
Enaminone-directed ruthenium(II)-catalyzed C–H activation and annulation of arenes with diazonaphthoquinones for polycyclic benzocoumarins†
Sudeshna
Mondal
,
Chandan Kumar
Giri
 and
Mahiuddin
Baidya
*
and
Mahiuddin
Baidya
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai 600 036, Tamil Nadu, India. E-mail: mbaidya@iitm.ac.in
First published on 9th October 2023
Abstract
The weakly coordinating enaminone functionality has been leveraged for a C–H bond activation strategy under ruthenium catalysis and employed in the regioselective annulative coupling of arenes with diazonaphthoquinones, offering polycyclic benzocoumarins in very high yields. The enaminone motif plays a dual role and the protocol operates through a Ru(II)/Ru(IV) catalytic pathway which is amenable to the diversification of various pharmacophore-coupled substrates.
Over the past two decades, transition-metal-catalyzed site-selective inert C–H bond activation and annulation strategies have revolutionized the field of synthetic chemistry, offering powerful tools for the construction of high-value carbocycles and heterocycles from simple molecular building blocks.1 In this context, Ru(II)-catalysis has emerged as a particularly compelling choice, garnering widespread attention for its operational simplicity, cost-effectiveness, and impressive efficiency facilitated by weak coordination.2 However, the major advancements in the C–H bond activation guided annulation reactions of Ru(II)-catalysts have been accomplished with alkynes and alkenes with key applications in the synthesis of bioactive compounds.2,3 In contrast, the progress of such annulation reactions engaging carbene species that require the involvement of high-valent ruthenium(IV) intermediates is very limited.4 This is especially true when a weakly coordinating directing group is considered. An early example was disclosed by the Ackermann group wherein aromatic acids were coupled with sulfoxonium ylides, leading to the formation of functionalized isocoumarins in high yields (Scheme 1a).5 More recently, Samanta et al. demonstrated an amide-directed annulative coupling of azaheterocycles, such as indoles, with diazonaphthoquinones to afford diverse azacoumestans (Scheme 1b).6 However, in the latter case, the lactonization event was performed in a subsequent step in the presence of AcOH at 130 °C. To the best of our knowledge, these two examples represent the sole instances of Ru(II)-catalyzed annulation reactions of carbenes operating under the guidance of weakly coordinating directing groups.
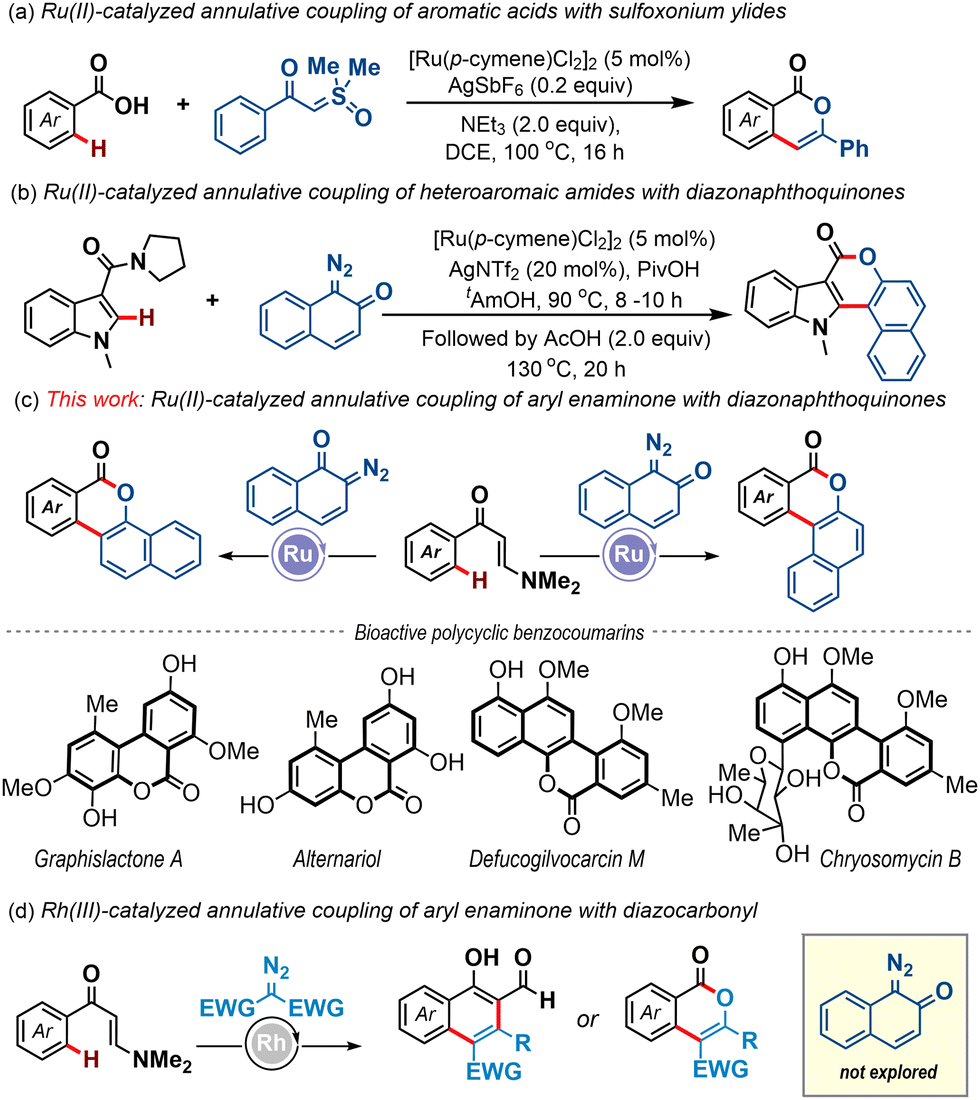 | ||
| Scheme 1 The C–H activation and annulative coupling reactions with carbene species assisted by weak coordination. | ||
Polycyclic benzocoumarins and derivatives thereof hold immense synthetic importance in light of their existence in natural products and pharmaceuticals.7,8 Consequently, the development of efficient synthetic protocols en route to these scaffolds is highly desirable.8 Herein, we report an annulative coupling of readily prepared aryl enaminones with diazonaphthoquinones as a concise route towards high-value polycyclic benzocoumarin motifs (Scheme 1c). Notably, the present work represents the first application of the enaminone functionality as a directing group in ruthenium-catalyzed C–H activation strategies and the enaminone group serves as an intriguing one-carbon synthon to effect the desired annulation event in one pot. Of note, Zhu et al. first introduced the enaminone motif as a directing group under Rh-catalysis to access functionalized naphthalenes and the major developments towards C–H activation reactions involving the enaminone directing group have been confined to require expensive Rh-catalysis (Scheme 1d).9 Also, our work showcased the reactivity of enaminone-bearing metallacycles in combination with quinoid carbene species, which has not been previously studied.9,10
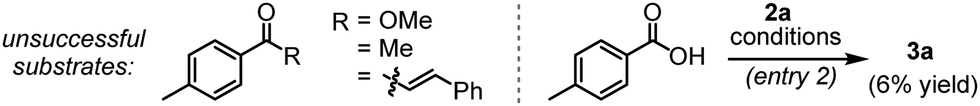
We commenced our investigation following the model reaction between (E)-3-(dimethylamino)-1-(p-tolyl)prop-2-en-1-one (1a) with diazonaphthoquinone 2a (Table 1). When the mixture of 1a and 2a was treated with a catalytic amount of [Ru(p-cymene)Cl2]2 in the presence of AgSbF6 (20 mol%), K2CO3 (0.5 equiv.) and 1-adamentanecarboxylic acid (1-AdCO2H, 0.2 equiv.) in MeOH at 100 °C, we did not detect any product formation with the recovery of 1a (entry 1). However, a substantial amount of tetracyclic benzocoumarin 3a was formed in CF3CH2OH solvent (entry 1) and the reaction yield improved significantly in hexafluoroisopropanol (HFIP) medium, furnishing the desired product 3a in 94% isolated yield (entry 2). On the contrary, the reaction was unsuccessful in aprotic solvents such as toluene, DCE, and THF (entry 3). Further screening of other bases gave inferior results (entry 4). The change of acid additive from 1-AdCO2H to AcOH or 2,4,6-trimethylbenzoic acid (MesCO2H) also reduced the productivity (entry 5). The reaction at a lower temperature (80 °C) delivered 3a only in 68% yield (entry 6). The amount of K2CO3 turned out to be critical as the reaction with a higher loading of K2CO3 (1 equiv.) resulted in poor conversion (entry 7). Nonetheless, control experiments suggested that both K2CO3 and 1-AdCO2H were crucial to achieving the high yield (entries 8 and 9) and the reaction completely shut down in the absence of AgSbF6 and ruthenium(II) catalyst (entries 10 and 11). Interestingly, the reaction was unproductive with Cp*Co(III)-catalyst, while we have found moderate reactivity with the Rh(III)-catalyst leading to 3a in 59% yield (entry 12). Furthermore, the enaminone motif is highly important for this C–H bond activation/annulation reaction, and our attempts to induce the coupling of 2a with other carbonyl compounds, which include aromatic ester, ketone, and chalcone derivatives, were so far unsuccessful, while the reaction with the aromatic acid gave the desired product 3a only in 6% yield (Table 1).6,9i
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yieldb (%) |
| a Reaction conditions: 1a (0.20 mmol), 2a (1.5 equiv.), solvent (1 mL) for 24 h. b Isolated yields. NR: no reaction with the recovery of 1a. | ||
| 1 | MeOH/CF3CH2OH instead of HFIP | NR/72 |
| 2 | None | 94 |
| 3 | Toluene/DCE/THF instead of HFIP | NR |
| 4 | Na2CO3/KOAc/K3PO4 instead of K2CO3 | 40/65/NR |
| 5 | AcOH/MesCO2H instead of 1-AdCO2H | 47/56 |
| 6 | At 80 °C | 68 |
| 7 | K2CO3 (1 equiv.) | 60 |
| 8 | Without K2CO3 | 41 |
| 9 | Without 1-AdCO2H | 43 |
| 10 | Without AgSbF6 | NR |
| 11 | Without Ru(II)-catalyst | NR |
| 12 | Cp*Co(CO)I2/[Cp*RhCl2]2 | NR/59 |
|
||
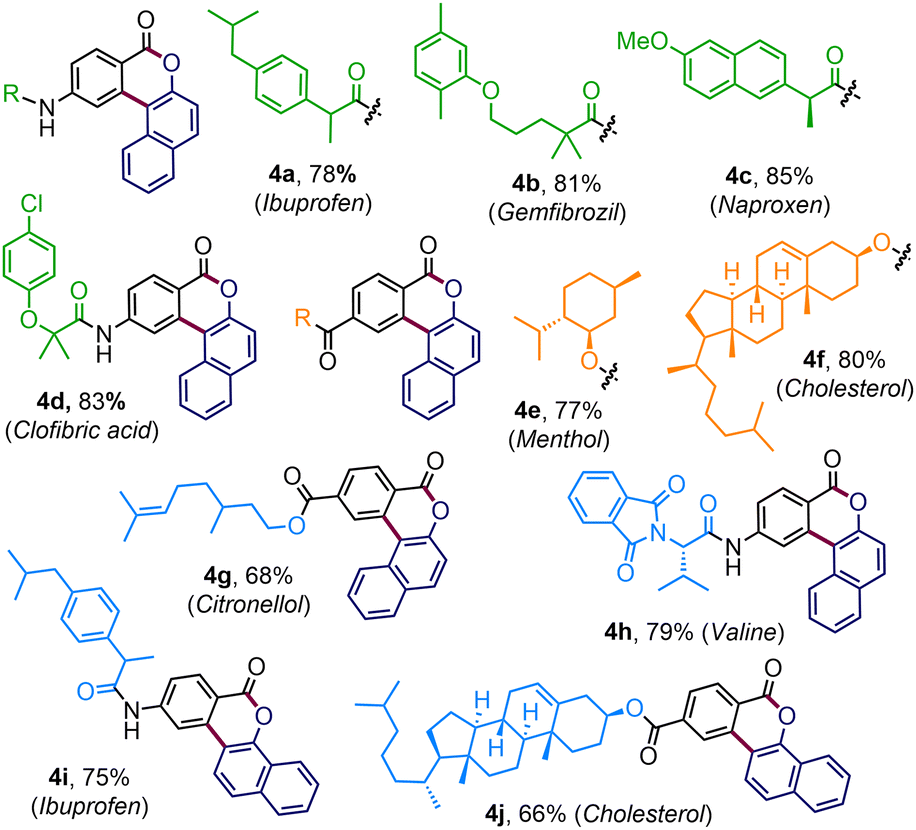
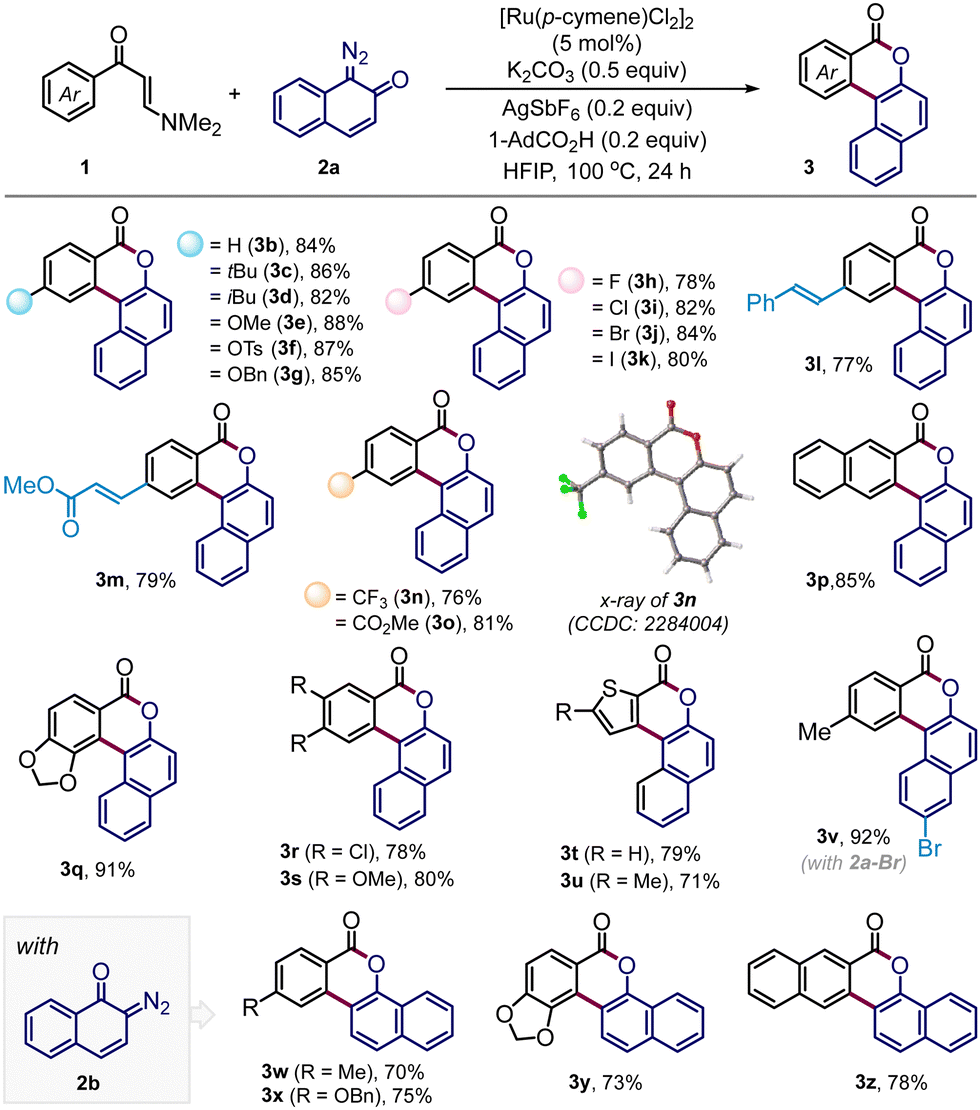
Having acquired the optimized conditions (Table 1, entry 2), we next investigated the substrate scope (Table 2). The protocol is quite general accommodating a wide range of arene-substituted enaminones. The reactions proceeded smoothly with substrates bearing electron-donating substituents such as alkyl (3b–d), methoxy (3e), tosyl (3f) and benzyloxy (3g) in the aryl ring to deliver the desired polycyclic benzocoumarins in very high yields (82–88%). The halogen functionalities such as fluoro (3h), chloro (3i), bromo (3j), and iodo (3k) were undisturbed under the reaction conditions and these halogen functionalities are useful synthetic handles for further functionalization. The presence of sensitive olefin functionalities also did not interfere with the annulation reaction, offering 3l and 3m in 77% and 79% yields, respectively. Satisfyingly, substrates with electron-withdrawing groups, which include trifluoromethyl and carboxylate ester, also effectively participated in this reaction, dispensing 3n and 3o in very high yields. The product 3n was crystalized and the presence of a polycyclic benzocoumarin framework was unambiguously confirmed through single crystal X-ray analysis. 2-Naphthyl-derived enaminone produced pentacyclic compound 3p in 85% yield where the reaction took place at the sterically less hindered site. On the other hand, annulation occurred at the more hindered site for the piperonyl-substituted enaminone to give 3q in 91% yield. This change in the regioselectivity can be attributed to the anchimeric assistance through the bridge oxygen.11 For other enaminones having dichloro or dimethoxy substituents, the reactions proceeded at the less hindered site, forging 3r–3s with high efficiency. Heterocyclic substrates such as thiophene enaminones were also compatible to afford 3t and 3u in 79% and 71% yields, respectively. Variation in the diazonaphthoquinone coupling partner was also considered. The reaction with 6-bromo-1-diazonaphthalen-2(1H)-one (2a-Br) furnished the desired coumarin 3v in excellent yield. Gratifyingly, under the standard reaction conditions, the annulation reaction was also fruitful with diazonaphthoquinone 2b, a regioisomer of 2a, and we have successfully prepared polycyclic benzocoumarins 3w–3z in high yields (Table 2).
|
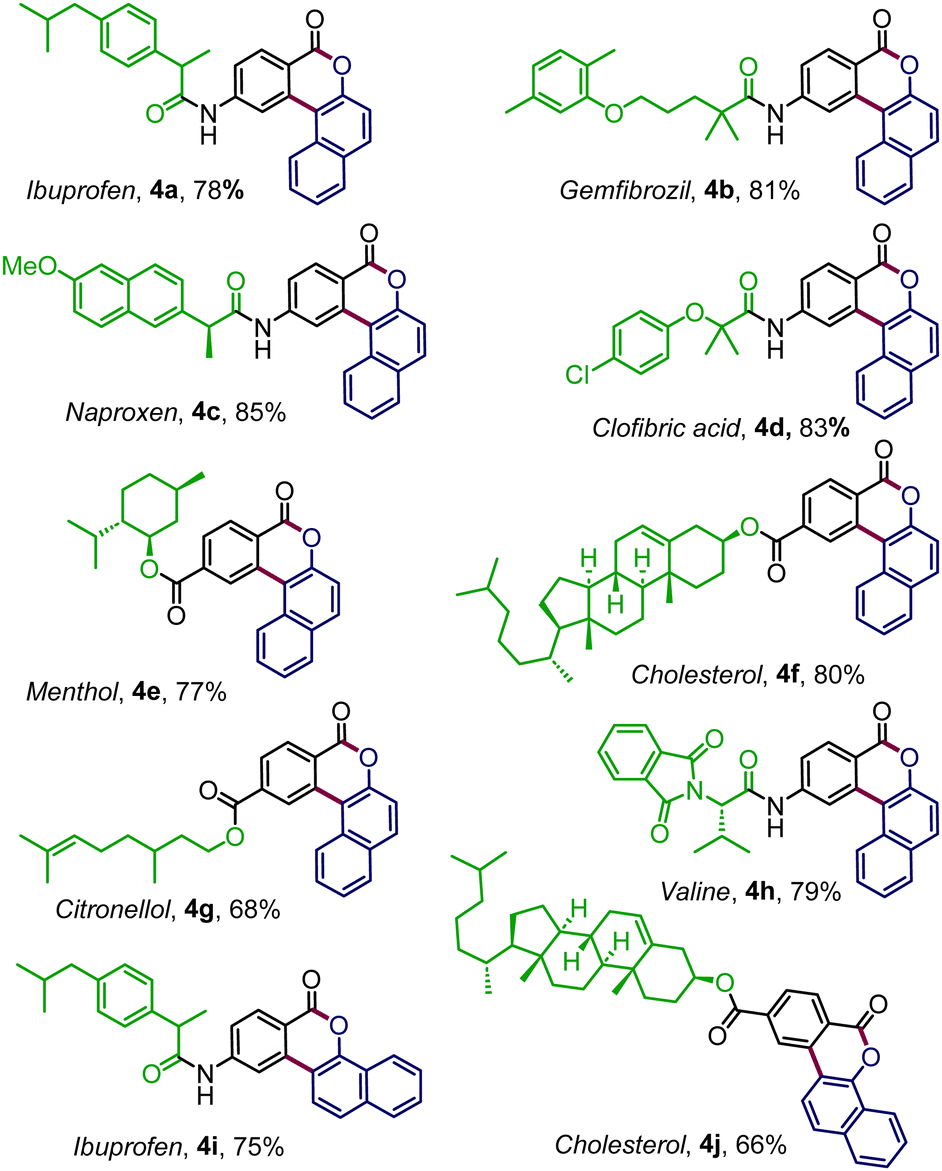
To expand the reaction scope further, enaminone embedded with various biologically relevant scaffolds was examined (Table 3). To our delight, enaminones coupled with commercial drugs, for instance, ibuprofen, gemfibrozil, clofibric acid, and naproxen, effortlessly afforded the desired products 4a–4d, in good to high yields. The standard conditions were also well suited for the enaminones derived from biorelevant motifs like menthol (4e), cholesterol (4f), and citronellol (4g). The strategy was equally productive with enaminone connected to valine amino acid where the polycyclic benzocoumarin 4h was obtained in 79% yield. Similarly, the pharmacophore-tethered annulated products 4i–4j were prepared in good yields by coupling diazonaphthoquione 2b (Table 3).
| a Reaction conditions: 5 (0.2 mmol), 2a or 2b (1.5 equiv.), [Ru(p-cymene)Cl2]2 (5 mol%), 1-AdCO2H (0.2 equiv.), AgSbF6 (0.2 equiv.), K2CO3 (0.5 equiv.) and HFIP (1 mL) at 100 °C for 24 h. |
|---|
|
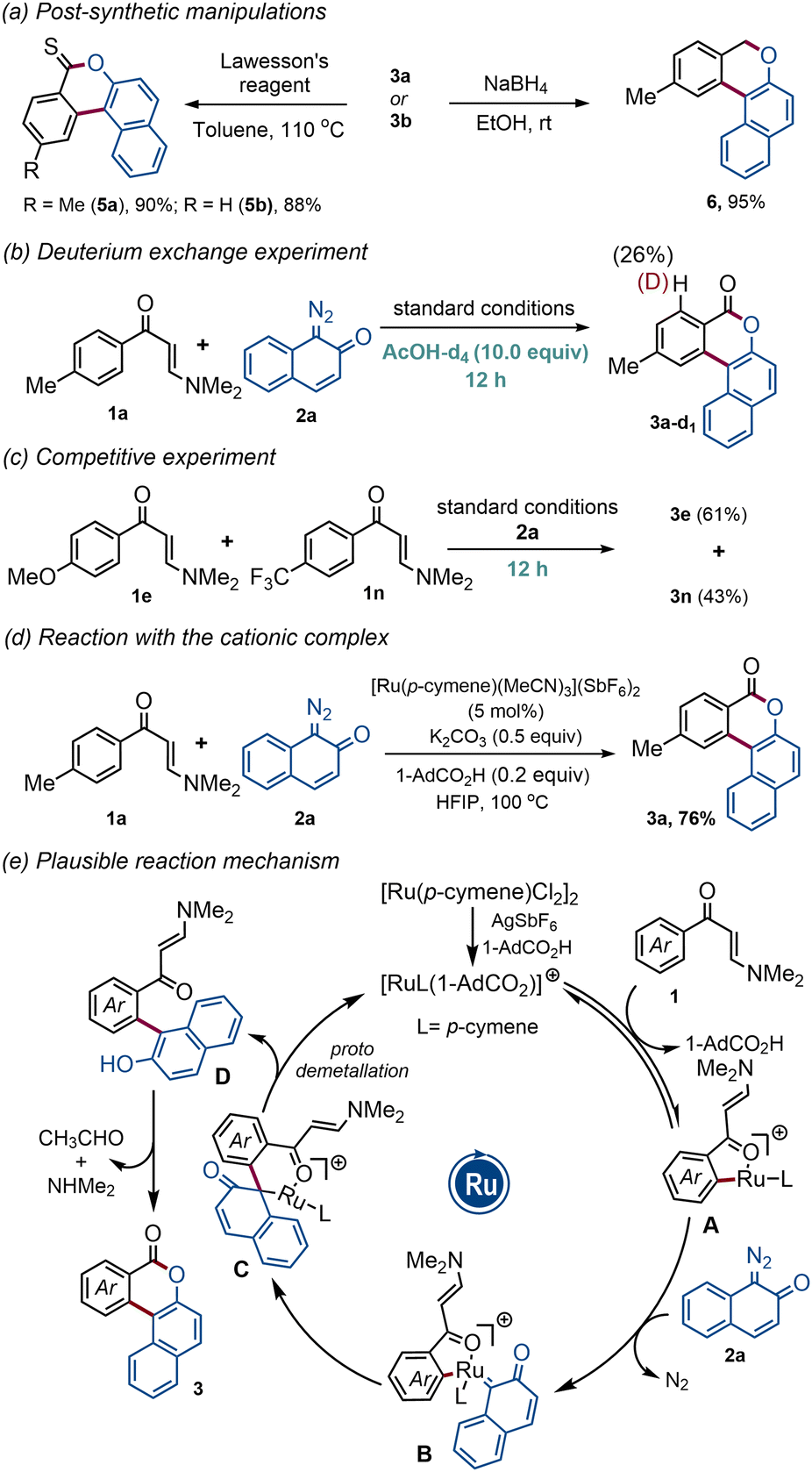
The synthetic utility was further showcased through post-synthetic manipulations (Scheme 2a). Satisfyingly, treatment of polycyclic benzocoumarins 3a–b with Lawesson's reagent furnished thiocoumarin analogs 5a–b in excellent yields. Product 3a was also transformed into polycyclic benzochromene 6 in 95% yield through NaBH4 reduction (Scheme 2a, right).
 | ||
| Scheme 2 Post-synthetic manipulations, mechanistic study, and reaction mechanism. | ||
To understand the nature of the C–H metallation step, deuterium incorporation experiments were performed. The reaction of enaminone 1a with 2a under standard reaction conditions for 12 h in the presence of deuterated acetic acid (10.0 equiv.) resulted in a significant amount of deuterium incorporation, indicating that the C–H ruthenation process is reversible (Scheme 2b). The competitive experiment with electron-rich and electron-deficient enaminones gave a 1.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of benzocoumarins 3e and 3n, signifying that the C–H metallation step most likely follows a concerted metallation deprotonation (CMD) pathway (Scheme 2c). Employing the Ru(II)-cationic complex {[Ru(p-cymene)(MeCN)3](SbF6)2} instead of a combination of [Ru(p-cymene)Cl2]2 dimer and AgSbF6, we have isolated the annulated product 3a in 76% yield, suggesting the indispensable role of the cationic Ru(II)-complex in this transformation (Scheme 2d). Based on these results and literature precedents, a plausible reaction mechanism has been proposed in Scheme 2e. First, a cationic ruthenium complex is formed through the ligand exchange event and it then initiates the C–H activation reaction with enaminone 1 to give five-membered ruthenacycle A. Next, intermediate A reacts with diazonaphthoquinone 2a to generate the carbenoid intermediate B and the subsequent migratory insertion provides six-membered intermediate C. Finally, protodemetallation followed by aromatization delivers D which cyclizes under the reaction conditions to produce the desired product 3.
:1 mixture of benzocoumarins 3e and 3n, signifying that the C–H metallation step most likely follows a concerted metallation deprotonation (CMD) pathway (Scheme 2c). Employing the Ru(II)-cationic complex {[Ru(p-cymene)(MeCN)3](SbF6)2} instead of a combination of [Ru(p-cymene)Cl2]2 dimer and AgSbF6, we have isolated the annulated product 3a in 76% yield, suggesting the indispensable role of the cationic Ru(II)-complex in this transformation (Scheme 2d). Based on these results and literature precedents, a plausible reaction mechanism has been proposed in Scheme 2e. First, a cationic ruthenium complex is formed through the ligand exchange event and it then initiates the C–H activation reaction with enaminone 1 to give five-membered ruthenacycle A. Next, intermediate A reacts with diazonaphthoquinone 2a to generate the carbenoid intermediate B and the subsequent migratory insertion provides six-membered intermediate C. Finally, protodemetallation followed by aromatization delivers D which cyclizes under the reaction conditions to produce the desired product 3.
In summary, we have exploited the versatile enaminone functionality as a directing group for C–H bond activation the reaction under ruthenium catalysis and successfully devised an annulation reaction with diazonaphthoquinones, offering biologically relevant polycyclic benzocoumarins in very high to excellent yields. The protocol is operationally simple, displays a broad substrate generality and functional group compatibility, and is also effective in the diversification of substrates bearing bioactive scaffolds and natural product motifs. The protocol leverages the dual role of the enaminone functionality and involves a ruthenium(II)/(IV) catalytic pathway involving quinoid carbene species.
We gratefully acknowledge DST-SERB, India (CRG/2019/001003) for the financial support. S. M. acknowledges IIT Madras for providing the HTRA fellowship and C. K. G. thanks UGC for the SRF.
Conflicts of interest
There are no conflicts to declare.References
- (a) M. Zhang, Adv. Synth. Catal., 2009, 351, 2243 CrossRef CAS; (b) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (c) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (d) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed; (e) P. Gandeepan and C. H. Cheng, Chem. – Asian J., 2016, 11, 448 CrossRef CAS PubMed; (f) M. Gulius and J. L. Mascarenas, Angew. Chem., Int. Ed., 2016, 55, 11000 CrossRef PubMed; (g) R. Y. Zhu, M. E. Farmer, Y. Q. Chen and J. Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578 CrossRef CAS PubMed; (h) L. Zheng and R. Hua, Chem. Rec., 2018, 18, 556 CrossRef CAS PubMed; (i) C.-Y. Huang, H. Kang, J. Li and C.-J. Li, J. Org. Chem., 2019, 84, 12705 CrossRef CAS PubMed; (j) A. Mandal, S. Dana, D. Chowdhury and M. Baidya, Chem. – Asian J., 2019, 14, 4074 CrossRef CAS PubMed; (k) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192 CrossRef CAS PubMed; (l) K. Ghosh, R. K. Rit, M. Shankar, K. Mukherjee and A. K. Sahoo, Chem. Rec., 2020, 20, 1017 CrossRef CAS PubMed; (m) P. Ghosh, D. Chowdhury, S. Dana and M. Baidya, Chem. Rec., 2021, 21, 3795 CrossRef CAS PubMed; (n) L. Song and E. V. Van der Eycken, Chem. – Eur. J., 2021, 27, 121 CrossRef CAS PubMed; (o) Y. Zhang and M. Szostak, Chem. – Eur. J., 2022, 28, 1 CAS; (p) R. Gramage-Doria and C. Bruneau, Coord. Chem. Rev., 2021, 428, 213602 CrossRef CAS.
- (a) S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461 CrossRef CAS; (b) G. Duarah, P. P. Kaishap, T. Begum and S. Gogoi, Adv. Synth. Catal., 2019, 361, 654 CrossRef CAS; (c) M. Pichette Drapeau and L. J. Gooßen, Chem. – Eur. J., 2016, 22, 18654 CrossRef CAS PubMed.
- For selected examples: (a) L. Ackermann and J. Pospech, Org. Lett., 2011, 13, 4153 CrossRef CAS PubMed; (b) L. Ackermann, A. V. Lygin and N. Hofmann, Angew. Chem., Int. Ed., 2011, 50, 6379 CrossRef CAS PubMed; (c) B. Li, J. Ma, N. Wang, H. Feng, S. Xu and B. Wang, Org. Lett., 2012, 14, 736 CrossRef CAS PubMed; (d) H. Tan, H. Li, J. Wang and L. Wang, Chem. – Eur. J., 2015, 21, 1904 CrossRef CAS PubMed; (e) J. Wu, W. Xu, Z. X. Yu and J. Wang, J. Am. Chem. Soc., 2015, 137, 9489 CrossRef CAS PubMed; (f) A. Mandal, G. Mehta, S. Dana and M. Baidya, Org. Lett., 2019, 21, 5879 CrossRef CAS PubMed; (g) Q. Lu, S. Mondal, S. Cembellín, S. Greßies and F. Glorius, Chem. Sci., 2019, 10, 6560 RSC; (h) D. Chowdhury, S. Dana, A. Mandal and M. Baidya, Chem. Commun., 2019, 55, 11908 RSC; (i) D. Chowdhury, S. Dana, S. Maity and M. Baidya, Org. Lett., 2020, 22, 6760 CrossRef CAS PubMed; (j) S. Dana, C. K. Giri and M. Baidya, Chem. Commun., 2020, 56, 13048 RSC; (k) C. K. Giri, S. Dana and M. Baidya, Chem. Commun., 2021, 57, 10536 RSC; (l) For more references, see, ESI† page S3.
- For selected examples with strong coordinating directing groups, see: (a) Y. Li, Z. Qi, H. Wang, X. Yang and X. Li, Angew. Chem., Int. Ed., 2016, 55, 11877 CrossRef CAS PubMed; (b) M. Wang, L. Kong, Q. Wu and X. Li, Org. Lett., 2018, 20, 4597 CrossRef CAS PubMed; (c) G. S. Kumar, N. P. Khot and M. Kapoor, Adv. Synth. Catal., 2019, 361, 73 CrossRef CAS; (d) H. Li, C. Wu, H. Liu and J. Wang, J. Org. Chem., 2019, 84, 13262 CrossRef CAS PubMed; (e) L. Kong, X. Han and X. Li, Chem. Commun., 2019, 55, 7339 RSC; (f) X. Wang, J. Zhang, Y. He, D. Chen, C. Wang, F. Yang, W. Wang, Y. Ma and M. Szostak, Org. Lett., 2020, 22, 5187 CrossRef CAS PubMed; (g) M. Zheng, J. Zhou, F. Fang, Z. Xu, F. Zheng, Z. Jiang, T. Liu, H. Liu, L. Zhao and Y. Zhou, Org. Chem. Front., 2023, 10, 62 RSC.
- Y.-F. Liang, L. Yang, T. Rogge and L. Ackermann, Chem. – Eur. J., 2018, 24, 16548 CrossRef CAS PubMed.
- S. Sarkar and R. Samanta, Org. Lett., 2022, 24, 4536 CrossRef CAS PubMed.
- (a) Z. Mao, W. Sun, L. Fu, H. Luo, D. Lai and L. Zhou, Molecules, 2014, 19, 5088 CrossRef PubMed; (b) Y. L. Garazd and M. M. Garazd, Chem. Nat. Compd., 2016, 52, 1 CrossRef CAS.
- (a) T. Hosoya, E. Takashiro, T. Matsumoto and K. Suzuki, J. Am. Chem. Soc., 1994, 116, 1004 CrossRef CAS; (b) I. Takemura, K. Imura, T. Matsumoto and K. Suzuki, Org. Lett., 2004, 6, 2503 CrossRef CAS PubMed; (c) S. Luo, F.-X. Luo, X.-S. Zhang and Z.-J. Shi, Angew. Chem., Int. Ed., 2013, 52, 10598 CrossRef CAS PubMed; (d) W. Yang, S. Wang, Q. Zhang, Q. Liu and X. Xu, Chem. Commun., 2015, 51, 661 RSC; (e) S. Pramanik, M. Josh, D. Mondal and C. Chowdhury, Adv. Synth. Catal., 2019, 361, 5223 CrossRef CAS; (f) C. C. Chen, C. L. Hsu and M. J. Wu, Tetrahedron, 2019, 75, 1034 CrossRef CAS.
- For previous works under Rh-catalysis, see: (a) S. Zhou, J. Wang, L. Wang, C. Song, K. Chen and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 9384 CrossRef CAS PubMed; (b) S. Zhou, B. W. Yan, S. X. Fan, J. S. Tian and T. P. Loh, Org. Lett., 2018, 20, 3975 CrossRef CAS PubMed; (c) C. Song, C. Yang, H. Zeng, W. Zhang, S. Guo and J. Zhu, Org. Lett., 2018, 20, 3819 CrossRef CAS PubMed; (d) Z. Wang and H. Xu, Tetrahedron Lett., 2019, 60, 664 CrossRef CAS; (e) Z. Jiang, J. Zhou, H. Zhu, H. Liu and Y. Zhou, Org. Lett., 2021, 23, 4406 CrossRef CAS PubMed; (f) W. Wu, X. Wu, S. Fan and J. Zhu, Org. Lett., 2022, 24, 7850 CrossRef CAS PubMed; (g) C. Yang, X. Zhang and X. Fan, Org. Chem. Front., 2023, 10, 4282 RSC; (h) Q. Wang, Y. Li, J. Sun, S. Chen, H. Li, Y. Zhou, J. Li and H. Liu, J. Org. Chem., 2023, 88, 5348 CrossRef CAS PubMed . For a spirocyclization with aromatic acids, see: ; (i) X. Han, L. Kong, J. Feng and X. Li, Chem. Commun., 2020, 56, 5528 RSC.
- For a book chapter: (a) H.-X. Wang, V. K.-Y. Lo and C.-M. Che, in Transition Metal-Catalyzed Carbene Transformations, ed. J. Wang, C.-M. Che and M. P. Doyle, Wiley-VCH GmbH, 2022, ch. 9, p. 269; For a recent review CrossRef; (b) S. Bera, S. Sarkar and R. Samanta, New J. Chem., 2021, 45, 10135 RSC.
- H. Shiota, Y. Ano, Y. Aihara, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 14952 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2284004. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc03999d |
| This journal is © The Royal Society of Chemistry 2023 |