
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBinding modes of high stoichiometry guest complexes with a Co8L12 cage uncovered by mass spectrometry†
Daniel L.
Stares‡
 a,
Cristina
Mozaceanu‡
b,
Michael D.
Ward
*b and
Christoph A.
Schalley
*a
a,
Cristina
Mozaceanu‡
b,
Michael D.
Ward
*b and
Christoph A.
Schalley
*a
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Arnimallee 20, Berlin, 14195, Germany. E-mail: c.schalley@fu-berlin.de
bDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: m.d.ward@warwick.ac.uk
First published on 13th September 2023
Abstract
We demonstrate how different modes of guest binding with a Co8L12 cubic cage can be determined using ESI-MS. High stoichiometry guest binding was observed, with the guests preferentially binding externally, but internal guest inclusion was also seen at higher guest loading.
Self-assembled metal–organic cages are some of the most sophisticated and complex synthetic host structures.1,2 Characterisation of these hosts typically relies on either NMR or X-ray crystallography with it being less common to utilise mass spectrometry (MS) for structural analysis. MS has the general benefits that an ion of interest can be mass-selected, so it does not require completely pure samples; it is highly sensitive and data can be obtained relatively quickly.3 For self-assembled systems more specifically, MS has the added advantage that it deals with isolated ions, free from any competitive solvent, and as such, any dynamic exchange processes which can complicate characterisation in solution are absent.4 Because of this, the mass-to-charge ratio (m/z) of an ion easily furnishes a system's stoichiometry which can be especially useful when studying host–guest systems capable of binding multiple guest molecules.5,6 Beyond stoichiometry, structural information such as guest binding mode can be gained through tandem MS techniques such as ion-mobility mass spectrometry (IMS) and collision-induced dissociation (CID).7–10 IMS separates ions based on their size and can thus distinguish isomers that vary due to different isomeric ligands, guest binding position and even diastereomeric composition of the cage.11–15 CID is a dissociation technique, where ions are collided with a neutral buffer gas to induce fragmentation and can be used to probe structural features of the cage as well as guest binding modes.16–18
The Co8L12 cubic cage (HWCo) (Fig. 1), based on hydroxymethyl-substituted bis-bidentate ligands (LW) which span the edges of an approximately cubic array of Co(II) ions, can bind guests internally and externally and is also capable of binding multiple guests.19–22 However, multiple guest binding has only been demonstrated using X-ray crystallography and NMR spectroscopy under forcing conditions with a large excess of guest.19–21 Detailed MS studies, which are well suited to studying higher stoichiometry guest binding, have currently not been performed. Herein, a combination of IMS and CID experiments were used to investigate the host–guest properties of HWCo, particularly to uncover its multiple guest binding capabilities. The results serve as a further case study of the benefit of MS in the study of host–guest chemistry of metal–organic cages especially by allowing observation of different binding modes.
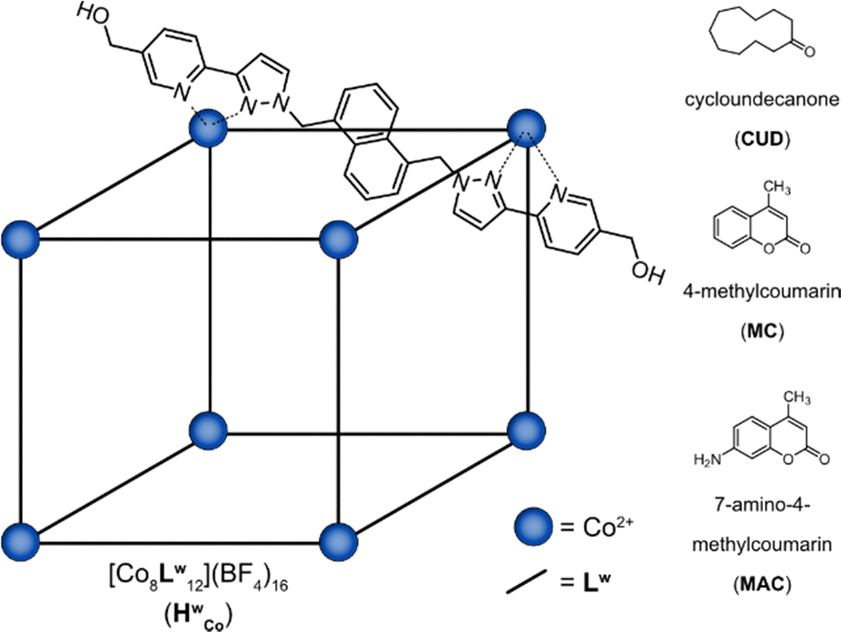 | ||
| Fig. 1 Structure of the HWCo cage and the guests used in the current study. | ||
HWCo ionises through loss of BF4− counterions generating charge states from 5+ to 10+ (Fig. 2). Additionally, some fragment ions were present and a loss of neutral HBF4 (likely as HF and BF3) was also observed for charge states 5+ to 9+. The charge state distribution could be shifted towards higher charges by increasing the capillary voltage, but cage fragmentation became more pronounced and charge states over 10+ were still not observed (Fig. S1, ESI†). This indicates that charge states over 10+ are not stable enough to survive in the instrument with the six counterions necessary to counterbalance the charge repulsion within the cage. X-ray crystal structures of HWCo show discrete binding of six counterions in the portals of the faces which will significantly reduce charge repulsion of the cage as these counterions are surrounded by four metal centres and their respective charges.23,24 The removal of these six counterions during ionisation would thus be more difficult than peripherally bound ones requiring higher capillary voltages to do so, and, if successfully removed, would lead to significant destabilisation.25 The interplay of these two factors can explain why charge states greater than 10+ are not observed. CID of the cage showed fragmentation into face, corner and edge fragments, (Fig. S2 and S3, ESI†) but overall, the cage is relatively stable, considering the charge state, with HWCo10+ surviving until a collision voltage of 8 V (Fig. S4, ESI†).
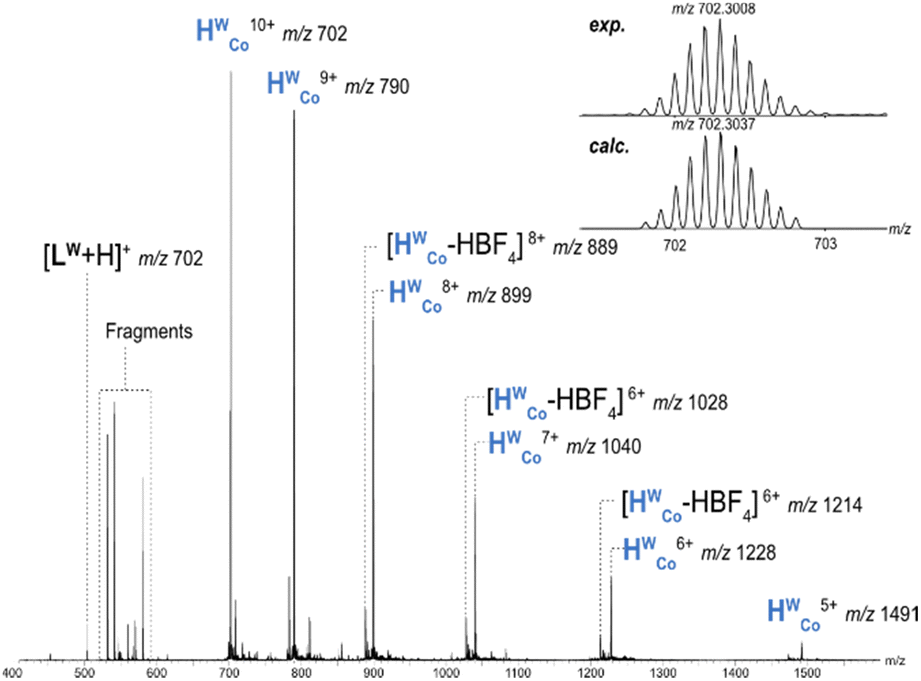 | ||
| Fig. 2 ESI-MS spectrum of 10 μM HWCo in H2O, the 10+ to 5+ cage charge states are marked. Fragmentation products and cage species resulting from the loss of HBF4 are also observed. The experimental vs. calculated m/z values for HWCo10+ are shown in the inset. | ||
The host–guest chemistry of HWCo was then explored with guests whose binding has been studied in solution and the solid state: cycloundecanone (CUD); 4-methylcoumarin (MC); and 7-amino-4-methylcoumarin (MAC) (Fig. 1). CUD has been shown to bind strongly inside the cavity of HWCo in water driven by the hydrophobic effect with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 HWCo/CUD stoichiometry.26 In contrast, it has been shown that multiple guest binding is possible when MC and MAC are used as guests.19 Accordingly, a 1:1 HWCo/CUD complex was observed in the mass spectrum with no evidence of multiple guests binding (Fig. S5, ESI†) whilst for MC, small signals for the 1:2 complex were seen in addition to the 1:1 HWCo/MC complex (Fig. S6, ESI†). With MAC, strong signals were observed for HWCo/(1–4)MAC complexes with even higher host:guest stoichiometries also seen albeit at lower signal intensities (Fig. 3a and Fig. S7, ESI†). The results for CUD and MC are consistent with what has previously been observed in solution and the solid state but the higher stoichiometries associated with MAC binding are observed for the first time.
:1 HWCo/CUD stoichiometry.26 In contrast, it has been shown that multiple guest binding is possible when MC and MAC are used as guests.19 Accordingly, a 1:1 HWCo/CUD complex was observed in the mass spectrum with no evidence of multiple guests binding (Fig. S5, ESI†) whilst for MC, small signals for the 1:2 complex were seen in addition to the 1:1 HWCo/MC complex (Fig. S6, ESI†). With MAC, strong signals were observed for HWCo/(1–4)MAC complexes with even higher host:guest stoichiometries also seen albeit at lower signal intensities (Fig. 3a and Fig. S7, ESI†). The results for CUD and MC are consistent with what has previously been observed in solution and the solid state but the higher stoichiometries associated with MAC binding are observed for the first time.
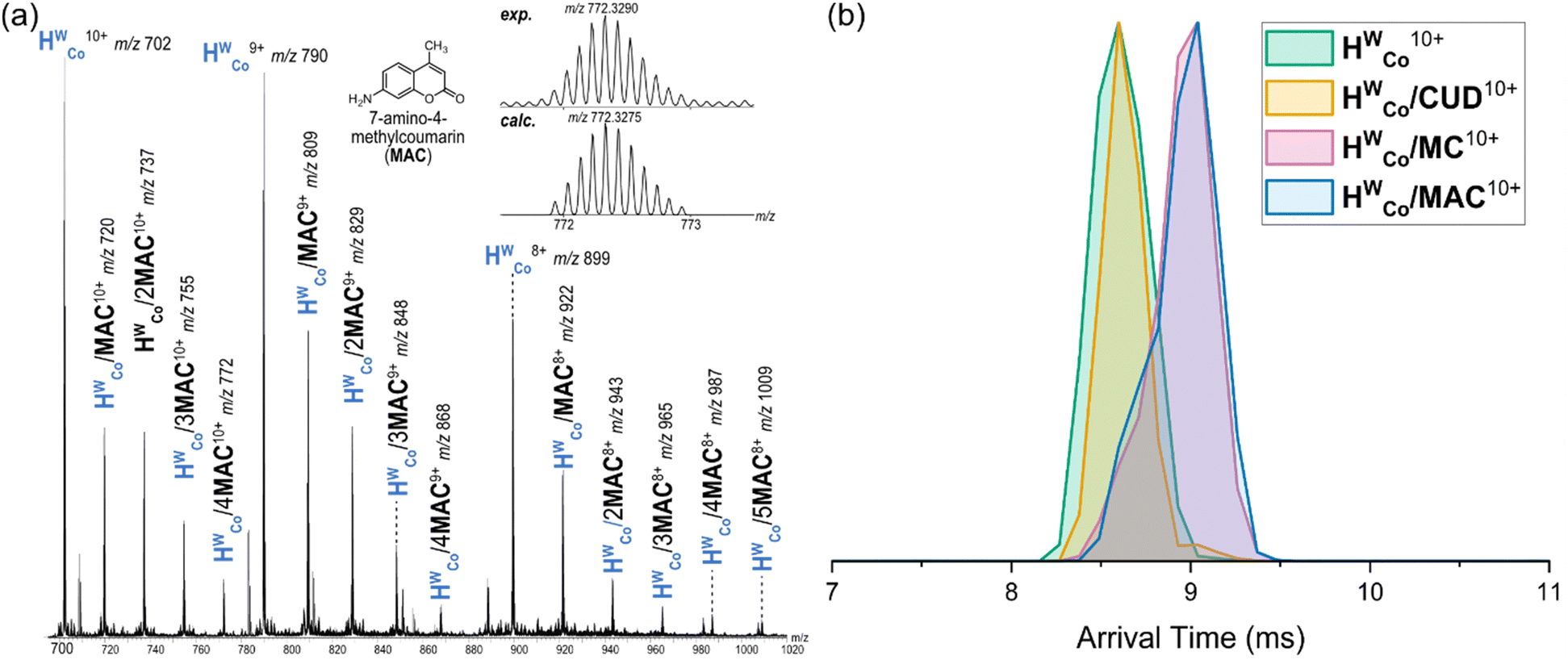 | ||
| Fig. 3 (a) ESI-HRMS spectrum of HWCo with MAC (10 μM/50 μM) in 5% CH3OH/H2O. Multiple guest binding stoichiometries with up to 5 MACs bound are observed. The experimental vs. calculated m/z values for the HWCo/4MAC10+ are shown in the inset (b) ATDs of HWCo10+, HWCo/CUD10+, HWCo/MC10+, HWCo/MAC10+. The ATDs of HWCo10+ and HWCo/CUD10+ overlap indicating encapsulation whilst HWCo/MC10+ and HWCo/MAC10+ have longer arrival times consistent with external binding. | ||
For all three guests, CID of the 1:1 HWCo/Guest (G)8+ species resulted in complete guest loss to the free HWCo8+ at low collision voltages before further fragmentation of HWCo8+ as seen for the free cage (Fig. S3, S4 and S8–S10, ESI†). Although a lower charge state was mass-selected to reduce Coulomb repulsion and instrument conditions were softened as much as possible, a portion of empty cage was still observed after mass selection of HWCo/G8+ ions without additional collision voltages applied. This means that some guest dissociation can occur just from transmission through the instrument. The relative intensities of HWCo/CUD8+ and HWCo/MAC8+ (71 and 77%, respectively) after mass selection are considerably higher than that for HWCo/MC8+ (47%) meaning that higher proportions of the CUD and MAC complexes survive transmission though the instrument (Fig. S11, ESI†). Complete guest loss was observed at low collision voltages with all guests but as the HWCo/G complexes do not represent 100% of the total intensity after mass selection, it is not possible to complete a full survival yield analysis to determine relative stabilities of the guests. Thus, no reasonable conclusion about the stabilities of the host:guest complexes can be drawn other than that all three guests are lost at similar collision voltages.
IMS of the different 1:1 HWCo/G complexes showed comparable arrival times for HWCo10+ and HWCo/CUD10+ with longer arrival times for HWCo/MC10+ and HWCo/MAC10+ (Fig. 3b). A straightforward explanation of these results is that CUD is encapsulated in the central cavity of HWCo as seen in crystal structures,26 and hence does not contribute significantly to the collisional cross section (CCS) of the cage ion, whereas both MC and MAC are bound to the external cage surface increasing the CCS and hence arrival times. Although a comparison of absolute CCS values is difficult due to a lack of suitable calibrants and issues with the theoretical calculations for metal–organic complexes,27 relative comparisons of theoretical CCS values are still insightful as any errors cancel each other out. Here, theoretical CCS values using the trajectory method (TMCCSN2) were calculated for universal force field (UFF) optimised structures of HWCo10+ and HWCo/G10+ with the different guests.28,29 They supported the above assessment by showing the same general results as the IMS with very similar TMCCSN2 values for HWCo10+ and HWCo/CUD10+ when the CUD was encapsulated; with increases in the TMCCSN2 values of HWCo/MC10+ and HWCo/MAC10+ with MAC and MC binding externally via hydrogen bonding with LW (Fig. S12, ESI†). Thus, the IMS experiments strongly support encapsulation of CUD in the host cavity, with external binding of MAC and MC for the 1:1 stoichiometries.
Encapsulation of CUD in solution is driven by the hydrophobic effect which is not present in the gas-phase and thus CUD complexes may be expected to not be observed via MS. However, CUD is capable of ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–C hydrogen bonds in the fac tris-chelate pocket of the cavity and guest loss would also be partially prevented by the counterions in the face-portals increasing the dissociation barrier and enabling a significant amount of the CUD complex to survive after ionisation.26 External binding of MC likely involves O⋯H–O hydrogen bonds with the hydroxy groups of LW. Compared to MC, the presence of the aniline in MAC would enable it to form an additional O–H⋯NH2 hydrogen bond with the corner of the cage. Computational models at the B97-3c level of theory implemented in the Orca program shows that both mer and fac corner units of HWCo are ideally suited to accommodate two hydrogen bonds with MAC (Fig. 4 and Fig. S13, ESI†).30–32 The aniline might also enable ion pairing by deprotonating the OH to give R–NH3+⋯−O–R′ interactions. These additional interactions available to MAC could potentially explain the higher stoichiometry complexes not observed for the other guests. Furthermore, the binding modes of MC and MACvia hydrogen bonding with the cage exterior also justify the difficulty in observing the complexes in solution as those measurements are performed in competitive solvents such as H2O where cavity binding dominates due to the hydrophobic effect.19
O⋯H–C hydrogen bonds in the fac tris-chelate pocket of the cavity and guest loss would also be partially prevented by the counterions in the face-portals increasing the dissociation barrier and enabling a significant amount of the CUD complex to survive after ionisation.26 External binding of MC likely involves O⋯H–O hydrogen bonds with the hydroxy groups of LW. Compared to MC, the presence of the aniline in MAC would enable it to form an additional O–H⋯NH2 hydrogen bond with the corner of the cage. Computational models at the B97-3c level of theory implemented in the Orca program shows that both mer and fac corner units of HWCo are ideally suited to accommodate two hydrogen bonds with MAC (Fig. 4 and Fig. S13, ESI†).30–32 The aniline might also enable ion pairing by deprotonating the OH to give R–NH3+⋯−O–R′ interactions. These additional interactions available to MAC could potentially explain the higher stoichiometry complexes not observed for the other guests. Furthermore, the binding modes of MC and MACvia hydrogen bonding with the cage exterior also justify the difficulty in observing the complexes in solution as those measurements are performed in competitive solvents such as H2O where cavity binding dominates due to the hydrophobic effect.19
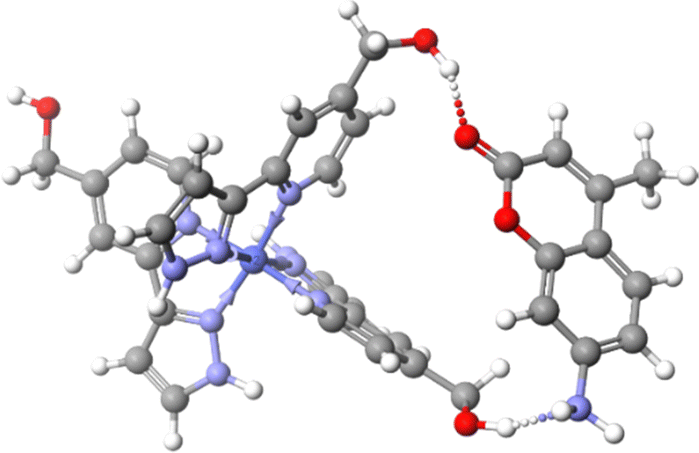 | ||
| Fig. 4 Binding mode of MAC with two LW in a mer tris-chelate corner of HWCo. Structure was optimised at the B97-3c level of theory. | ||
The arrival time distributions (ATD) of the HWCo/nMAC10+ complexes show increasing arrival times with increasing n but also show shoulder regions at earlier arrival times which increase in magnitude with additional MACs until clear double peaks are seen in the ATDs of HWCo/3MAC10+ and HWCo/4MAC10+ (Fig. 5a). This can be explained by two concurrent binding modes: one where all MAC are bound externally and the other where one MAC has been encapsulated. The increasing proportion of encapsulated MAC at higher guest loading indicates that it preferentially binds externally but at high enough guest loading, probability will dictate that one will be encapsulated. The ATDs show that the contribution coming from the binding mode with one MAC encapsulated is slightly larger than the lower stoichiometry with all guests bound externally (e.g.HWCo/3MAC10+ with one MAC encapsulated is slightly larger than the HWCo/2MAC10+ with all MAC bound externally), whereas HWCo10+ and HWCo/CUD10+ overlap (Fig. 3b). This could reflect the better fit of the conformationally flexible CUD, whereas encapsulation of the more rigid MAC and MC likely leads to some expansion of the cage which is reflected in the longer arrival times.
It should be noted that due to the facile guest loss, the observed ions could well be derived from higher guest stoichiometry complexes which have subsequently undergone guest loss in the instrument. Energy-resolved IMS can study this by performing CID in the trap cell prior to IMS and monitoring the changes in the ATDs allowing an assessment of the stability of different binding modes.33 Measurements with mass selected HWCo/4MAC10+ led to dissociation of MAC to form HWCo/3MAC10+. When comparing the ATD of the ‘free’ HWCo/3MAC10+ (mass selected from full spectrum) it showed that the HWCo/3MAC10+ formed purely from dissociation of the HWCo/4MAC10+ only had one major contribution in the ATD which correlated to the earlier contribution of that of the ‘free’ HWCo/3MAC10+ (Fig. 5b). This means that externally bound guests dissociate more easily than internally bound ones even though encapsulation occurs less readily.
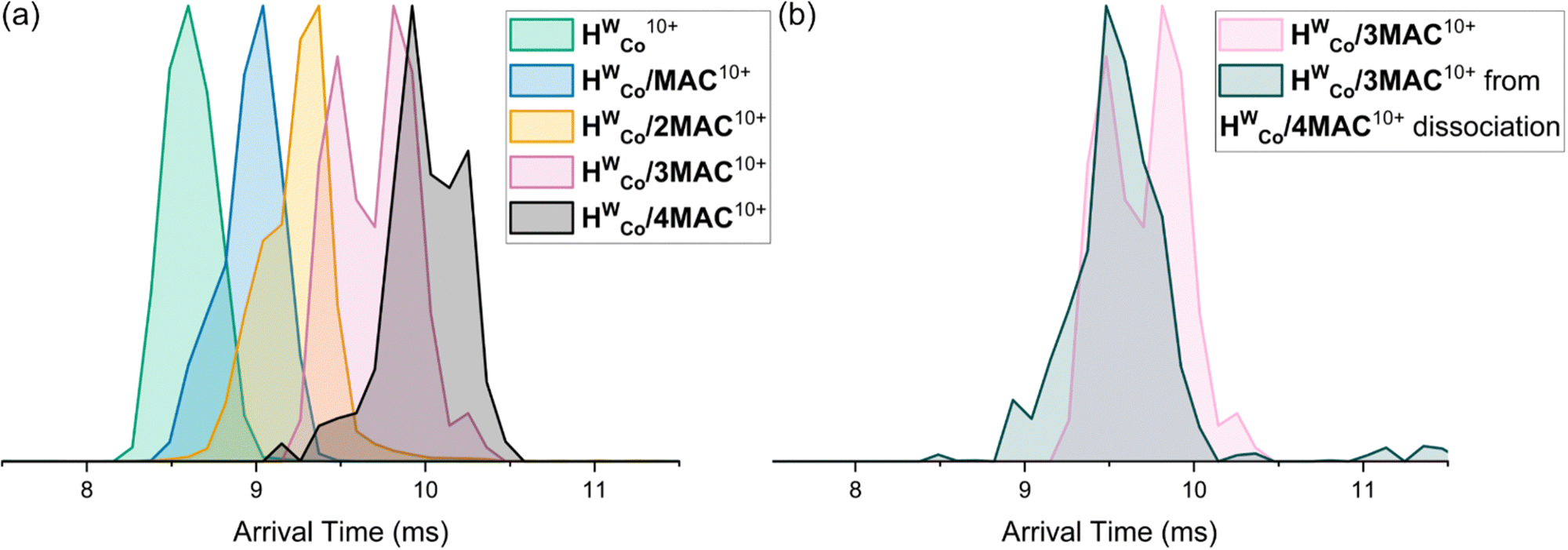 | ||
| Fig. 5 (a) ATDs of HWCo/nMAC10+ with n = 1–4. (b) ATDs of ‘free’ HWCo/3MAC10+ and HWCo/3MAC10+ formed purely from dissociation of HWCo/4MAC10+. | ||
In conclusion, the MS techniques of IMS and CID were used to study a Co8L12 cubic cage and its host–guest properties with three different guests. The studies demonstrated a major stabilising effect of the counterions binding in the faces of the portals. The host–guest properties of the cage showed that guests had different preferences for external or internal binding. Higher stoichiometries of guest binding, which have not been seen with other techniques, were observed and investigated. These showed that MAC preferentially binds externally but could also be driven inside the cavity at high guest loading. These results demonstrate the ability of MS to investigate metal–organic hosts and probe guest binding modes.
We thank the European Union through the NOAH project (H2020-MSCA-ITN project Ref. 765297) and FU Berlin for funding. Support for measurements by the BioSupraMol core facility at FU Berlin is gratefully acknowledged. Thank you to those at Warwick for help with cage synthesis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- F. J. Rizzuto, L. K. S. von Krbek and J. R. Nitschke, Nat. Rev. Chem., 2019, 3, 204–222 CrossRef.
- D. Fujita, Y. Ueda, S. Sato, N. Mizuno, T. Kumasaka and M. Fujita, Nature, 2016, 540, 563–566 CrossRef CAS PubMed.
- C. A. Schalley, Mass Spectrom. Rev., 2001, 20, 253–309 CrossRef CAS PubMed.
- Z. Qi, T. Heinrich, S. Moorthy and C. A. Schalley, Chem. Soc. Rev., 2015, 44, 515–531 RSC.
- A. J. McConnell, Chem. Soc. Rev., 2022, 51, 2957–2971 RSC.
- W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2011, 50, 3479–3483 CrossRef CAS PubMed.
- H. Wang, C. Guo and X. Li, CCS Chem., 2021, 4, 785–808 CrossRef.
- E. Kalenius, M. Groessl and K. Rissanen, Nat. Rev. Chem., 2019, 3, 4–14 CrossRef CAS.
- C. Vicent, V. Martinez-Agramunt, V. Gandhi, C. Larriba-Andaluz, D. G. Gusev and E. Peris, Angew. Chem., Int. Ed., 2021, 60, 15412–15417 CrossRef CAS PubMed.
- K. J. Endres, K. Barthelmes, A. Winter, R. Antolovich, U. S. Schubert and C. Wesdemiotis, Rapid Commun. Mass Spectrom., 2020, 34, e8717 CrossRef CAS PubMed.
- K. E. Ebbert, L. Schneider, A. Platzek, C. Drechsler, B. Chen, R. Rudolf and G. H. Clever, Dalton Trans., 2019, 48, 11070–11075 RSC.
- G. Carroy, V. Lemaur, C. Henoumont, S. Laurent, J. De Winter, E. De Pauw, J. Cornil and P. Gerbaux, J. Am. Soc. Mass Spectrom., 2018, 29, 121–132 CrossRef CAS PubMed.
- H. Lee, J. Tessarolo, D. Langbehn, A. Baksi, R. Herges and G. H. Clever, J. Am. Chem. Soc., 2022, 144, 3099–3105 CrossRef CAS PubMed.
- T. R. Schulte, J. J. Holstein and G. H. Clever, Angew. Chem., Int. Ed., 2019, 58, 5562–5566 CrossRef CAS PubMed.
- M. C. Pfrunder, D. L. Marshall, B. L. J. Poad, E. G. Stovell, B. I. Loomans, J. P. Blinco, S. J. Blanksby, J. C. McMurtrie and K. M. Mullen, Angew. Chem., Int. Ed., 2022, 61, e202212710 CrossRef CAS PubMed.
- E. Taipale, J. S. Ward, G. Fiorini, D. L. Stares, C. A. Schalley and K. Rissanen, Inorg. Chem. Front., 2022, 9, 2231–2239 RSC.
- L. Sleno and D. A. Volmer, J. Mass Spectrom., 2004, 39, 1091–1112 CrossRef CAS PubMed.
- S. Ibáñez, C. Vicent and E. Peris, Angew. Chem., Int. Ed., 2022, 61, e202112513 CrossRef PubMed.
- C. G. P. Taylor, S. P. Argent, M. D. Ludden, J. R. Piper, C. Mozaceanu, S. A. Barnett and M. D. Ward, Chem. – Eur. J., 2020, 26, 3054–3064 CrossRef CAS PubMed.
- C. Mozaceanu, C. G. Taylor, J. R. Piper, S. P. Argent and M. D. Ward, Chemistry, 2020, 2, 22–32 CrossRef CAS.
- C. G. Taylor, J. S. Train and M. D. Ward, Chemistry, 2020, 2, 510–524 CrossRef CAS.
- C. Mozaceanu, A. B. Solea, C. G. P. Taylor, B. Sudittapong and M. D. Ward, Dalton Trans., 2022, 51, 15263–15272 RSC.
- W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Nat. Chem., 2016, 8, 231–236 CrossRef CAS PubMed.
- M. D. Ward, C. A. Hunter and N. H. Williams, Acc. Chem. Res., 2018, 51, 2073–2082 CrossRef CAS PubMed.
- B. Hasenknopf, J.-M. Lehn, B. O. Kneisel, G. Baum and D. Fenske, Angew. Chem., Int. Ed. Engl., 1996, 35, 1838–1840 CrossRef CAS.
- S. Turega, W. Cullen, M. Whitehead, C. A. Hunter and M. D. Ward, J. Am. Chem. Soc., 2014, 136, 8475–8483 CrossRef CAS PubMed.
- N. Geue, T. S. Bennett, A.-A.-M. Arama, L. A. I. Ramakers, G. F. S. Whitehead, G. A. Timco, P. B. Armentrout, E. J. L. McInnes, N. A. Burton, R. E. P. Winpenny and P. E. Barran, J. Am. Chem. Soc., 2022, 144, 22528–22539 CrossRef CAS PubMed.
- V. Shrivastav, M. Nahin, C. J. Hogan and C. Larriba-Andaluz, J. Am. Soc. Mass Spectrom., 2017, 28, 1540–1551 CrossRef CAS PubMed.
- A. K. Rappé, C. J. Casewit, K. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef.
- J. G. Brandenburg, C. Bannwarth, A. Hansen and S. Grimme, J. Chem. Phys., 2018, 148, 064104 CrossRef PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- X. Li, Y.-T. Chan, G. R. Newkome and C. Wesdemiotis, Anal. Chem., 2011, 83, 1284–1290 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Sample preparation, instrumental parameters, computational details and additional figures. See DOI: https://doi.org/10.1039/d3cc04291j |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |