liu
*a
aDepartment of Materials Science and Engineering, City University of Hong Kong, Hong Kong SAR 999077, China. E-mail: junli@tsinghua.edu.cn; bliu48@cityu.edu.hk
bDepartment of Chemistry, Southern University of Science and Technology, Shenzhen 518055, China
cNational Synchrotron Radiation Research Center, Hsinchu, 30076, Taiwan
dSchool of Materials and Energy, Yunnan University, 650091, China
eDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore
fDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
gDepartment of Chemistry and Key Laboratory of Organic Optoelectronics and Molecular Engineering of Ministry of Education, Tsinghua University, Beijing 100084, China
First published on 13th July 2023
Abstract
Heterogeneous single-atom catalysts (SACs) show maximum atom utilization efficiency and great product selectivity in various catalytic reactions. To date, SACs have been intensively investigated with various metal active centers, while non-metal active centers are rarely elucidated, owing to the challenges in identifying non-metal active sites and reaction pathways. Herein, we develop a non-metal single-atom catalyst composed of high-density, isolated phosphorus atoms anchored at the edge of graphene as a robust electrocatalyst for the CO2 reduction reaction. The CO2 molecular activation and the electrochemical CO2 reduction pathways at P atomic sites are identified by combining both in situ/ex situ experiments and quantum-chemical theoretical study. Our work not only extends the definition of SACs to a non-metal catalytic center, but also offers unique insights into the non-metal active site geometric and structural transformation during the CO2 reduction reaction.
Broader contextHeterogeneous single-atom catalysts (SACs) refer to the catalysts that contain merely atomically dispersed catalytic centers on a support. SACs, by definition, can be categorized into metal SACs (M-SACs) and non-metal SACs (NM-SACs) based on the type of catalytic center. To date, SACs with only metal active centers ranging from noble metals to transition and main group metals have been extensively investigated in a wide range of catalytic reactions, while non-metal active centers in SACs are poorly elucidated, owing to the challenges in confirming the active sites and identifying the reaction intermediates and reaction mechanisms. In this context, we developed a non-metal single-atom catalyst consisting of isolated phosphorus atoms anchored at the edge of graphene (P-SAC-NG) as an efficient electrocatalyst for CO2 reduction to CO. The CO2 molecular activation and electrochemical reduction pathways at the P atomic sites at the edge of graphene with the 2C–P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(OH) configuration are clearly identified by in situ/ex situ experiments together with density functional theory (DFT) calculations. Our work extends the definition of SACs to a non-metal catalytic center, and offers insights critical for the development of a family of non-metal SACs. O(OH) configuration are clearly identified by in situ/ex situ experiments together with density functional theory (DFT) calculations. Our work extends the definition of SACs to a non-metal catalytic center, and offers insights critical for the development of a family of non-metal SACs.
|
Introduction
Heterogeneous single-atom catalysts (SACs) refer to the catalysts that contain merely atomically dispersed catalytic centers on a support. SACs integrate many advantageous characteristics of both homogeneous and heterogeneous catalysts, which include high activity, selectivity, stability, and atomic efficiency.1–4 The catalytic performance of SACs could be rationally tuned by controlling the electronic states of the central active atoms and the characteristics of the support.5–8 The stability of SACs and metal atom–support covalent interaction were rationalized theoretically.9,10 The SACs, by definition, can be categorized into metal SACs (M-SACs) and non-metal SACs (NM-SACs) based on the type of catalytic center. Generally speaking, nonmetallic atom catalytic centers interact with reactant molecules through covalent bonds, which distinguishes them from metallic atom catalytic centers. This difference results in variations in the adsorption mode and adsorption strength of reactants on the catalysts.11,12 To date, only SACs with metal active centers ranging from noble metals to transition and main group metals3,13 have been extensively investigated in a wide range of catalytic reactions, while non-metal active centers in SACs are poorly elucidated,11,12 owing to the challenges in confirming the active sites and identifying the reaction intermediates and reaction mechanisms, which are generally interfered with by the metal element(s) in the catalysts.14,15Herein, we have developed a non-metal single-atom catalyst consisting of isolated phosphorus atoms anchored at the edge of graphene (P-SAC-NG) as an efficient electrocatalyst for CO2 reduction to CO. The developed phosphorus SAC has a large surface area (1544 m2 g−1) and pore volume (2.5 cc g−1) with P atom content as high as 2.1 at%, and exhibits outstanding CO2 reduction reaction (CO2RR) catalytic activity, reaching a specific current density of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mA cm−2 (geometric area) at a moderate overpotential of 0.62V for CO conversion with 93% selectivity. The CO2 molecular activation and electrochemical reduction pathways at P atomic sites at the edge of graphene with the 2C–PO(OH) configuration are clearly identified by aberration-corrected high-angle annular dark-field scanning transmission electron microscopy, X-ray absorption spectroscopy and X-ray photoelectron spectroscopy techniques. Combining in situ/ex situ experiments together with density functional theory (DFT) calculations, it is revealed that the electrochemical reduction of CO2 to CO at single P atomic sites starts from a structural transformation from 2C–PO(OH) to 2C–PO via proton coupled electron transfer, which effectively lowers the activation energy of CO2 at the P atomic sites to form the 2C–P(CO2)δ−O intermediate. Our work extends the definition of SACs to a non-metal catalytic center, and offers insights critical for the development of a family of non-metal SACs.
mA cm−2 (geometric area) at a moderate overpotential of 0.62V for CO conversion with 93% selectivity. The CO2 molecular activation and electrochemical reduction pathways at P atomic sites at the edge of graphene with the 2C–PO(OH) configuration are clearly identified by aberration-corrected high-angle annular dark-field scanning transmission electron microscopy, X-ray absorption spectroscopy and X-ray photoelectron spectroscopy techniques. Combining in situ/ex situ experiments together with density functional theory (DFT) calculations, it is revealed that the electrochemical reduction of CO2 to CO at single P atomic sites starts from a structural transformation from 2C–PO(OH) to 2C–PO via proton coupled electron transfer, which effectively lowers the activation energy of CO2 at the P atomic sites to form the 2C–P(CO2)δ−O intermediate. Our work extends the definition of SACs to a non-metal catalytic center, and offers insights critical for the development of a family of non-metal SACs.
Results
Synthesis and structural characterization
The P-SAC-NG catalyst was prepared by pyrolyzing a porous layered hierarchical precursor, which was self-assembled from melamine and glyphosate as shown in Fig. 1(a), with a molar ratio of 8:1, via the hydrogen bond and Cl− ion as the linker. Fig. S1–S4 (ESI†) describe the detailed synthesis information of P-SAC-NG. Self-assembly of melamine and glyphosate into a porous layered hierarchical structure before pyrolysis, is essential to obtain the P-SAC-NG catalyst with large porosity (Table S1, ESI†) and high content of P atoms (Table S2, ESI†). Both scanning electron microscopy (SEM) and transmission electron microscopy (TEM) (Fig. 1(b), (c) and Fig. S5, S6, ESI†) images reveal a 3D interconnected architecture built from two-dimensional (2D) porous graphene nanosheets, which is further confirmed by atomic force microscopy (AFM) measurements (Fig. 1(d)).
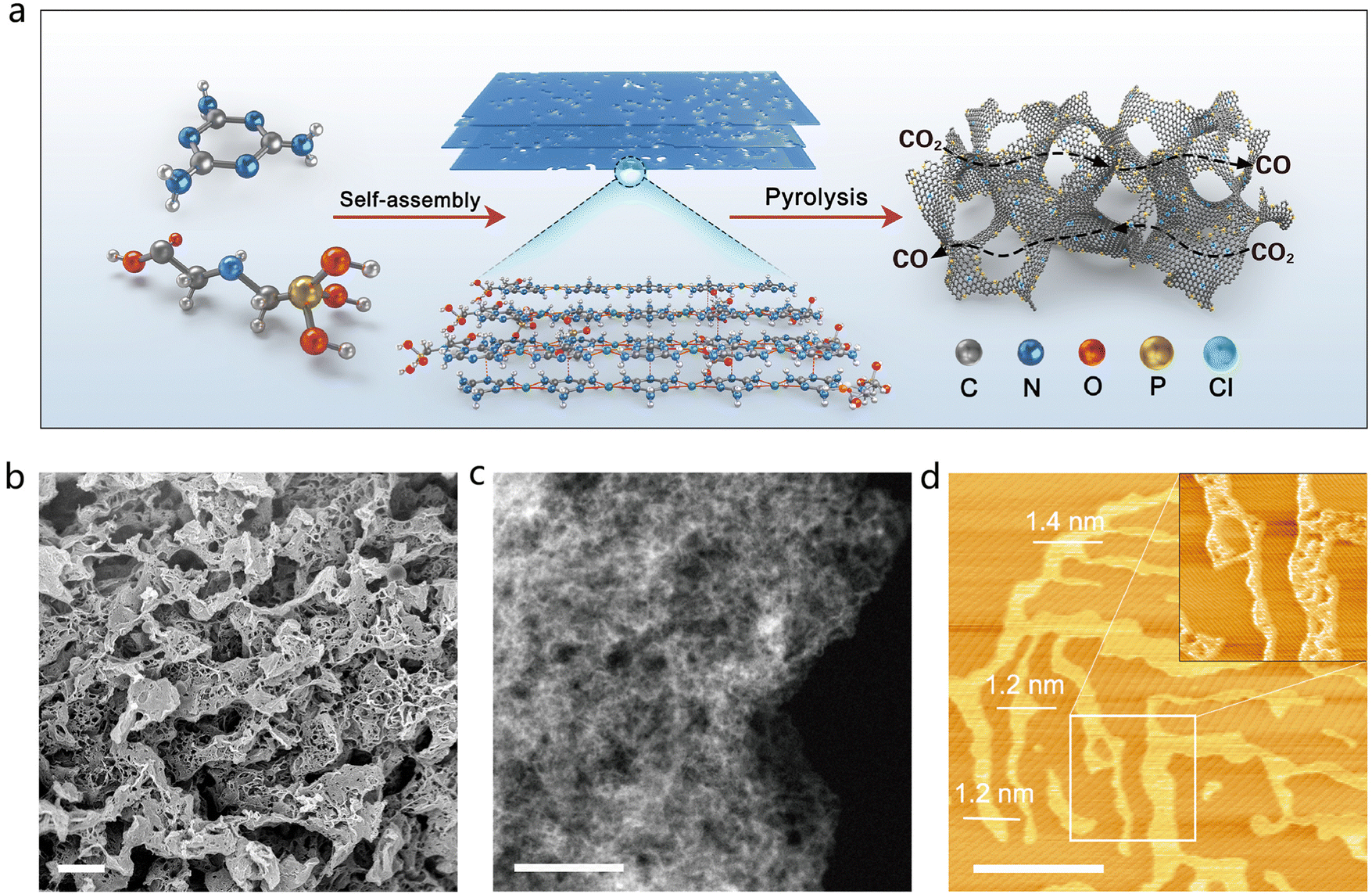 | ||
| Fig. 1 Preparation and structural characterization of P-SAC-NG. (a) Schematic illustration showing the preparation process of P-SAC-NG. (b) SEM, (c) dark-field TEM and (d) AFM images of P-SAC-NG, scale bars: 1 μm, 100 nm and 1 μm for the SEM, TEM and AFM images, respectively. | ||
Fig. 2(a) compares the XRD patterns of the P-SAC-NG and N-doped graphene (denoted as NG) that was prepared by pyrolyzing a mixture of melamine and L-alanine, in which only (002) and (100) carbon diffraction peaks are observable. A similar (100) peak at 43.9° indicates a similar structure in the carbon plane, while the broadened and weakened (002) peak of P-SAC-NG suggests a lower degree of crystallization, which is further confirmed by a higher ID/IG ratio in the Raman spectrum of P-SAC-NG (Fig. S7, ID and IG are intensities of the Raman D and G band, respectively, ESI†). The interplanar spacing, determined by the (002) diffraction for P-SAC-NG and NG, is 0.356 and 0.337 nm, respectively, and both are larger than that for graphite (0.335 nm), agreeing well with the HRTEM results (Fig. S8 and S9, ESI†), which can be ascribed to the larger size of P and N atoms in comparison to C atoms (covalent atomic radius: 106 pm for P, 75 pm for N, and 73 pm for C(sp2)). The elemental mapping images show uniform distributions of C, N, O, and P in P-SAC-NG and C, N, and O in NG (Fig. S10 and S9, ESI†). As shown in the HRTEM image in Fig. 2(b) (more images can be found in Fig. S8, ESI†), some dark dots appear at the edge of the porous graphene sheets, highlighted in Fig. S8b (ESI†) by red circles, which are related to the P atoms, due to the larger atomic mass of P than that of N and C. The dispersion of P atoms on graphene is confirmed by the aberration-corrected high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) (Fig. 2(c), (d), and Fig. S8, ESI†), where most of the bright spots corresponding to the P atoms are distributed at the edge of graphene (Fig. 2(d), Fig. S8c, d and E, ESI†). The sizes of the bright spots (Fig. S11, ESI†) are <0.15 nm, which are obviously smaller than those of the single metal atoms on the carbon matrix (typical value is in the range of 0.18–0.22 nm16–18). The electron energy loss spectroscopy (EELS) spectrum, collected from the circled area as shown in Fig. 2(f), displays a weak signal at around 130 eV, which originates from the energy loss of the P L2,3-edge (inset of Fig. 1(e));19 moreover, no signals from metal elements are detectable. The specific surface area of the P-SAC-NG catalyst measured by the Brunauer–Emmett–Teller (BET) method is around 1544 m2 g−1, which is much larger than that of NG (375 m2 g−1). Besides, P-SAC-NG has a pore volume of 2.53 cm3 g−1 (Table S1, ESI†), comprising both micropores (∼1.0 nm) and mesopores (3–8 nm) (Fig. 1(f)), whereas most pores of NG fall in the category of mesopores with an average pore size of ∼11.8 nm.
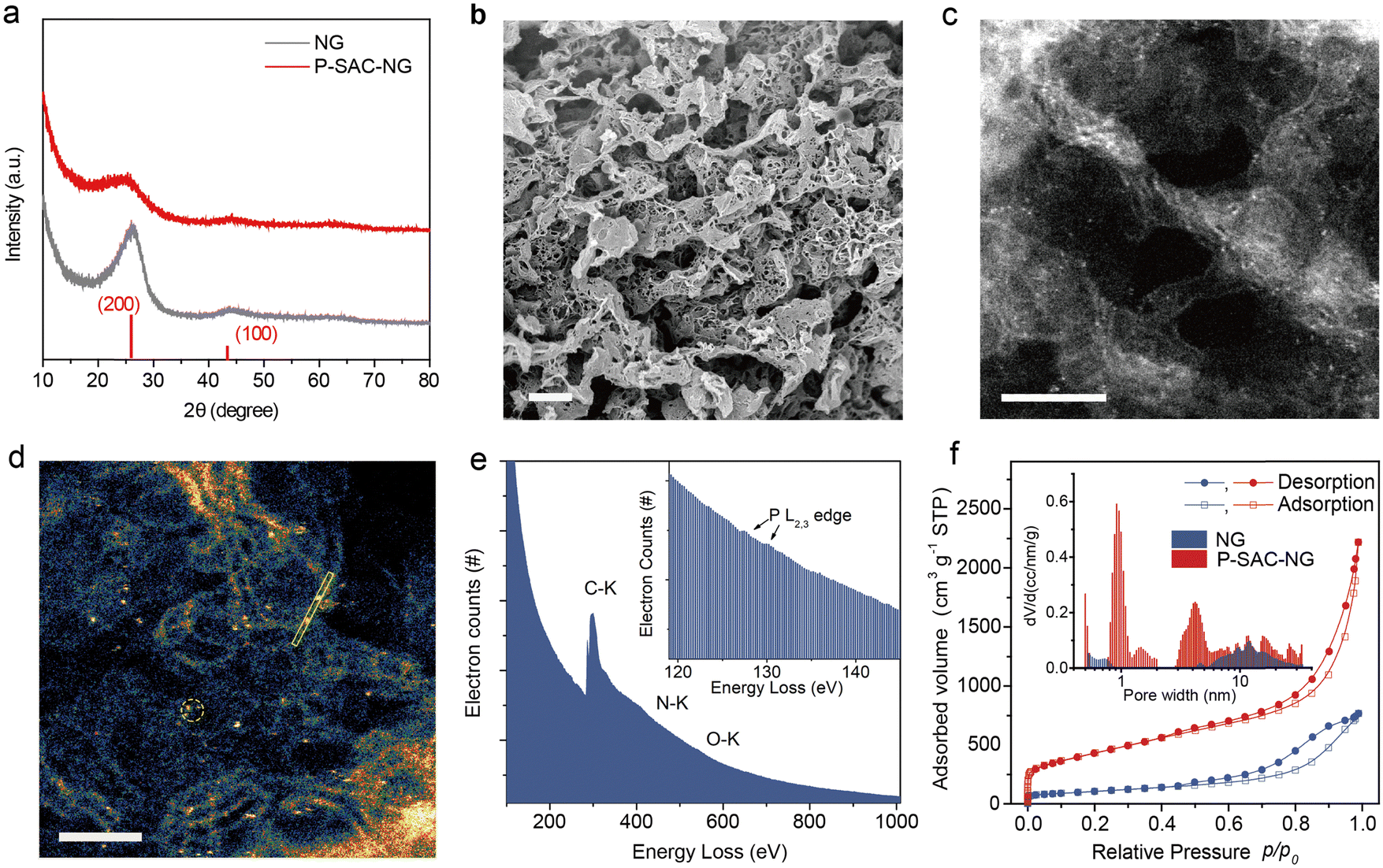 | ||
| Fig. 2 Characterization of single P atoms dispersed on graphene. (a) X-ray diffraction patterns of P-SAC-NG and NG. The JCPDS #41-1487 profile of graphite (red) is displayed for reference. (b) HR-TEM and (c), (d) HAADF-STEM images of P-SAC-NG. Scale bars in b, c and d are 5, 5 and 2 nm, respectively. (e) EELS spectrum of P-SAC-NG. Inset shows the EELS signal of P for P-SAC-NG. (f) Nitrogen adsorption isotherms of P-SAC-NG and NG measured at 77 K. Inset displays the pore size distribution of P-SAC-NG and NG. | ||
CO2 adsorption on single P atoms
The chemical composition and elemental states of P-SAC-NG and NG were investigated by XPS (Fig. S12 and Table S2, ESI†). As shown in Fig. S12a (ESI†), no signal from metal elements was detectable in both samples, while two peaks at ∼133 eV (P 2p) and 190 eV (P 2s) of P-SAC-NG suggest the successful introduction of the P heteroatoms into the carbon structure via pyrolysis, matching well with the results from HAADF-STEM and EDX measurements. The metal-free feature of P-SAC-NG was further verified by ICP-MS and high resolution XPS measurements (Fig. S13 and S14, ESI†). The P-SAC-NG contains 3.0 at% N, 9.3 at% O and 2.1 at% P, whereas NG has a higher content of N (5.9 at%) and a lower content of O (4.4 at%). The higher O content in P-SAC-NG suggests that the introduced additional O atoms in P-SAC-NG might connect to P atoms. This conclusion is further supported by comparing the difference of the FTIR spectra of P-SAC-NG and NG as shown in Fig. S15 (ESI†). There appear two absorption bands at ∼1025 and 1230 cm−1 for P-SAC-NG, which can be ascribed to the P–OH and PO stretching modes, respectively.20 By deconvoluting the N 1s XPS spectrum, it shows that P-SAC-NG and NG samples have similar percentages of graphitic nitrogen occupying about ∼50 and 54% of the total N, but the P-SAC-NG has lower pyridinic N (9%) than NG (19%), as shown in Table S2 (ESI†). Fig. 3(a) displays the high-resolution P 2p XPS spectrum; the bonding energy (133.0 eV) of P 2p is higher than that of P–P (∼130 eV) and C3–P (∼131 eV) bonds, but lower than that of the high oxidation species (such as C–O–P at 134.0 eV).21,22 The P 2p spectrum can be deconvoluted into two P species with different P oxidation states, at 131.9 eV and 133.4 eV, belonging to C3–PO and C2–PO2, respectively.23,24 Considering the low content of N and P atoms in P-SAC-NG, contribution from P–N species to the P 2p spectrum can be neglected. To further confirm the atomic dispersion of P atoms on carbon support and their chemical states, solid-state nuclear magnetic resonance (NMR) spectroscopy measurements were performed. As shown in Fig. S12e (ESI†), the 31P solid-state NMR spectrum of P-SAC-NG shows the presence of two main peaks appearing at −5.3, and −16.1 ppm, respectively. These 31P peaks can be assigned to P atoms analogous to triphenylphosphine (PPh3, expected value at −5 ppm)25,26 and diphenylphosphate (OP(OH)Ph2, expected value at −13 ppm),27 which are in good agreement with the deconvolution of the P 2p XPS spectrum, indicating two types of P species with different P oxidation states, belonging to C3–P(O) and C2–PO2, respectively. Overall, 31P solid state NMR and XPS results indicate that P atoms are atomically dispersed in P-SAC-NG.
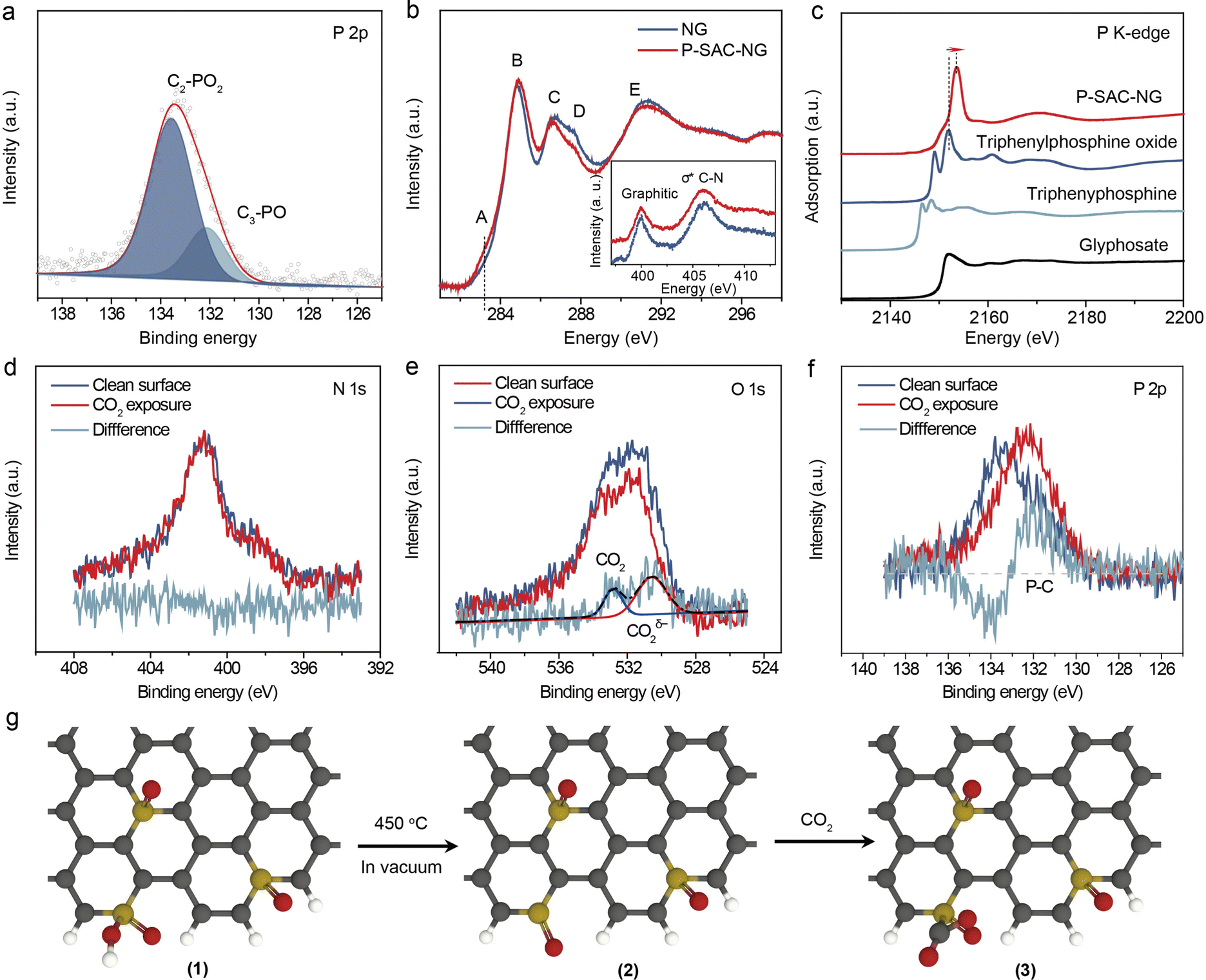 | ||
| Fig. 3 Chemical states of P atoms in P-SAC-NG and CO2 activation on single P atoms. (a) High resolution XPS P 2p spectrum. (b) Carbon and nitrogen (inset) K-edge XANES spectra of P-SAC-NG and NG catalysts. In the carbon K-edge XANES spectra, A: defects, B: π*CC, C: π*C–OH, D: π*C–O–C, C–N, CO, and COOH, and E: σ*C–C. (c) Phosphorus K-edge XANES spectra of P-SAC-NG and various references. (d) N 1s, (e) O 1s, and (f) P 2p XPS spectra before and after CO2 adsorption on P-SAC-NG. (g) Schematic illustration showing the structural transformation of P-SAC-NG during degassing and exposure to CO2, (1) 2C–PO(OH) to (2) 2C–PO and (3) interaction with CO2. Black, gray, red and yellow spheres represent C, H, O, and P, respectively. | ||
The bonding characteristics of P-SAC-NG were further investigated by C, N and P K-edge X-ray absorption spectroscopy (XAS). As shown in Fig. 3(b), C and N K-edge X-ray absorption near edge structure (XANES) spectra of P-SAC-NG and NG display similar characteristics; the slight intensity difference in the C K-edge XANES of P-SAC-NG in the energy range from A to E indicates richer defects in P-SAC-NG and a higher C–N content in NG. Moreover, the N K-edge XANES spectra of the two samples show nearly identical contents of the N species, further ruling out the P–N species in P-SAC-NG, in agreement with the XRD, Raman, and XPS data. Fig. 3(c) displays the P K-edge XANES spectra. The spectral shape and energy position of the references are consistent with the reported data.28–31 Two well-defined absorption bands in triphenylphosphine and triphenylphosphine oxide arise from three-coordinated P–C with a lone electron pair and three-coordinated P–C and one P–O, respectively. For the P-SAC-NG, the white-line position at 2153.6 eV results from the dipole allowed transition from 1s → 3p, where the 3p orbital is empty for the P in the +5 highest oxidation state.31 The higher absorption energy of the white-line for the P-SAC-NG as compared to the reference samples, in which the oxidation state of the P atom is +5, is due to more electron-withdrawing ability of the doped nitrogen atom than the carbon atom in the N-doped graphene matrix, which results in more electron withdrawing, thus leading to a lower electron density on phosphorus with a higher excitation energy. The P K-edge XANES spectra of P-SAC-NG can be fitted by two absorption bands (Fig. S16, ESI†), corresponding to the transition from 1s to two 3p orbitals related to the P–C (σ*) and P–O (π*) coordination.30,31 Based on the physical and chemical state characterization (Fig. 1 and 2), it is concluded that most of the P atoms are with the 2C–PO(OH) or 3C–PO configuration, as shown in Fig. 2(g) (1).
Before investigating the electrochemical CO2RR, the interaction between the as-prepared catalysts and CO2 molecules was studied by photoelectron spectroscopy. Prior to exposing the catalyst to the CO2 gas, the catalyst was thermally treated in a vacuum to remove the adsorbed oxygen and water species. As shown in Fig. S17 (ESI†), after thermal treatment, the intensity variation of the C 1s, N 1s and P 2p (P 2s) spectra for NG and P-SAC-NG is not obvious, while the intensity of the O 1s spectrum dramatically decreases due to the decrease of O content from ∼4.4 at% to 1.8 at% for NG and ∼9.3 at% to 4.5 at% for P-SAC-NG. Furthermore, the O 1s spectrum displays similar components of O species for NG before and after thermal treatment (Fig. S18, ESI†). However, the content of the –OH group (P–OH and aliphatic C–OH) decreases from 50% to 39% (from 4.6 at% to 1.8 at%) for P-SAC-NG, implying unstable P–OH bonds (bond energy of P–OH: 335 vs. PO: 544 (kJ mol−1)) and also confirming the high content of –OH group in P-SAC-NG (Fig. S17e, ESI†). Fig. S17c (ESI†) shows a slight shift of the P 2p peak to higher binding energies after thermal treatment, which can be ascribed to the stronger electrophilic attack on P by the carbon ring via conjugated π bonds than by the hydroxy groups, as shown in Fig. 3g (2).
The thermally treated NG and P-SAC-NG were exposed to CO2 gas in the XPS chamber, and the behaviors of CO2 adsorption were studied by analyzing the difference of the XPS spectrum for each element between the two states (before and after CO2 exposure). Both C 1s and N 1s spectra of P-SAC-NG and NG did not show any noticeable change (Fig. 3(d) and Fig. S19, ESI†). The differential XPS spectrum of O 1s for NG (Fig. S19d, ESI†), induced by the CO2 exposure, is very weak, indicating the low amount of chemically adsorbed CO2 and the weak interaction between CO2 and NG. For P-SAC-NG, as shown in Fig. 3(e), after CO2 gas exposure, the increase in the O 1s intensity is obvious and the differential O 1s spectrum before and after CO2 exposure can be divided into two contributions: physical adsorption at ∼531 eV and chemical adsorption at ∼534 eV. The chemically adsorbed CO2δ– anion on P-SAC-NG is confirmed by an increase in the work function of P-SAC-NG after exposure to CO2 gas (Fig. S20e, ESI†), which was determined from the shift of the second electron cut-off edge (∼0.12 eV) to lower binding energies. The results from the photoelectron spectra show similar interaction between CO2 molecules and P atoms to that of the metal center in metal-SAC.16
The bonding of CO2 at the P atomic sites was further studied by analyzing the XPS P 2p spectrum. Fig. 3(f) shows the differential XPS spectrum of P 2p induced by CO2 adsorption, which displays an increase in intensity at the low binding energy side and a decrease in intensity at the high binding energy side, which can be assigned to the increased P–C species. Therefore, the photoelectron spectroscopy results could be rationally explained by the chemical CO2 adsorption on the P atom of P-SAC-NG via forming a P–C bond as shown in Fig. 3g (3).
Evaluation of the CO2RR performance
The electrochemical CO2RR performance was first evaluated on a rotating disc electrode by linear sweep voltammetry (LSV) and cyclic voltammetry (CV, see Fig. S21, ESI†). The solution resistance was determined by electrochemical impedance spectroscopy (EIS) measurements (Fig. S22, ESI†). Fig. 4(a) shows the LSV curves of the catalysts acquired in CO2-saturated 0.5 M KHCO3 solution, in which the P-SAC-NG exhibits larger polarization current density and lower overpotential, ∼100 mV lower than that of NG. The LSV curves of P-SAC-NG recorded at a rotation speed from 400 to 2025 r.p.m., as shown in the inset of Fig. 4(a), indicate the CO2 mass transfer-controlled reaction process. The CO2RR products were analyzed using an on-line gas chromatograph with an H-type electrochemical cell separated by a Nafion® 117 membrane. CO, H2 and trace CH4 were detected in the gas phase, while no liquid products were detectable in the potential range from −0.25 to −1.04 V (versus RHE). Fig. S23 and S24 (ESI†) display the typical current profiles of the catalysts and carbon paper support for analyzing the faradaic efficiency of products. Fig. 4(b) compares the faradaic efficiency of CO and H2 between the P-SAC-NG and NG catalysts; P-SACs-NG exhibits a lower onset overpotential towards CO formation (∼120 mV, Fig. S25, ESI†). Furthermore, the P-SAC-NG electrode exhibits a maximum CO faradaic efficiency of around 93% at an overpotential of 0.62 V, and larger partial current density for CO formation (Fig. S26, ESI†), as compared to NG. Using isotope 13CO2 as the feedstock for the reaction in a carbon free electrolyte (0.2 M Na2SO4) (Fig. S27, ESI†), it is confirmed that CO is indeed formed via CO2RR. The Tafel slope of CO formation for P-SAC-NG is 140 mV dec−1 (Fig. S28, ESI†), suggesting that the first electron transfer to CO2 to form a surface adsorbed anionic CO2δ− intermediate is the rate-determining step in the CO2RR.32 We performed in situ attenuated total reflectance (ATR) surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS, Fig. S29, ESI†) to monitor the surface species during the electrochemical CO2RR33,34 over the NG and P-SAC-NG catalysts. During the CO2RR, as the HER is the major competing reaction, *H and *CO would occupy most of the reaction sites. As shown in Fig. 4(c) and (d), the surface coverage of *CO on P-SAC-NG is much higher than that on NG. Fig. 4(e) shows the potential dependent intensity of *CO for NG and P-SAC-NG, displaying a similar trend to the potential dependent FECO (Fig. 4(b)), verifying the higher selectivity and catalytic activity of P-SAC-NG toward the CO2RR. Moreover, P-SAC-NG maintains a high *CO coverage even at a very high applied cathodic potential (ca. −0.9 V vs. RHE) (Fig. 4(e)) with an FECO of only ∼50%, suggesting that the declined FECO may not result from the direct competition between the CO2RR and HER at the same catalytic sites, but due to activation of new catalytic sites for the HER at high applied cathodic potentials (ca. over −0.8 V vs. RHE). Fig. 4(f) displays the CO2RR stability of P-SAC-NG, which could maintain a stable CO2 to CO conversion with a CO faradaic efficiency of 88% at an overpotential of ∼0.51 V. Overall, it can be concluded that although the performance of P-SAC-NG is lower than those of most transition metal single-atom catalysts,16,17 the above experimental results unambiguously indicate the P atoms in P-SAC-NG as the active sites that catalyzed CO2 reduction to CO.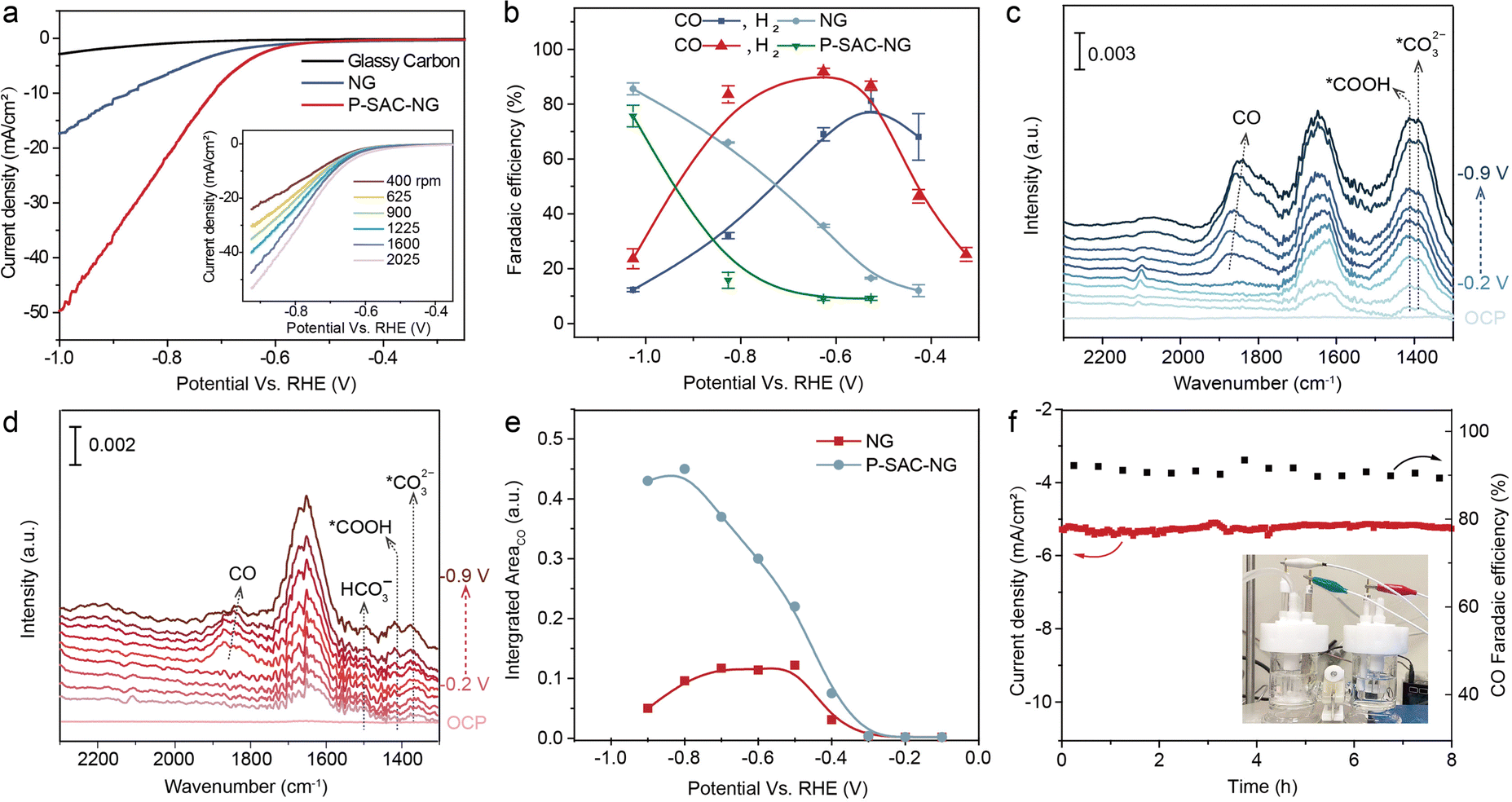 | ||
| Fig. 4 CO2 reduction reaction. (a) LSV curves for the catalysts acquired in CO2-saturated 0.5 M KHCO3 solution on a rotating disc electrode at a rotation speed of 1600 rpm and a scan rate of 5 mV s−1. Catalyst loading: 0.15 mg cm−2. The inset of A shows the LSV curves of the P-SAC-NG electrode recorded at different rotation speeds. (b) CO and H2 faradaic efficiencies recorded at various applied potentials. The error bars represent the standard deviation from three independent tests. In situ ATR-SEIRAS spectra of (c) P-SAC-NG and (d) NC collected in CO2-saturated 0.5 M KHCO3 in the bias potential region from OCV to −1.1 V vs. RHE. (e) The potential dependence of *CO band intensity (integrated from 1750 to 1950 cm−1) over NG and P-SACs-NG, potential range: from −0.2 to −0.9 V (vs. RHE). (f) Stability of P-SAC-NG on carbon fiber paper for the CO2RR recorded at a potential of −0.625 V vs. RHE. All measurements were performed under the conditions of 1 atm CO2 and room temperature in 0.5 M KHCO3 (pH = 7.3). | ||
Discussion
To identify the catalytic site for the CO2RR, the carbon, nitrogen, oxygen, and phosphorus K-edge XANES spectra for P-SAC-NG and NG catalysts were recorded before and after the CO2RR. All XANES spectra were normalized to the post edge with background subtracted. The variations of the C and N K-edge XANES spectra (Fig. S30, ESI†) for the two catalysts are unobvious before and after the CO2RR, consistent with the near-ambient pressure XPS results. However, as shown in Fig. 5(a), the variation of the O K-edge XANES spectra before and after the CO2RR is distinct. The difference in the O K-edge XANES spectra before the CO2RR between P-SAC-NG and NG catalysts results from the high content of CO and –OH species in P-SAC-NG, which is also indicated in the O 1s XPS spectra (Fig. S12d, ESI†). The A to E peaks in the O K-edge XANES spectra can be assigned to A: π* CO, (COOH), B: π* C–O–C, C: hydroxyl-derived state σ* C–OH, D: σ* C–OH and C–O–C, and E: σ* CO based on previous reports.35–37 The evolutions of the O K-edge XANES spectra after the CO2RR for P-SAC-NG and NG catalysts are highlighted in the differential spectra of Fig. 5(b). The decreased intensity of peak D (σ* C–OH and C–O–C) for the two catalysts corresponds to a reduction of C–OH (or P–OH) and C–O–C functional groups under a negative applied potential on P-SAC-NG and NG. The increased peak intensity of A, B and E (π* (–COOH) and σ* of C–O) after the CO2RR, for the two catalysts, results from the adsorbed –COOH intermediate on the catalyst surface, highlighting the carboxyl (*COOH) pathway of the CO2 reduction to CO on the P-SAC-NG and NG catalysts. Furthermore, the increase in the amplitude of peaks A and B for P-SAC-NG is much larger than that for NG, which is consistent with the higher CO2δ− species intensity in the NA-XPS O 1s spectrum, in situ ATR-SEIRAS and larger cathodic current for CO production in the electrochemical CO2RR. The mode of CO2 bonding to the P-SAC-NG catalyst was further disclosed by the change of the P K-edge XANES spectra before and after the CO2RR, as shown in Fig. 5(c). The differential spectrum shows that the intensity increases at the low energy side and decreases at the high energy side, which results from transition of 1s to antibonding orbitals of P–C (σ*) and P–O (π*), indicating the adsorption of the CO2RR intermediate on the P site with a P–CO2δ− configuration.
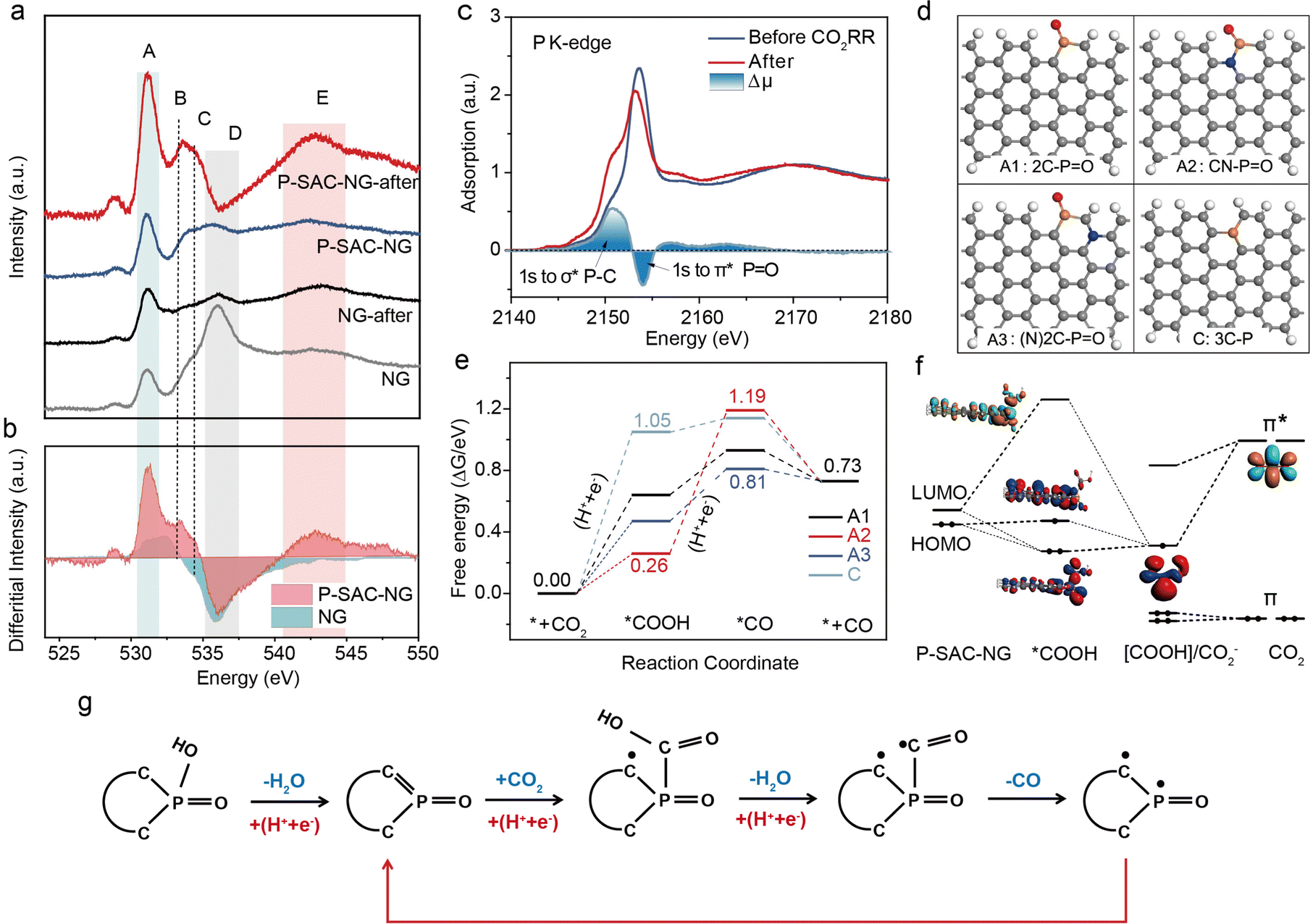 | ||
| Fig. 5 CO2RR mechanism. (a) O K-edge XANES spectra of P-SAC-NG and NG catalysts acquired before and after the CO2RR. A: π* CO, (COOH), B: π* C–O–C, C: hydroxyl-derived state σ* C–OH, D: σ* C–OH and C–O–C, and E: σ* CO. The differential spectra were obtained via subtracting the spectra before the reaction from those after the reaction. (b) Changes in intensity of the O K-edge XANES spectra for P-SAC-NG and NG catalysts after the CO2RR. (c) P K-edge XANES spectra of P-SAC-NG before and after the CO2RR (P 1s → σ* of P–C and P 1s → π* of P–O). (d) SDifferent configurations of P atoms optimized with DFT calculations, A1: 2C–PO, A2: CN–PO, A3: (N)2C–PO, and C: 3C–P. Gray, white, 7/24/2023Sred, yellow and blue spheres represent C, H, O, P and N, respectively. (e) Gibbs free energy profile for the CO2RR on P-SAC-NG at U = 0 V (for U = −0.5 V, Fig. S35, ESI†). (f) The orbital interaction between the P-SAC-NG and *COOH intermediate. Schematic energy-level correlation diagram of the *COOH intermediate in the cluster model of A1 configuration. (g) Schematic of the CO2RR process on P-SAC-NG. | ||
To explore the origin of P single-atoms as the active center, and the reaction mechanism of the CO2RR on P-SAC-NG, density functional theory (DFT)38–40 calculations were performed based on the computational hydrogen electrode (CHE) model41 (see the ESI†). Various geometric configurations were examined by considering the position of P atoms (at the armchair or the basal plane), the influence of N, O incorporation and the coordination interaction of P atoms, as shown in Fig. S31 (ESI†). Based on the above structural characteristics and the experimental results, the configurations with P coordinated by two carbon atoms, one O atom and one OH group (A: 2C–PO(OH)) and by three carbon atoms, one O atom (B: 3C–PO) were first considered. However, for the first step of the CO2RR as shown in Fig. S32 (ESI†), the adsorption of *COOH on the P site with 2C–PO(OH) and 3C–PO configurations is energetically unfavorable, with a free energy barrier of 3.14 and 2.45 eV, respectively. Moreover, the 3C–P(COOH)O intermediate state is thermodynamically unstable; one of the P–C bonds would dissociate and oxidize to CO, and transfer to the 2C–P(COOH) configuration. Competing with direct CO2 adsorption and hydrogenation to form *COOH on 2C–PO(OH), the 2C–PO(OH) site transfers to the 2C–PO configuration via a proton-coupled electron transfer (PCET) process,42 which is energetically more favorable with an energy barrier of merely 0.93 eV (Fig. S33, ESI†). Protonation of 2C–PO(OH) by the PCET process is akin to that in the oxygen reduction reaction of the Fe–N–C catalyst.43 In the PCET step, the –OH group acts as a proton acceptor; meanwhile the 2C–P O(OH) accepts an electron, causing the 2C–PO(OH) transfer to 2C–PO (named as A1), in which the P atom has an unsaturated coordination (featured by one unoccupied 3pz orbital). This process matches well with the XPS and XANES results.
Furthermore, to probe the influence of N and O incorporation on the reaction mechanism, the A2–A6 configurations were also constructed. Gibbs free energy of the CO2RR at all possible P, C and N sites for A1–A3 was calculated, as shown in Fig. 5(d) and Fig. S34 (ESI†). Fig. 5(e) shows the free energy profiles of the CO2RR at the P sites; the potential limiting step (PLS) of the A1 and A3 configurations is the first electron transfer step to generate the *COOH intermediate, in good accordance with the Tafel slope obtained from the experiment. The energy barrier of A1 in the PLS is 0.64 eV at U = 0 V and 0.14 eV at an applied potential of −0.5 V versus RHE (Fig. S35, ESI†). Compared to A1, N incorporation (the A3 configuration) can facilitate the CO2RR by reducing the energy barrier of the PLS to 0.47 eV at U = 0. For the A2 and A4 configurations, the PLS is the second electron transfer step for the formation of the *CO intermediate, which contradicts with the experimentally determined Tafel slope on P-SAC-NG. Considering the low doping concentration of N (∼3 at%) and P (∼2 at%), the direct bonding between N and P (the A2 and A4 configurations) should be rare in P-SAC-NG. For O introduction in graphene, the P–O bonding is not stable in the basal plane and shows an energy barrier of more than 1.0 eV for the adsorption of *COOH, as can be seen from the optimized structures and free energies for A5 and A6 configurations in Fig. S31 and S34 (ESI†). We also investigated the reaction mechanisms at the C and N sites (Fig. S34, ESI†), which show that the CO2RR at the N and C sites is less active than that at the P site (e.g., A(*C), A1-3(*C) and A2, 3(*N)) because of the difficulty in the valence state change for N and C sites. In addition, the catalytic properties of P atoms in the basal plane of graphene were compared by generating C and E models with 3C–P configurations. It is found that P atoms of all these structures are less active than the P sites at the edge and the adsorption of *CO on the P atoms in plane is also not favored, even when considering the introduction of N and O (the C1–C3 and E1 configurations, as shown in Fig. S31, ESI†). Moreover, the effects of the solvent (water molecules) on the CO2RR for different configurations of P atoms (A1, A2, A3 and C) were investigated.44 The results show that under both conditions, with (Fig. S36, ESI†) and without (Fig. 5(e)) considering solvent effects, the P sites with 2C–PO feature are identified as the catalytic sites for the CO2RR to CO. The Gibbs free energies of competitive hydrogen evolution on P sites for A1–A3 and C configurations were also calculated (Fig. S37, ESI†). Owing to the higher H* adsorption energy than that of *COOH formation at P sites of the A3 configuration, the high selectivity of CO2 to CO at P sites with the A3 configuration is guaranteed.
To elucidate the nature of the interactions between P-SAC-NG (A1: 2C–PO) and COOH adsorbates, the orbital interactions were investigated qualitatively in a simplified cluster model45,46 (Fig. S38, ESI†) as shown in Fig. 5(f). It can be seen that when CO2 adsorbs at the P site, the vacant degenerate π* orbitals of CO2 will be activated and stabilized by gaining one electron via a proton coupled electron transfer process (* + CO2 + H+ + e− → *COOH) and contributes to one vacant orbital and one singly occupied orbital with one unpaired electron at the COOH adsorbate. Then the highest occupied molecular orbital (HOMO) of P-SAC-NG can further transfer electrons to the adsorbate and form P-COOH bonds through strong orbital interactions. In this way, the single electron will diffuse and delocalize at the carbon atoms of P-SAC-NG. With another proton coupled electron transfer step, the C–OH bond of the COOH adsorbate will be broken by forming H2O, leaving one unpaired electron at the C atom of the CO adsorbate (*COOH + H+ + e− → *CO + H2O). Then this unpaired electron transfers to the P atom immediately, releasing the CO molecule and forming one P–C bond with the other unpaired electron localized at C atoms of P-SAC-NG at the same time. Consequently, the P-SAC-NG catalyst recovers to complete the catalytic cycle, as detailed in the CO2RR pathway (Fig. 5(g)). The understanding of the activation process of CO2 on P-SAC-NG from orbital interactions is consistent with the results from Bader charge and projected density of states (pDOS) analyses as presented in Fig. S39 (ESI†). Overall, it can be concluded that P-SAC-NG exhibits excellent catalytic performance for the CO2RR. The optimal catalytic site of P-SAC-NG is the phosphorus atomic site at the armchair of graphene. The generation of the *COOH intermediate is the potential limiting step.
In summary, we have developed a non-metal single-atom catalyst consisting of high density, isolated phosphorus atoms anchored at the edge of porous graphene as an electrocatalyst for the CO2 reduction reaction. The activation and electrochemical reduction processes of CO2 on single P atomic sites are unambiguously probed by X-ray absorption spectroscopy, photoelectron spectroscopy and DFT studies. Our work not only extends the definition of SACs to a non-metal catalytic center but also offers a unique insight into the non-metal active site geometric and structural transformation during the CO2 reduction reaction. Due to the intrinsic difference between non-metal atoms and metal atoms, SACs with non-metal centers will also have unique characteristics. We expect that with the insights provided here it is viable to develop a family of non-metal SACs and non-metal/metal dual site catalysts for advanced catalytic applications.
Author contributions
H. Y., T. Z. and B. L. conceived and designed the project. Y. L. performed the X-ray absorption experiments. H. Y. performed the electrochemical experiments and analyzed the electrochemical data. C. G. and W. C. conducted photoelectron spectroscopy measurements. W. L., J. D., Q. W., J. Z., Y. H. and W. C. contributed to the structure characterization. C. X., S. B. and J. L. carried out the DFT calculations and theoretical analyses. H. Y. and B. L. analyzed the experimental data and prepared the manuscript. All authors reviewed and contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the funds from the City University of Hong Kong start up fund, CAS Project for Young Scientists in Basic Research (YSBR-022), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB36030200), the National Natural Science Foundation of China (Grant 22075195 and 22033005), and Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002). Computational resources are supported by the Center for Computational Science and Engineering (SUSTech) and Tsinghua National Laboratory for Information Science and Technology.References
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- M. K. Samantaray, V. D’Elia, E. Pump, L. Falivene, M. Harb, S. Ould Chikh, L. Cavallo and J.-M. Basset, Chem. Rev., 2020, 120, 734–813 CrossRef CAS PubMed.
- S. Mitchell and J. Pérez-Ramírez, Nat. Commun., 2020, 11, 4302 CrossRef CAS PubMed.
- Z. Chen, E. Vorobyeva, S. Mitchell, E. Fako, M. A. Ortuño, N. López, S. M. Collins, P. A. Midgley, S. Richard, G. Vilé and J. Pérez-Ramírez, Nat. Nanotechnol., 2018, 13, 702–707 CrossRef CAS PubMed.
- M. S. Frei, C. Mondelli, R. García-Muelas, K. S. Kley, B. Puértolas, N. López, O. V. Safonova, J. A. Stewart, D. Curulla Ferré and J. Pérez-Ramírez, Nat. Commun., 2019, 10, 3377 CrossRef PubMed.
- R. Qin, K. Liu, Q. Wu and N. Zheng, Chem. Rev., 2020, 120(21), 11810–11899 CrossRef CAS PubMed.
- H.-Y. Zhuo, X. Zhang, J.-X. Liang, Q. Yu, H. Xiao and J. Li, Chem. Rev., 2020, 120, 12315–12341 CrossRef CAS PubMed.
- J.-C. Liu, Y. Tang, Y.-G. Wang, T. Zhang and J. Li, Natl. Sci. Rev., 2018, 5, 638–641 CrossRef CAS.
- C. Liu, Q. Li, C. Wu, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Am. Chem. Soc., 2019, 141, 2884–2888 CrossRef CAS PubMed.
- X. Yu, P. Han, Z. Wei, L. Huang, Z. Gu, S. Peng, J. Ma and G. Zheng, Joule, 2018, 2, 1610–1622 CrossRef CAS.
- F. Luo, A. Roy, L. Silvioli, D. A. Cullen, A. Zitolo, M. T. Sougrati, I. C. Oguz, T. Mineva, D. Teschner, S. Wagner, J. Wen, F. Dionigi, U. I. Kramm, J. Rossmeisl, F. Jaouen and P. Strasser, Nat. Mater., 2020, 19, 1215–1223 CrossRef CAS PubMed.
- W. Fu, Y. Wang, W. Tian, H. Zhang, J. Li, S. Wang and Y. Wang, Angew. Chem., Int. Ed., 2020, 59, 23791 CrossRef CAS PubMed.
- Y. Zhao, T. Ling, S. Chen, B. Jin, A. Vasileff, Y. Jiao, L. Song, J. Luo and S.-Z. Qiao, Angew. Chem., Int. Ed., 2019, 58, 12252 CrossRef CAS PubMed.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- S. Liu, H. B. Yang, S.-F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang, Y. Huang, T. Zhang and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS PubMed.
- J. Gao, H. b Yang, X. Huang, S.-F. Hung, W. Cai, C. Jia, S. Miao, H. M. Chen, X. Yang, Y. Huang, T. Zhang and B. Liu, Chem, 2020, 6, 658–674 CAS.
- C. Su, M. Tripathi, Q.-B. Yan, Z. Wang, Z. Zhang, C. Hofer, H. Wang, L. Basile, G. Su, M. Dong, J. C. Meyer, J. Kotakoski, J. Kong, J.-C. Idrobo, T. Susi and J. Li, Sci. Adv., 2019, 5, eaav2252 CrossRef CAS PubMed.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, in Introduction to Infrared and Raman Spectroscopy, ed. N. B. Colthup, L. H. Daly and S. E. Wiberley, Academic Press, San Diego, 3rd edn, 1990, pp.355–385 DOI:10.1016/B978-0-08-091740-5.50015-6.
- Y. Li, S. Li, Y. Wang, J. Wang, H. Liu, X. Liu, L. Wang, X. Liu, W. Xue and N. Ma, Phys. Chem. Chem. Phys., 2017, 19, 11631–11638 RSC.
- S. Liu, H. Yang, X. Su, J. Ding, Q. Mao, Y. Huang, T. Zhang and B. Liu, J. Energy Chem., 2019, 36, 95–105 CrossRef.
- M. J. Valero-Romero, E. M. Calvo-Muñoz, R. Ruiz-Rosas, J. Rodríguez-Mirasol and T. Cordero, Ind. Eng. Chem. Res., 2019, 58, 4042–4053 CrossRef CAS.
- M. J. Valero-Romero, F. J. García-Mateos, J. Rodríguez-Mirasol and T. Cordero, Fuel Process. Technol., 2017, 157, 116–126 CrossRef CAS.
- M. Tan, Y. Ishikuro, Y. Hosoi, N. Yamane, P. Ai, P. Zhang, G. Yang, M. Wu, R. Yang and N. Tsubaki, Chem. Eng. J., 2017, 330, 863–869 CrossRef CAS.
- J. Shin, J. Bertoia, K. R. Czerwinski and C. Bae, Green Chem., 2009, 11, 1576–1580 RSC.
- J. Albero, A. Vidal, A. Migani, P. Concepción, L. Blancafort and H. García, ACS Sustainable Chem. Eng., 2019, 7, 838–846 CrossRef CAS.
- C. Engemann, R. Franke, J. Hormes, C. Lauterbach, E. Hartmann, J. Clade and M. Jansen, Chem. Phys., 1999, 243, 61–75 CrossRef CAS.
- A. V. Blake, H. Wei, C. M. Donahue, K. Lee, J. M. Keith and S. R. Daly, J. Synchrotron Radiat., 2018, 25, 529–536 CrossRef CAS PubMed.
- C. Tsukada, T. Tsuji, K. Matsuo, T. Nomoto, G. Kutluk, M. Sawada, S. Ogawa, T. Yoshida and S. Yagi, IOP Conf. Ser.: Mater. Sci. Eng., 2015, 76, 012001 Search PubMed.
- I. Persson, W. Klysubun and D. Lundberg, J. Mol. Struct., 2019, 1179, 608–611 CrossRef CAS.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS.
- M. Dunwell, X. Yang, B. P. Setzler, J. Anibal, Y. Yan and B. Xu, ACS Catal., 2018, 8, 3999–4008 CrossRef CAS.
- M. Dunwell, X. Yang, Y. Yan and B. Xu, J. Phys. Chem. C, 2018, 122, 24658–24664 CrossRef CAS.
- H. K. Jeong, H. J. Noh, J. Y. Kim, M. H. Jin, C. Y. Park and Y. H. Lee, EPL, 2008, 82, 67004 CrossRef.
- A. Ganguly, S. Sharma, P. Papakonstantinou and J. Hamilton, J. Phys. Chem. C, 2011, 115, 17009–17019 CrossRef CAS.
- R. P. Gandhiraman, D. Nordlund, C. Javier, J. E. Koehne, B. Chen and M. Meyyappan, J. Phys. Chem. C, 2014, 118, 18706–18712 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse, J. Non-Cryst. Solids, 1995, 192–193, 222–229 CrossRef.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- S. C. Jensen, S. Bettis Homan and E. A. Weiss, J. Am. Chem. Soc., 2016, 138, 1591–1600 CrossRef CAS PubMed.
- Y. Wang, Y.-J. Tang and K. Zhou, J. Am. Chem. Soc., 2019, 141, 14115–14119 CrossRef CAS PubMed.
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00156c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |