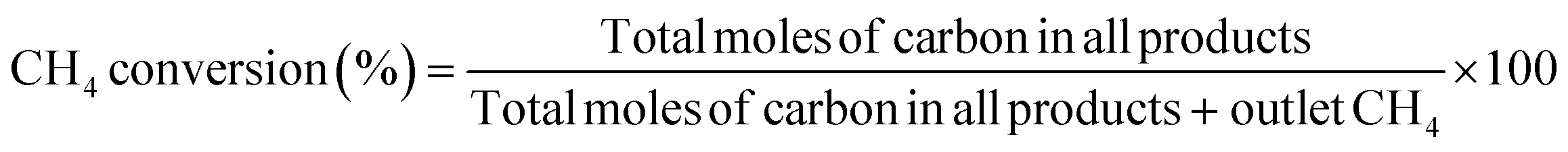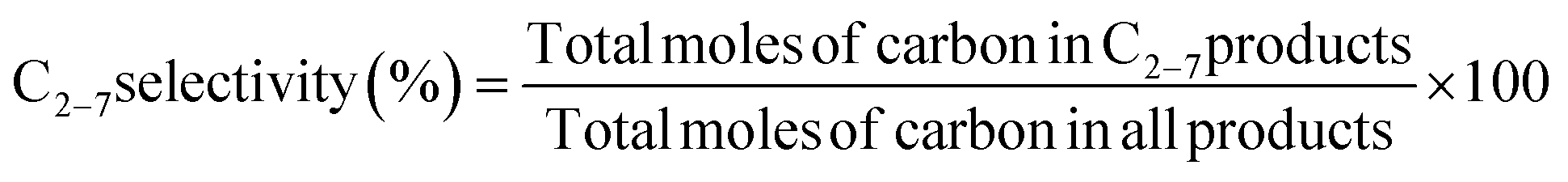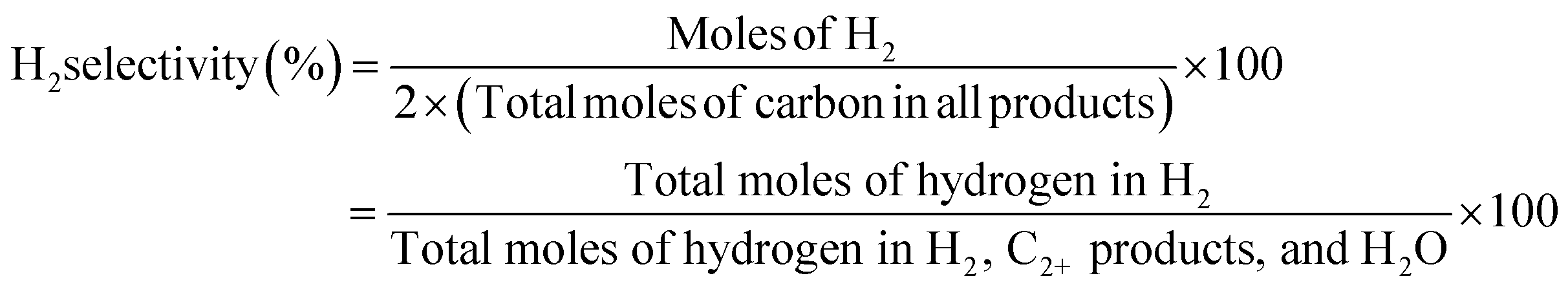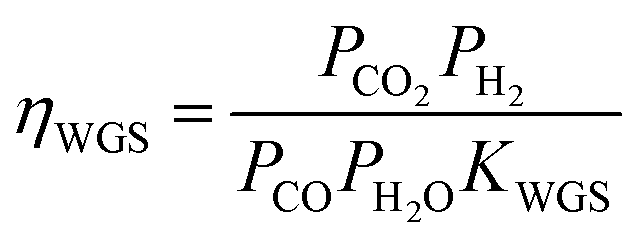Consequence of products from oxidative coupling of methane in a non-oxidative high temperature environment†
Received
21st December 2022
, Accepted 18th February 2023
First published on 20th February 2023
Abstract
Oxidative coupling of methane (OCM) is a direct process that converts methane to higher hydrocarbons, such as ethylene. For several decades, various catalysts and their reaction mechanisms have been investigated to obtain high selectivity for the target products. However, the consequences of OCM products after O2 depletion at high temperatures, which is generated by an exothermic reaction, have been often overlooked. In the present study, a two-stage reactor that mimics an industrial reactor was used to study the successive reactions of OCM products. Gas phase homogeneous and heterogeneous reactions on the surface of catalysts and supports have been systematically investigated. Dehydrogenation of OCM products to acetylene and the following condensation occurs in the gas phase. Meanwhile, steam reforming of OCM products concurrently followed by water gas shift reactions was observed on various catalysts and supports and led to the loss of C2 yield. Based on the investigations, a design guideline for the OCM reactor is proposed.
Introduction
Olefins, such as ethylene, propylene, and butadiene, are important chemical feedstocks in the manufacturing process of various functional compounds, such as plastics and rubbers. Currently, olefins are obtained by refining crude oil followed by their cracking, and their demand is expected to increase continuously. Due to concerns about crude oil depletion, however, it is necessary to develop synthetic routes using other carbon sources. Methane is considered an alternative hydrocarbon source due to the recent shale gas revolution. The general synthetic route for obtaining olefins from methane involves at least three steps: (1) synthesis gas (syngas) generation through a methane reforming reaction, (2) methanol synthesis from syngas, and (3) methanol-to-olefin reaction.1 Syngas is a mixture of H2, CO, and some CO2, which are the major intermediates in the industrial methane conversion process. The methods to obtain syngas include steam reforming, partial oxidation, and carbon dioxide reforming using H2O, O2, and CO2 as reactants, respectively. The reaction equations for each reforming process are shown below.| | | CH4 + H2O → CO + 3H2ΔH0298K = 206 kJ mol−1 | (1) |
| | | CH4 + 1/2O2 → CO + 2H2ΔH0298K = −36 kJ mol−1 | (2) |
| | | CH4 + CO2 → 2CO + 2H2ΔH0298K = 247 kJ mol−1 | (3) |
The indirect conversion process of CH4via syngas is an attractive process that is widely used at an industrial level. On the other hand, the syngas production process itself is costly and energy-intensive. In addition, the formation of hydrocarbons from synthesis gas requires either CO or H2 to remove the oxygen from CO leading to a loss of carbon atom utilization efficiency or consumption of valuable H2.2 In short, the indirect route is a process involving oxygen addition to and removal from hydrocarbons. In contrast, oxidative coupling of methane (OCM), which can yield C2 or higher olefins from methane in one step in the presence of oxygen,2–4 has been attracting great attention for further improving the efficiency of the CH4 conversion process. The advantage of the OCM reaction over conventional industrialized reactions is that OCM is an exothermic reaction (eqn (4)) and has no thermodynamic barriers due to negative changes in both enthalpy and Gibbs energy.
| | | 2CH4 + 1/2O2 → C2H6 + H2OΔH0298K = −177 kJ mol−1 | (4) |
Unlike indirect conversion
via synthesis gas, OCM has the potential to reduce conversion costs because of a reduction in the number of steps in the conversion process and the energy input.
Research concerning the OCM reaction started as early as the 1980s by Keller and Bhasin,5 and the reaction is known to go through a complex homogeneous–heterogeneous reaction network between the intermediates on the surface of catalysts and gas phase radicals.6,7 The widely accepted reaction mechanism of the OCM reaction is initiated by the dissociative adsorption of O2 on the catalyst surface to form surface oxygen species O*. The surface oxygen species abstract hydrogen atom from CH4 to form CH3 radicals, which dimerize in the gas phase to produce C2H6 as the primary product. However, because the resulting hydrocarbons can be easily activated compared to CH4 due to their weaker C–H bond strength, their overoxidation to COx is a challenge, which results in a loss of selectivity and the resulting yield at high CH4 conversion.8–10 Various catalysts, such as Li/MgO-, La2O3-, and Sm2O3-based catalysts, were investigated to improve the process's selectivity and reaction rate,11–15 One of the most studied catalysts is alkali metal-based tungstate, such as Na2WO4, due to its high C2 selectivity. The unique feature of the tungstate catalysts is that the addition of H2O in the reactant stream improves the CH4 conversion rate and the selectivity to C2 products.16,17 We have proposed that the OH radical is generated from H2O and O2 on the catalyst surface and that the resulting highly reactive OH radical readily abstracts the hydrogen atom from stable CH4.16–20 On the surface of the catalyst, alkali metal peroxides/superoxides were observed,18 and these species are suggested to be involved in catalyzing the OH radical formation. The reported single-pass C2 yield reached 25% using Mn/Na2WO4/SiO2 catalysts at 850 °C,21 which is close to the industrial target of ∼30%. The C2+ yield in a sequential dual reactor with Na2WO4/SiO2 reached 27.6% at 850–880 °C.22
Since OCM is an exothermic reaction as shown in eqn (4), the use of an adiabatic reactor is considered necessary to make the process more efficient via utilization of the reaction heat as thermal energy. However, the resulting gas mixture is predicted to reach a high temperature even above 1000 °C after complete depletion of O2.23 Although various studies have been devoted to developing selective catalysts and revealing their reaction mechanisms,24–30 the consequences of OCM products exposed under non-oxidative high temperature conditions have often been overlooked. For example, ethane generated from the OCM reaction is reported to undergo dehydrogenation to ethylene in the later stages at high temperatures after O2 depletion.31,32
| | | C2H6 → C2H4 + H2ΔH0298K = 136 kJ mol−1 | (5) |
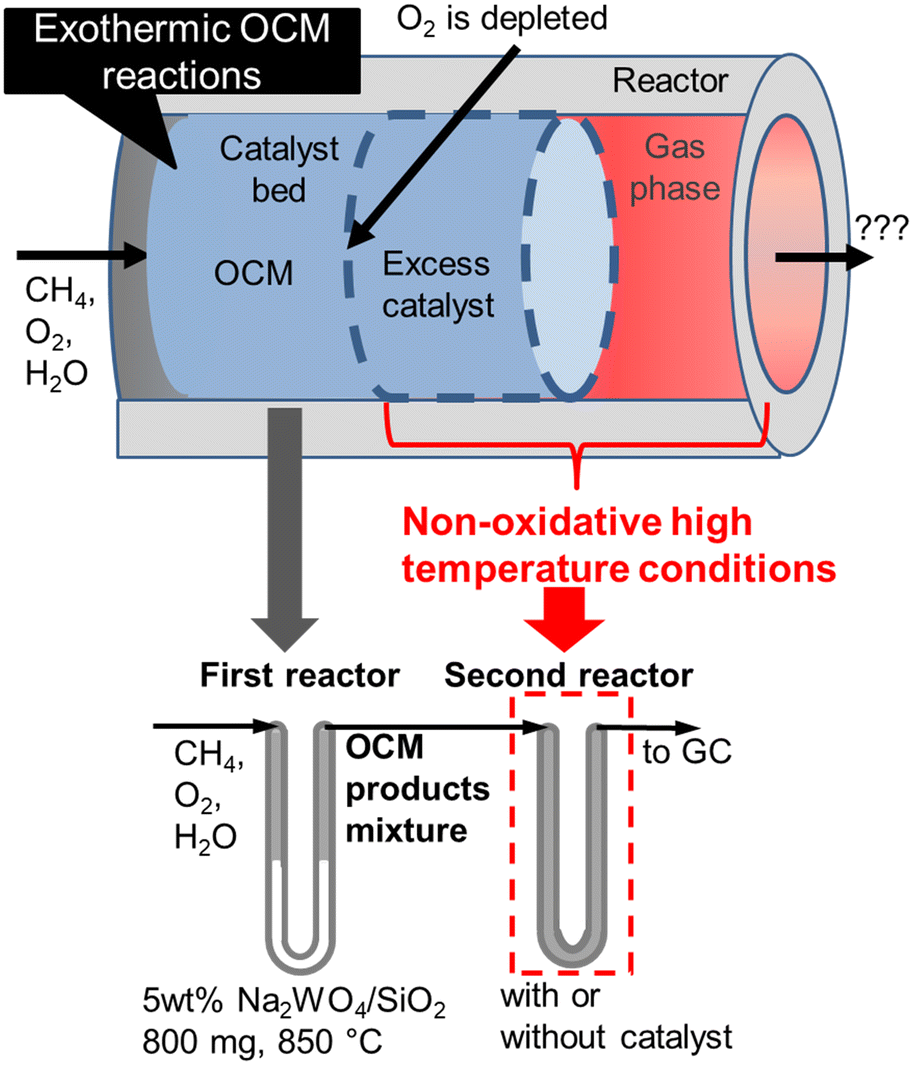
Nevertheless, undesired reactions involving OCM products have not been previously discussed. In an industrial reactor, excess amounts of catalysts need to be introduced to compensate for the potential loss of activity, a process that would result in the exposure of the OCM products to the extra catalysts and supports at high temperatures due to their exothermicity. In this study, we systematically investigated successive reactions of OCM products under high temperature non-oxidative conditions using a two-stage reactor that mimics an industrial reactor (Fig. 1). The OCM reaction is completed in the first stage reactor, while the OCM product mixture is exposed to the high temperature non-oxidative conditions in the second stage so that the successive reactions can be investigated. Both the homogeneous gas phase and heterogeneous reactions on various supports and catalysts after O2 depletion have been studied independently. Based on the present investigation, a design guideline for an OCM reactor that can achieve a high C2 yield is proposed.
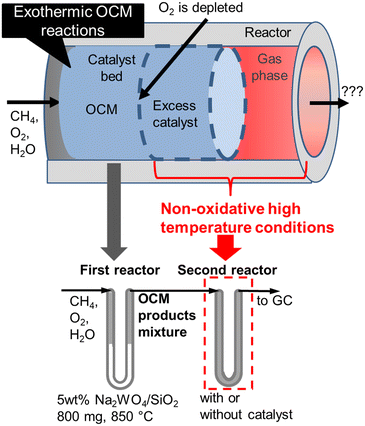 |
| | Fig. 1 Schematic representation of the two-stage reactor that mimics industrial reactors. | |
Experimental
Catalyst preparation
A wet impregnation method was used to prepare 5 wt% Na2WO4/SiO2 using Na2WO4·2H2O (≥99%, Sigma-Aldrich) as a precursor on a SiO2 support (Sigma-Aldrich, Silica gel, Davisil Grade 643, pore size 150 Å, 200–425 mesh), followed by calcination in air at 900 °C for 8 h. Al2O3 (Sigma-Aldrich, α-phase, −100 mesh) and ZrO2 (RC-100, 99.5%, Daiichi Kigenso Kagaku Kogyo) supports were used as received. Cristobalite-phase SiO2 was obtained by heating the above-mentioned SiO2 at 900 °C in air for 8 h. All samples were sieved after pelletization to obtain aggregates in a size range of 0.25 to 0.5 mm.
Catalytic performance evaluation
All activity tests were performed using a U-shaped quartz flow reactor (6 mm O.D., 4 mm I.D.) heated using an electric furnace (temperature distribution is shown in Fig. S1†). The catalyst bed was held between two plugs of quartz wool, and the thermocouple was located outside the quartz tube close to the catalyst bed to control the furnace temperature. CH4 (>99.999%) and O2 (20.0% diluted by Ar) were used as reactants, and Ar (>99.9999%) was used as a diluent. Mass flow controllers (Brooks) were used to obtain the desired gas composition and flow rate. A saturator with a temperature-controlled water jacket (15 °C) was used to introduce 1.7 kPa H2O in the reactant stream. The concentrations of the reactants and products were measured using an online gas chromatograph (Shimadzu GC-2014) equipped with a flame ionization detector ([FID] for hydrocarbon products) and a thermal conductivity detector ([TCD] for H2, O2, CO, and CO2), which are connected to GS-Gaspro and Shincarbon columns, respectively. All reactants and products passed through a water vapor trap that was located before the GC. The schematic image of the two-stage reactor is shown in Fig. 1, in which the OCM gas products from the first reactor were directly introduced into the second reactor. The reactor's furnace temperatures were controlled independently.
The conversion of methane and the selectivity to each carbon product and H2 were calculated as shown below.
| | 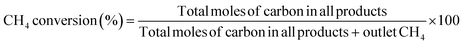 | (6) |
| | 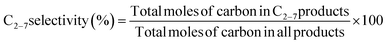 | (7) |
| |  | (8) |
The H2 selectivity in this study defines the percentage of the measured H2 relative to the hydrogen in the converted CH4 or relative to all the hydrogen in the products containing hydrogen.
Simulation of homogeneous gas phase reactions
Only the gas phase reactions of the OCM products are simulated without surface reactions. Simulations were performed using a plug-flow reactor model with the CHEMKIN software [ANSYS CHEMKIN-PRO v. 17.2, 2018] by varying the residence times and isothermal temperatures. The gas-phase chemical kinetic model is KAUST-Aramco PAH Mech 1-GS (KAM1-GS), which contains 574 species and 3379 reactions.33–35 OCM product mixtures, with experimentally obtained gas composition from the outlet of the first reactor, were fed to the inlet in the simulation.
Results and discussion
Selectivity under various O2 depletion conditions
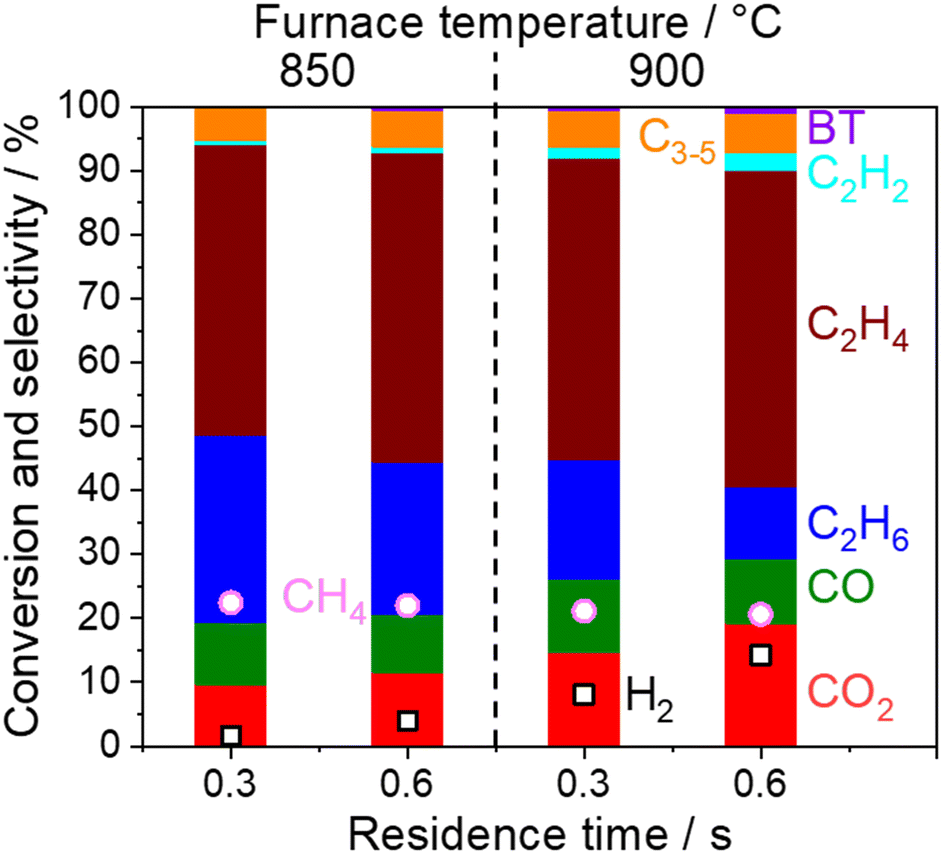
In the present study, 5 wt% Na2WO4/SiO2 is used as a model OCM catalyst due to its high C2 selectivity. C2 selectivity in a single reactor after O2 depletion was firstly investigated at various temperatures and residence times under dilute conditions (PCH4 = 10 kPa, PO2 = 1.7 kPa, and PH2O = 1.7 kPa), as shown in Fig. 2.
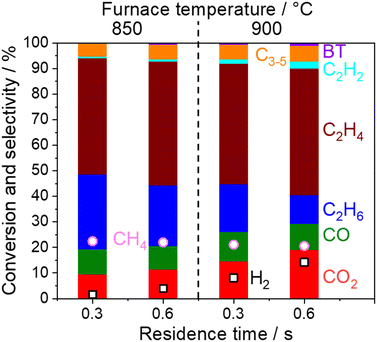 |
| | Fig. 2 Product distribution and CH4 conversion measured with a single quartz tube reactor using 800 mg of 5 wt% Na2WO4/SiO2 under O2 depletion conditions (850 or 900 °C, CH4 10 kPa, O2 1.7 kPa, H2O 1.7 kPa, total pressure 101 kPa, Ar balance, 40 or 80 mL min−1). The residence time was calculated by using the volume occupied by the catalysts in the quartz tube. BT denotes benzene and toluene. | |
CH4 conversion remained constant at approximately 22% under the studied conditions. C2+ selectivity was 80% at a furnace temperature of 850 °C and a residence time of 0.3 s, which is comparable to the results from a previous report.19 However, the H2, C2H2, C2H4, and COx selectivities increased while the C2H6 selectivity decreased at higher furnace temperatures and residence times, which indicates the presence of successive reactions of OCM products. Homogeneous gas phase reactions and heterogeneous surface reactions may occur at high temperatures. To clarify their contributions in the successive reactions of OCM products, a two-stage reactor, which mimics an industrial reactor, was used as shown in Fig. 1. The OCM reaction was first completed, which means that O2 was depleted in the first reactor using 800 mg of the model catalyst, 5 wt% Na2WO4/SiO2. The product gas mixture was further introduced to the second reactor in which the temperature could be controlled independently from that in the first reactor. The difference in gas compositions between the outlets from the first and second reactors allowed us to investigate the successive reactions of OCM products in non-oxidative environments. The homogeneous gas phase reactions were investigated without loading catalysts in the second quartz tube reactor, while the heterogeneous surface reactions were studied by loading various catalysts and supports into the reactor.
Contributions from homogenous gas phase reactions
Homogeneous reactions of the product gas mixture from the first stage OCM reactor were investigated using an empty quartz tube in the second reactor at high temperatures as shown in Fig. 3.
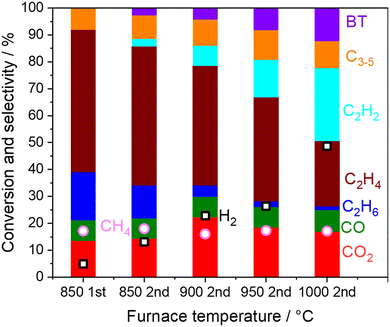 |
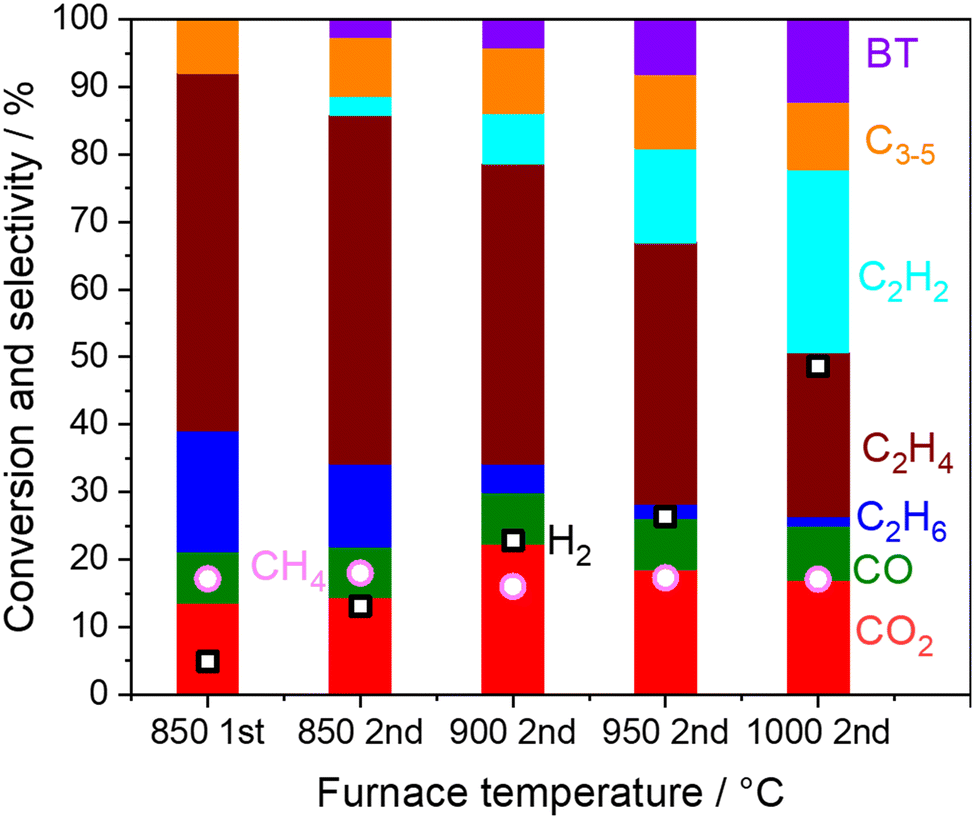
| | Fig. 3 Product distribution and CH4 conversion from the outlets of the first and the second reactor. Inlet gas; CH4 10 kPa, O2 1.4 kPa, H2O 1.7 kPa, total pressure 101 kPa, Ar balance, 40 mL min−1. The first reactor; 800 mg of 5 wt% Na2WO4/SiO2 at 850 °C. The second reactor; without catalysts at 850–1000 °C. O2 is depleted under all the conditions. | |
First, the result on the left side in Fig. 3 is the selectivity and conversion at the outlet from the first reactor. Full conversion of O2 was confirmed under the reaction at 850 °C, where the first reactor is shown in Fig. S2.† When the outlet gas from the first-stage reactor was introduced into the second reactor, the C2H6 and C2H4 selectivities decreased, while the C2H2 selectivity increased while increasing the furnace temperature. Considering that H2 selectivity also increased after O2 was already depleted, the dehydrogenation of C2 products seemed to have occurred in the gas phase as shown in eqn (5) and (9).
| | | C2H4 → C2H2 + H2ΔH0298K = 175 kJ mol−1 | (9) |
In general, gas phase reactions first require the initiation of radical formation from gas molecules. These radicals successively react with the gas phase molecules and reach a steady-state condition
via radical propagation reactions. The radical initiation reactions in the present OCM product gas mixture would be the following equations.
36| | | 2C2H4 → C2H3˙ + C2H5˙ | (11) |
CH
4 conversion did not change significantly. Since CH
4 is a stable hydrocarbon, pyrolysis reactions under non-oxidative conditions have been reported to proceed only at temperatures above 1200 °C.
37,38 The selectivity to C
3–5 also did not change significantly, while that to benzene and toluene increased with an increase in temperature in the second reactor, which was due to condensation of C
2H
2.
| | | 3C2H2 → C6H6ΔH0298K = −600 kJ mol−1 | (12) |
Graphitic carbon, which is produced after further condensation, was also observed on the quartz tube wall after the reaction. Formation of graphitic carbon is not considered in the calculation of the selectivity due to its negligible amount (<0.1% C selectivity). Since the dehydrogenation and condensation reactions are endothermic and exothermic reactions, respectively, these reactions are expected to be balanced. In the present study, the gas mixture diluted by Ar was investigated. The homogeneous reactions are expected to play an important role in determining the selectivity for the operation at industrially relevant high pressure (>1.0 MPa) in which intense reaction heat is also generated.
In the present experimental study, gas phase reactions are limited to the volume in the quartz tube microreactor (I.D. 4 mm and length approximately 8 cm) whose residence time is approximately 0.6 s. Although our microreactor system has the limitation of varying residence times, in large-scale industrial reactors, the product gas mixture would be exposed to high temperature, potentially for an extended time.39 Homogeneous gas phase reactions were further simulated under isothermal conditions with an extended residence time in a single plug-flow reactor (PFR) according to the reported gas-phase chemical kinetic model by feeding the OCM products, with experimentally obtained outlet composition from the first reactor. This simulation clarifies the reaction pathways in the gas phase without the contribution from the reactor wall, which leads to the change in product distributions.
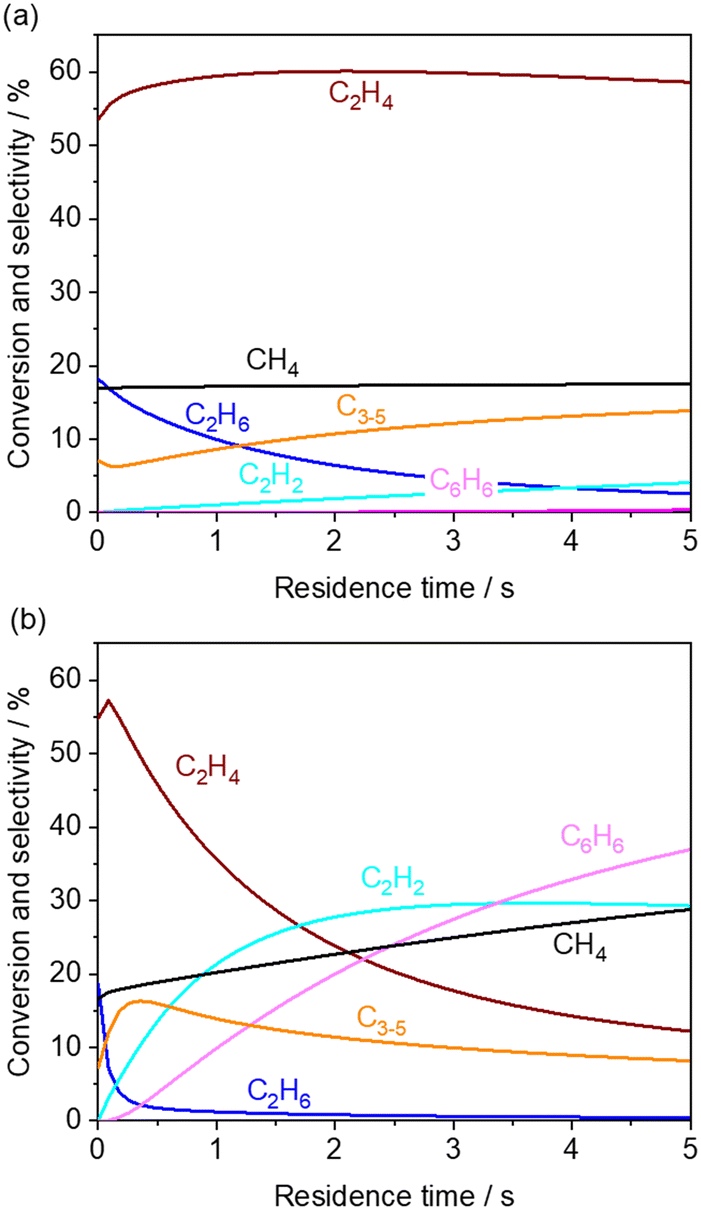
First, the simulated CH4 conversion did not change at 850 °C, while it gradually increased at 1000 °C due to pyrolysis (Fig. 4a and b).23,40 Meanwhile, in our measurement at the residence time of approximately 0.6 s, the CH4 conversion did not change significantly (Fig. 3). From the rate of production (ROP) analysis in the simulation, the H radical was found to be a major source in the reaction with CH4. In our experiments using a microreactor, quenching of H radical on the wall of the quartz tube may have occurred, which limited the conversion of CH4. Alternatively, CH4 was also formed from the CH3 radical after abstracting hydrogen from hydrocarbons. These processes of formation and consumption of CH4 were balanced; thus CH4 conversion was minimally affected.
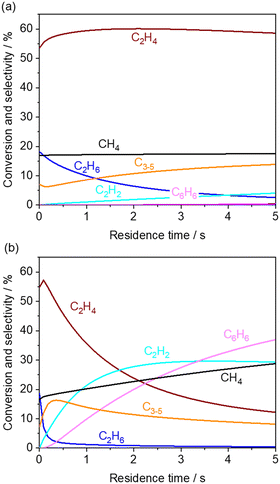 |
| | Fig. 4 Simulated product distribution and CH4 conversion at (a) 850 and (b) 1000 °C using KAM1-GS gas phase kinetics. Inlet gas composition was given by the experimentally obtained values from the outlet of the first reactor. The corresponding mole fractions are shown in Fig. S3.† | |
The simulated C2H6 selectivity decreases rapidly, while the C2H2 and C6H6 selectivity increases at higher temperatures, which agrees with our experimental observations. ROP analysis at the residence time of 0 s revealed that C2H6 is the major source in the radical initiation reactions to produce a radical compared to other hydrocarbons and H2O in the gas phase. A CH3 radical was produced from C2H6 through eqn (10) with the help of a third body, which leads to the following radical propagation reactions. The third body including inert gas itself is not consumed during the gas phase reactions but influences the reaction rate via collision either by providing the energy to form excited intermediates or by dissipating the energy from the excited intermediates after radical recombinations.41,42 The sharp decay of C2H6 selectivity is due to its weak C–H bond strength (423 kJ mol−1) compared to other hydrocarbons (CH4: 439 kJ mol−1; C2H4: 463 kJ mol−1).43 As the residence time increases, the C2 products transform to C2H2 and C6H6 in the simulation. In the present microreactor experimentally used, its volume is limited. However, these transformations may turn out to be significant in industrial reactors with a huge volume and generated heat. Mole fractions of CO and CO2 did not change significantly (Fig. S3†) by the gas phase kinetics at the simulated temperature and in the residence time range, which is in contrast to the experimental observation shown in Fig. 3. The deviation may be caused by heterogeneous steam reforming concurrently followed by the water gas shift reaction on the wall of the quartz tube, SiO2, which is discussed in the next section.
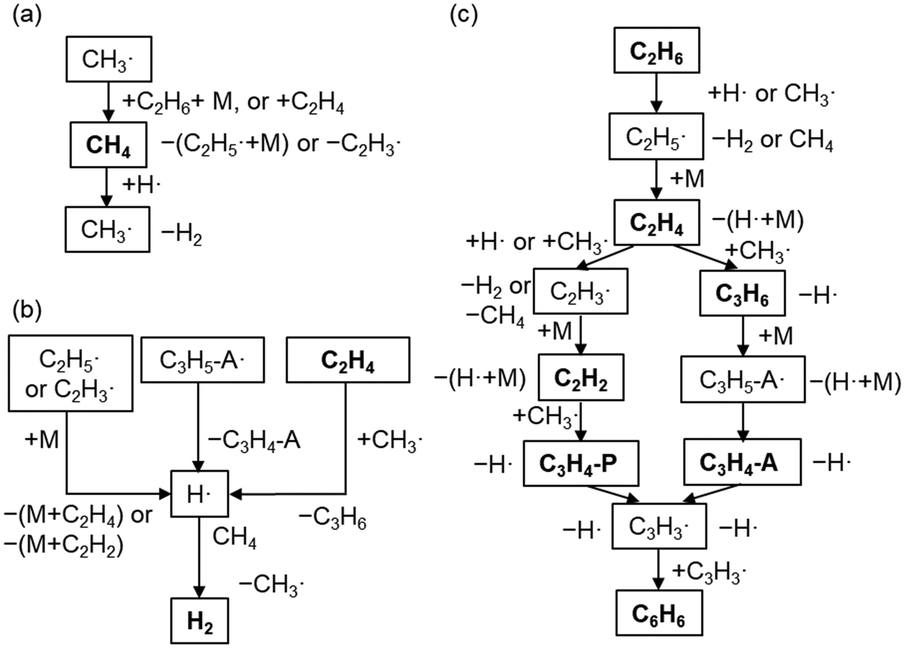
The major pathways obtained from ROP analysis in the simulation at 1000 °C and a residence time of 1 s are shown in Fig. 5. Fig. 5a shows the consumption and production cycle of CH4 during which CH4 is consumed by an H radical, and CH4 is regenerated through hydrogen abstraction from the major hydrocarbons via the CH3 radical. This cycle was most likely balanced in our experimental setup resulting in the maintenance of CH4 conversion. The H radical is formed from the decomposition of unstable hydrocarbon radicals (Fig. 5b). H radical can readily react with CH4, which is the primary component in the gas phase. Combination of pathways in Fig. 5a and b results in the dehydrogenation of C2 products to form H2. H2 hardly reacts in the gas phase. Hydrogen abstraction by H and CH3 radicals is the dominant reaction involved in consuming C2H6 and the formation of C2H5 radicals, which are further converted into C2H4 (Fig. 5c). The majority of C2H4 was found to have reacted with the CH3 radical to form C2H3 radicals and C3H6. Both of them are converted to C3H3 through a couple of radical reactions, which then finally form benzene. C2H2 and C3 products are the intermediate products in this pathway.
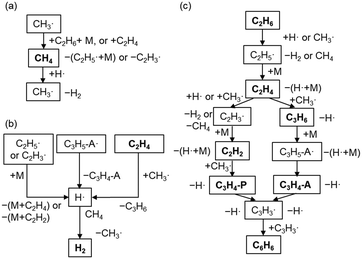 |
| | Fig. 5 Major pathways based on the ROP analysis at 1000 °C and a residence time of 1 s. (a) The production and consumption of CH4, (b) the formation of H radicals and H2, and (c) the transformation from C2 products to benzene. M, P, and A represent the third body, propene, and allene, respectively. | |
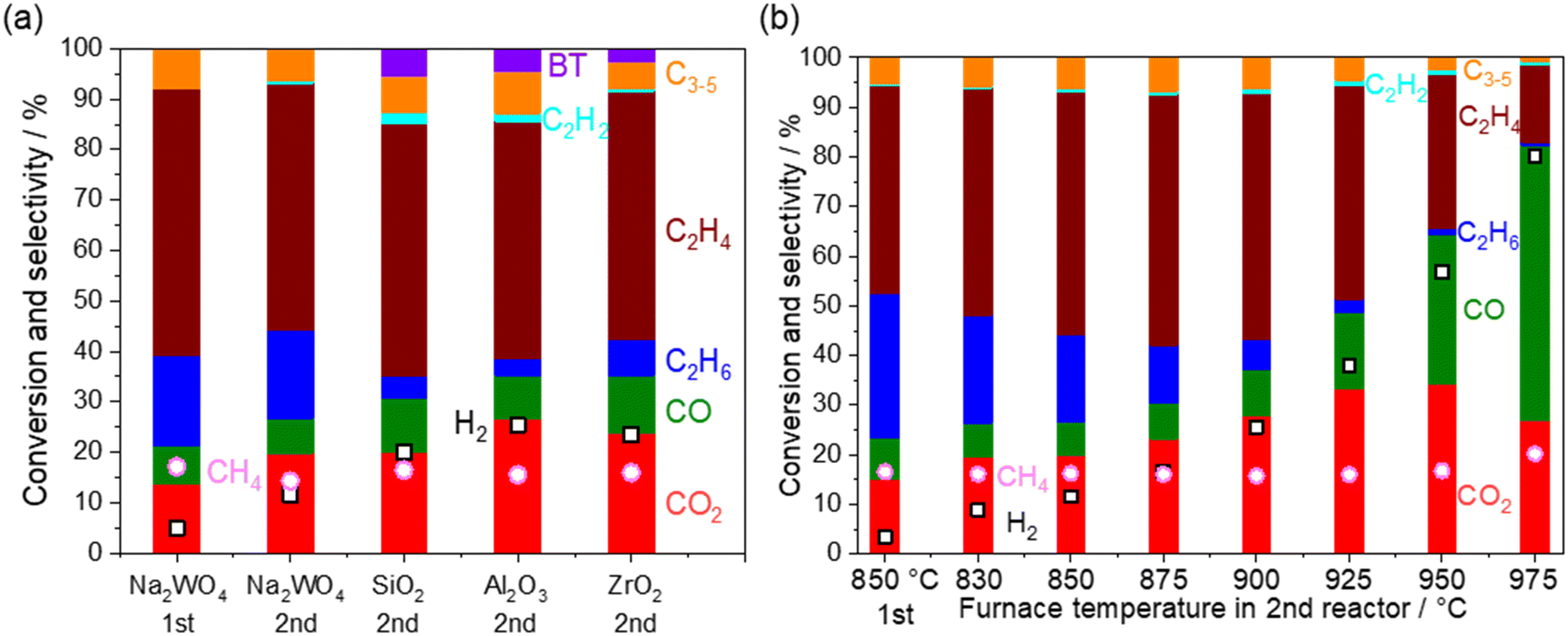
Heterogeneous reactions on various catalysts
Heterogeneous reactions on the catalysts and supports after O2 depletion were further investigated at 850 °C by loading various catalysts and supports (5 wt% Na2WO4/SiO2, SiO2, ZrO2, and Al2O3) in the second reactor (Fig. 6a). The cristobalite phase, SiO2, was selected because the SiO2 support is known to transform into cristobalite at the operating temperature in the presence of Na2WO4.21,28 First, CH4 conversion remained constant after going through the second reactor, indicating that CH4 hardly reacts under non-oxidative conditions even in the presence of these materials. After going through the second reactor, the selectivity to COx and H2 was increased at the expense of the C2 selectivity. Considering that the OCM product mixture also contained water vapor, steam reforming of C2 products and water gas shift reaction seemed to have occurred.| | | C2H6 + 2H2O → 2CO + 5H2ΔH0298K = 347 kJ mol−1 | (13) |
| | | C2H4 + 2H2O → 2CO + 4H2ΔH0298K = 210 kJ mol−1 | (14) |
| | | CO + H2O → CO2 + H2ΔH0298K = −41 kJ mol−1 | (15) |
Because the increase in the COx selectivity was not observed at 850 °C when the second reactor did not contain any catalyst and support (Fig. 3), these reactions seem to have occurred heterogeneously. Surprisingly, the above mentioned reactions occurred even on SiO2 (cristobalite), ZrO2, and Al2O3, which are often assumed to be inert and commonly used for reactor materials and supports. COx formation was slightly suppressed when 5 wt% of Na2WO4 was loaded onto the SiO2. This may be because molten Na2WO4 (melting temperature 697 °C) partially covered the surface of SiO2.20,25,44,45
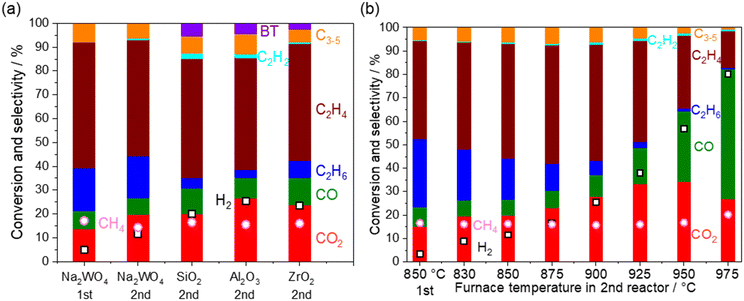 |
| | Fig. 6 Product distribution and CH4 conversion from the outlets of the first and the second reactor. Inlet gas; CH4 10 kPa, O2 1.4 kPa, H2O 1.7 kPa, total pressure 101 kPa, Ar balance, 40 mL min−1. O2 is depleted under all the conditions. The first reactor: 800 mg of 5 wt% Na2WO4/SiO2 at 850 °C. (a) The second reactor: 800 mg of the catalysts (5 wt% Na2WO4/SiO2) or 500 mg of the supports (SiO2, Al2O3, and ZrO2) at 850 °C. (b) The second reactor: 800 mg of the catalysts (5 wt% Na2WO4/SiO2) at 830–975 °C. | |
Regarding the reported steam reforming and water gas shift reactions on oxides, Fe2O3/Cr2O3 and CuO/ZnO are used industrially for water gas shift reactions below 500 °C.46,47 Their common research direction is to lower the reaction temperature, a process that is thermodynamically preferred for obtaining a high H2 yield. Steam reforming of ethylene using oxide materials without supported metals has been rarely reported. NiAl2O4 and MnCr2O4 spinel oxides, which are candidate barrier layers in hydrocarbon steam crackers to prevent coke formation, were evaluated for the steam reforming of ethylene below 700 °C.48,49 The present study revealed a unique challenge on oxides after OCM, which operates at high temperature (>800 °C).
The temperature in the second reactor with 5 wt% Na2WO4/SiO2 as the model catalyst was further varied to mimic the extra amount of catalyst exposed at high temperature generated by the exothermic OCM reaction (Fig. 6b). Again, CH4 conversion remained constant even at 975 °C, indicating the difficulty in activating the stable CH4 in a non-oxidative environment. COx and H2 selectivities increased significantly after increasing the temperature, whereas C2 selectivity decreased. Notably, the loss of C2 selectivity was also observed even at 830 °C, which is below the operating temperature for OCM. A closer look at the results reveals that the consumption of C2H6 was greater than the production of COx in the temperature range below 950 °C. The selectivity to C2H4 increased from the outlet of the first reactor in this temperature range. These observations suggest that the dehydrogenation of C2H6 to C2H4 (eqn 5) dominates the consumption of C2H6 compared to the steam reforming of C2H6 to CO (eqn 13). Therefore, the COx formation should originate from the steam reforming of C2H4 (eqn 14). Similar interpretations have been previously reported during the steam reforming of C2H4 and C2H6 using a Mn0.5Cr2.5O4 spinel catalyst operated below 700 °C.49 The measured H2 formation in the second reactor agrees with the one estimated from the consumption of C2H6 and COx formation (see Fig. S4 and ESI† note 1). The steam reforming and the water gas shift reactions seem to be the major H2 source, particularly at high temperatures.
The water gas shift reaction on the commonly used metal catalysts, such as Ni, Ru, Rh, Pt, and Ir, is known to be reversible and readily reaches equilibrium.47,50,51 The outlet gas composition from the second reactor was compared with the equilibrium constant of the water gas shift reaction, KWGS, at various temperatures using the equation shown below (Fig. S5†):
| | 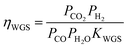 | (16) |
ηWGS showed a gradual increase by increasing the furnace temperature due to the enhanced kinetics till its equilibrated value of unity above 900 °C was reached. This result further clarifies that the excess heat needs to be effectively removed after O
2 depletion to avoid the formation of undesired CO
2.
The product gas mixture from the first reactor was directly introduced in the second reactor in this study. Investigations on the heterogeneous reactions for the individual hydrocarbons in the presence of steam would help to further clarify their reaction rates, which can be input parameters when designing industrial reactors.
Overall, the present study revealed significant contributions from the gas phase, catalysts, and reactor materials towards the successive reactions of OCM products under high temperature non-oxidative conditions. Particularly, the catalytic steam reforming and water gas shift reactions, which would occur on an excess amount of catalysts and reactor materials especially at increased temperatures from exothermic reaction, need to be suppressed to prevent undesired COx formation. Because OCM is an exothermic reaction, measures to effectively dissipate the excess heat would be needed to prevent any undesired reactions. For example, microchannel reactors with coated catalyst walls are expected to have a high heat transfer coefficient, which is inversely dependent on the tube diameter.52,53 A co- or counter flow of a coolant can be also introduced within the channels.54 Alternatively, a monolith structure may facilitate the heat and mass transfer compared to the packed bed reactor.55 Metallic foams are considered thermally conductive monolith supports.56–58 Membrane reactors, which provide O2 gradually along the reaction zone, may prevent the formation of strong hot spots.59–61 Cooling by endothermic reactions is also proposed, for example, successive steam reforming of unconverted CH4 to syngas using the reaction heat from OCM,62 although these reactions may not fully compensate the strong exothermicity from OCM. Additionally, since the steam reforming reaction was also observed below the operation temperature of OCM (Fig. 6b), the residence time in the catalyst bed needs to be carefully controlled. Efforts to improve the stability of the catalyst are highly needed to reduce the excess amount of the catalyst. Since the steam reforming reaction was also observed using ZrO2 and Al2O3, which are commonly used supports as well as reactor materials, the choice of those materials should be avoided or its surface decoration to make it inert is necessary. Operando temperature measurements within the catalyst bed also helps to develop efficient OCM reactors.63–66
Conclusions
The present study investigated the successive reactions of OCM products under non-oxidative conditions at high temperatures, which would be generated via exothermic heat after depletion of O2 in industrial reactors. The developed two-stage reactor successfully revealed the contributions from the gas-phase and heterogeneous reactions. In the absence of catalysts and supports, C2 products dehydrogenate to C2H2 followed by condensation to benzene, toluene, and graphitic carbon deposits, which would cause the pressure drop and suppression of heat transfer in OCM reactors. Notably, the steam reforming of the C2 products and water gas shift reactions were observed to lead to the loss of C2 yield in the presence of the model catalysts and support materials, which are commonly considered to be inactive. Research efforts should be directed to design an efficient OCM reactor to remove the excess heat generated to prevent the undesired reactions. The residence time also needs be carefully controlled to minimize the extra contact of the C2 products with catalysts and reactor materials at increased temperature.
Author contributions
Conceptualization, K. T.; investigation H. K., K. O., and D. L.; writing – original draft, H. K. and K. O.; writing – review & editing, K. O., S. M. S. and K. T.; supervision, K. T.; funding acquisition, K. T.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by MHI Innovation Accelerator LLC.
References
- M. J. da Silva, Fuel Process. Technol., 2016, 145, 42–61 CrossRef.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- J. H. Lunsford, Angew. Chem., Int. Ed. Engl., 1995, 34, 970–980 CrossRef CAS.
- J. T. Grant, J. M. Venegas, W. P. McDermott and I. Hermans, Chem. Rev., 2018, 118, 2769–2815 CrossRef CAS PubMed.
- G. Keller and M. M. Bhasin, J. Catal., 1982, 73, 9–19 CrossRef CAS.
- M. Y. Sinev, Z. T. Fattakhova, V. I. Lomonosov and Y. A. Gordienko, J. Nat. Gas Chem., 2009, 18, 273–287 CrossRef CAS.
- M. Y. Sinev, J. Catal., 2003, 216, 468–476 CrossRef CAS.
- S. Pak, P. Qiu and J. H. Lunsford, J. Catal., 1998, 179, 222–230 CrossRef CAS.
- J. A. Labinger and K. C. Ott, J. Phys. Chem., 1987, 91, 2682–2684 CrossRef CAS.
- J. A. Labinger, Catal. Lett., 1988, 1, 371–375 CrossRef CAS.
- J. S. Lee and S. T. Oyama, Catal. Rev.: Sci. Eng., 1988, 30, 249–280 CrossRef CAS.
- C. Batiot and B. K. Hodnett, Appl. Catal., A, 1996, 137, 179–191 CrossRef CAS.
- J. Song, Y. Sun, R. Ba, S. Huang, Y. Zhao, J. Zhang, Y. Sun and Y. Zhu, Nanoscale, 2015, 7, 2260–2264 RSC.
- D. Noon, A. Seubsai and S. Senkan, ChemCatChem, 2013, 5, 146–149 CrossRef CAS.
- H. Wang, C. Yang, C. Shao, S. Alturkistani, G. Magnotti, J. Gascon, K. Takanabe and S. M. Sarathy, ChemCatChem, 2022, 14, e202200927 CAS.
- K. Takanabe and E. Iglesia, Angew. Chem., Int. Ed., 2008, 47, 7689–7693 CrossRef CAS PubMed.
- K. Takanabe and E. Iglesia, J. Phys. Chem. C, 2009, 113, 10131–10145 CrossRef CAS.
- D. Li, W. S. Baslyman, S. M. Sarathy and K. Takanabe, Energy Technol., 2020, 8, 1900563 CrossRef CAS.
- D. Li, S. Yoshida, B. Siritanaratkul, A. T. Garcia-Esparza, D. Sokaras, H. Ogasawara and K. Takanabe, ACS Catal., 2021, 11, 14237–14248 CrossRef CAS.
- K. Takanabe, A. M. Khan, Y. Tang, L. Nguyen, A. Ziani, B. W. Jacobs, A. M. Elbaz, S. M. Sarathy and F. F. Tao, Angew. Chem., Int. Ed., 2017, 56, 10403–10407 CrossRef CAS PubMed.
- A. Palermo, J. Holgadovazquez, A. Lee, M. Tikhov and R. Lambert, J. Catal., 1998, 177, 259–266 CrossRef CAS.
- Y. Liang, Z. Li, M. Nourdine, S. Shahid and K. Takanabe, ChemCatChem, 2014, 6, 1245–1251 CAS.
- D. Li, W. S. Baslyman, B. Siritanaratkul, T. Shinagawa, S. M. Sarathy and K. Takanabe, Ind. Eng. Chem. Res., 2019, 58, 22884–22892 CrossRef CAS.
- S. Sourav, Y. Wang, D. Kiani, J. Baltrusaitis, R. R. Fushimi and I. E. Wachs, Angew. Chem., Int. Ed., 2021, 60, 21502–21511 CrossRef CAS PubMed.
- M. J. Werny, Y. Wang, F. Girgsdies, R. Schlögl and A. Trunschke, Angew. Chem., Int. Ed., 2020, 59, 14921–14926 CrossRef CAS PubMed.
- Z. C. Jiang, C. J. Yu, X. P. Fang, S. B. Li and H. L. Wang, J. Phys. Chem., 1993, 97, 12870–12875 CrossRef CAS.
- S. Ji, T. Xiao, S. Li, L. Chou, B. Zhang, C. Xu, R. Hou, A. P. E. York and M. L. H. Green, J. Catal., 2003, 220, 47–56 CrossRef CAS.
- D. Kiani, S. Sourav, W. Taifan, M. Calatayud, F. Tielens, I. E. Wachs and J. Baltrusaitis, ACS Catal., 2020, 10, 4580–4592 CrossRef CAS.
- D. J. Wang, M. P. Rosynek and J. H. Lunsford, J. Catal., 1995, 155, 390–402 CrossRef CAS.
- D. Kiani, S. Sourav, J. Baltrusaitis and I. E. Wachs, ACS Catal., 2019, 9, 5912–5928 CrossRef CAS.
- C. Guéret, M. Daroux and F. Billaud, Chem. Eng. Sci., 1997, 52, 815–827 CrossRef.
- F. Larkins and A. Khan, Aust. J. Chem., 1989, 42, 1655 CrossRef CAS.
- S. M. Burke, W. Metcalfe, O. Herbinet, F. Battin-Leclerc, F. M. Haas, J. Santner, F. L. Dryer and H. J. Curran, Combust. Flame, 2014, 161, 2765–2784 CrossRef CAS.
- W. K. Metcalfe, S. M. Burke, S. S. Ahmed and H. J. Curran, Int. J. Chem. Kinet., 2013, 45, 638–675 CrossRef CAS.
- D. Darcy, H. Nakamura, C. J. Tobin, M. Mehl, W. K. Metcalfe, W. J. Pitz, C. K. Westbrook and H. J. Curran, Combust. Flame, 2014, 161, 65–74 CrossRef CAS.
- K. M. Sundaram and G. F. Froment, Ind. Eng. Chem. Fundam., 1978, 17, 174–182 CrossRef CAS.
- A. Holmen, O. Olsvik and O. A. Rokstad, Fuel Process. Technol., 1995, 42, 249–267 CrossRef CAS.
- U. P. M. Ashik, W. M. A. W. Daud and H. F. Abbas, Renewable Sustainable Energy Rev., 2015, 44, 221–256 CrossRef CAS.
-
G. Radaelli, G. Chachra and D. Jonnavittula, Low-Energy, Low-Cost Production of Ethylene by Low- Temperature Oxidative Coupling of Methane, United States, 2017 Search PubMed.
- H. B. Palmer, J. Lahaye and K. C. Hou, J. Phys. Chem., 1968, 72, 348–353 CrossRef CAS.
- M. Y. Sinev, Y. P. Tulenin, O. V. Kalashnikova, V. Y. Bychkov and V. N. Korchak, Catal. Today, 1996, 32, 157–162 CrossRef CAS.
- M. Sinev, V. Arutyunov and A. Romanets, Adv. Chem. Eng., 2007, 32, 167–258 CAS.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- Z. C. Jiang, C. J. Yu, X. P. Fang, S. B. Li and H. L. Wang, J. Phys. Chem., 1993, 97, 12870–12875 CrossRef CAS.
- Z. Q. Yu, X. M. Yang, J. H. Lunsford and M. P. Rosynek, J. Catal., 1995, 154, 163–173 CrossRef CAS.
- C. Rhodes, G. J. Hutchings and A. M. Ward, Catal. Today, 1995, 23, 43–58 CrossRef CAS.
- S. Hla, D. Park, G. Duffy, J. Edwards, D. Roberts, A. Ilyushechkin, L. Morpeth and T. Nguyen, Chem. Eng. J., 2009, 146, 148–154 CrossRef CAS.
- L. Yang, M. P. Bukhovko, A. Malek, L. Li, C. W. Jones, P. K. Agrawal and R. J. Davis, Appl. Catal., A, 2020, 603, 117739 CrossRef CAS.
- L. Yang, M. P. Bukhovko, G. Brezicki, A. Malek, L. Li, C. W. Jones, P. K. Agrawal and R. J. Davis, J. Catal., 2019, 380, 224–235 CrossRef CAS.
- J. Wei and E. Iglesia, J. Phys. Chem. B, 2004, 108, 4094–4103 CrossRef CAS.
- A. Yamaguchi and E. Iglesia, J. Catal., 2010, 274, 52–63 CrossRef CAS.
- R. Knitter and M. A. Liauw, Lab Chip, 2004, 4, 378 RSC.
- T. Serres, L. Dreibine and Y. Schuurman, Chem. Eng. J., 2012, 213, 31–40 CrossRef CAS.
- I. Tezcan and A. K. Avci, J. Chem. Technol. Biotechnol., 2015, 90, 1827–1838 CrossRef CAS.
- G. Groppi and E. Tronconi, Chem. Eng. Sci., 2000, 55, 2161–2171 CrossRef CAS.
- M. Bracconi, M. Ambrosetti, M. Maestri, G. Groppi and E. Tronconi, Chem. Eng. Process., 2018, 129, 181–189 CrossRef CAS.
- L. Giani, G. Groppi and E. Tronconi, Ind. Eng. Chem. Res., 2005, 44, 9078–9085 CrossRef CAS.
-
C. G. Visconti, A. Montebelli, G. Groppi, E. Tronconi and S. Kohler, in Methanol, Elsevier, 2018, pp. 519–538 Search PubMed.
- H. R. Godini, S. Xiao, M. Kim, N. Holst, S. Jašo, O. Görke, J. Steinbach and G. Wozny, J. Ind. Eng. Chem., 2014, 20, 1993–2002 CrossRef CAS.
- M.-S. Salehi, M. Askarishahi, H. R. Godini, O. Görke and G. Wozny, Ind. Eng. Chem. Res., 2016, 55, 3287–3299 CrossRef CAS.
- N. Holst, S. Jašo, H. R. Godini, S. Glöser, H. Arellano-Garcia, G. Wozny and J. Steinbach, Chem. Eng. Technol., 2012, 35, 294–301 CrossRef CAS.
- T. P. Tiemersma, T. Kolkman, J. A. M. Kuipers and M. van Sint Annaland, Chem. Eng. J., 2012, 203, 223–230 CrossRef CAS.
- S. Pak and J. H. Lunsford, Appl. Catal., A, 1998, 168, 131–137 CrossRef CAS.
- B. Zohour, D. Noon and S. Senkan, ChemCatChem, 2013, 5, 2809–2812 CrossRef CAS.
- D. Noon, B. Zohour and S. Senkan, J. Nat. Gas Sci. Eng., 2014, 18, 406–411 CrossRef CAS.
- O. Korup, S. Mavlyankariev, M. Geske, C. F. Goldsmith and R. Horn, Chem. Eng. Process.: Process Intesif., 2011, 50, 998–1009 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Duanxing
Li
a,
Duanxing
Li