
Mechanistic studies on catalytic alkane oxidation by Murahashi's O2/copper(II)/aldehyde system†
Kohei
Yamaguchi
,
Yuya
Uemura
 ,
Hideki
Sugimoto
,
Rin
Ito
,
Yuma
Morimoto
and
Shinobu
Itoh
*
,
Hideki
Sugimoto
,
Rin
Ito
,
Yuma
Morimoto
and
Shinobu
Itoh
*
Department of Molecular Chemistry, Division of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: shinobu@chem.eng.osaka-u.ac.jp
First published on 25th August 2023
Abstract
Mechanistic studies on catalytic alkane hydroxylation by Murahashi's O2/copper(II)/aldehyde system have been conducted to show that the autoxidation of an aldehyde (RCHO) by an O2 generating acyl radical intermediate (RC(O)˙) is involved as an initiation step of the catalytic cycle. The generated RC(O)˙ is trapped by O2 to give an acylperoxyl radical intermediate RC(O)OO˙, which may react with another RCHO to generate an adduct intermediate RC(O)OOC(R)(H)O˙. The following O–O bond homolytic cleavage of this intermediate will give acyloxyl intermediate RC(O)O˙ and RCOOH, in which the former acts as a reactive species for hydrogen atom abstraction (HAA) from alkane substrates (R1R2CH2; R1 and R2 are alkyl groups or hydrogen atoms), giving R1R2CH˙. The generated R1R2CH˙ reacts with O2 to generate alkylperoxyl radical intermediate R1R2CHOO˙, which then undergoes the Russell reaction to give R1R2CHOH (alcohol) and R1R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (ketone) in a 1
O (ketone) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio as the oxidation products. The acyloxyl intermediate RC(O)O˙ also reacts with RCHO to give carboxylic acid RC(O)OH and RC(O)˙, constructing the catalytic cycle. The role of copper(II) ions in the above catalytic process is also investigated using a series of copper(II) complexes. Furthermore, Murahashi's system was adopted in the catalytic oxidation of methane.
:1 ratio as the oxidation products. The acyloxyl intermediate RC(O)O˙ also reacts with RCHO to give carboxylic acid RC(O)OH and RC(O)˙, constructing the catalytic cycle. The role of copper(II) ions in the above catalytic process is also investigated using a series of copper(II) complexes. Furthermore, Murahashi's system was adopted in the catalytic oxidation of methane.
1. Introduction
Alkanes are abundant chemicals widely found in petroleum and natural gas. Selective C–H bond functionalization of inert alkane substrates is of great importance for converting them into value-added and easily usable organic compounds.1,2 In particular, selective oxidation of methane, the main component of natural gas and shale gas as well as methane hydrate, into C1 oxygenates such as methanol, formaldehyde and formic acid has been recently attracting much attention in industrial chemistry.3–8 Therefore, the development of selective oxidation of alkanes including gaseous alkanes has become an important research topic in industrial chemistry as well as in synthetic organic chemistry.9,10Selective oxidation of organic compounds with molecular oxygen (O2) is one of the most economically and environmentally desirable reactions, since O2 is abundant and a clean oxidant generating no toxic by-products.10,11 In this respect, Murahashi's alkane oxidation reaction using O2 as an oxidant, copper(II) acetate (Cu(OAc)2) as a catalyst, and acetaldehyde as a sacrificial reductant is an attractive reaction system, where high conversion of alkanes to alcohols and ketones was achieved (Scheme 1).12–14 Such an O2/transition-metal/aldehyde catalytic system has also been adopted in the alkene epoxidation reaction, the so-called Mukaiyama epoxidation, where peracids (RC(O)OOH) and/or acyl peroxyl radicals (RC(O)OO˙) produced by aldehyde-autoxidation were proposed as reactive oxidants.15–19 However, mechanistic details of Murahashi's alkane oxidation reaction have yet to be clarified.
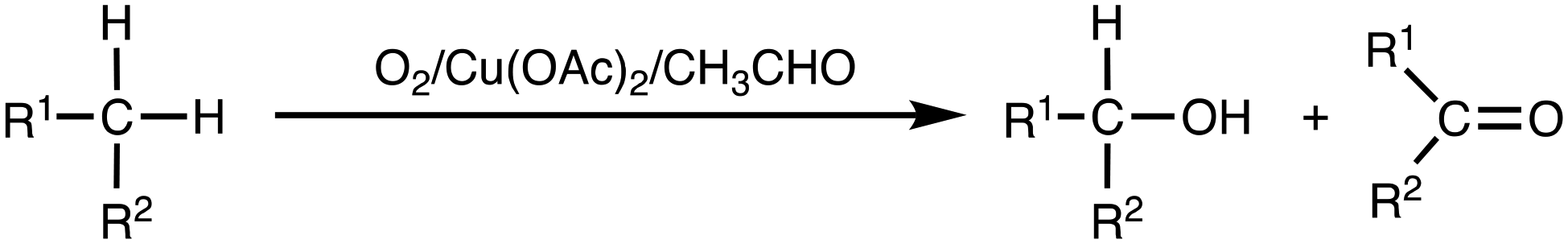 | ||
| Scheme 1 Murahashi's alkane oxidation reaction. | ||
In this study, we have conducted detailed mechanistic studies on Murahashi's alkane oxidation reaction to obtain insights into reactive oxidants as well as the catalytic role of metal ions using a series of CuII-complexes. Furthermore, the catalytic system was adopted in the oxidation of methane.
2. Results and discussion
2.1. Catalytic alkane oxidation with the O2/complex 1/PhCHO system
Murahashi and co-workers reported an efficient alkane oxidation reaction in the presence of acetaldehyde using Cu(OAc)2 as a catalyst (Scheme 1).12 In this study, we revisited this reaction system to obtain mechanistic insights into the catalytic alkane oxidation reaction. First, we examined the catalytic oxidation of cyclohexane (CyH) by using [CuII(Me6tren)(CH3CN)](ClO4)2 (1, Me6tren = tris[2-(dimethylamino)ethyl]amine, see Fig. 1) and benzaldehyde (PhCHO) instead of Cu(OAc)2 and acetaldehyde, respectively. Adoption of the copper(II) complexes enables us to tune the geometry and redox potential of the copper catalyst and the usage of PhCHO allows us to detect the products derived from the aldehyde.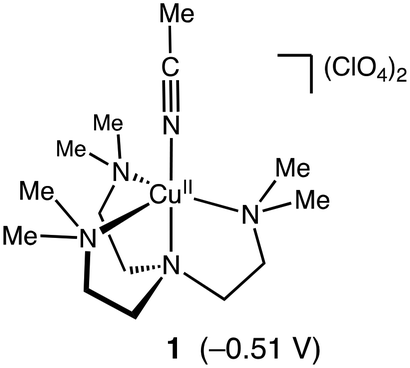 | ||
| Fig. 1 Structure of complex 1. Reduction potential of 1 (vs. Fc/Fc+ in acetonitrile) is shown in parentheses. The cyclic voltammogram of 1 is presented in Fig. S2.† | ||
The reaction was started by adding PhCHO (0.2 M) to a CH3CN/CH2Cl2 (v/v = 3/2) mixed solvent solution containing CyH (2.0 M) and a catalytic amount of 1 (0.2 mM) at 40 °C under an O2 atmosphere (Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Time/h | Yield/mM | ||||
| A | K | L | CyCl | PhH | |
| Reaction conditions: [1] = 0.2 mM, [PhCHO] = 0.2 M, [cyclohexane] = 2.0 M in CH3CN/CH2Cl2 (v/v = 3/2) at 40 °C under O2. 51 mM benzoic acid (PhCO2H) was obtained after 5 h. | |||||
| 1 | 4.0 | 2.6 | 0 | 0.1 | 0.7 |
| 2 | 9.8 | 7.8 | 0.2 | 0.5 | 2.3 |
| 3 | 11 | 9.5 | 0.4 | 0.6 | 2.9 |
| 4 | 15 | 15 | 1.2 | 0.8 | 3.2 |
| 5 | 17 | 16 | 1.2 | 1.5 | 5.2 |
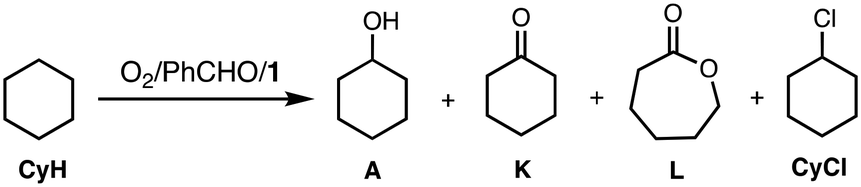
Table 1 shows a time-dependent formation of cyclohexanol (A), cyclohexanone (K), ε-caprolactone (L) and chlorocyclohexane (CyCl) monitored by GC-FID. L may be generated from Kvia the Baeyer–Villiger oxidation by perbenzoic acid, PhC(O)OOH (see below discussion). In this reaction, benzene (PhH) was also obtained. Formation of PhH and CyCl suggests involvement of benzoyloxyl radical (PhC(O)O˙) and cyclohexyl radical (Cy˙) intermediates, respectively (also see below).
To confirm the formation of the cyclohexyl radical (Cy˙) intermediate, the reaction was carried out in a CH3CN/CCl4 mixed solvent (v/v = 3/2) instead of CH3CN/CH2Cl2. As seen in Table S1,† the formation ratio of CyCl increased from 3% to 17%, clearly demonstrating the formation of Cy˙, since CCl4 has been demonstrated to act as an organic radical trapping agent to give the chlorinated product CyCl.20
From the early stage of the catalytic reaction (1 h), A and K were formed in an almost 1:1 ratio and this ratio was maintained during the reaction as shown in Table 1. If K was formed via over-oxidation of A, A should be the main product at the initial stage of the reaction, and the ratio of K should increase gradually afterwards. Thus, the results indicate that the reaction involves the “Russell rearrangement” of the tetroxide intermediate (Cy–O–O–O–O–Cy) as shown in Scheme 2.21 Namely, the cyclohexyl radical intermediate (Cy˙) formed by hydrogen atom abstraction (HAA) from CyH by a reactive oxidant rapidly reacts with O2 at a rate of 109 M−1 s−1 to produce the cyclohexyl–peroxyl radical species (CyOO˙).22 Radical coupling of two CyOO˙ produces the tetroxide intermediate Cy–O–O–O–O–Cy, from which the Russell rearrangement takes place to give an equal amount of A and K (Scheme 2). The fact that the amount of A slightly exceeded that of K in the early stages of the reaction suggests that A is also formed directly via the reaction of Cy˙ and an oxidant such as perbenzoic acid.
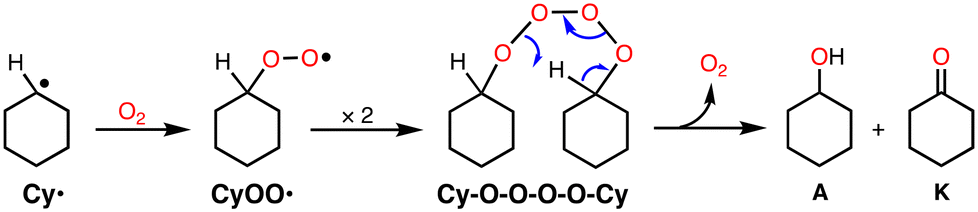 | ||
| Scheme 2 Russell rearrangement. | ||
The product distribution pattern was almost the same when the reaction mixture was quenched by PPh3 (Table S2†). Since alkylhydroperoxides (ROOH) are known to be quantitatively converted to the corresponding alcohols (ROH) by the reaction with PPh3,23 the result indicates that most CyOO˙ was converted to the tetroxide intermediate but not to cyclohexyl hydroperoxide (CyOOH).
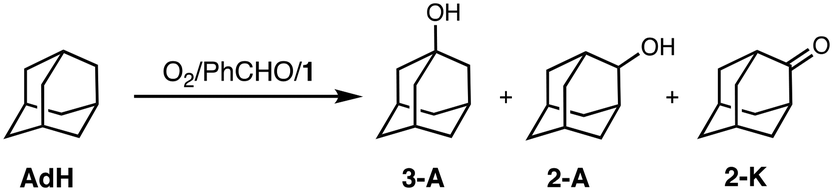
To obtain information about the active oxidant, the oxidation reaction of adamantane (AdH) was also examined (Table 2). Oxidation of AdH with 1 and PhCHO under O2 also proceeded efficiently to give the corresponding alcohols (3-A and 2-A) together with a small amount of ketone (2-K). It should be noted that the regioselectivity between the secondary and the tertiary carbons was quite high (3°/2° = 12), suggesting that the active intermediate is not a free hydroxy radical (˙OH), since a Fenton-type autoxidation reaction containing ˙OH as a reactive oxidant is known to exhibit almost no regioselectivity (3°/2° ∼ 1).24
|
|
||||
|---|---|---|---|---|
| Reaction conditions: [1] = 0.2 mM, [PhCHO] = 0.2 M, [adamantane] = 0.25 M in CH3CN/CH2Cl2 (v/v = 3/2) at 40 °C under O2.a The amounts of the products are determined by GC-FID using calibration curves of the products.b [3-A]/([2-A] + [2-K]) multiplied by 3. | ||||
| Substrate | Yielda/mM | 3°/2°b | ||
| 3 A | 2 -K | 2 -K | 12 | |
| Adamantane | 16 | 1.7 | 2.2 | |
2.2. Reactive oxidants
To explore the reactive oxidants, we focused on the autoxidation of aldehydes. Scheme 3 shows a possible mechanism of the autoxidation of PhCHO.25–29 First of all, acyl radical intermediate a (PhCO˙) is produced by a certain initiating step, which is converted to acyl peroxyl radical intermediate b (PhC(O)OO˙) by the reaction with O2. Then, the generated acyl peroxyl radical intermediate b abstracts a hydrogen atom from another molecule of PhCHO, resulting in the formation of peracid d (PhC(O)OOH) and a, constructing a radical chain cycle. On the other hand, Hutchings and co-workers recently reported that acyloxyl radical intermediate c (PhC(O)O˙) is formed through radical adduct formation between b and PhCHO and following fragmentation (Scheme 3).30 Judging from the bond dissociation energy (BDE) of the C–H bond of benzaldehyde (Ph(O)C–H, 88.7 ± 2.6 kcal mol−1), the O–H bond of perbenzoic acid (PhC(O)OO–H, 96.5 kcal mol−1), and the O–H bond of benzoic acid (PhC(O)O–H, 111 ± 4),31 acyloxy radical c is the most plausible reactive oxidant for the C–H bond activation of cyclohexane (Cy–H, 99.5 kcal mol−1).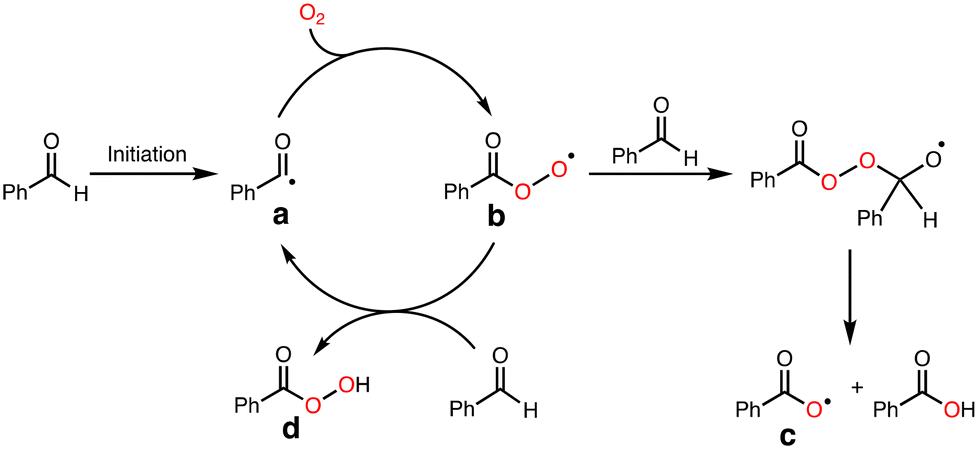 | ||
| Scheme 3 Autoxidation mechanism of benzaldehyde. | ||
It was reported that acyloxy radical c is capable of abstracting a hydrogen atom from CyH with a rate constant of about 106 M−1 s−1.32 This reaction competes with decarboxylation from c with a rate constant of 106 s−1 to give benzene (PhH) (Scheme 4).32 Thus, the ratio of the oxidation products (P = A + K + L + CyCl) against benzene (PhH, the decarboxylation product of c), [P]/[PhH], is expected to increase as the concentration of the substrate increased, since the hydrogen atom abstraction (HAA) process and decarboxylation reaction are competing.
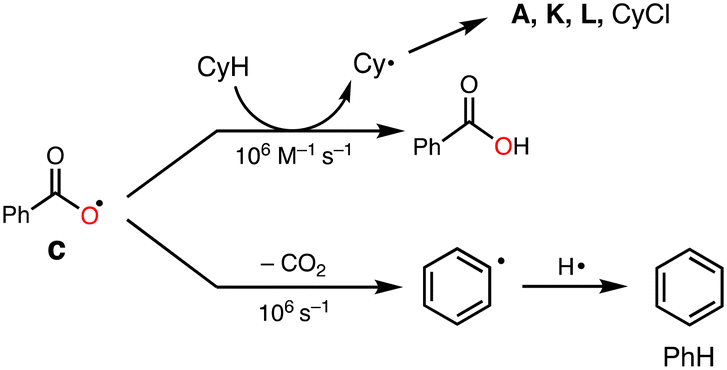 | ||
| Scheme 4 Competition of HAA with decarboxylation from c. | ||
As clearly seen in Fig. 2, [P]/[PhH] increased as [CyH] is increased. Thus, we concluded that acyloxy radical c is a reactive oxidant for the HAA from CyH. The amount of benzoic acid (PhCO2H) formed after 5 h was 51 mM that was larger than the total amount of products (35.7 mM, Table 1). This is reasonable since PhCO2H is formed both via the substrate oxidation by PhC(O)O˙ (c) (Scheme 4) and via the autooxidation of PhCHO (Scheme 3).
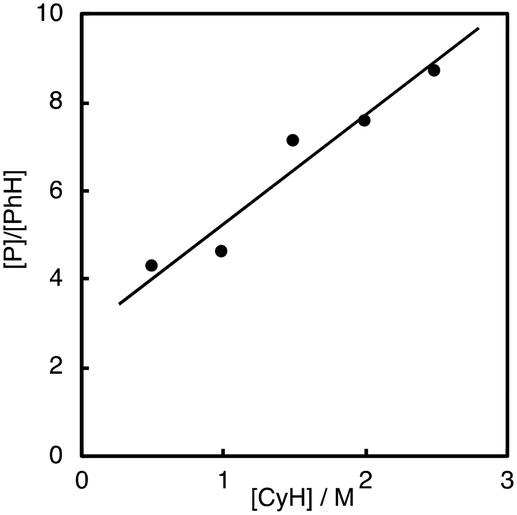 | ||
| Fig. 2 Dependence of [P]/[PhH] on [CyH]. | ||
2.3. Catalytic activity of other CuII-complexes
Fig. 3 compares the catalytic activity of a series of CuII-complexes 1–5 supported by tripodal tetradentate ligands. Their crystal structures are shown in Fig. S1† together with the crystallographic data in Tables S3–S5.† Among the CuII-complexes with a similar trigonal bipyramidal structure, complex 1 exhibited the highest initial reaction rate, and the induction period became longer going from 1 to 5. These results suggest that the CuII-complexes play an important role in the alkane oxidation reaction.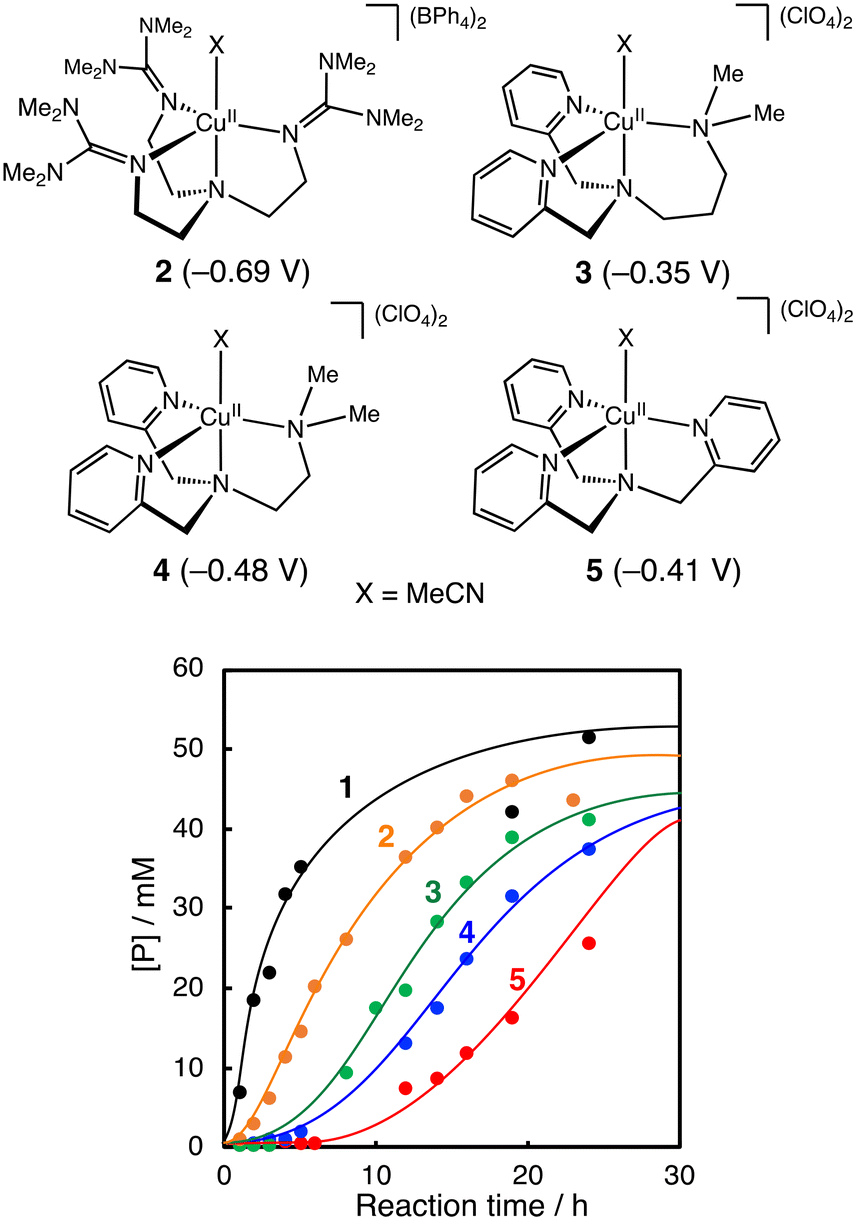 | ||
| Fig. 3 Time courses of product formation in the oxidation of cyclohexane (2.0 M) catalysed by CuII-complexes (1–5, 0.2 mM) with PhCHO (0.2 M) in CH3CN/CH2Cl2 (v/v = 3/2) at 40 °C under O2. [P] = [A] + [K] + [L] + [CyCl]. Reduction potentials of the CuII-complexes (vs. Fc/Fc+ in acetonitrile) are shown in parentheses. The cyclic voltammograms of the CuII-complexes are presented in Fig. S2.† | ||
2.4. Possible role of CuII-complexes in the initiation step of aldehyde autoxidation
Generation of acyl radical intermediate a (Scheme 3) by the reaction of aldehydes and transition-metal complexes has been proposed as the initiation step of aldehyde autoxidation reactions.33–35 However, little has been examined on the direct reaction between aldehydes and transition-metal complexes.36,37 To evaluate such a possibility, we tried to trap acyl radical intermediate a in CCl4 (Table S6†). Thus, the reaction of complex 1 (2 mM) with an excess amount of PhCHO (0.2 M) was examined in CCl4 under anaerobic conditions (to prevent the reaction of a with O2) and then the resulting reaction mixture was poured into methanol. If acyl radical a was generated by the reaction of PhCHO and 1, a would be trapped by CCl4 to generate acyl chloride (PhC(O)Cl),36,38 which will be converted to methyl benzoate (PhC(O)OMe), a more easily detectable product, in methanol (Scheme 5). The same reaction was conducted by using [CuII(TEPA)OTf](OTf) instead of 1, since [CuII(TEPA)OTf](OTf) has a much higher reduction potential (+0.05 V vs. Fc/Fc+) as compared to 1 (−0.51 V). In other words, [CuII(TEPA)OTf](OTf) is a more powerful oxidant than 1. In both cases, however, no significant difference was observed in the yield of PhC(O)OMe from that of a blank experiment (without the copper complex).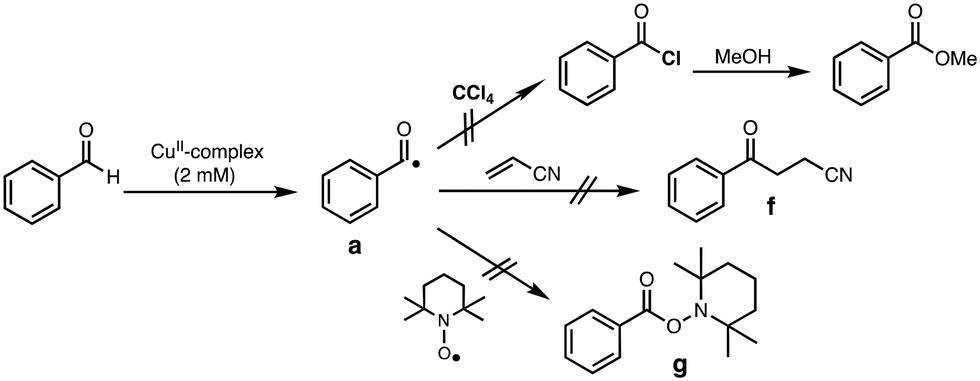 | ||
| Scheme 5 Acyl radical trapping experiments. | ||
We also tried to trap a using acrylonitrile (Fig. S3†) or TEMPO (Fig. S4†), both of which are known to react with the acyl radical intermediate.39,40 However, neither the acrylonitrile-adduct f nor the TEMPO-adduct g were obtained in the presence of complex 1 (Scheme 5).
Furthermore, if the reaction had played a significant role in the initiation step, the length of the induction period in the catalytic oxidation should have a negative correlation with the redox potential of the CuII-complexes. However, no such correlation was observed (Fig. 3).
All these results strongly suggest that the direct oxidation of benzaldehyde by the CuII-complex to generate acyl radical intermediate a is negligible under the reaction conditions examined in this study.
2.5. Role of CuII-complexes in peracid activation
In the oxidation of CyH (2.0 M) with PhCHO (0.2 M) and the copper(II) complex under O2, a distinct induction period was observed in the early stage of the reaction (Fig. 3). Even in the case of complex 1 showing the highest initial rate, the induction period became prominent when the concentration of 1 was reduced from 0.2 mM to 0.025 mM (black line in Fig. 4). On the other hand, addition of 10 eq. (based on 1) of m-CPBA (m-chloroperbenzoic acid) at the beginning of the reaction significantly shortened the induction period as shown in Fig. 4 (red line), indicating the important role of peracid d generated in the catalytic cycle (see Scheme 3).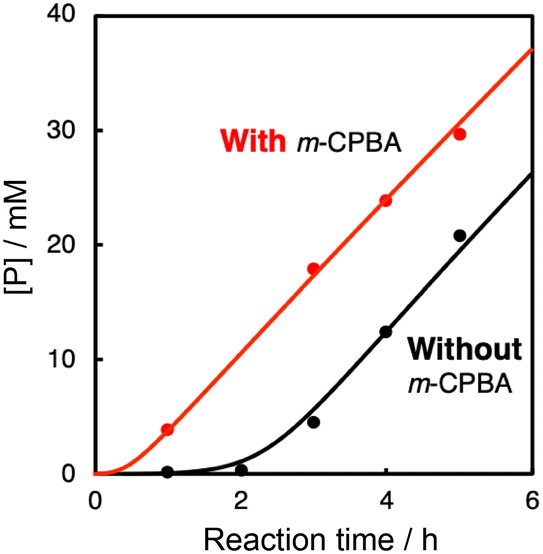 | ||
| Fig. 4 Time courses of the cyclohexane oxidation by the O2/1/PhCHO system with and without m-CPBA (10 mM). Reaction conditions: [1] = 0.025 mM, [PhCHO] = 0.2 M, [cyclohexane] = 2.0 M at 40 °C under O2. | ||
We assumed that activation of peracid d by the CuII-complex contributes to the shortening of the induction period. To test this possibility, a direct reaction of 1 and m-CPBA was examined spectroscopically (Scheme 6). When 10 eq. of m-CPBA was added to an acetonitrile solution of 1 (2 mM), the spectrum changed from black (λmax = 845 nm) to red (λmax = 700 nm) in about 30 min at 40 °C (Fig. 5A), which was different from the spectrum of the m-CBA (m-chlorobenzoic acid, the decomposition product of m-CPBA) adduct of 1 (see Fig. S5†). The generated complex exhibited an ESI-mass spectrum showing a peak cluster, whose peak positions as well as isotope distribution pattern are consistent with the chemical formula of the m-CPBA adduct complex of 1 (Fig. S6†). The m-CPBA adduct complex was relatively stable at this temperature, but immediately decomposed when 2,4,6-tri-tert-butylphenol (10 equiv.) was added to the solution to give a 2,4,6-tri-tert-butylphenoxyl radical (the blue spectrum in Fig. 5B).41 On the other hand, the formation of the phenoxy radical was negligible in the absence of 1. These results suggest that an adduct formation reaction takes place between the CuII-complex and peracid d generated in situ in the catalytic oxidation of CyH by the O2/CuII-complex/PhCHO system (Scheme 3).
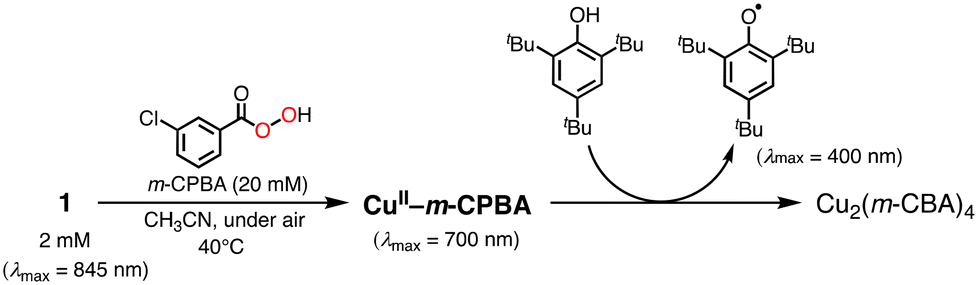 | ||
| Scheme 6 Reaction of 1 and m-CPBA. | ||
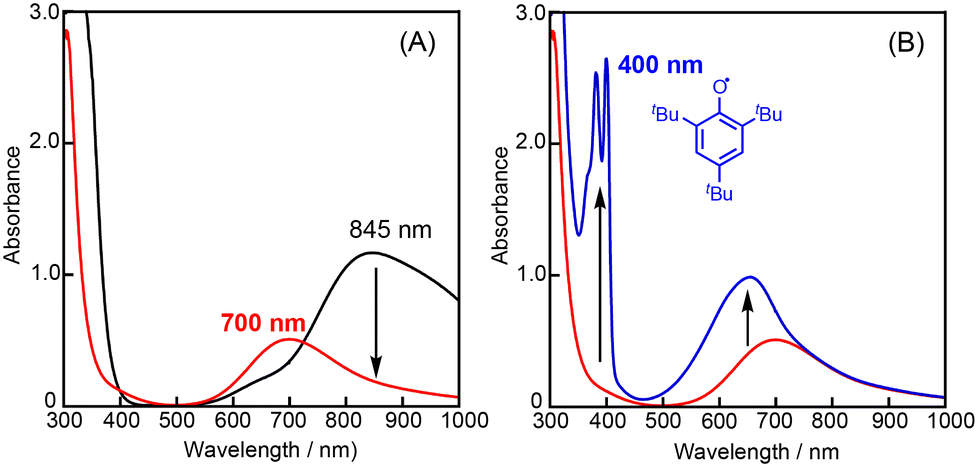 | ||
| Fig. 5 (A) UV-vis spectral change observed upon an addition of m-CPBA (10 eq.) to 1 (2 mM) at 40 °C in acetonitrile and (B) UV-vis spectral change of the reaction between the m-CPBA adduct CuII-complex with 2,4,6-tri-tert-butylphenol (10 eq.). | ||
To further investigate the role of CuII-complexes in the peracid activation process, the catalytic oxidation of CyH was performed under anaerobic (N2) conditions in the presence of 1 and m-CPBA. When the catalytic oxidation of CyH (2.0 M) by m-CPBA (0.2 M) was carried out in the presence of a catalytic amount of 1 (0.2 mM) in a CH3CN/CH2Cl2 (v/v = 3/2) mixed solvent, A was obtained as the major product together with K, L and CyCl as the minor products (Fig. S7†). The catalytic activity of the CuII-complexes 2–5 was also examined under the same conditions (Fig. S7†). Apparently, the CuII-complexes showed different catalytic activities, which are correlated with the length of the induction period observed in the O2/CuII-complex/PhCHO system except 2 as shown in Fig. 6. Namely, the higher the catalytic activity in the m-CPBA system, the shorter the induction period in the O2/CuII-complex/PhCHO system. Therefore, it can be concluded that the peracid activation by the CuII-complex accelerates the catalytic reaction of CyH in the early stages of the catalytic reaction. Deviation of complex 2 from the straight line shown in Fig. 6 may be due to steric hindrance of the TMG substituents around the metal center, which prohibits the substrate access to the metal centre. Deactivation of complex 2 by intramolecular hydroxylation of the methyl group of TMG substituents may also cause its lower catalytic activity.42
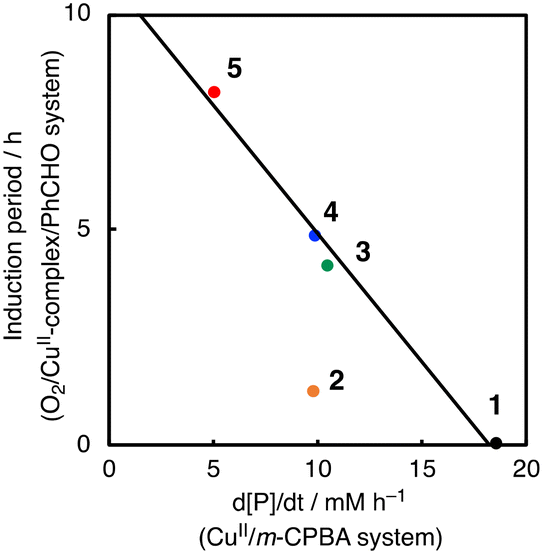 | ||
| Fig. 6 Correlation between the length of the induction period in the O2/PhCHO/CuII-complex system and the catalytic activity of CuII-complexes in the oxidation of cyclohexane by m-CPBA. | ||
Given the linear correlation shown in Fig. 6 and that the aldehyde autoxidation produces peracids (Scheme 3), the induction phase in the O2/CuII-system/PhCHO is thought to involve formation of a CuII-peracid complex, which cleaves the C–H bond of PhCHO to generate acyl radical intermediate a, accelerating the autooxidation reaction of PhCHO (Scheme 3). To obtain mechanistic insights into the peracid activation process in the early stages of the catalytic reaction, we further investigated the induction period phase as below.
As shown in Fig. 7 (black line), there was a prominent induction period of about 2 h, when the lower concentration (0.025 mM) of 1 was used as a catalyst. If the reaction started without adding 1 at the beginning and then 1 was added to the reaction solution after 2 h, there was almost no induction period after the addition of 1 (Fig. 7, blue line). In other words, 1 was not involved in the induction process (first 2 h). Considering that aldehydes are sensitive to air, the formation of peracids by the oxidation of aldehydes may be the true initiating reaction. Indeed, iodometry revealed that peroxide formation increased with time when PhCHO was stirred in acetonitrile in an O2 atmosphere under conditions that minimized the influence of metal particles, using an aqua regia rinsed vial and stirrer (Fig. S8†). Thus, we concluded that the CuII-complex played an important role in initiating the aldehyde autoxidation reaction by reacting with the peracid produced by the direct reaction of PhCHO and O2.
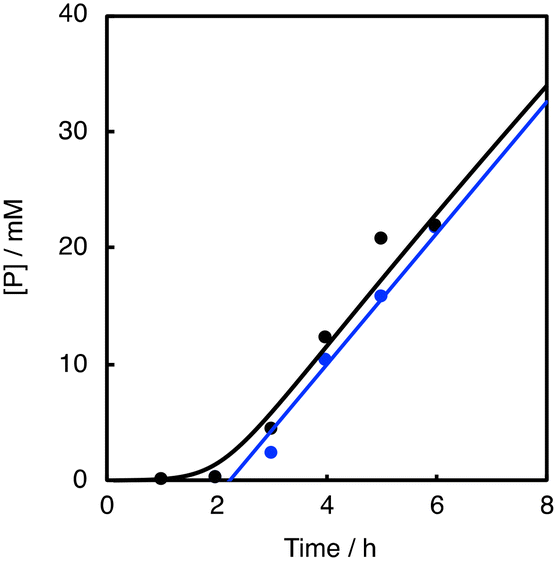 | ||
| Fig. 7 Time courses of the product formation in the O2/1/PhCHO system (black line) and that of the reaction where 1 was added after 2 h (blue line). Reaction conditions: [PhCHO] = 0.2 M, [cyclohexane] = 2.0 M, [1]0 = 0.025 mM at 40 °C under O2. | ||
2.6. Application to methane oxidation
Finally, oxidation of methane was briefly examined using Murahashi's system. In this reaction, Cu(OAc)2 was used as a catalyst to avoid intramolecular and/or intermolecular ligand hydroxylation (catalyst degradation). Thus, the oxidation of methane was carried out under pressurized conditions (CH4 = 3.0 MPa, O2 = 1.0 MPa) in the presence of Cu(OAc)2 (0.040 μmol) and PhCHO (2.0 mmol) at ambient temperature (25 °C) for 8 h in CH3CN. GC-MS analysis of the final reaction solution indicated the formation of 2.0 μmol methanol as shown in Fig. S9.† Formation of a trace amount of formaldehyde was also noted. The low yield of the oxidation products is apparently due to the higher BDE of methane (105 kcal mol−1) compared to that of CyH (99.5 kcal mol−1) and the lower concentration of the gaseous substrate.3. Conclusions
In this study, we have conducted a mechanistic study on the catalytic alkane oxidation by an O2/CuII-complex/RCHO system (Murahashi's reaction). The reaction of RCHO and O2 generates acyl–peroxyl radical RC(O)OO˙ intermediate bvia the formation of acyl radical intermediate a (Scheme 3). The generated RC(O)OO˙ (b) abstracts a hydrogen atom from another RCHO to give a and peracid d, constructing a radical chain cycle. Intermediate b also undergoes adduct formation with RCHO to generate the RC(O)OOC(O)(H)O˙ intermediate, from which acyloxyl radical c and RC(O)OH are produced. Judging from the high BDE of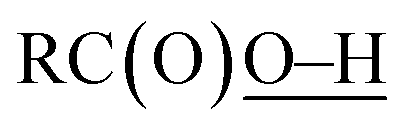 (111 ± 4 kcal mol−1), acyloxyl radical c is the most powerful oxidant for the hydrogen atom abstraction (HAA) from the alkane substrate (Scheme 4). Then, the generated alkane radical intermediate undergoes the Russell reaction to give alcohol (A) and ketone (K) products (Scheme 2).
(111 ± 4 kcal mol−1), acyloxyl radical c is the most powerful oxidant for the hydrogen atom abstraction (HAA) from the alkane substrate (Scheme 4). Then, the generated alkane radical intermediate undergoes the Russell reaction to give alcohol (A) and ketone (K) products (Scheme 2).
Regarding the role of the copper(II) complex, we expected that the higher the oxidation ability of the copper(II) complexes, the larger the activation rate of PhCHO, shortening the induction period. However, there was no such correlation between the redox potential of the copper(II) complexes and the length of the induction period (Fig. 3). This is one of the pieces of evidence that the copper(II) complexes do not participate directly to the oxidation of PhCHO to generate acyl radical intermediate a. On the other hand, there was a good linear correlation (except complex 2) between the length of the induction period and the catalytic activity of the copper(II) complexes in the m-CPBA system (Fig. 6). This result indicates that the activation of peracid by the copper(II) complex is involved in the induction period of the catalytic oxidation. This assumption is consistent with the result shown in Fig. 7. Thus, we concluded that the significant role of the copper(II) complex is the activation of peracid d to generate a copper(II)–peracid adduct intermediate, which may also generate a reactive oxidant to accelerate the autoxidation of RCHO.
Oxidation of methane was briefly examined using Murahashi's system. Judging from the BDEs (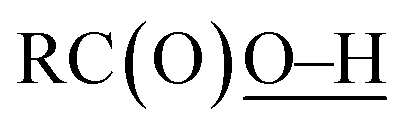 : 111 ± 4 kcal mol−1vs.
: 111 ± 4 kcal mol−1vs.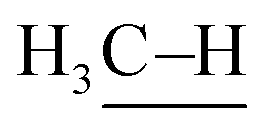 : 105 kcal mol−1), the presumed reactive oxidant, acyloxyl radical c, has enough power to induce the HAA from methane. However, the available concentration of methane (3.0 MPa) was too low to compete with the oxidation of PhCHO. Nonetheless, the present study gives important information for the development of efficient catalytic systems for gaseous alkane oxidation reactions.
: 105 kcal mol−1), the presumed reactive oxidant, acyloxyl radical c, has enough power to induce the HAA from methane. However, the available concentration of methane (3.0 MPa) was too low to compete with the oxidation of PhCHO. Nonetheless, the present study gives important information for the development of efficient catalytic systems for gaseous alkane oxidation reactions.
4. Experimental section
4.1. General
The reagents and the solvents used in this study, except the ligands and the copper complexes, were commercial products of the highest available purity and were purified by the standard methods,43 if necessary. m-Chloroperbenzoic acid (m-CPBA) was purified by recrystallization from CH2Cl2 in a refrigerator kept at −20 °C, and the purity was determined to be 80–90% by redox titration using NaI. Ligands (TMPA,44 TEPA,45 Me2-uns-penp,46 TMG3tren47 and Me2-pp3 (ref. 48)) and CuII-complexes ([CuII(Me6tren)(CH3CN)](ClO4)2 (1),49 [CuII(TMPA)(CH3CN)](ClO4)2 (5)50 and [CuII(TEPA)(OTf)](OTf) (6)51) were prepared according to reported procedures. Anaerobic reactions were carried out under a N2 atmosphere using a glovebox (Miwa DB0-1KP or KK-011-AS, KOREA KIYON product, [O2] < 1 ppm). UV-visible spectra were taken on a Jasco V-570 or a Hewlett Packard 8453 photodiode array spectrophotometer equipped with a Unisoku thermostated cryostat cell holder USP-203. Near IR spectra were taken on a Jasco V-670. 1H-NMR spectra were recorded on a JEOL ECP400. Elemental analyses were performed on a Yanaco New Science Inc. CHN order MT-5 or a J-SCIENCE LAB Co., Ltd. MICRO CORDER JM10. ESI-MS (electrospray ionization-mass spectrometry) measurements were performed on a BRUKER cryospray microTOFII. Gas chromatography (flame ionization detector) measurements were performed on a Shimadzu GC-2010 equipped with a GL Science InertCapWAX capillary column (30 m × 0.25 mm), an AOC-20s auto sampler, and an AOC-20i auto injector. Gas chromatography-mass spectrometry (GC-MS) measurements were performed on a Shimadzu GCMS-QP2010 Plus equipped with a RESTEK Rtx-VMS column (30 m × 0.25 mm), an AOC-20s auto sampler, and an AOC-21i auto injector.4.2. Synthesis
4.3. X-ray crystallographic analysis
Single crystals of the copper(II) complexes were mounted on a DT-MicroLoop (MiTegan, LLC) with mineral oil. X-ray diffraction data were collected on a Rigaku R-AXIS RAPID diffractometer using filtered MoKα radiation (λ = 0.71075 Å). Direct methods with SHELXT were used for the structure solution of the crystals. All calculations were performed with the observed reflections [I > 2σ(I)] with the Olex2 crystallographic software packages except for refinement which was performed using SHELXL. Hydrogen atoms were refined using a riding model. CCDC 2262940–2262944 contain the supplementary crystallographic data (CIF files) for the copper(II) complexes (1–5).4.4. Catalytic oxidation of the O2/Cu(II)-complex/aldehyde system
All procedures of the catalytic oxidation reactions were carried out under an O2 atmosphere using an O2 balloon unless otherwise noted. The reaction was started by adding an aldehyde to a mixed solvent (CH3CN/CH2Cl2, v/v = 3/2) containing a copper(II) complex and a substrate. After quenching the reaction by passing the reaction mixture through an alumina-column, products were analysed by GC-FID. All peaks of interest were identified by comparing the retention times with those of the authentic samples. The products were quantified by comparing their peak areas with that of an internal standard (nitrobenzene) using calibration curves consisting of plots of molar ratio (moles of organic compound/moles of internal standard) versus area ratio (area of organic compound/area of standard).4.5. Catalytic oxidation of the m-CPBA/Cu(II) system
The reaction was started by adding m-CPBA to a mixed solvent (CH3CN/CH2Cl2, v/v = 3/2) containing a copper(II) complex and a substrate under N2. After quenching the reaction by passing the reaction mixture through an alumina-column, products were analysed by GC-FID. All peaks of interest were identified by comparing the retention times with those of the authentic samples. The products were quantified by comparing their peak areas with that of an internal standard (nitrobenzene) using calibration curves consisting of plots of molar ratio (moles of organic compound/moles of internal standard) versus area ratio (area of organic compound/area of standard).4.6. Acyl radical trapping experiment by CCl4
PhCHO (100 eq.) was added to an CH3CN/CCl4 (v/v = 3/2) solution (2.5 mL) of a copper(II)-complex (0.2 mM). The reaction mixture was stirred for 9 days at room temperature under N2. After quenching the reaction by adding 1 mL of CH3OH into the reaction mixture, products were analysed by GC–MS and GC-FID.4.7. Oxidation of methane
The oxidation of methane was conducted under pressurized conditions using a methane/oxygen gas mixture in a high-pressure reactor. After quenching the reaction using a silica column, the products were analysed by using GC-MS. All peaks of interest were identified by comparing the retention times and mass spectra with those of the authentic samples. The products were quantified by comparing their peak areas with that of an internal standard (nitrobenzene) using a calibration curve consisting of a plot of mole ratio (moles of organic compound/moles of internal standard) versus area ratio (area of organic compound/area of internal standard).Author contributions
S. I. conceived the idea and designed the project. K. Y. and Y. U. performed most of the experiments. H. S. contributed to X-ray crystallographic analysis. Y. M. was involved in the discussion of the reaction mechanism. S. I. and K. Y. wrote the manuscript. All the authors commented on the final draft of the manuscript and contributed to the analysis and interpretation of the data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST-CREST (JPMJCR16P1 to SI).Notes and references
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed.
- V. K. Ahluwalia, Oxidation in organic synthesis, CRC Press, Boca Raton, 2012 Search PubMed.
- R. H. Crabtree, Chem. Rev., 1995, 95, 987–1007 CrossRef CAS.
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521–8573 CrossRef CAS PubMed.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- V. C. C. Wang, S. Maji, P. P. Y. Chen, H. K. Lee, S. S. F. Yu and S. I. Chan, Chem. Rev., 2017, 117, 8574–8621 CrossRef CAS PubMed.
- B. E. R. Snyder, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Chem. Rev., 2018, 118, 2718–2768 CrossRef CAS PubMed.
- Y. Tang, Y. Li and F. Tao, Chem. Soc. Rev., 2022, 51, 376–423 RSC.
- C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Chem. Soc. Rev., 2011, 40, 1950–1975 RSC.
- C. Tang, X. Qiu, Z. Cheng and N. Jiao, Chem. Soc. Rev., 2021, 50, 8067–8101 RSC.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2364 CrossRef CAS PubMed.
- S.-I. Murahashi, Y. Oda, T. Naota and N. Komiya, J. Chem. Soc., Chem. Commun., 1993, 139–140 RSC.
- Y. Hayashi, N. Komiya, K. Suzuki and S.-I. Murahashi, Tetrahedron Lett., 2013, 54, 2706–2709 CrossRef CAS.
- H. Sterckx, B. Morel and B. U. W. Maes, Angew. Chem., Int. Ed., 2019, 58, 7946–7970 CrossRef CAS PubMed.
- K. R. Lassila, F. J. Waller, S. E. Werkheiser and A. L. Wressell, Tetrahedron Lett., 1994, 35, 8077–8080 CrossRef CAS.
- N. Mizuno, H. Weiner and R. G. Finke, J. Mol. Catal. A: Chem., 1996, 114, 15–28 CrossRef CAS.
- W. Nam, H. J. Kim, S. H. Kim, R. Y. N. Ho and J. S. Valentine, Inorg. Chem., 1996, 35, 1045–1049 CrossRef CAS PubMed.
- B. B. Wentzel, P. A. Gosling, M. C. Feiters and R. J. M. Nolte, J. Chem. Soc., Dalton Trans., 1998, 2241-2246, 10.1039/A801175C.
- B. B. Wentzel, P. L. Alsters, M. C. Feiters and R. J. M. Nolte, J. Org. Chem., 2004, 69, 3453–3464 CrossRef CAS PubMed.
- T. Shinke, M. Itoh, T. Wada, Y. Morimoto, S. Yanagisawa, H. Sugimoto, M. Kubo and S. Itoh, Chem. – Eur. J., 2021, 27, 14730–14737 CrossRef CAS PubMed.
- G. A. Russell, J. Am. Chem. Soc., 1957, 79, 3871–3877 CrossRef CAS.
- B. Maillard, K. U. Ingold and J. C. Scaiano, J. Am. Chem. Soc., 1983, 105, 5095–5099 CrossRef CAS.
- I. Garcia-Bosch and M. A. Siegler, Angew. Chem., Int. Ed., 2016, 55, 12873–12876 CrossRef CAS PubMed.
- L. Cermenati, D. Dondi, M. Fagnoni and A. Albini, Tetrahedron, 2003, 59, 6409–6414 CrossRef CAS.
- H. R. Cooper and H. W. Melville, J. Chem. Soc., 1951, 1984–1993 RSC.
- H. R. Cooper and H. W. Melville, J. Chem. Soc., 1951, 1994–2002 RSC.
- W. A. Waters and C. Wickham-Jones, J. Chem. Soc., 1951, 812–823 RSC.
- J. R. McNesby and T. W. Davis, J. Am. Chem. Soc., 1954, 76, 2148–2152 CrossRef CAS.
- G. E. Zaikov, J. A. Howard and K. U. Ingold, Can. J. Chem., 1969, 47, 3017–3029 CrossRef CAS.
- M. Sankar, E. Nowicka, E. Carter, D. M. Murphy, D. W. Knight, D. Bethell and G. J. Hutchings, Nat. Commun., 2014, 5, 3332 CrossRef PubMed.
- Y.-R. Luo, Conprehensive Handbook of Chemical Bond Energy, CRC Press, Boca Raton, London, New York, 2007 Search PubMed.
- J. Chateauneuf, J. Lusztyk and K. U. Ingold, J. Am. Chem. Soc., 1988, 110, 2877–2885 CrossRef CAS.
- C. E. H. Bawn and J. B. Williamson, Trans. Faraday Soc., 1951, 47, 721–734 RSC.
- C. E. H. Bawn and J. E. Jolley, Proc. R. Soc. London, Ser. A, 1956, 237, 297–312 CAS.
- C. H. E. Bawn, T. P. Hobin and L. Raphael, Proc. R. Soc. London, Ser. A, 1956, 237, 313–324 CAS.
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991–2070 CrossRef CAS PubMed.
- I. V. Alabugin, L. Kuhn, M. G. Medvedev, N. V. Krivoshchapov, V. A. Vil', I. A. Yaremenko, P. Mehaffy, M. Yarie, A. O. Terent'ev and M. A. Zolfigol, Chem. Soc. Rev., 2021, 50, 10253–10345 RSC.
- S. Winstein and F. H. Seubold Jr., J. Am. Chem. Soc., 1947, 69, 2916–2917 CrossRef CAS.
- G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, Org. Lett., 2015, 17, 4830–4833 CrossRef CAS PubMed.
- W. Liu, Y. Li, K. Liu and Z. Li, J. Am. Chem. Soc., 2011, 133, 10756–10759 CrossRef CAS PubMed.
- V. W. Manner, T. F. Markle, J. H. Freudenthal, J. P. Roth and J. M. Mayer, Chem. Commun., 2008, 256–258 RSC.
- F. F. Pfaff, F. Heims, S. Kundu, S. Mebs and K. Ray, Chem. Commun., 2012, 48, 3730–3732 RSC.
- D. D. Perrin, W. L. F. Armarego and D. R. Perrin, Purification of Laboratory Chemicals, Pergamon Press, Elmsford, NY, 4th edn, 1996 Search PubMed.
- J. W. Canary, Y. Wang, R. Roy Jr. , J. L. Que and H. Miyake, in Inorganic Syntheses, 1998, pp. 70–75, DOI:10.1002/9780470132630.ch11.
- K. D. Karlin, J. C. Hayes, J. P. Hutchinson, J. R. Hyde and J. Zubieta, Inorg. Chim. Acta, 1982, 64, L219–L220 CrossRef CAS.
- T. Brückmann, J. Becker, C. Würtele, M. T. Seuffert, D. Heuler, K. Müller-Buschbaum, M. Weiß and S. Schindler, J. Inorg. Biochem., 2021, 223, 111544 CrossRef PubMed.
- H. Wittmann, V. Raab, A. Schorm, J. Plackmeyer and J. Sundermeyer, Eur. J. Inorg. Chem., 2001, 2001, 1937–1948 CrossRef.
- F. Mehlich, A. E. Roberts, M. Kerscher, P. Comba, G. A. Lawrance, C. Würtele, J. Becker and S. Schindler, Inorg. Chim. Acta, 2019, 486, 742–749 CrossRef CAS.
- Y. J. Choi, K.-B. Cho, M. Kubo, T. Ogura, K. D. Karlin, J. Cho and W. Nam, Dalton Trans., 2011, 40, 2234–2241 RSC.
- J. Wang, M. P. Schopfer, S. C. Puiu, A. A. N. Sarjeant and K. D. Karlin, Inorg. Chem., 2010, 49, 1404–1419 CrossRef CAS PubMed.
- V. V. Smirnov and J. P. Roth, J. Am. Chem. Soc., 2006, 128, 3683–3695 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2262940–2262944. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cy00944k |
| This journal is © The Royal Society of Chemistry 2023 |