
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Contact with soil impacts ferrihydrite and lepidocrocite transformations during redox cycling in a paddy soil†
Katrin
Schulz
 a,
Luiza
Notini
a,
Andrew R. C.
Grigg
a,
L. Joëlle
Kubeneck
a,
Worachart
Wisawapipat
b,
Laurel K.
ThomasArrigo
*ac and
Ruben
Kretzschmar
a
a,
Luiza
Notini
a,
Andrew R. C.
Grigg
a,
L. Joëlle
Kubeneck
a,
Worachart
Wisawapipat
b,
Laurel K.
ThomasArrigo
*ac and
Ruben
Kretzschmar
a
aSoil Chemistry Group, Institute of Biogeochemistry and Pollutant Dynamics, Department of Environmental Systems Science, ETH Zurich, Universitätstrasse 16, CHN, CH-8092 Zurich, Switzerland
bSoil Chemistry and Biogeochemistry Group, Department of Soil Science, Faculty of Agriculture, Kasetsart University, Bangkok 10900, Thailand
cEnvironmental Chemistry Group, Institute of Chemistry, University of Neuchâtel, Avenue de Bellevaux 51, 2000, Neuchatel, Switzerland. E-mail: laurel.thomas@unine.ch
First published on 16th November 2023
Abstract
Iron (Fe) oxyhydroxides can be reductively dissolved or transformed under Fe reducing conditions, affecting mineral crystallinity and the sorption capacity for other elements. However, the pathways and rates at which these processes occur under natural soil conditions are still poorly understood. Here, we studied Fe oxyhydroxide transformations during reduction–oxidation cycles by incubating mesh bags containing ferrihydrite or lepidocrocite in paddy soil mesocosms for up to 12 weeks. To investigate the influence of close contact with the soil matrix, mesh bags were either filled with pure Fe minerals or with soil mixed with 57Fe-labeled Fe minerals. Three cycles of flooding (3 weeks) and drainage (1 week) were applied to induce soil redox cycles. The Fe mineral composition was analyzed with Fe K-edge X-ray absorption fine structure spectroscopy, X-ray diffraction analysis and/or 57Fe Mössbauer spectroscopy. Ferrihydrite and lepidocrocite in mesh bags without soil transformed to magnetite and/or goethite, likely catalyzed by Fe(II) released to the pore water by microbial Fe reduction in the surrounding soil. In contrast, 57Fe-ferrihydrite in mineral-soil mixes transformed to a highly disordered mixed-valence Fe(II)–Fe(III) phase, suggesting hindered transformation to crystalline Fe minerals. The 57Fe-lepidocrocite transformed to goethite and small amounts of the highly disordered Fe phase. The extent of reductive dissolution of minerals in 57Fe-mineral-soil mixes during anoxic periods increased with every redox cycle, while ferrihydrite and lepidocrocite precipitated during oxic periods. The results demonstrate that the soil matrix strongly impacts Fe oxyhydroxide transformations when minerals are in close spatial association or direct contact with other soil components. This can lead to highly disordered and reactive Fe phases from ferrihydrite rather than crystalline mineral products and promoted goethite formation from lepidocrocite.
Environmental significanceIron mineral dynamics are closely coupled to nutrient and contaminant cycling in intermittently or permanently flooded soils, where sub- or anoxic conditions establish frequently. In turn, interactions of Fe minerals with these elements can influence the mineral stability. We show that a close spatial association with the soil matrix drastically impacts Fe mineral transformations. This leads to less crystalline transformation products and increased reductive dissolution with repeated redox cycles. The findings contribute to an improved understanding of Fe mineral dynamics in redox-active soils, such as paddy soils and are important to predicting the biogeochemical cycling of nutrients. |
Introduction
When oxygen is depleted under flooded soil conditions, ferric iron (Fe(III)) can act as an alternative electron acceptor during microbial respiration.1,2 This leads to the reductive dissolution of Fe(III) oxyhydroxides,1 such as lepidocrocite and ferrihydrite, and the release of ferrous iron (Fe(II)) to the soil solution. Since Fe oxyhydroxides are important sorbents for, for example, organic matter, nutrients and trace elements, these compounds can be released into the soil porewater during reductive dissolution.3–5 When the soil water content decreases due to drainage or evapotranspiration, the soil becomes aerated and dissolved and/or sorbed Fe(II) is oxidized. This results in the precipitation of mixed-valence or Fe(III) oxyhydroxides and the immobilization of components which coprecipitate or sorb to these phases.4,6,7 Therefore, Fe mineral dynamics can control the cycling of, for example, phosphate,5,8 arsenic,3 or zinc4 in soils and sediments that are intermittently flooded, such as rice paddy soils.9,10Under Fe-reducing conditions, Fe(III) oxyhydroxides (hereafter Fe oxyhydroxides) and Fe(II) can coexist. Sorption of Fe(II) to Fe oxyhydroxide surfaces leads to the oxidation of Fe(II) and, through electron transfer, the reduction of structural Fe(III), which is subsequently released as dissolved Fe(II).11 Since ferrihydrite and lepidocrocite are thermodynamically metastable, this process can accelerate their transformations to more crystalline Fe minerals, such as goethite or magnetite.12 In mineral suspensions, interactions of minerals with Fe(II) result in abiotic ferrihydrite transformation to lepidocrocite, goethite, or magnetite.12 Lepidocrocite often transforms to magnetite13–15 or goethite,12,16 or undergoes recrystallization14 during the interaction with Fe(II). The trajectory of abiotic Fe(II)-catalyzed ferrihydrite and lepidocrocite transformation depends on the Fe(II)–Fe(III) ratio12,14,17 and the pH of the solution.15,18,19 For example, magnetite formation from Fe(II)-catalyzed ferrihydrite transformation is favored at high Fe(II)–Fe(III) ratios and pH > 7,12,14 while slightly acidic pH conditions (6–7) favor lepidocrocite and goethite formation.12,14 When transformations of ferrihydrite and lepidocrocite are mediated by dissimilatory Fe-reducing bacteria, the biotic mineral transformations can lead to the formation of ferrous carbonate minerals, such as siderite (FeCO3),20 and green rust21 in addition to magnetite and goethite.20–22 Abiotic and biotic transformations of Fe oxyhydroxides can be hindered or altered by the presence of, for example, organic matter,23–25 trace metals,26 or other oxyanions.14,27–29 For example, the presence of organic matter23,24 and silicate14,27 can hinder the abiotic Fe(II)-catalyzed transformation of ferrihydrite to goethite and magnetite.
Model studies, like those mentioned above, used mineral suspensions or flow-through columns to demonstrate the effect of individual components on Fe mineral transformations. Yet, Fe oxyhydroxide transformations in natural systems are influenced by soil porewater, which comprises complex mixtures of dissolved organic matter, nutrients and trace elements. Previous works aimed at studying mineral transformations in soil and incubated ferrihydrite in flooded soils using permeable bags30,31 or diffusive gel samplers.32 In contrast to these studies, where ferrihydrite was spatially separated from the soil matrix and dissolved soil components, including Fe(II), were required to diffuse into the permeable bags or sample holders,30–32 Fe minerals in natural systems are part of the soil matrix and thus, are more closely associated to or in direct contact with organic matter, other minerals, and microorganisms. This likely impacts the trajectory of mineral transformations.33
Redox conditions in intermittently flooded soils may alternate between oxic and sub- or anoxic conditions. Therefore, several studies have investigated Fe mineral dynamics in soils during redox cycles,34,35 mainly using soil slurries36 purged with N2(g) or O2(g).37,38 After exposure to one or multiple redox cycles, both an overall increase34,35,37 and decrease35,37,38 in short-range-ordered Fe minerals in the soil has been reported. Changes in the short-range-ordered Fe mineral fraction in soils can strongly impact the bioavailability of Fe for microbial reduction and thus the Fe(II) production upon the onset of Fe reducing conditions.39 Short-range-ordered Fe phases, such as ferrihydrite, are regularly precipitated during oxic periods of redox cycles36–38 and are highly susceptible to mineral transformation.12,21 During redox cycles, reductive Fe mineral transformations are interrupted by oxic periods, which may lead to different mineral transformation extents compared to mineral transformations under permanently sub- or anoxic conditions.
Due to the importance of Fe minerals for element cycling, we need to improve our understanding of how Fe oxyhydroxides transform in soils. In comparison with mixed soil slurries used in previous studies,36–38 undisturbed soils are characterized by higher soil-to-solution ratios, chemical and physical heterogeneity at the pore- and aggregate scale,40–44 and pronounced diffusion limitations of chemical processes.40,41 These factors likely influence Fe mineral transformations, for example, by impacting the local concentrations of Fe(II) and organic matter. To better understand Fe mineral dynamics in soils, recent approaches combined the use of 57Fe-labeled solutions or minerals in soil experiments with 57Fe Mössbauer spectroscopy, which is only sensitive to 57Fe.33,45,46 For example, Tishchenko et al.46 reacted 57Fe(II) with a soil slurry and used Mössbauer spectroscopy to study the Fe atom exchange between dissolved Fe(II) and structural Fe(III) and Fe mineral crystallization. Chen and Thompson45 added dissolved 57Fe(II) to soils, oxidized the soil slurries, and analyzed the oxidation products with Mössbauer spectroscopy. Recently, Notini et al.33 demonstrated a new approach for studying the transformations of synthetic Fe minerals in close contact with soil by using 57Fe-labeled minerals and following their transformation with Mössbauer spectroscopy. Adopting this approach in the current study, we follow the transformation of 57Fe-labeled ferrihydrite and lepidocrocite in a soil matrix exposed to redox fluctuations.
The objectives of this study were to (i) investigate how redox fluctuations affect the transformations of ferrihydrite and lepidocrocite in a paddy soil at a natural soil-to-solution ratio and (ii) to assess how the close spatial association between the added Fe minerals and the soil matrix affects the Fe mineral transformations. We incubated ferrihydrite and lepidocrocite in soil mesocosms exposed to three flooding–drainage cycles. As a control, an additional mesocosm remained permanently flooded. Ferrihydrite and lepidocrocite were incubated in mesh bags either containing the minerals without soil or 57Fe-labeled minerals mixed with soil to create a close spatial association with the soil matrix. The mineral transformation products in the incubated samples were analyzed with Fe K-edge X-ray absorption fine structure spectroscopy and/or X-ray diffraction and 57Fe Mössbauer spectroscopy.
Materials and methods
Soil sampling and characterization
A rice paddy soil was sampled at the Ubon Ratchathani Rice Research Center in Thailand during the dry season in February 2020. A soil profile of 2 m depth was established in the dry rice field and classified as a Hydragric Loamic Anthrosol on sandstone (details in the ESI, Section S1†) after the World Reference Base for Soil Resources.47 Surface soil (0–15 cm depth, 100 kg) was sampled for the incubation experiments and was oven-dried at 30 °C until constant weight and homogenized by mixing and sieving (<2 mm). The texture of the sieved soil was silty sand (3% clay, 12% silt, 85% sand). For chemical and mineralogical analyses, representative subsamples of the sieved soil were milled with a vibratory disc mill. The total element contents were measured with X-ray fluorescence spectrometry (XRF; XEPOS, Spectro, Germany) in the milled soil (Fe = 3.3 g kg−1, aluminum = 14.0 g kg−1, silicon = 417.4 g kg−1, Table S1†). Carbon (4.0 g kg−1) and nitrogen (0.5 g kg−1) contents were determined using a CN elemental analyzer (vario MAX cube, Elementar, Germany). The pH of the soil was weakly acidic (pH 5.5 in 0.01 M CaCl2).Mineral synthesis
Ferrihydrite and lepidocrocite with natural abundance (NA) Fe isotope composition (NAFe-Fh, NAFe-Lp) were synthesized by adapting the methods of Schwertmann and Cornell.48 Briefly, for NAFe-Fh synthesis, a 0.2 M ferric nitrate (Fe(NO3)3·9H2O, Merck) solution was titrated (836 Titrando, Metrohm) with 1 M NaOH (Titrisol®, Merck) to pH 7.5 ± 0.1 under vigorous stirring at room temperature. For NAFe-Lp, a 0.2 M Fe(II) solution prepared from FeCl2 (Merck) was titrated to pH 6.7–6.9 with 1 M NaOH at room temperature, then oxidized under vigorous stirring and gentle purging with air (approx. 100 mL min−1) while the pH was maintained by the further addition of 1 M NaOH. The isotopically labeled ferrihydrite (57Fe-Fh) and lepidocrocite (57Fe-Lp) were prepared by dissolving 57Fe(0) (96.14% 57Fe, Isoflex USA) in 2 M HCl (NORMATOM®, 34–37%, VWR) overnight, resulting in a 0.18 M 57Fe(II) solution. For ferrihydrite synthesis, the 57Fe(II) solution was oxidized to 57Fe(III) by adding 35% H2O2 (Merck). The 57Fe(II) and 57Fe(III) solutions were passed through a 0.45 μm nylon filter before 57Fe-Fh and 57Fe-Lp were prepared as described for NAFe-Fh and NAFe-Lp. All precipitates were repeatedly washed by resuspending the precipitates in ultra-pure water (UPW, ≥18.2 MΩ·cm, Milli-Q, Millipore), centrifuging the suspensions (3438 g for 15 min, 20 °C) and decanting the supernatant. After the washing procedure, the precipitates were again resuspended in UPW, shock frozen dropwise in liquid N2, freeze-dried, gently homogenized with mortar and pestle, and stored in amber glass bottles in a desiccator. All minerals were characterized by X-ray diffraction (XRD, Bruker D8 Advance), which confirmed the expected mineral composition and showed no crystalline mineral impurities. However, Mössbauer spectra of the NAFe- and 57Fe-lepidocrocites collected at 77 K and 5 K indicated the presence of minor amounts (7% in NAFe-Lp and 16% in 57Fe-Lp) of nano-goethite (ESI Section S2†). Mineral characterization details, including XRD patterns (Fig. S3†) and Mössbauer spectra (Fig. S4†) with fit components (Table S2†) are provided in ESI Section S2.†Preparation of mesh bags and sample holders
For the incubation of the minerals, mesh bags (internal dimensions ∼1 × 3 × 0.2 cm) were prepared from a polyethylene terephthalate (PETE) mesh fabric (SEFAR, Switzerland). The mesh fabric had a pore size of 51 μm to allow the free exchange of water, solutes and bacteria. The mesh fabric sheets were triple-layered, folded, and heat-sealed on two sides before filling. For the NAFe-minerals, 100 mg of NAFe-Fh or NAFe-Lp were filled inside each mesh bag before the third side was carefully heat-sealed. For the 57Fe-minerals, 8 mg of 57Fe-Fh or 57Fe-Lp were thoroughly mixed with 800 mg of the sieved soil in a 2 mL Eppendorf tube using a plastic rod before being transferred to the mesh bags, as described above. The mineral-to-soil ratio in the mineral-soil mixes was chosen to achieve minimal addition of Fe to the soil while ensuring a sufficiently high Mössbauer signal contribution from the 57Fe-labeled minerals, as discussed in Notini et al.33 In this experiment, the addition of the Fe minerals tripled the total soil Fe content while >98% of 57Fe in the mineral-soil mixes originated from the added 57Fe-labeled minerals. Sample holders were designed and 3D-printed (Clear Photopolymer Resin, Formlabs) to have a pointy tip to enable the insertion of mesh bags into the soil. The sample holders had large vertical openings on the side to maximize the contact between the mesh bag and the surrounding soil. The mesh bags were placed in the sample holders and a threaded plastic rod was attached to facilitate the insertion into the soil (ESI Section S3†).Mesocosm setup and flooding of the soil
The soil mesocosms were designed to mimic flooding and drainage cycles in rice paddy soils. Each mesocosm was made from a rectangular plastic box (polypropylene, 37.6 × 26 × 28.3 cm, 20 L). The box had holes (diameter 0.5 cm, n = 5) in the bottom to allow the drainage of the mesocosms. A layer of mesh fabric, similar to the mesh fabric used for the mesh bags but with a larger pore size (105 μm), was placed on the bottom of the box to prevent loss of soil during the drainage. The mesh fabric was then covered by a ∼1 cm thick sand layer (grain size 0.1–0.5 mm, acid-washed, 1.5 kg sand), followed by 12 kg of sieved soil, resulting in a soil depth of 12 cm. Each mesocosm was contained within a second rectangular plastic box equipped with a drainage port to control the water level in the soil. More details on the mesocosm setup are provided in ESI Section S3.†The experiment was conducted in a climate chamber at 30 °C air temperature and 50% relative humidity. Each mesocosm was flooded with 8 L of a solution prepared from CaCl2 and NaCl (1 mM Ca, 4 mM Na, 6 mM Cl) using a peristaltic pump and Tygon tubes (Saint-Gobain Tygon R3607, inner diameter 1.85 mm), with four tubes per mesocosm. The solution composition was chosen based on analyses of porewaters from a similar paddy field at the Ubon Ratchathani Rice Research Center (1.2 mM Ca, 6.8 mM Na). The mesocosms were flooded dropwise (∼7 mL min−1 per mesocosm) over 20 hours, during which the position of the tubes was changed multiple times to enable homogenous wetting of the soil and to minimize the formation of air pockets. Four identical mesocosms were prepared, of which three were designated to undergo water table fluctuations (three weeks flooded, one week drained) to induce soil redox cycles. The fourth mesocosm remained permanently flooded for the duration of the experiment (12 weeks) to compare mineral transformation processes under permanently sub- or anoxic soil conditions to transformations during redox cycles. Flooding-drainage cycles will be referred to as redox cycles and flooded and drained periods will be referred to as anoxic and oxic periods, respectively.
After the first flooding, each mesocosm with fluctuating water table was equipped with one gel-filled redox glass electrode (Mettler Toledo; 3 M KCl, Ag/AgCl reference electrode), one tensiometer (METER group), and three Rhizon porewater samplers (Rhizosphere Research Products, The Netherlands, 0.6 μm pore cut-off) installed at ∼8 cm depth. The oxidation–reduction potential was recorded hourly using a mobile logger system (pH Meter 913, Metrohm, built-in data logger) and converted (+209 mV, 30 °C) to redox potentials relative to the standard hydrogen electrode (Eh). The soil matric potential was recorded every 10 min (ZL6, METER Group). The sample holders were installed in the soil at 8 cm depth. In each mesocosm with fluctuating water table, six sample holders of each mineral (NAFe-Fh, 57Fe-Fh, NAFe-Lp, 57Fe-Lp) were installed, resulting in 24 samples per mesocosm. In the permanently flooded mesocosm, three sample holders of each mineral (NAFe-Fh, 57Fe-Fh, NAFe-Lp, 57Fe-Lp) were installed (total: 12 samples). A schematic setup of the experiment is provided in Fig. S5.†
Anoxic sampling and drainage
After soil flooding, the porewater of all mesocosms was sampled at 1, 3, 7, 14 and 21 days, using the three permanently installed Rhizons in each mesocosm. Each Rhizon was connected to a 10 mL pre-acidified plastic syringe to stabilize Fe species immediately. Approximately 10 mL of porewater was sampled and subsequently passed through a 0.45 μm nylon filter for DOC analysis (DIMATOC 2000, Dimatec) or a 0.22 μm nylon filter for total element analysis by inductively coupled plasma optical emission spectroscopy (ICP-OES, Agilent 5100). Porewater element concentrations will be shown as the average of the triplicate porewater samples for each mesocosm in figures and reported as the average of the three redox fluctuating mesocosms (i.e. 9 porewater samples) in the text. The pH in the mesocosms was measured manually in the wet soil at sample depth (8 cm) at each porewater sampling with a pH glass electrode (Metrohm).At 21 days after flooding, one set of mineral samples (NAFe-Fh, 57Fe-Fh, NAFe-Lp, 57Fe-Lp) was removed from each redox fluctuating mesocosm. The threaded plastic rod was unscrewed, and the sample holder was placed into a 50 mL plastic centrifuge tube. The tube was immediately flushed with N2(g) and closed to prevent sample oxidation. All samples were transferred to an anoxic glovebox (N2 atmosphere; MBraun, <5 ppm O2) after sampling. In the glovebox, the mesh bags were removed from the sample holders and air-dried for two days before the samples were homogenized with a mortar and pestle. After the mineral sampling, all redox fluctuating mesocosms were drained by opening the valve of the drainage tube at the bottom of the mesocosms. The drainage of the soil to field capacity (i.e. matric potential = −6 kPa) was complete within three days, while residual water remained in the lower boxes of the mesocosms (∼0.9 L).
Oxic sampling and re-flooding
Seven days after drainage, another set of mineral samples was extracted from each redox fluctuating mesocosm, representing the end of one complete redox cycle. The mesh bags were removed from the sample holders, and the samples were air-dried in the climate chamber for two days before the samples were homogenized using a mortar and pestle. To make sure that air drying did not affect the Fe speciation in the samples, we ran a test incubation where 57Fe-Fh-soil mixes were incubated in the experimental soil for one redox cycle (3 weeks flooded, 1 week drained) and dried under ambient air or in the glovebox. The test samples were analyzed with Mössbauer spectroscopy and showed similar solid-associated Fe(II) fractions (Fig. S7†). After the mineral sampling, all redox fluctuating mesocosms were re-flooded as described above. Due to residual water in the soil and in the lower plastic box, the re-flooding only required 4.8 L of solution per mesocosm to achieve the same water level as in the previous flooded period. The entire procedure, including the anoxic and oxic sampling described above, was repeated two additional times, resulting in redox cycles I–III over 12 weeks.Anoxic sampling in the permanently flooded mesocosm
The porewater in the permanently flooded mesocosm was sampled on the same days as in the redox fluctuating mesocosms. The pH was measured as described above and the oxidation–reduction potential was measured manually with a redox electrode (Mettler Toledo) at each porewater sampling. The mesh bags in the permanently flooded mesocosm were sampled only at the end of the experiment (12 weeks) following the same procedure as described above for anoxic mineral samplings in the redox fluctuating mesocosms. In the permanently flooded mesocosm, soil surrounding one of the three replicates of 57Fe-Fh and 57Fe-Lp was additionally sampled with a plastic Humax core sleeve (diameter 5 cm) by taking a soil core containing the sample holders. All samples and the soil cores were immediately flushed with N2 and transferred to the glovebox after sampling. The soil cores were set up vertically in the glovebox, and the water was allowed to drain for one hour before the plastic sleeve was opened. The cores were sampled circularly around the sample holder (three layers of ∼0.5 cm) using a spatula, and samples were dried in the glovebox. After sampling the soil in the cores, the mesh bags were removed from the sample holders, and the samples were dried and homogenized in the glovebox as described above. Aqua regia digestions of soil core samples combined with Fe isotope analysis of aqua regia extracts (methods described below) allowed us to follow potential changes in the Fe concentration and Fe isotope composition of the soil surrounding the mesh bags.Solid phase analysis
Solid samples from NAFe-mineral mesh bags without soil were analyzed by bulk Fe K-edge (7112 eV) X-ray absorption spectroscopy (XAS) at the SAMBA beamline at the SOLEIL synchrotron (St. Aubin, France), and by powder XRD. X-ray absorption near-edge structure and extended X-ray absorption fine structure (EXAFS) spectra were recorded as transmission spectra. Linear combination fit (LCF) analyses of k3-weighted EXAFS spectra were performed in Athena.49 Reference compounds for Fe-minerals comprised ferrihydrite, lepidocrocite, magnetite, goethite, siderite, and green rust carbonate. Rietveld quantitative phase analysis (QPA)50 of XRD patterns was used to obtain the mineral fractions of transformation products in the samples. Ferrihydrite was included in the QPA as a mass-calibrated PONKCS51 phase, as described in earlier studies.14,18,23 Detailed information on sample preparation, measurements, fitting of XAS spectra and Rietveld analyses are found in ESI Sections S7 and S8.†The mineral transformation in 57Fe-Fh- and 57Fe-Lp-soil mixes during the incubation was investigated by 57Fe Mössbauer spectroscopy, which is only sensitive to 57Fe in the sample. With 8 mg of 57Fe-labeled mineral (57Fe fraction = 95.08%) mixed with 800 mg of soil (Fe = 3.3 g kg−1) with NAFe isotope composition (2.12% 57Fe; ref. 52), >98% of the Mössbauer signal came from the introduced mineral Fe, while ≤2% of the signal originated from the soil Fe. Additionally, NAFe-mineral samples collected at the end of anoxic (11 weeks) and oxic (12 weeks) periods of redox cycle III in the redox fluctuating mesocosms and at 12 weeks from the permanently flooded mesocosm were also analyzed with Mössbauer spectroscopy. The Mössbauer spectra were collected at 77 K and 5 K in transmission mode and fitted using the Recoil software.53 The 57Fe-mineral-soil mixes sampled after redox cycle III (12 weeks) were additionally measured at room temperature (295 K) to consider potential magnetite formation. Details to sample preparation and fitting of Mössbauer spectra are presented in ESI Section S11.†
Mineral dissolution and aqua regia digestions
The elemental composition of initial and incubated NAFe-mineral samples was determined after dissolution in concentrated HCl (NORMATOM®, VWR) at room temperature by ICP-OES. The C content of NAFe-Fh and NAFe-Lp at the end of the last redox cycle (12 weeks) was determined using a CHN analyzer (LECO TruSpec Micro). To measure changes in the Fe content and Fe isotope composition, initial and reacted 57Fe-mineral-soil mixes and soil samples from Humax cores were digested with aqua regia. Aliquots (∼150 mg) of the soil and/or mineral-soil mixes were weighed into 15 mL centrifuge tubes and 10 mL of freshly prepared aqua regia (HNO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HCl ratio 1:3, HNO3 (distilled; Merck), HCl (NORMATOM®, VWR)) was added to each vial. The samples were digested at 120 °C for 90 min and passed through a 0.45 μm PTFE filter. The total Fe concentration in the filtrates was measured by ICP-OES. For Fe isotope analyses, all samples were diluted to ∼50 ppb Fe and analyzed with triple-quadrupole inductively coupled plasma mass spectrometry (ICP-MS, Agilent 8800 Triple Quad), and 57Fe isotope fractions (f57Fe) were calculated relative to the sum (counts per second) of 54Fe, 56Fe, 57Fe, and 58Fe.14,23
:HCl ratio 1:3, HNO3 (distilled; Merck), HCl (NORMATOM®, VWR)) was added to each vial. The samples were digested at 120 °C for 90 min and passed through a 0.45 μm PTFE filter. The total Fe concentration in the filtrates was measured by ICP-OES. For Fe isotope analyses, all samples were diluted to ∼50 ppb Fe and analyzed with triple-quadrupole inductively coupled plasma mass spectrometry (ICP-MS, Agilent 8800 Triple Quad), and 57Fe isotope fractions (f57Fe) were calculated relative to the sum (counts per second) of 54Fe, 56Fe, 57Fe, and 58Fe.14,23
Results and discussion
Soil conditions during the experiment
Matric potentials (ψ) showed that the soil was water saturated during flooded periods (ψ = 0 kPa), while the water content dropped to slightly below field capacity during drained periods (ψ = −9 kPa, field capacity = −6 kPa; Fig. S8†). The pH dropped from circumneutral conditions (pH 6.5–6.9) during flooded periods to slightly acidic conditions (pH 5.7–6.0) during drained periods (Fig. S9†). The pH was slightly higher in the last, compared to the first flooded period. The soil Eh in the mesocosms with fluctuating water table indicated Fe reducing soil conditions (<100 mV at pH 7; ref. 9 and 54) during flooded periods (−163 ± 22 mV, mean Eh at 3, 7 and 11 weeks ± standard error of the mean, Fig. S8†), while the Eh was well above conditions expected for Fe reduction during the drained periods (+386 ± 25 mV, mean Eh at 4, 8 and 12 weeks). After anoxic conditions had established in the permanently flooded mesocosm, the Eh remained stable at −141 ± 7 mV (mean Eh at 2–12 weeks) throughout the experiment (Fig. S8†).Porewater element concentrations
In the redox fluctuating mesocosms, Fe was released from the solid phase to the porewater within one day after soil flooding (Fig. 1A). The dissolved Fe concentration was highest one week after flooding (1.1 mM), followed by a slight decrease to 0.9 mM Fe at the end of the anoxic period of redox cycle I (3 weeks). In the following two redox cycles (II and III), dissolved Fe dynamics were similar to redox cycle I, but Fe release was slower and peak Fe concentrations were lower. Combined with measured changes in Eh and pH (Fig. S8 and S9†), the Fe release to the porewater indicates that solid phase Fe(III) was reductively dissolved during anoxic periods.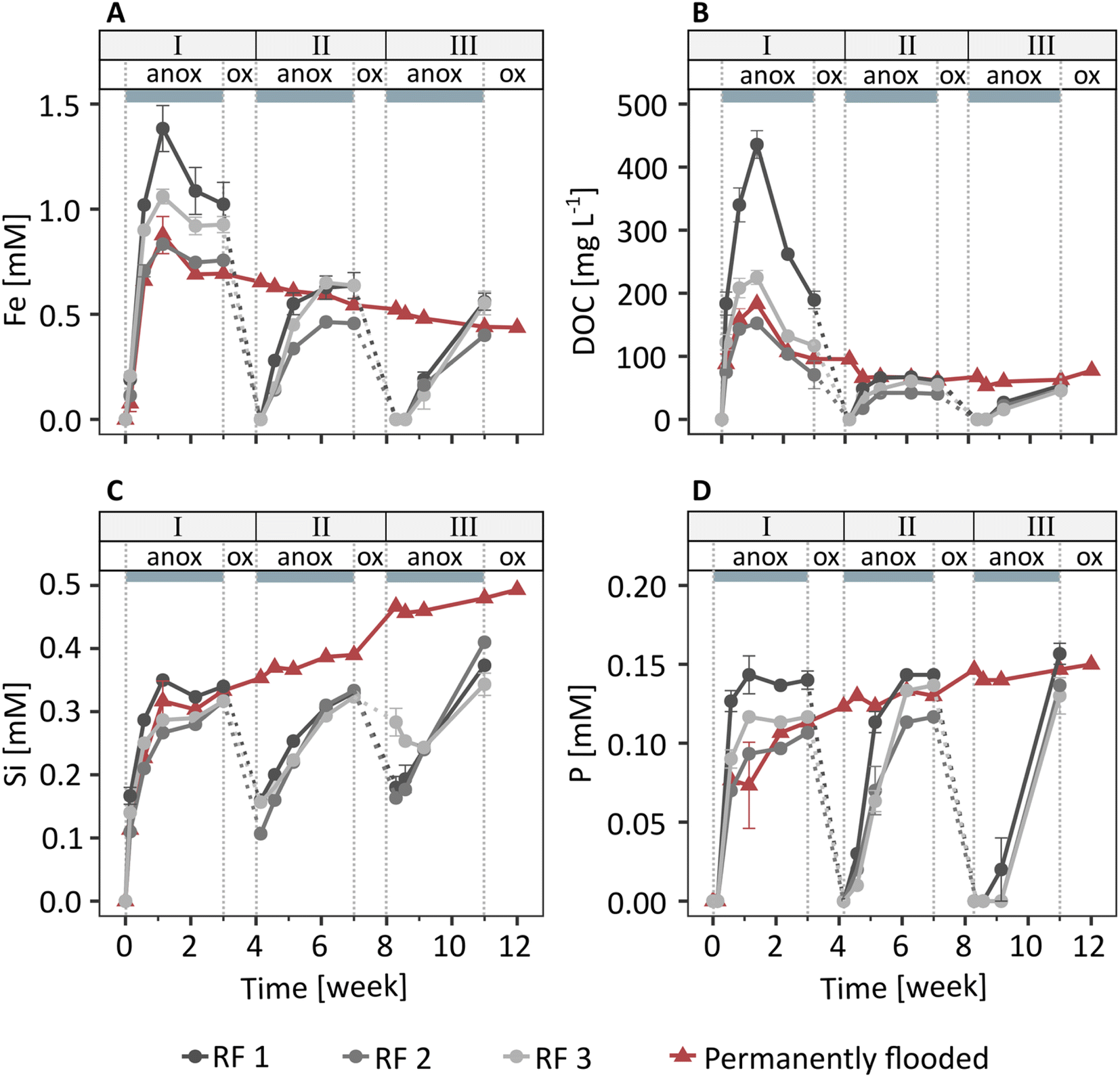 | ||
| Fig. 1 Total dissolved concentrations of (A) iron (Fe), (B) organic carbon (DOC), (C) silicon (Si), and (D) phosphorus (P) in porewaters of soils in the redox fluctuating (RF 1–3) and the permanently flooded mesocosms. Labels above grey boxes indicate anoxic (anox, grey boxes) and oxic (ox) periods in redox cycles I–III. Error bars show the standard error of triplicate porewater samples in each mesocosm. Dashed lines are included for visual aid. | ||
Dissolved organic C and dissolved Si and P were present in the porewater at one week (249 mg L−1 DOC, 0.3 mM Si, 0.1 mM P, Fig. 1B–D), along with major base cations (3.8 mM Ca, 8.0 mM Na; initial flooding water contained 1 mM Ca, 5 mM Na) and small amounts of K and Mg (Fig. S10†). Concentrations of other elements were below detection limit. Since major base cations, such as Ca and Mg, have a relatively low affinity for adsorption to ferrihydrite,55 the direct interactions between Ca, Mg, K and Na with Fe oxyhydroxides were likely much weaker than for DOC56 and negatively charged oxyanions, such as phosphate57 and silicate.57,58 Therefore, here we will focus on the element dynamics of Fe, DOC, Si, and P. The concentrations of DOC, Si, and P showed trends similar to dissolved Fe during redox cycles, with low concentrations at the start of anoxic periods followed by an increase in concentrations in the following days. The similar trends in Fe compared to DOC, Si, and P release during anoxic periods suggest that these components were associated with Fe minerals and released to solution upon Fe mineral reductive dissolution. Additional DOC, Si, and P may have been released from metabolized organic matter.
In the permanently flooded mesocosm, the element concentrations in the porewater in the first three weeks were similar to the concentrations observed during the anoxic period of redox cycle I in the redox fluctuating mesocosms (Fig. 1). Dissolved Fe and DOC concentrations reached their maximums at one week (0.9 mM Fe, 183 mg L−1 DOC), followed by a steady decrease to 0.4 mM Fe and 78 mg L−1 DOC at the end of the experiment (12 weeks, Fig. 1A and B). Si and P were released within one week after the flooding of the soil in the permanently flooded mesocosm (0.3 mM Si and 0.1 mM P at 1 week) and subsequently increased up to 0.5 mM Si and 0.15 mM P at the end of the experiment (12 weeks, Fig. 1C and D).
The dissolved Fe, DOC and P concentrations reached a similar level in the permanently flooded mesocosm compared to the end of each anoxic period in the redox fluctuating mesocosms (Fig. 1A, B and D). This suggests that the soil replenished any C and P that was lost with the drainage water in the redox fluctuating mesocosms upon the next flooding. The dissolved Si concentrations at the end of the anoxic periods of redox cycles II and III were slightly lower compared to the permanently flooded mesocosm. This may be explained by interrupted silicate dissolution during oxic periods in the redox fluctuating mesocosms. Additionally, Si polymerization may have been enhanced due to lower pH and the relative increase in Si concentrations59,60 during oxic periods in the redox fluctuating mesocosms.
NAFe-mineral transformations in mesh bags without soil
The NAFe-Fh and NAFe-Lp in mesh bags without soil transformed to more crystalline Fe minerals over the three redox cycles, as shown by linear combination fitting of Fe K-edge EXAFS spectra (Fig. 2 and S15†). During the anoxic period of redox cycle I, NAFe-Fh transformed to goethite (52% of total Fe) and magnetite (30%) (Fig. 2A). During the following oxic period, fractions of ferrihydrite increased in NAFe-Fh, while goethite fractions remained relatively stable and magnetite fractions decreased. At the end of redox cycle I (4 weeks), NAFe-Fh samples contained 33% ferrihydrite, 51% goethite and 16% magnetite. In comparison, NAFe-Lp transformed to magnetite (26%) during the anoxic period of redox cycle I (Fig. 2B) and the (nano-)goethite that was present in the initial NAFe-Lp (ESI Section S2†) disappeared. With the subsequent oxic period, fractions of lepidocrocite increased and magnetite fractions decreased, leading to 94% lepidocrocite and 6% magnetite in NAFe-Lp samples after redox cycle I (4 weeks). Similar trends in Fe mineral fractions were observed during redox cycles II and III, but changes in mineral fractions between anoxic and oxic periods were smaller.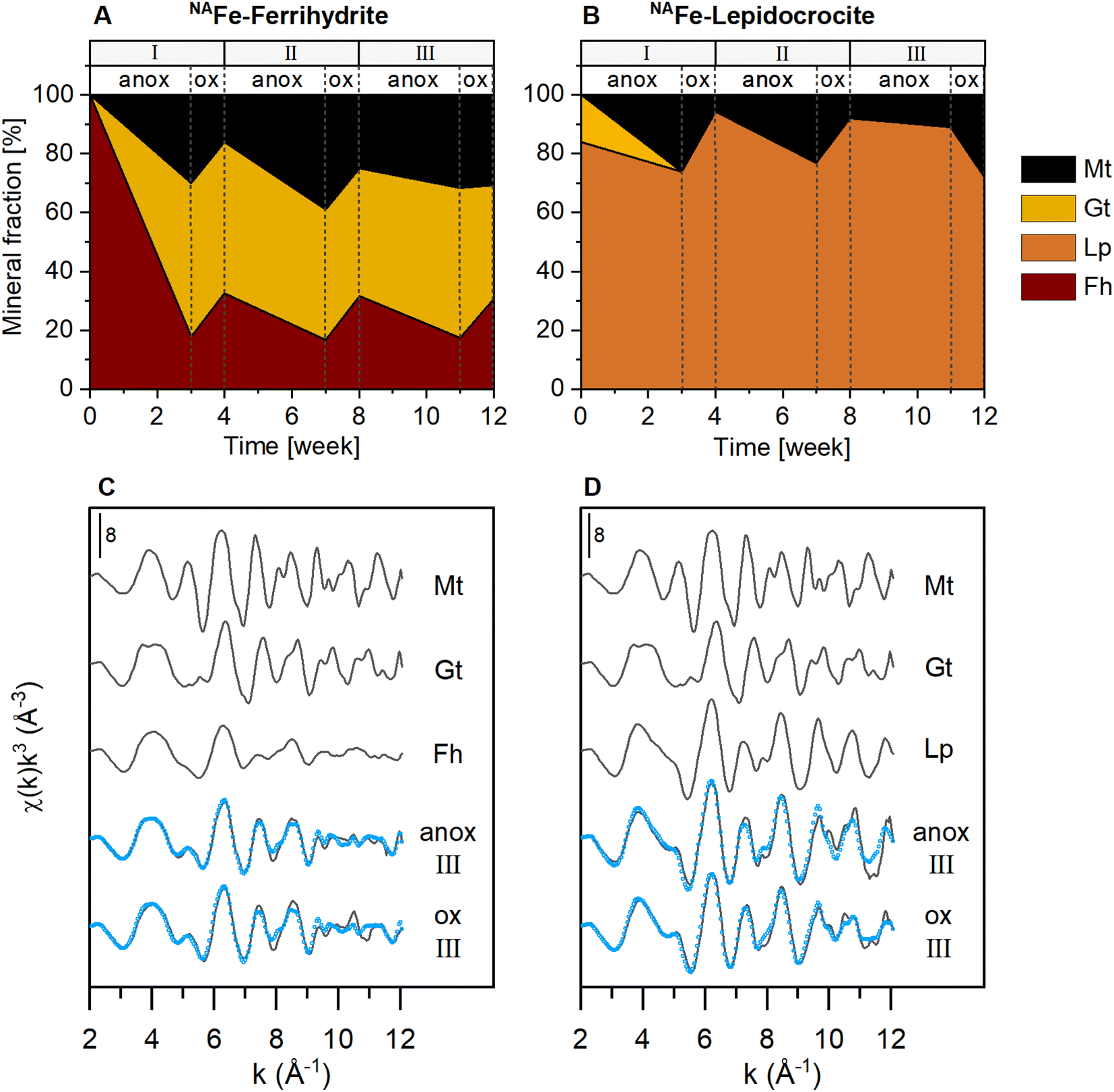 | ||
| Fig. 2 Iron mineral fractions as determined by linear combination fitting of Fe K-edge EXAFS spectra in NAFe-mineral samples from mesh bags without soil for (A) NAFe-ferrihydrite and (B) NAFe-lepidocrocite incubated for 12 weeks in the redox fluctuating mesocosms, with (C and D) reference spectra and spectra from redox cycle III. According to EXAFS data, the initial NAFe-lepidocrocite contained minor fractions (16%) of goethite which was likely nano-crystalline (ESI Section S2†). All spectra and fits are presented in Fig. S15.† Fractions of fit components and reduced χ2 are presented in Table S4.† Abbreviations: Mt = magnetite, Gt = goethite, Fh = ferrihydrite, Lp = lepidocrocite, anox = anoxic, ox = oxic. | ||
The presence of goethite and/or magnetite in the incubated NAFe-Fh and NAFe-Lp samples was also confirmed by Mössbauer spectroscopy (Fig. 3 and Table 1) and XRD (Fig. S12 and S13†). Mössbauer spectra (5 K) of samples from redox cycle III showed sextets with parameters matching reported values of Fe in magnetite.61 Fitting of magnetite in Mössbauer spectra at low temperature is challenging since magnetite contributes up to five sextets at 5 K.61 We fit the spectra following the approach of Doriguetto et al.61 (Fig. S19A and B†). Since two of the five sextets covered ≥80% of the sextet area attributed to magnetite, we continued with fitting only the two dominant sextets (Fig. S19C and D†). These sextets correspond to sextets S4 and S5 in Fig. 3 and in the following text. According to Doriguetto et al.,61 the parameters of sextets S4 and S5 represent octahedral Fe2.67+ and tetrahedral Fe3+ in magnetite, respectively. In addition to Fe mineral phases detected by EXAFS and Mössbauer, XRD identified small fractions (≤5%) of siderite in NAFe-Fh and NAFe-Lp samples from anoxic and oxic periods (Fig. S12†). The applied methods for the identification and quantification of mineral phases (Fe K-edge EXAFS, Mössbauer spectroscopy, XRD) generally agreed. A comparison of quantified mineral fractions from all methods is presented for samples from redox cycle III and the permanently flooded mesocosm (12 weeks) in Table S13.†
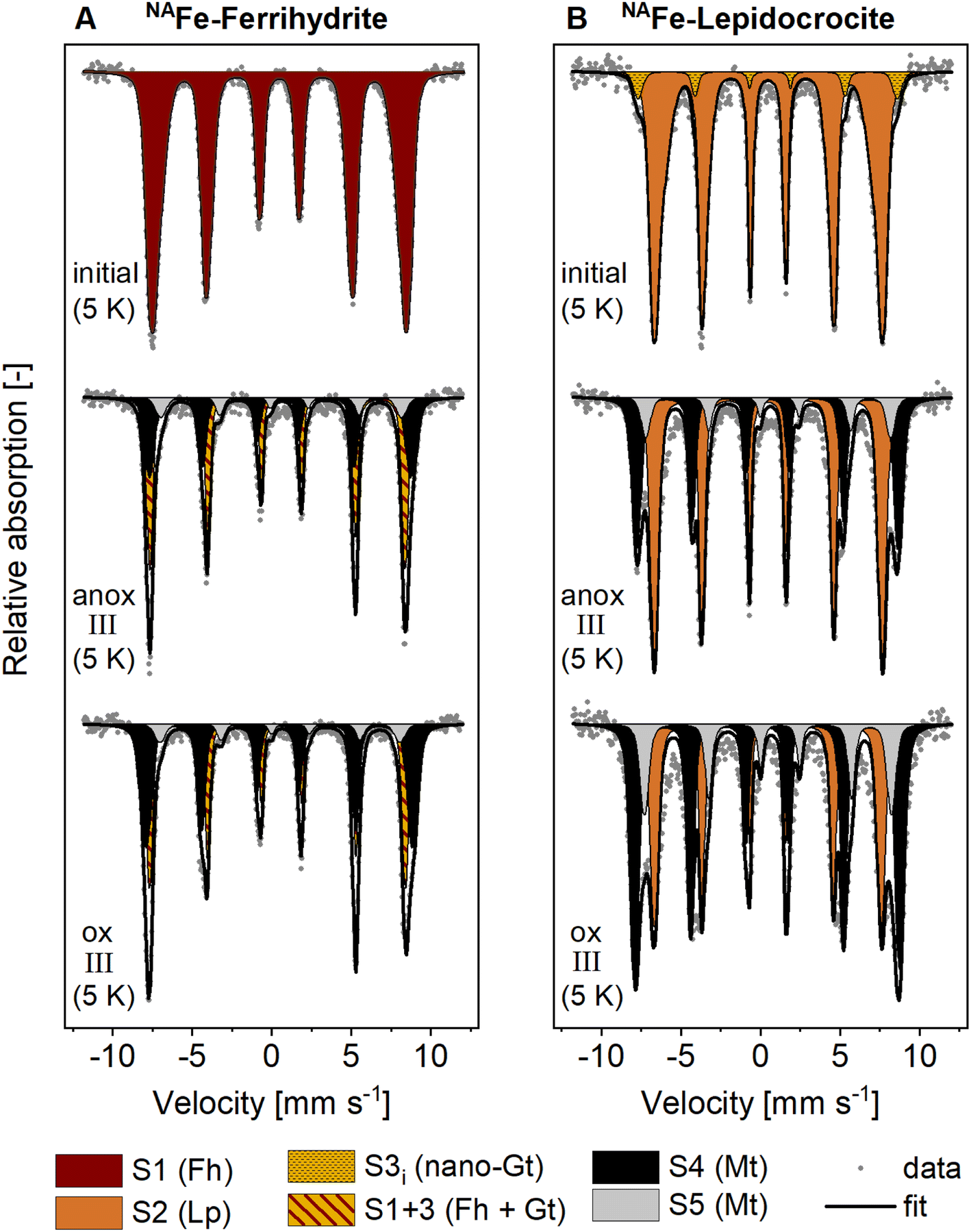 | ||
| Fig. 3 Mössbauer spectra collected at 5 K from (A) NAFe-ferrihydrite and (B) NAFe-lepidocrocite mesh bags without soil before the incubation (initial) and after the incubation in the redox fluctuating mesocosms (redox cycle III anoxic and oxic period). Averaged fitting parameters and interpretations of Mössbauer fit components are given in Table 1, all fitting parameters are presented in Tables S11 and S12.† Abbreviations: anox = anoxic, ox = oxic, S = sextet. | ||
| Fit component | CSa [mm s−1] | QSb or εc [mm s−1] | H [T] | Interpretation | Ref. |
|---|---|---|---|---|---|
| a CS: center shift. b QS: quadrupole splitting (for doublets). c ε: quadrupole shift (for sextets). d H: hyperfine field. Abbreviations: D = doublet, S = sextet, CF = collapsed feature. | |||||
| D1 | 0.47 | 0.84 | — | Complexed and silicate-associated Fe(III) | 35 |
| D2 | 1.22 | 2.62 | — | Adsorbed Fe(II) or Fe(II) in primary minerals or silicates | 35 and 45 |
| S1 | 0.47 | <−0.01 | 48.84 | Fe(III) in ferrihydrite | 67 |
| S2 | 0.48 | 0.01 | 42.68 | Fe(III) in lepidocrocite | 68 |
| S3 | 0.48 | −0.13 | 49.89 | Fe(III) in goethite | 45 |
| S3i | 0.51 | −0.06 | 50.45 | Nano-crystalline goethite | 45 |
| S1+3 | 0.48 | −0.06 | 47.15 | Mixture of ferrihydrite and goethite | — |
| S4 | 0.44 | 0.02 | 51.18 | Octahedral Fe(II) and Fe(III) in magnetite | 61 |
| S5 | 0.85 | −0.34 | 47.47 | Tetrahedral Fe(III) in magnetite | 61 |
| CF | 0.81 | 0.00 | 26.31 | (Mixed-valence) disordered Fe phase | 35, 37 and 45 |
The decrease in magnetite fractions observed in NAFe-Fh and NAFe-Lp during the oxic periods may be related to the oxidation of solid-associated Fe(II) and its precipitation as ferrihydrite, lepidocrocite or goethite. This may have led to a relative decrease in magnetite fractions. Magnetite also underwent changes in the Fe(II):Fe(III) stoichiometry during redox cycles, especially in NAFe-Fh samples. The Mössbauer fitting results indicate a decrease in the ratio between areas of sextets S4 (Fe(II) and Fe(III)) and S5 (only Fe(III)) during the oxic period (Fig. 3) which suggests that the Fe(II):Fe(III) ratio in magnetite was higher during anoxic compared to oxic periods. This is supported by Rietveld fitting results from XRD patterns, which show a larger unit cell length (a) in magnetite in anoxic (8.381 Å, redox cycle I) compared to oxic (8.365 Å, redox cycle I) periods (Fig. S14†). Similar decreases in the magnetite unit cell length have previously been related to a decreased Fe(II):Fe(III) ratio in magnetite62 (ESI Section S7†).
Since the NAFe-mineral samples were spatially separated from the soil, a large part of the mineral transformations were likely caused by the interactions with Fe(II) from the porewater diffusing into the mesh bags, as suggested by a recent study using confocal Raman microspectroscopy.30 In turn, microbial reduction of ferrihydrite and lepidocrocite inside the mesh bags likely played only a minor role in mineral transformation. That Fe(II) catalyzed the transformation of ferrihydrite and lepidocrocite in NAFe-mineral mesh bags without soil is consistent with the fact that goethite is a frequent product of Fe(II)-catalyzed ferrihydrite transformations in model experiments with mineral suspensions spiked with aqueous Fe(II).12–17 The formation of goethite from ferrihydrite has also been observed in similar incubation studies using mineral mesh bags in flooded soils.30,31 However, goethite only formed from NAFe-Fh, but not from NAFe-Lp samples, whereas magnetite formed from both NAFe-Fh and NAFe-Lp. This observation agrees with recent reports from Fe(II)-catalyzed mineral transformation experiments, where goethite nucleated from ferrihydrite but not from lepidocrocite, while magnetite formed from both minerals.14,63 Considering the slightly acidic conditions of the soil in the current study (Fig. S9†) as well as the presence of dissolved (Fig. 1) and sorbed (Fig. S11†) C, Si and P, the transformation to magnetite was somewhat unexpected. Magnetite has been reported as a product of Fe(II)-catalyzed ferrihydrite and lepidocrocite transformation at higher pH (≥7)12,18 and the presence of organic matter or Si can hinder the formation of magnetite.14,23 However, solid concentrations of C, Si and P were low (molar ratios of C/Fe; up to 0.1%, Si/Fe; up to 1.3%, and P/Fe; up to 0.9%) compared to element ratios reported in other studies in which magnetite formed (e.g., C/Fe ratio of 0.7 (ref. 23) and P/Fe ratios of ∼0.05 (estimated from the reported value of 646 mmol P kg−1 ferrihydrite)64). Additionally, C, Si, and P may have accumulated in the outermost mineral layer of the NAFe-Fh and NAFe-Lp mesh bags,30 as the interaction of NAFe-Fh and NAFe-Lp with dissolved elements in the porewater was controlled by the diffusion of these elements into the mineral mesh bags. However, despite the accumulation of sorbed elements, electron transfer between dissolved Fe(II) and structural Fe(III) can still occur.14,23,65 This likely enabled mineral transformation to crystalline Fe minerals inside the mesh bags.30 Magnetite formation during anoxic periods would have been supported by locally high Fe(II) concentrations. Locally high Fe(II) concentrations may have been established due to a Fe(II) diffusion gradient towards the mineral mesh bags, caused by the large sorption capacity of ferrihydrite and lepidocrocite for Fe(II) in mineral mesh bags. Considering the similar use of mineral mesh bags, it is intriguing that Grigg et al.30 observed goethite but no magnetite formation. A potential reason for the formation of magnetite in the present study is the 5-times lower amount of ferrihydrite in the mesh bags. Mesh bags with a lower amount of ferrihydrite contain less Fe(III) and thus require smaller amounts of Fe(II) to achieve sufficiently high Fe(II):Fe(III) ratios for magnetite formation. Once magnetite nucleates, its accumulation quickly captures additional Fe(II) from solution.14,21,66
Interpretation of Mössbauer fit components for 57Fe-mineral-soil mixes
In contrast to the NAFe-mineral mesh bags without soil, the synthetic ferrihydrite and lepidocrocite in the 57Fe-mineral-soil mixes constituted only a small fraction (1% w/w) of the samples (8 mg of 57Fe-Fh or 57Fe-Lp added to 800 mg of soil). Therefore, XRD could not be used to follow the transformations of the added minerals. Instead, we used Mössbauer spectroscopy, which selectively detects 57Fe in the samples, of which >98% originated from the added 57Fe-labeled minerals. In the initial 57Fe-Fh-soil mix (Fig. 4C), the fitting parameters of the sextet (center shift, CS = 0.47 mm s−1; quadrupole shift, ε = −0.007 mm s−1; hyperfine field, H = 48.84 T), matched with parameters reported for ferrihydrite.67 Since fitting ferrihydrite in Mössbauer spectra is challenging67 and our samples also contain multiple other sextets at 5 K, we decided to fix the ferrihydrite parameters, as derived from the initial 57Fe-Fh-soil mix, for subsequent fits. The initial 57Fe-Lp-soil mix (Fig. 4D) showed a sextet with a CS of 0.49 mm s−1, an ε of 0.03 mm s−1 and a H of 44.35 T, which agrees with parameters reported for lepidocrocite.68 In the initial 57Fe-Lp, a second sextet (S3i) with parameters (CS = 0.49 mm s−1, ε = −0.05 mm s−1, H = 50.37 T), indicated that nano-goethite was present in this sample (more details are presented in ESI Section S2†).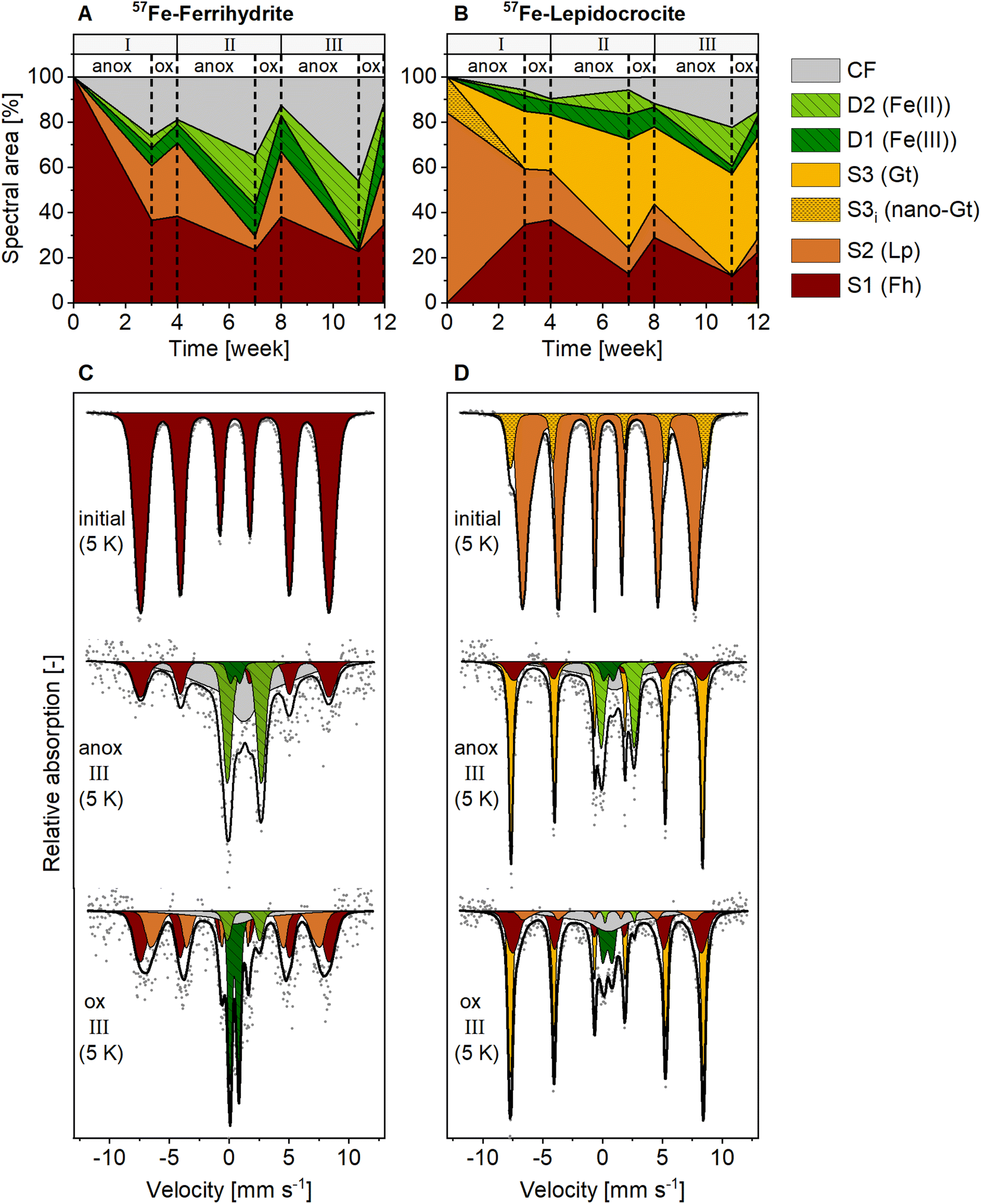 | ||
| Fig. 4 Spectral area of fit components in 5 K Mössbauer spectra for (A) 57Fe-ferrihydrite-soil mixes, and (B) 57Fe-lepidocrocite-soil mixes incubated in the redox fluctuating mesocosms. Lower panels show Mössbauer spectra collected at 5 K from initial mineral-soil mixes containing (C) 57Fe-ferrihydrite or (D) 57Fe-lepidocrocite and corresponding samples after incubation in the redox fluctuating mesocosms, sampled in the anoxic (11 weeks) and oxic (12 weeks) period of redox cycle III. Abbreviations: anox = anoxic, ox = oxic, D = doublet, S = sextet, CF = collapsed feature, Fh = ferrihydrite, Lp = lepidocrocite, Gt = goethite. Goethite in initial 57Fe-lepidocrocite likely was nano-crystalline. Interpretations of Mössbauer fit components are given in Table 1, fitting parameters are presented in Tables S9 and S10.† | ||
The fit components for Mössbauer spectra of the incubated 57Fe-Fh and 57Fe-Lp-soil mixes collected at 5 K, averaged fitting parameters and corresponding interpretations are summarized in Table 1. Results from 77 K spectra are presented in Fig. S20.† All fitted parameters are reported in Tables S7–S10.† The Fe(III) doublet (D1) parameters at 5 K are similar to parameters indicating pyrite.69 However, pyrite formation is unlikely since total dissolved S concentrations in this experiment were below the detection limit. Therefore, in this study the Fe(III) doublet (D1) is interpreted as complexed and silicate-associated Fe(III), in agreement with previous studies.35,45 The fitting of 5 K spectra collected from 57Fe-Fh and 57Fe-Lp-soil mixes required a collapsed feature. In this work, we interpreted the collapsed feature as a highly disordered Fe phase,35,37,45 which in our case was a mixed-valence Fe(II)–Fe(III) phase with large fractions of Fe(II), as seen from differences in Fe(II) fractions in 77 K and 5 K spectra (Fig. S23†). A detailed discussion on the nature of the collapsed feature, including results from sequential Fe extractions (ESI Section S10†), and fitting results from 77 K spectra (ESI Section S11†) are found in the ESI.†
Over the experiment duration, we observed increased noise in Mössbauer spectra from incubated 57Fe-mineral-soil mixes (compare spectra of initial and incubated mineral-soil mixes in Fig. 4C and D). The results from aqua regia digestions of the 57Fe-mineral-soil mixes showed decreasing Fe contents and decreasing 57Fe fractions over time (Fig. S16A and B†). Based on this, we infer that the increased spectral noise was due to the reductive dissolution of 57Fe-minerals and the loss of 57Fe(II) from the mesh bags through diffusion and water flow during drainage. This is supported by the higher loss of 57Fe from 57Fe-Fh compared to 57Fe-Lp-soil mixes, which agrees with the lower stability of ferrihydrite during reductive dissolution.20 Despite the increased spectral noise, the fits were robust, and we do not expect a major change in the interpretation of the Mössbauer spectra. Regardless of the loss of 57Fe from the mesh bags, the majority of the Mössbauer signal still originated from the added 57Fe-labeled ferrihydrite (>71%) or lepidocrocite (>84%, Fig. S16C†). Nevertheless, the identification and quantification of minor fit components (<10% of fitted spectral area) should be interpreted with caution due to the spectral noise. The stated 57Fe phase fractions in the following text are derived from the fitting of Mössbauer spectra and thus represent relative fractions of the 57Fe that remained in the mesh bags. The results from aqua regia digestions of the initial and incubated 57Fe-mineral-soil mixes and the surrounding soil are presented in ESI Section S9.†
57Fe-mineral transformations in mesh bags with soil
Mineral transformations in 57Fe-Fh-soil mixes resulted in less crystalline mineral transformation products and a larger variety of Fe phases compared to NAFe-Fh samples without soil (compare Fig. 3 with Fig. 4). During the anoxic period of redox cycle I (3 weeks), the fraction of ferrihydrite in the 57Fe-Fh-soil mix decreased from 100% to 37% of total 57Fe, while the fraction of lepidocrocite (24%) and the mixed-valence disordered Fe phase (26%) increased (Fig. 4A). Additionally, small fractions (each ≤8%) of adsorbed/clay-associated Fe(II) and organically-complexed/silicate Fe(III) formed. Upon the drainage of the soil, the fraction of lepidocrocite (32%) increased and the fraction of the mixed-valence disordered Fe phase (19%) decreased. The fraction of ferrihydrite remained relatively stable (39%). Overall, these results indicate that the 57Fe-Fh was reductively dissolved and partially transformed to lepidocrocite and the mixed-valence disordered Fe phase during the first anoxic period. During the subsequent oxic period, part of the adsorbed Fe(II) and Fe(II) in the mixed-valence disordered Fe phase was oxidized and precipitated as lepidocrocite or ferrihydrite. The formation of lepidocrocite and the mixed-valence disordered Fe phase in the 57Fe-Fh-soil mixes contrasts the results from NAFe-Fh samples without soil, where goethite and magnetite formed.In the following redox cycles (II and III), the fractions of Fe(II)-bearing phases (adsorbed/clay-associated Fe(II), mixed-valence disordered Fe phase) in the 57Fe-Fh-soil mixes increased during anoxic and decreased during oxic periods. Over the three redox cycles, the contribution of Fe(II)-bearing phases in 57Fe-Fh-soil mixes generally increased (cf. fractions of Fe(II) and the mixed-valence disordered Fe phase in anoxic periods of redox cycles I–III, Fig. 4A). As opposed to trends in Fe(II)-bearing phases, the fractions of Fe(III)-phases (ferrihydrite, lepidocrocite, organically-complexed/silicate-associated Fe(III)) decreased during anoxic and increased during oxic periods, with most pronounced changes for lepidocrocite fractions. Lepidocrocite in the 57Fe-Fh-soil mixes disappeared quickly during the anoxic periods in redox cycles II and III, indicating that the newly formed lepidocrocite was highly susceptible to reductive dissolution. The fast reductive dissolution of lepidocrocite was likely promoted by coprecipitation with other dissolved components in the porewater, which can lead to a decreased crystallinity of lepidocrocite.70,71
In 57Fe-Lp-soil mixes, the lepidocrocite fraction decreased from 84% to 25% during the anoxic period of redox cycle I (3 weeks, Fig. 4B). In contrast to magnetite formation in NAFe-Lp mesh bags without soil, the main mineral transformation products in the 57Fe-Lp-soil mixes were ferrihydrite (35%) and goethite (25%). The nano-goethite that was detected in initial 57Fe-Lp (ESI Section S2†) was not found in the incubated 57Fe-Lp-soil mixes. Also, small contributions (each ≤7%) from adsorbed/clay-associated Fe(II), organically-complexed/silicate-associated Fe(III) and the mixed-valence disordered Fe phase were present. Upon the soil drainage in redox cycle I, the oxidation state and speciation of 57Fe atoms in 57Fe-Lp-soil mixes were relatively stable. However, in the following redox cycles (II and III), fractions of the Fe(II)-bearing phases (adsorbed/clay-associated Fe(II), mixed-valence disordered Fe phase) increased slightly during anoxic periods and decreased during oxic periods, but changes were smaller compared to the 57Fe-Fh-soil mixes. The contribution of lepidocrocite in 57Fe-Lp-soil mixes generally decreased over the redox cycles and had almost completely disappeared (≤6%) at the end of redox cycle III (12 weeks, Fig. 4D). In contrast to lepidocrocite fractions, the goethite fraction increased during anoxic periods and remained stable during oxic periods. This led to a general increase of the goethite fraction over the redox cycles, with goethite accounting for up to 45% of 57Fe at the end of redox cycle III (12 weeks). This suggests that lepidocrocite and ferrihydrite in the 57Fe-Lp-soil mixes were preferentially reductively dissolved during anoxic periods, promoting the formation of more goethite. Goethite present in the soil or nano-goethite in initial 57Fe-Lp may have additionally supported the formation of goethite in these samples.63,72 In contrast to magnetite accumulation in the NAFe-mineral mesh bags without soil, no indication of magnetite was found in the 57Fe-mineral-soil mixes. The absence of magnetite in incubated 57Fe-mineral-soil mixes was further tested by collecting room temperature Mössbauer spectra, where magnetite would form two sextets.61,73 No sextet was present in the room temperature Mössbauer spectra of 57Fe-Fh and 57Fe-Lp-soil mixes (redox cycle III, oxic, Fig. S22†).
The fraction of the mixed-valence disordered Fe phase increased during anoxic periods in both the 57Fe-Fh and 57Fe-Lp-soil mixes. During oxic periods, most of the Fe(II) in the mixed-valence disordered Fe phase was oxidized and its fraction decreased. Especially in 57Fe-Fh-soil mixes, fractions of the mixed-valence disordered Fe phase showed drastic changes between anoxic and oxic periods (Fig. 4A). The formation of the mixed-valence disordered Fe phase in the 57Fe-mineral-soil mixes during anoxic periods was likely related to the close association of Fe minerals with the soil matrix. This likely hindered the recrystallization of ferrihydrite and lepidocrocite through the direct exposure to, for example, DOC and dissolved Si and P. Assuming a dissolution-reprecipitation mechanism for mineral recrystallization,63,74,75 these findings agree with the hindered crystallization of Fe oxyhydroxides in the presence of organic matter,76 Si,77–79 and P.57,71,78 Hindered mineral recrystallization likely also supported ferrihydrite formation in 57Fe-Lp samples. Similarly, a recent study that incubated 57Fe-labeled ferrihydrite in soil microcosms reported ferrihydrite transformation to a green rust-like phase with no sign of lepidocrocite or goethite formation.33 Despite the use of a similar soil as in the study of Notini et al.,33 in the present study, no green rust was found, which may be related to the lower pH80 (pH 6.9 in this study, compared to pH 7.6 in Notini et al.33). However, we cannot exclude small contributions of green rust in the collapsed feature observed in Mössbauer spectra.
The changes in solid-phase 57Fe speciation in the 57Fe-Fh and 57Fe-Lp-soil mixes induced by the onset of anoxic and oxic soil conditions became larger with every redox cycle. For example, the fractions of Fe(II)-bearing phases (adsorbed/clay-associated Fe(II), mixed-valence disordered Fe phase) during anoxic periods increased with every redox cycle (Fig. 4A and B). The increasingly reduced state of solid-associated 57Fe in 57Fe-Fh and 57Fe-Lp samples after incubation observed in 5 K Mössbauer spectra was also supported by 77 K spectra collected from anoxic samples. In 77 K spectra, the fraction of a doublet (CS = 1.29 mm s−1;quadrupole splitting, QS = 2.62 mm s−1), which corresponds to Fe(II),45 increased with every redox cycle up to 76% and up to 32% of total 57Fe in 57Fe-Fh- and 57Fe-Lp-soil mixes, respectively (redox cycle III, anoxic period, Fig. S20†). This can likely be explained by the precipitation of more easily reducible Fe phases during the oxic phases, facilitating faster microbial Fe reduction during the subsequent anoxic periods.39,81 The increasing fractions of Fe(II)-bearing phases and the increasingly pronounced changes in the 57Fe oxidation state demonstrate that the susceptibility of Fe phases to reductive dissolution increased with repeated soil redox cycles.
Studies using soils from contrasting locations found that fractions of short-range-ordered Fe phases can increase34,35,37 or decrease35,37,38 during soil redox fluctuations. For example, Thompson et al.38 studied highly weathered tropical forest soils, where Fe mineralogy was dominated by nano- and micro-crystalline goethite (determined by Mössbauer spectroscopy) and found increasing Fe crystallinity after multiple complete redox cycles (4 cycles within 8 weeks). In comparison, ThomasArrigo and Kretzschmar36 investigated organic matter-rich soils from Iceland, where ferrihydrite, goethite, and some lepidocrocite were the main Fe mineral phases (determined by Fe K-edge XAS) and reported similar Fe mineral composition before and after the exposure to one complete redox cycle with varying duration (1–5 weeks anoxic, 2–8 days oxic). These studies shed light on the redox transformation of native soil minerals, which included multiple Fe phases. With the use of 57Fe-labeled minerals, the present study particularly demonstrated the transformation of ferrihydrite and lepidocrocite during three redox cycles in a sandy and weakly acidic paddy soil which was poor in organic matter. The outcomes of this study suggest that the stability of Fe oxyhydroxides during mineral transformation and reductive dissolution strongly impact mineral transformations towards lower or higher crystallinity during soil redox fluctuations.
Mineral transformations in the permanently flooded mesocosm
For both the NAFe-mineral samples (Fig. 5A) and the 57Fe-mineral-soil mixes (Fig. 5B), the same mineral phases formed from minerals incubated in the permanently flooded (12 weeks) and the redox fluctuating mesocosms (redox cycle III, anoxic period). However, the relative fractions of Fe phases varied. The NAFe-Fh in the permanently flooded mesocosm transformed to similar amounts of goethite (48%) but more magnetite (46%) compared to NAFe-Fh in the redox fluctuating mesocosms (51% goethite, 32% magnetite, Fig. 5A). Thus, the transformation extent of NAFe-Fh was higher when incubated under permanently flooded conditions. Similarly, for NAFe-Lp, the incubation in the permanently flooded mesocosm resulted in a larger extent of lepidocrocite transformation and higher magnetite fractions (30%) compared to the redox fluctuating mesocosms (11% magnetite).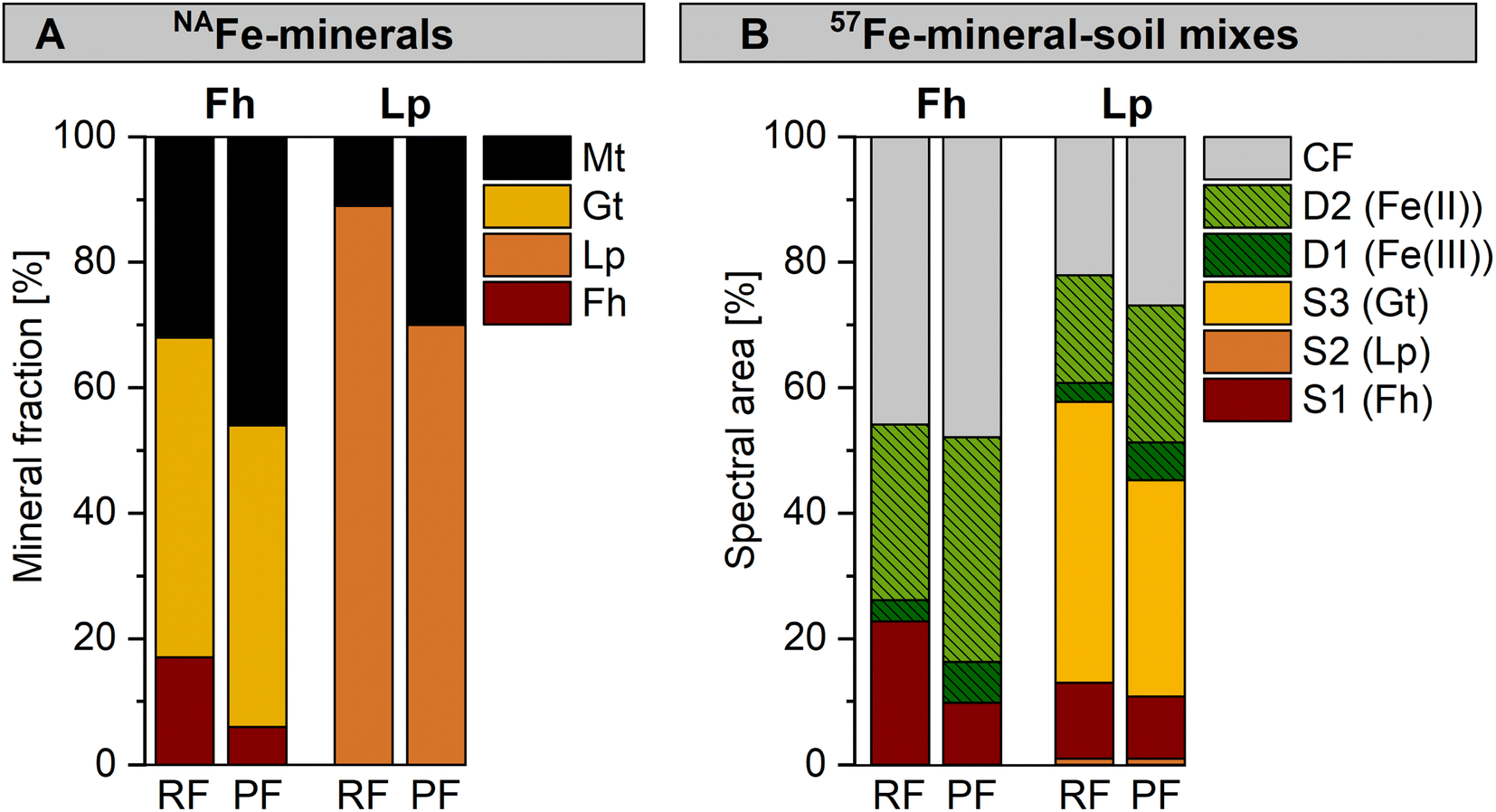 | ||
| Fig. 5 Comparison between samples incubated in the redox fluctuating (RF, anoxic period of redox cycle III) and permanently flooded (PF, 12 weeks) mesocosms with fractions of fitted components in (A) Fe K-edge EXAFS spectra collected from NAFe-minerals and (B) Mössbauer spectra collected from 57Fe-mineral-soil mixes at 5 K, with results for ferrihydrite (Fh) and lepidocrocite (Lp). Abbreviations: S = sextet, D = doublet, CF = collapsed feature, Gt = goethite, Mt = magnetite. Interpretation of Mössbauer fit components for panel B are presented in Table 1. | ||
For 57Fe-mineral-soil mixes, Mössbauer spectroscopy showed that in the 57Fe-Fh-soil mixes, the ferrihydrite fraction was slightly lower after the incubation in the permanently flooded mesocosm (10%, 12 weeks) compared to the redox fluctuating mesocosms (23%, redox cycle III, anoxic period, Fig. 5B). In turn, the solid-associated Fe(II) fraction was slightly higher after the incubation under permanently flooded conditions (36% in permanently flooded, 28% in redox fluctuating mesocosms). This may reflect the regular precipitation of short-range-ordered Fe phases during oxic periods in the redox fluctuating mesocosms. In 57Fe-Lp-soil mixes, slightly less goethite formed when incubated under permanently flooded conditions (34%) compared to redox fluctuating conditions (45%). Thus, the intermittent oxic periods may have supported goethite formation in 57Fe-Lp-soil mixes.
The differences in overall mineral transformations in 57Fe-mineral-soil mixes after the incubation in the permanently flooded or the redox fluctuating mesocosms were smaller than in NAFe-mineral mesh bags without soil. The higher transformation extent in NAFe-mineral mesh bags without soil incubated in the permanently flooded mesocosm is likely related to the continuous supply of Fe(II) from the Fe reducing soil matrix. In contrast, the Fe(II) supply to the NAFe-minerals in the mesh bags in the redox fluctuating mesocosms was likely interrupted during oxic drained periods, stalling mineral transformations. Since the minerals in the 57Fe-mineral-soil mixes were exposed to in situ reductive dissolution, the short-range-ordered Fe phases that formed during oxic periods likely were quickly reductively dissolved upon the onset of Fe reducing conditions. Accordingly, the drastic shifts in Fe crystallinity that occurred during oxic periods were largely reversed during the following anoxic periods. This allowed the progression of mineral transformations to continue as though the oxic interruption had not occurred.
The effect of close association with the soil on mineral transformations
Iron oxyhydroxide dynamics in soils are controlled by a balance between microbial reductive dissolution and mineral transformation or recrystallization, which determines the fate of the mineral. Our study demonstrates how this balance is impacted by the close association between minerals and the soil matrix and by initial mineral crystallinity. During the incubation of NAFe-mineral mesh bags without soil, magnetite and/or goethite were formed (Fig. 2 and 3). In contrast, when Fe minerals were closely associated with the soil in 57Fe-mineral-soil mixes, minerals were reductively dissolved while mineral transformation was hindered. The hindered mineral transformation resulted in the formation of a mixed-valence disordered Fe phase (Fig. 4), the formation of which was promoted by fast reductive dissolution. This is supported by the fact that large fractions of the mixed-valence disordered Fe phase formed from poorly crystalline ferrihydrite in the 57Fe-Fh-soil mixes (up to 46%, Fig. 4A). The higher stability of lepidocrocite during reductive dissolution20 likely led to slower mineral dissolution in 57Fe-Lp-soil mixes. Thus, the accumulation of the mixed-valence disordered Fe phase was lower (up to 22%) and goethite formed (Fig. 4B). It is likely that the minerals in close contact with soil in the mineral-soil mixes were exposed to geochemical conditions more similar to those measured in the pore water outside the mesh bags compared to the geochemical conditions within the pure mineral mesh bags. As such, it is likely that the mineral transformations in the mineral soil mixes were strongly influenced by the presence of dissolved DOC, Fe and P, which ultimately hindered the transformation to crystalline mineral products. The formation of the mixed-valence disordered Fe phase in 57Fe-mineral-soil mixes suggests that similar highly disordered and reactive Fe phases may also occur in natural soils.82Conclusions
This study investigated the transformation of Fe(III) oxyhydroxides during three soil redox cycles and the role of close contact between minerals and the soil. The transformation and recrystallization of NAFe-minerals in mesh bags without soil were likely mainly catalyzed by Fe(II) from the surrounding soil diffusing into the mesh bags and resulted in goethite and/or magnetite formation. Since Fe minerals in soils are part of the complex soil matrix, the results from 57Fe-mineral-soil mixes may be more environmentally relevant. The presented findings highlight the complex interplay of biogeochemical factors directing Fe mineral transformations in redox-active soils. During the incubation as mineral-soil mixes, ferrihydrite was less stable during mineral transformation and reductive dissolution, compared to lepidocrocite. Thus, ferrihydrite was more strongly affected by redox cycles. Further, mixing ferrihydrite with soil hindered mineral transformation compared to ferrihydrite samples that were not mixed with soil. For lepidocrocite, the close association with the soil enabled, and potentially even promoted, the transformation of lepidocrocite to goethite. Further, the close association of minerals with the soil promoted the formation of a highly disordered mixed-valence Fe(II)–Fe(III) phase. This was likely through a combination of in situ Fe reduction, Fe(II)-catalyzed mineral recrystallization and interactions with other dissolved soil components. It is possible that such highly disordered Fe phases serve as highly reactive sorbents for trace elements in paddy soils, impacting the availability of these elements for the uptake by the rice plants.With repeated redox cycles, the production of solid-associated Fe(II) during anoxic periods increased likely due to the recurring formation of easily reducible short-range-ordered Fe phases during oxic periods. This highlights how the susceptibility of redox-affected soils to Fe reduction is influenced by the crystallinity of Fe phases in the soil and if or how frequently the soil has undergone redox fluctuations recently. Despite drastic changes in Fe crystallinity between anoxic and oxic periods, the redox cycling itself may only have a small impact on overall mineral transformations compared with permanent flooding of the same soil. In this respect, also the activity and composition of microbial communities and the availability of carbon sources need to be considered. Regarding coupled element cycles, the recurring formation of short-range-ordered Fe phases during redox cycles can provide a large sorption potential for trace elements. However, with repeated redox cycles the increased susceptibility of Fe to reductive dissolution during anoxic periods comes with an increasing risk of releasing mineral-associated elements to the porewater. Thus, the duration and frequency of flooded periods in paddy fields may impact the abundance of short-range-ordered Fe oxyhydroxides, their stability under Fe-reducing conditions and their potential to release associated trace-elements. These outcomes contribute to an improved understanding of Fe dynamics and potential implications for coupled element cycles during soil redox cycles.
Author contributions
Katrin Schulz: conceptualization, methodology, validation, investigation, formal analysis, writing – original draft, visualization; Luiza Notini: formal analysis, writing – review & editing; Andrew R. C. Grigg: methodology, writing – review & editing; L. Joëlle Kubeneck: investigation, writing – review & editing; Worachart Wisawapipat: methodology, writing – review & editing; Laurel K. ThomasArrigo: conceptualization, methodology, writing – review & editing, supervision; Ruben Kretzschmar: conceptualization, methodology, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are grateful to Kurt Barmettler (ETH Zürich) for assisting with the soil sampling and laboratory analyses. We further thank the staff of the Ubon Ratchathani Rice Research Center for their support during the soil sampling, and the NRCT for granting the research permit (permit no. no. 0002/1164). Our thanks also goes to SOL-CONSEIL (Gland, France) for conducting the soil texture analysis, and to the MoBiAS laboratory (ETH Zürich, Switzerland), especially to Peter Kälin, for the analysis of carbon contents in NAFe-mineral samples. We acknowledge SOLEIL (proposal no. 20221355) for the provision of synchrotron radiation facilities and thank Gautier Landrot (SAMBA) for his support during the synchrotron measurements. This work received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 788009-IRMIDYN-ERC-2017-ADG).References
- D. R. Lovley and E. J. P. Phillips, Novel mode of microbial energy metabolism: organic carbon oxidation coupled to dissimilatory reduction of iron or manganese, Appl. Environ. Microbiol., 1988, 54, 1472–1480 CrossRef CAS PubMed.
- D. R. Lovley, J. D. Coates, E. L. Blunt-Harris, E. J. P. Phillips and J. C. Woodward, Humic substances as electron acceptors for microbial respiration, Nature, 1996, 382, 445–448 CrossRef CAS.
- F. A. Weber, A. F. Hofacker, A. Voegelin and R. Kretzschmar, Temperature dependence and coupling of iron and arsenic reduction and release during flooding of a contaminated soil, Environ. Sci. Technol., 2010, 44, 116–122 CrossRef CAS PubMed.
- N. Sharma, Z. Wang, J. G. Catalano and D. E. Giammar, Dynamic responses of trace metal bioaccessibility to fluctuating redox conditions in wetland soils and stream sediments, ACS Earth Space Chem., 2022, 6, 1331–1344 CrossRef CAS.
- W. H. Patrick and R. A. Khalid, Phosphate release and sorption by soils and sediments: Effect of aerobic and anaerobic conditions, Science, 1974, 186, 53–55 CrossRef CAS PubMed.
- G. Lee, J. M. Bigham and G. Faure, Removal of trace metals by coprecipitation with Fe, Al and Mn from natural waters contaminated with acid mine drainage in the Ducktown Mining District, Tennessee, Appl. Geochem., 2002, 17, 569–581 CrossRef CAS.
- M. Sodano, C. Lerda, R. Nisticò, M. Martin, G. Magnacca, L. Celi and D. Said-Pullicino, Dissolved organic carbon retention by coprecipitation during the oxidation of ferrous iron, Geoderma, 2017, 307, 19–29 CrossRef CAS.
- C. P. Szilas, O. K. Borggaard and H. C. B. Hansen, Potential iron and phosphate mobilization during flooding of soil material, Water, Air, Soil Pollut., 1998, 106, 97–109 CrossRef CAS.
- I. Kögel-Knabner, W. Amelung, Z. H. Cao, S. Fiedler, P. Frenzel, R. Jahn, K. Kalbitz, A. Kölbl and M. Schloter, Biogeochemistry of paddy soils, Geoderma, 2010, 157, 1–14 CrossRef.
- L. C. Roberts, S. J. Hug, A. Voegelin, J. Dittmar, R. Kretzschmar, B. Wehrli, G. C. Saha, A. B. M. Badruzzaman and M. A. Ali, Arsenic dynamics in porewater of an intermittently irrigated paddy field in Bangladesh, Environ. Sci. Technol., 2011, 45, 971–976 CrossRef CAS PubMed.
- A. G. B. Williams and M. M. Scherer, Spectroscopic evidence for Fe(II)-Fe(III) electron transfer at the iron oxide-water interface, Environ. Sci. Technol., 2004, 38, 4782–4790 CrossRef CAS PubMed.
- C. M. Hansel, S. G. Benner and S. Fendorf, Competing Fe(II)-induced mineralization pathways of ferrihydrite, Environ. Sci. Technol., 2005, 39, 7147–7153 CrossRef CAS PubMed.
- H. D. Pedersen, D. Postma, R. Jakobsen and O. Larsen, Fast transformation of iron oxyhydroxides by the catalytic action of aqueous Fe(II), Geochim. Cosmochim. Acta, 2005, 69, 3967–3977 CrossRef CAS.
- K. Schulz, L. K. ThomasArrigo, R. Kaegi and R. Kretzschmar, Stabilization of ferrihydrite and lepidocrocite by silicate during Fe(II)-catalyzed mineral transformation: Impact on particle morphology and silicate distribution, Environ. Sci. Technol., 2022, 56, 5929–5938 CrossRef CAS PubMed.
- Y. Tamaura, K. Ito and T. Katsura, Transformation of γ-FeO(OH) to Fe304 by adsorption of iron(II) on γ-FeO(OH), J. Chem. Soc., Dalton Trans., 1983, 189–194 RSC.
- W. Yan, H. Liu, R. Chen, J. Xie and Y. Wei, Dissolution and oriented aggregation: Transformation from lepidocrocite to goethite by the catalysis of aqueous Fe(II), RSC Adv., 2015, 5, 106396–106399 RSC.
- D. D. Boland, R. N. Collins, C. J. Miller, C. J. Glover and T. D. Waite, Effect of solution and solid-phase conditions on the Fe(II)-accelerated transformation of ferrihydrite to lepidocrocite and goethite, Environ. Sci. Technol., 2014, 48, 5477–5485 CrossRef CAS PubMed.
- M. Aeppli, R. Kaegi, R. Kretzschmar, A. Voegelin, T. B. Hofstetter and M. Sander, Electrochemical analysis of changes in iron oxide reducibility during abiotic ferrihydrite transformation into goethite and magnetite, Environ. Sci. Technol., 2019, 53, 3568–3578 CrossRef CAS PubMed.
- H. Liu, P. Li, M. Zhu, Y. Wei and Y. Sun, Fe(II)-induced transformation from ferrihydrite to lepidocrocite and goethite, J. Solid State Chem., 2007, 180, 2121–2128 CrossRef CAS.
- E. J. O'Loughlin, M. I. Boyanov, C. A. Gorski, M. M. Scherer and K. M. Kemner, Effects of Fe(III) oxide mineralogy and phosphate on Fe(II) secondary mineral formation during microbial iron reduction, Minerals, 2021, 11, 1–43 Search PubMed.
- C. M. Hansel, S. G. Benner, J. Neiss, A. Dohnalkova, R. K. Kukkadapu and S. Fendorf, Secondary mineralization pathways induced by dissimilatory iron reduction of ferrihydrite under advective flow, Geochim. Cosmochim. Acta, 2003, 67, 2977–2992 CrossRef CAS.
- R. S. Cutting, V. S. Coker, J. W. Fellowes, J. R. Lloyd and D. J. Vaughan, Mineralogical and morphological constraints on the reduction of Fe(III) minerals by Geobacter sulfurreducens, Geochim. Cosmochim. Acta, 2009, 73, 4004–4022 CrossRef CAS.
- L. K. ThomasArrigo, J. M. Byrne, A. Kappler and R. Kretzschmar, Impact of organic matter on iron(II)-catalyzed mineral transformations in ferrihydrite–organic matter coprecipitates, Environ. Sci. Technol., 2018, 52, 12316–12326 CrossRef CAS PubMed.
- C. Chen, R. K. Kukkadapu and D. L. Sparks, Influence of coprecipitated organic matter on Fe2+(aq)-catalyzed transformation of ferrihydrite: Implications for carbon dynamics, Environ. Sci. Technol., 2015, 49, 10927–10936 CrossRef CAS PubMed.
- C. Chen and D. L. Sparks, Fe(II)-induced mineral transformation of ferrihydrite-organic matter adsorption and co-precipitation complexes in the absence and presence of As(III), ACS Earth Space Chem., 2018, 2, 1095–1101 CrossRef CAS.
- J. M. Zachara, R. K. Kukkadapu, J. K. Fredrickson, Y. A. Gorby and S. C. Smith, Biomineralization of poorly crystalline Fe(III) oxides by dissimilatory metal reducing bacteria (DMRB), Geomicrobiol. J., 2002, 19, 179–207 CrossRef CAS.
- A. M. Jones, R. N. Collins, J. Rose and T. D. Waite, The effect of silica and natural organic matter on the Fe(II)-catalysed transformation and reactivity of Fe(III) minerals, Geochim. Cosmochim. Acta, 2009, 73, 4409–4422 CrossRef CAS.
- T. Borch, Y. Masue, R. K. Kukkadapu and S. Fendorf, Phosphate imposed limitations on biological reduction and alteration of ferrihydrite, Environ. Sci. Technol., 2007, 41, 166–172 CrossRef CAS PubMed.
- Y. Masue-Slowey, R. H. Loeppert and S. Fendorf, Alteration of ferrihydrite reductive dissolution and transformation by adsorbed As and structural Al: Implications for As retention, Geochim. Cosmochim. Acta, 2011, 75, 870–886 CrossRef CAS.
- A. R. C. Grigg, L. K. ThomasArrigo, K. Schulz, K. A. Rothwell, R. Kaegi and R. Kretzschmar, Ferrihydrite transformations in flooded paddy soils: rates, pathways, and product spatial distributions, Environ. Sci.: Processes Impacts, 2022, 24, 1867–1882 RSC.
- V. Vogelsang, S. Fiedler, R. Jahn and K. Kaiser, In-situ transformation of iron-bearing minerals in marshland-derived paddy subsoil, Eur. J. Soil Sci., 2016, 67, 676–685 CrossRef CAS.
- P. Kraal, C. M. Van Genuchten, W. K. Lenstra and T. Behrends, Coprecipitation of phosphate and silicate affects environmental iron (oxyhydr)oxide transformations: A gel-based diffusive sampler approach, Environ. Sci. Technol., 2020, 54, 12795–12802 CrossRef CAS PubMed.
- L. Notini, K. Schulz, L. J. Kubeneck, A. R. C. Grigg, K. A. Rothwell, G. Fantappiè, L. K. ThomasArrigo and R. Kretzschmar, A new approach for investigating iron mineral transformations in soils and sediments using 57Fe-labeled minerals and 57Fe Mössbauer spectroscopy, Environ. Sci. Technol., 2023, 57, 10008–10018 CrossRef CAS PubMed.
- V. Vogelsang, K. Kaiser, F. E. Wagner, R. Jahn and S. Fiedler, Transformation of clay-sized minerals in soils exposed to prolonged regular alternation of redox conditions, Geoderma, 2016, 278, 40–48 CrossRef CAS.
- A. Thompson, D. G. Rancourt, O. A. Chadwick and J. Chorover, Iron solid-phase differentiation along a redox gradient in basaltic soils, Geochim. Cosmochim. Acta, 2011, 75, 119–133 CrossRef CAS.
- L. K. ThomasArrigo and R. Kretzschmar, Iron speciation changes and mobilization of colloids during redox cycling in Fe-rich, Icelandic peat soils, Geoderma, 2022, 428, 116217 CrossRef CAS.
- P. Winkler, K. Kaiser, A. Thompson, K. Kalbitz, S. Fiedler and R. Jahn, Contrasting evolution of iron phase composition in soils exposed to redox fluctuations, Geochim. Cosmochim. Acta, 2018, 235, 89–102 CrossRef CAS.
- A. Thompson, O. A. Chadwick, D. G. Rancourt and J. Chorover, Iron-oxide crystallinity increases during soil redox oscillations, Geochim. Cosmochim. Acta, 2006, 70, 1710–1727 CrossRef CAS.
- B. R. Ginn, C. Meile, J. Wilmoth, Y. Tang and A. Thompson, Rapid iron reduction rates are stimulated by high-amplitude redox fluctuations in a tropical forest soil, Environ. Sci. Technol., 2017, 51, 3250–3259 CrossRef CAS PubMed.
- S. C. Ying, Y. Masue-Slowey, B. D. Kocar, S. D. Griffis, S. Webb, M. A. Marcus, C. A. Francis and S. Fendorf, Distributed microbially- and chemically-mediated redox processes controlling arsenic dynamics within Mn-/Fe-oxide constructed aggregates, Geochim. Cosmochim. Acta, 2013, 104, 29–41 CrossRef CAS.
- C. Pallud, Y. Masue-Slowey and S. Fendorf, Aggregate-scale spatial heterogeneity in reductive transformation of ferrihydrite resulting from coupled biogeochemical and physical processes, Geochim. Cosmochim. Acta, 2010, 74, 2811–2825 CrossRef CAS.
- K. U. Totsche, W. Amelung, M. H. Gerzabek, G. Guggenberger, E. Klumpp, C. Knief, E. Lehndorff, R. Mikutta, S. Peth, A. Prechtel, N. Ray and I. Kögel-Knabner, Microaggregates in soils, J. Plant Nutr. Soil Sci., 2018, 181, 104–136 CrossRef CAS.
- E. Jasinska, H. Wetzel, T. Baumgartl and R. Horn, Heterogeneity of physico-chemical properties in structured soils and its consequences, Pedosphere, 2006, 16, 284–296 CrossRef CAS.
- C. W. Mueller, C. Hoeschen, M. Steffens, H. Buddenbaum, K. Hinkel, J. G. Bockheim and J. Kao-Kniffin, Microscale soil structures foster organic matter stabilization in permafrost soils, Geoderma, 2017, 293, 44–53 CrossRef CAS.
- C. Chen and A. Thompson, The influence of native soil organic matter and minerals on ferrous iron oxidation, Geochim. Cosmochim. Acta, 2021, 292, 254–270 CrossRef CAS.
- V. Tishchenko, C. Meile, M. M. Scherer, T. S. Pasakarnis and A. Thompson, Fe2+ catalyzed iron atom exchange and re-crystallization in a tropical soil, Geochim. Cosmochim. Acta, 2015, 148, 191–202 CrossRef CAS.
- Food and Agriculture Organization of the United Nations, World Reference Base for Soil Resources 2014: International Soil Classification Systems for Naming Soils and Creating Legends for Soil Maps (Update 2015), 2014 Search PubMed.
- U. Schwertmann and R. M. Cornell, Iron Oxides in the Laboratory: Synthesis and Preparation, John Wiley & Sons, 1991 Search PubMed.
- B. Ravel and M. Newville, ATHENA , ARTEMIS , HEPHAESTUS : Data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- D. L. Bish and S. A. Howard, Quantitative phase analysis using the Rietveld method, J. Appl. Crystallogr., 1988, 21, 86–91 CrossRef CAS.
- N. V. Y. Scarlett and I. C. Madsen, Quantification of phases with partial or no known crystal structures, Powder Diffr., 2006, 21, 278–284 CrossRef CAS.
- P. D. P. Taylor, R. Maeck and P. De Bièvre, Determination of the absolute isotopic composition and Atomic Weight of a reference sample of natural iron, Int. J. Mass Spectrom. Ion Processes, 1992, 121, 111–125 CrossRef CAS.
- D. G. Rancourt and J. Y. Ping, Voigt-based methods for arbitrary-shape static hyperfine parameter distributions in Mössbauer spectroscopy, Nucl. Instrum. Methods Phys. Res., Sect. B, 1991, 58, 85–97 CrossRef.
- S. Fiedler, M. J. Vepraskas and J. L. Richardson, in Advances in Agronomy, Elsevier Masson SAS, 2007, vol. 94, pp. 1–54 Search PubMed.
- J. C. Mendez and T. Hiemstra, High and low affinity sites of ferrihydrite for metal ion adsorption: Data and modeling of the alkaline-earth ions Be, Mg, Ca, Sr, Ba, and Ra, Geochim. Cosmochim. Acta, 2020, 286, 289–305 CrossRef CAS.
- J. Chorover and M. K. Amistadi, Reaction of forest floor organic matter at goethite, birnessite and smectite surfaces, Geochim. Cosmochim. Acta, 2001, 65, 95–109 CrossRef CAS.
- P. Kraal, C. M. van Genuchten, T. Behrends and A. L. Rose, Sorption of phosphate and silicate alters dissolution kinetics of poorly crystalline iron (oxyhydr)oxide, Chemosphere, 2019, 234, 690–701 CrossRef CAS PubMed.
- P. J. Swedlund and J. G. Webster, Adsorption and polymerisation of silicic acid on ferrihydrite, and its effect on arsenic adsorption, Water Res., 1999, 33, 3413–3422 CrossRef CAS.
- P. J. Swedlund, R. D. Hamid and G. M. Miskelly, Insights into H4SiO4 surface chemistry on ferrihydrite suspensions from ATR-IR, Diffuse Layer Modeling and the adsorption enhancing effects of carbonate, J. Colloid Interface Sci., 2010, 352, 149–157 CrossRef CAS PubMed.
- P. J. Swedlund, G. M. Miskelly and A. J. McQuillan, Silicic acid adsorption and oligomerization at the ferrihydrite - water interface: Interpretation of ATR-IR spectra based on a model surface structure, Langmuir, 2010, 26, 3394–3401 CrossRef CAS PubMed.
- A. C. Doriguetto, N. G. Fernandes, A. I. C. Persiano, E. N. Filho, J. M. Grenèche and J. D. Fabris, Characterization of a natural magnetite, Phys. Chem. Miner., 2003, 30, 249–255 CrossRef CAS.
- C. A. Gorski and M. M. Scherer, Determination of nanoparticulate magnetite stoichiometry by Mössbauer spectroscopy, acidic dissolution, and powder X-ray diffraction: A critical review, Am. Mineral., 2010, 95, 1017–1026 CrossRef CAS.
- J. Liu, A. Sheng, X. Li, Y. Arai, Y. Ding, M. Nie, M. Yan and K. M. Rosso, Understanding the importance of labile Fe(III) during Fe(II)-catalyzed transformation of metastable iron oxyhydroxides, Environ. Sci. Technol., 2022, 56(6), 3801–3811 CrossRef CAS PubMed.
- T. Borch and S. Fendorf, Phosphate interactions with iron (hydr)oxides: Mineralization pathways and phosphorus retention upon bioreduction, Dev. Earth Environ. Sci., 2007, vol. 7, pp. 321–348 Search PubMed.
- Z. Zhou, D. E. Latta, N. Noor, A. Thompson, T. Borch and M. M. Scherer, Fe(II)-catalyzed transformation of organic matter-ferrihydrite coprecipitates: A closer look using Fe isotopes, Environ. Sci. Technol., 2018, 52, 11142–11150 CrossRef CAS PubMed.
- C. M. Hansel, S. G. Benner, P. Nico and S. Fendorf, Structural constraints of ferric (hydr)oxides on dissimilatory iron reduction and the fate of Fe(II), Geochim. Cosmochim. Acta, 2004, 68, 3217–3229 CrossRef CAS.
- J. M. Byrne and A. Kappler, A revised analysis of ferrihydrite at liquid helium temperature using Mössbauer spectroscopy, Am. Mineral., 2022, 107, 1643–1651 CrossRef.
- J. M. Byrne and A. Kappler, in Analytical Geomicrobiology: A Handbook of Instrumental Techniques, Cambridge University Press, Cambridge, 2019, pp. 314–338 Search PubMed.
- J. Thiel, J. M. Byrne, A. Kappler, B. Schink and M. Pester, Pyrite formation from FeS and H2S is mediated through microbial redox activity, Proc. Natl. Acad. Sci., 2019, 116, 6897–6902 CrossRef CAS PubMed.
- A. C. Senn, R. Kaegi, S. J. Hug, J. G. Hering, S. Mangold and A. Voegelin, Composition and structure of Fe(III)-precipitates formed by Fe(II) oxidation in water at near-neutral pH: Interdependent effects of phosphate, silicate and Ca, Geochim. Cosmochim. Acta, 2015, 162, 220–246 CrossRef CAS.
- R. Kaegi, A. Voegelin, D. Folini and S. J. Hug, Effect of phosphate, silicate, and Ca on the morphology, structure and elemental composition of Fe(III)-precipitates formed in aerated Fe(II) and As(III) containing water, Geochim. Cosmochim. Acta, 2010, 74, 5798–5816 CrossRef CAS.
- L. Notini, L. K. ThomasArrigo, R. Kaegi and R. Kretzschmar, Coexisting goethite promotes Fe(II)-catalyzed transformation of ferrihydrite to goethite, Environ. Sci. Technol., 2022, 56, 12723–12733 CrossRef CAS PubMed.
- J. M. Byrne, N. Klueglein, C. Pearce, K. M. Rosso, E. Appel and A. Kappler, Redox cycling of Fe(II) and Fe(III) in magnetite by Fe-metabolizing bacteria, Science, 2015, 347, 1473–1476 CrossRef CAS PubMed.
- A. Sheng, J. Liu, X. Li, L. Luo, Y. Ding, C. Chen, X. Zhang, C. Wang and K. M. Rosso, Labile Fe(III) supersaturation controls nucleation and properties of product phases from Fe(II)-catalyzed ferrihydrite transformation, Geochim. Cosmochim. Acta, 2021, 309, 272–285 CrossRef CAS.
- O. Qafoku, L. Kovarik, M. Bowden, E. Nakouzi, A. Sheng, J. Liu, C. I. Pearce and K. M. Rosso, Nanoscale Observations of Fe(II)-Induced Ferrihydrite Transformation, Environ. Sci. Nano, 2020, 7–16 Search PubMed.
- K. Eusterhues, F. E. Wagner, W. Häusler, M. Hanzlik, H. Knicker, K. U. Totsche, I. Kögel-Knabner and U. Schwertmann, Characterization of ferrihydrite-soil organic matter coprecipitates by X-ray diffraction and Mössbauer spectroscopy, Environ. Sci. Technol., 2008, 42, 7891–7897 CrossRef CAS PubMed.
- A. C. Cismasu, F. M. Michel, A. P. Tcaciuc and G. E. Brown, Properties of impurity-bearing ferrihydrite III. Effects of Si on the structure of 2-line ferrihydrite, Geochim. Cosmochim. Acta, 2014, 133, 168–185 CrossRef CAS.
- A. Voegelin, R. Kaegi, J. Frommer, D. Vantelon and S. J. Hug, Effect of phosphate, silicate, and Ca on Fe(III)-precipitates formed in aerated Fe(II)- and As(III)-containing water studied by X-ray absorption spectroscopy, Geochim. Cosmochim. Acta, 2010, 74, 164–186 CrossRef CAS.
- U. Schwertmann and H. Thalmann, The Influence of [Fe(II)], [Si], and pH on the formation of lepidocrocite and ferrihydrite during oxidation of aqueous FeCl2 solutions, Clay Miner., 1976, 11, 189–200 CrossRef CAS.
- J.-M. R. Génin, G. Bourrié, F. Trolard, M. Abdelmoula, A. Jaffrezic, P. Refait, V. Maitre, B. Humbert and A. Herbillon, Thermodynamic Equilibria in Aqueous Suspensions of Synthetic and Natural Fe(II)−Fe(III) Green Rusts: Occurrences of the Mineral in Hydromorphic Soils, Environ. Sci. Technol., 1998, 32, 1058–1068 CrossRef.
- D. Barcellos, K. T. Cyle and A. Thompson, Faster redox fluctuations can lead to higher iron reduction rates in humid forest soils, Biogeochemistry, 2018, 137, 367–378 CrossRef CAS.
- A. C. Cismasu, F. M. Michel, A. P. Tcaciuc, T. Tyliszczak and G. E. Brown, Jr, Composition and structural aspects of naturally occurring ferrihydrite, C. R. Geosci., 2011, 343, 210–218 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3em00314k |
| This journal is © The Royal Society of Chemistry 2023 |