
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePredicting and analyzing organic reaction pathways by combining machine learning and reaction network approaches†
Tomonori
Ida
 *a,
Honoka
Kojima
a and
Yuta
Hori
b
*a,
Honoka
Kojima
a and
Yuta
Hori
b
aDivision of Material Chemistry, Graduate School of Natural Science and Technology, Kanazawa University, Kanazawa 920-1192, Japan. E-mail: ida@se.kanazawa-u.ac.jp
bCenter for Computational Sciences, University of Tsukuba, Tsukuba 305-8577, Japan
First published on 29th September 2023
Abstract
A learning model is proposed that predicts both products and reaction pathways by combining machine learning and reaction network approaches. By training 50 fundamental organic reactions, the learning model predicted the products and pathways of 35 test reactions with a top-5 accuracy of 68.6%. The model identified the key fragment structures of the intermediates and could be classified as several basic reaction rules in the context of organic chemistry, such as the Markovnikov rule.
The generation of novel functional molecules using existing compounds is challenging because the vastness of organic chemical space should be explored.1,2 For hundreds of years, chemists synthesized novel compounds using only their knowledge and creativity,3,4 but various theoretical methods of identifying potential routes, such as quantum chemical calculations, have emerged. In addition, recent developments in terms of computational and machine-learning techniques have rapidly expanded the field of chemoinformatics, which has been successful in various respects, such as in elucidating quantitative structure–property(–activity) relationships,5,6 identifying novel functional materials,7,8 and predicting the products of chemical reactions.9–13 Recent machine-learning models based on deep neural networks have predicted the major products using organic reactants, reagents, and solvent species with a probability of 90%, and the prediction accuracy can exceed that of a human.12 As the prediction accuracy improves, the learning model becomes a black box, making it impossible to understand or explain why such predictions are generated. In terms of the required capacities of a machine-learning tool, Ferguson stated, “Comprehensible explanation can be absolutely critical for particular tasks to ensure that we are getting the right answer for the right reasons.”14 Thus, a model that can explain the reasons for product selection is essential for scientific prediction.
The simplest method of explaining the prediction of a product is to display the reaction pathway. In the past 50 years, novel computational approaches have been developed to identify reaction pathways.15–21 In the case of a basic pathway search, the structural formula of a compound is considered a graph, and a reaction network is constructed by iteratively forming and dissociating chemical bonds. Among these methods, the Chematica system developed by Grzybowski et al.22,23 was pivotal in advancing computer-aided organic synthesis. This system generates multiple reaction pathways toward a target compound based on several reaction templates (rules). For most target compounds, the final pathway is determined by researchers based on their knowledge and experience, as the combination of numerous reaction rules obscures the reasons for pathway selection.
In our previous work, we developed a method for efficiently generating reaction networks using simple reaction rules,24 and it was applied to formic acid decomposition to discuss the reaction mechanism. The reaction network approach can generate some favorable reaction pathways, not only a unique chemical reaction pathway. Thus, in a product search using the reaction network approach, multiple correct and incorrect reaction pathways can be obtained, even for a single reaction. Recently, some studies have been reported on the use of machine learning to predict reaction products by regarding the formation and dissociation of chemical bonds as a combination of electron flows.25,26 In these studies, the electron flows were learned from the structural differences between reactants and products. Thus, it is expected that the chemical knowledge required to predict the products and reaction pathways could be learned from the reaction networks constructed by simple reaction rules.
We herein propose a method that can predict the product and reaction pathway of organic chemical reactions by combining machine learning and reaction network approaches. This study focuses on fundamental organic chemical reactions as found in organic chemistry textbooks. By training the reaction network using simple organic reactions containing several molecular structures and connections, we construct a model that can predict the reaction pathways. A simple machine-learning model is employed to avoid black boxing and facilitate the analyses of the selection criteria. After training, the learning model is validated by predicting the products and pathways of named and relatively complex reactions. The used program codes are available on GitHub.
The training reactions used in this study were fundamental organic reactions with clearly understood reaction pathways.27–30 Note that the correct reaction pathway used for the learning process does not always correspond to the chemically correct reaction pathway. For computation, the selected reactions were limited to those in which all atoms obeyed the octet (duet) rule, with no radical state or catalyst required. For the selected 50 reactions as shown in Fig. S1 (ESI†), the reaction networks generated 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 753 reaction pathways as a training dataset. The details of reaction network construction are presented in Fig. 1.
753 reaction pathways as a training dataset. The details of reaction network construction are presented in Fig. 1.
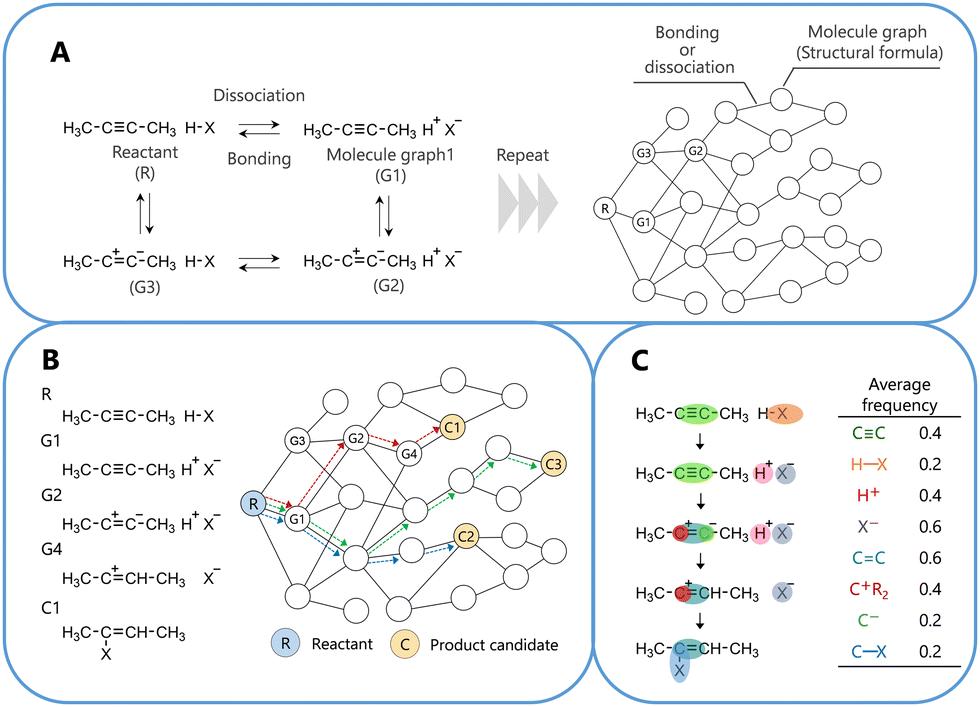 | ||
| Fig. 1 Schematic diagrams outlining the construction and analysis of the organic reaction network. (A) Construction of the network, (B) selection of a reaction pathway within the network, and (C) analysis of a feature of the reaction pathway. See the computational method in the ESI.† | ||
The obtained pathways were learned using pairwise logistic regression. During training, the proposed model adjusted the points of the fragment structures in the molecular graphs to ensure that those on the correct pathway displayed a higher number of points than those on the incorrect pathways. The points of the fragment structures in the molecular graphs were obtained after learning, and these values are shown in Table S3 (ESI†). The details of the learning method and conditions are described in the computational method24,27–31 of the ESI.†
To verify that our model can be applied to reactions other than the learned examples, reaction pathways were predicted using test data with higher numbers of reaction steps than those in the training reactions. The test data, comprising 35 reactions, were selected from textbooks.29,30 In each reaction network, molecular graphs were generated for steps with >3000 nodes because of the numerous reaction steps in the test data.
Pathway predictions were performed using only the structural formulae of the reactants as inputs. In all reaction networks, the total number of reaction pathways was 3902558, with only 2107 correct pathways. The pathways of each reaction were ranked by the average number of points on the molecular graphs of each reaction pathway, and the five highest pathways were designated as the top 5. Therefore, the probability of randomly selecting the top 1 is 2107/3902558 ≈ 0.05%. In contrast, in the proposed model, the respective probabilities of predicting the top 1 and within the top 5 were 45.7% and 68.6%. Although these values are lower than those recently reported for learning models that predict reaction products,12,13 the proposed learning model predicted the products and their reaction pathways, thereby advancing chemical knowledge. All predicted reaction pathways of the test data corresponding to the top 1–5 are shown in Fig. S2 (ESI†), wherein all pathways are purely the result of computer predictions following learning of the fundamental reactions.
Fig. 2 shows the reaction pathways predicted by the learning model for the aldol reaction and the halogen addition to butadiene as correct and incorrect predictions, respectively. Although the reaction steps are redundant as general steps, this is due to the simplified construction rule of the reaction network, multiple steps are considered as one step in actual reactions. In the aldol reaction, the proposed learning model predicts the correct product within the top 1, 2, and 5 (Fig. 2(A) and Fig. S2–5 in ESI†), where the only difference in the reaction pathways is the order of protonation. Notably, the learning model can predict pinacol rearrangement reactions (Fig. S2–7 in ESI†), although the training data contain no rearrangement reactions, and thus, the proposed model is a good predictor of fundamental reactions with multiple steps. Conversely, in the halogen addition to butadiene, all products predicted within the top 5 are incorrect (Fig. 2(B) and Fig. S2–6 in ESI†). As butadiene contains two double bonds, predicting the reaction pathways is challenging owing to the presence of multiple carbon reaction centers. These results suggest that the proposed learning model yields criteria for determining reaction directions.
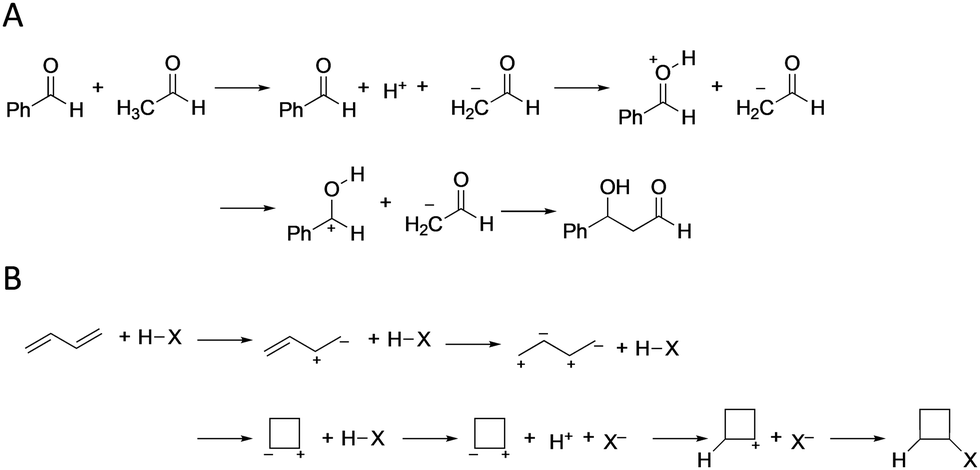 | ||
| Fig. 2 Top predicted reaction pathways in (A) the aldol reaction and (B) halogen (X) addition to butadiene as respective examples of correctly and incorrectly predicted reaction pathways. | ||
Following the successful prediction of the correct pathways of various types of reactions using the proposed model, the evaluation of the molecular graphs in the reaction pathway using this model was considered. For this purpose, the molecular graphs in the pathways of the addition, elimination, and substitution reactions of the training data were evaluated, and the points of the graphs in the various reaction steps were examined. Fig. 3(A) shows the evaluated points of molecular graphs in each reaction step, where step 0 corresponds to the reactant and the final step corresponds to product generation. The evaluated points of the reactants and products are low, and those of the reaction intermediates are high. Although the highest values of the intermediates appear to be related to the activation energy of the reaction, the points generated via learning do not contain energy data. In contrast, this model learns to increase the points of the intermediates. The values of fragment structures that appear more frequently in the intermediates are higher, and thus, the average points of the overall reaction increase, i.e., the higher the points associated with the intermediates are, the more likely the reaction is to proceed. The proposed model can thus predict the pathways of different reactions by focusing on the fragment structures of the intermediates. This perspective differs significantly from those of other product prediction methods, which focus only on the various reactants and products of a reaction.
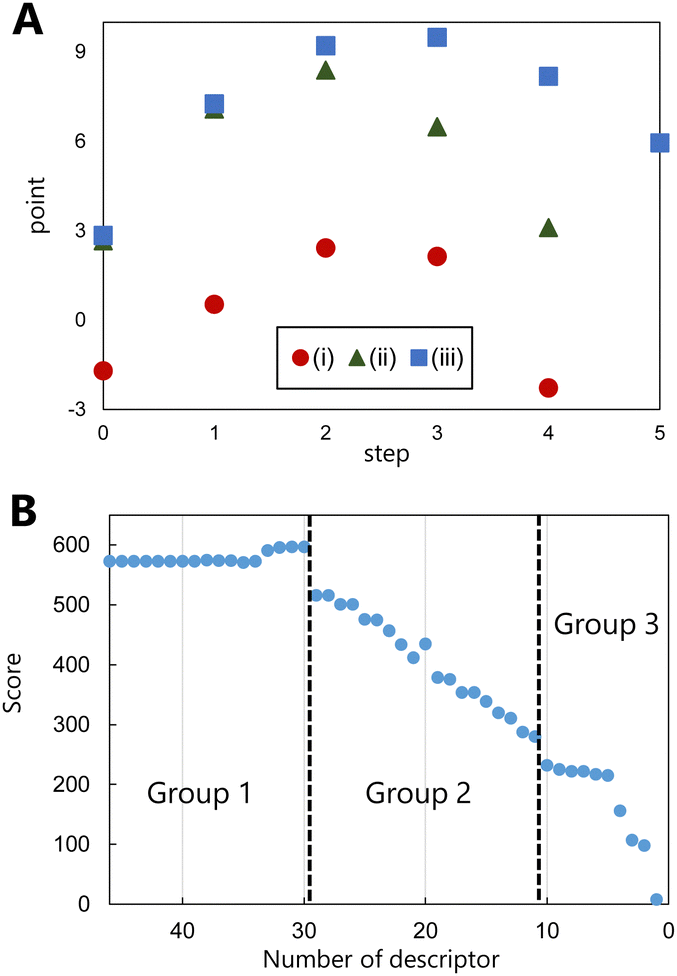 | ||
| Fig. 3 (A) Points of each reaction step in the molecular graphs of the following reactions: (i) addition of a halogen to an alkene, (ii) substitution of a halogen, and (iii) elimination of an alkyl halide. (B) Variation in the prediction accuracy with the number of the fragment structures (descriptors) in the feature. The prediction accuracies were estimated by scoring the correct ranking. | ||
The correlation between fragment structure and prediction accuracy was examined to determine which fragment structure was considered by the learning model as critical in an organic reaction. To clarify the correlation, pathway predictions were performed using the training data by omitting descriptors in order of decreasing absolute points of the fragment structure, while maintaining the other calculation conditions unchanged. The greater the positive point of fragments of a structure, the more likely the structure is to be selected as an intermediate; the lesser the negative point of fragments, the more likely the structure is to be avoided. Therefore, we considered that, regardless of the sign, a large absolute point contributes significantly to the prediction. In these predictions by omitting descriptors, the prediction accuracy was estimated by scoring the correct ranking instead of using the top 1 correct probability. The detailed method of the scoring correct ranking is in the computational methods of the ESI.† The estimated score based on the descriptors used is shown in Fig. 3(B), which indicates the contributions of the omitted descriptors (fragment structures) in pathway prediction. The fragment structures are divided roughly into three groups based on the change in scores. The fragment structures for which the scores do not change when they are omitted are defined as group 1, while those for which the scores rapidly decrease are defined as group 2. After the scores rapidly decrease (group 2) depending on the number of omitted descriptors, they decrease gradually. The structures are defined as group 3.
The fragment structures in group 1 include ionic states, such as O− and X−, and structures containing phenyl groups, such as Ph![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and PhC, which are generated only in specific reactions. All fragment structures are shown in Table S3 (ESI†). When the fragment structures in group 1 are not used in the prediction, the prediction accuracy remains constant. Therefore, the fragment structures in group 1 do not contribute to determining the reaction pathway, and thus, considering whether they are generated is unnecessary.
N and PhC, which are generated only in specific reactions. All fragment structures are shown in Table S3 (ESI†). When the fragment structures in group 1 are not used in the prediction, the prediction accuracy remains constant. Therefore, the fragment structures in group 1 do not contribute to determining the reaction pathway, and thus, considering whether they are generated is unnecessary.
Most fragment structures in group 3 had large negative points. The structures containing CC and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds, in particular, are considered unsuitable intermediates because unsaturated bonds are reaction centers in numerous reactions. However, this large negative point of the unsaturated bond causes the ring-closing reaction to be preferred in the prediction for the addition of a halogen to butadiene, as shown in Fig. 2(B). The other species in group 3 are structures that should be avoided as intermediates in all reactions, such as C+H3 and H2. The structure in this group with a positive point value is H+. Although conventional learning models ignore protons in product prediction, these results suggest that proton behavior is crucial in predicting organic reaction pathways.
C bonds, in particular, are considered unsuitable intermediates because unsaturated bonds are reaction centers in numerous reactions. However, this large negative point of the unsaturated bond causes the ring-closing reaction to be preferred in the prediction for the addition of a halogen to butadiene, as shown in Fig. 2(B). The other species in group 3 are structures that should be avoided as intermediates in all reactions, such as C+H3 and H2. The structure in this group with a positive point value is H+. Although conventional learning models ignore protons in product prediction, these results suggest that proton behavior is crucial in predicting organic reaction pathways.
In the proposed learning model, removing the fragment structures in group 2 significantly reduces the prediction accuracy, because the scores rapidly decrease depending on the number of omitted descriptors. Therefore, these structures are critical in determining the forward direction of a reaction network. Focusing on these points in group 2 (see Table S3 in ESI†), the proposed learning model identifies three types of rules in the context of organic chemistry, as shown in Fig. 4. The detailed relationships between the points and rules are as follows.
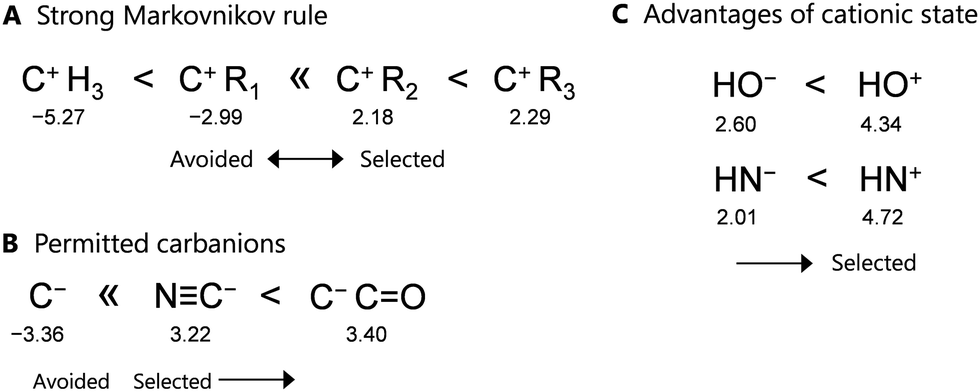 | ||
| Fig. 4 Three rules identified by our learning model: (A) strong Markovnikov, (B) permitted carbanions, and (C) advantages of the cationic states. The numbers under the fragment structures represent their points on a molecular graph. | ||
First, focusing on the carbocations, a tertiary carbocation attached to three alkyl groups (C+R3) provides higher addition points, whereas secondary and primary carbocations (C+HR2 and C+H2R) provide lower points. These results suggest that tertiary and secondary carbocations are more likely to form as intermediates based on this learning model, whereas primary and zero-degree carbocations (belonging to group 3) are unlikely to form. This trend is consistent with the Markovnikov rule in the context of organic chemistry, and thus, the developed learning model obeys this rule (Fig. 4(A)).
In addition, only the carbanion (C−) in group 3 is avoided as an intermediate, whereas the two carbanions in group 2, i.e., NC− and C−(CO), are acceptable as intermediates due to their higher positive points. This result is also consistent with the reaction rules of organic chemistry, where the stable base NC− and the presence of an anion at the α-position of a carbonyl group are favorable (Fig. 4(B)).
As a slightly unique rule, HN+ and HO+ exhibit higher positive points than their anions, i.e., the learning model selects the pathway with HO+ as the intermediate rather than that with HO−. This is consistent with the formation of HO+ during the second step of the aldol reaction, as shown in Fig. 2(A). Thus, the proposed model suggests that protons initially react with not only unsaturated bonds but also N and O atoms in the functional groups (Fig. 4(C)). However, this rule may have been influenced so that some reactions used as training were acid-catalyzed reactions. The generality of the rule needs further investigation.
In conclusion, we proposed a learning model that predicts both products and reaction pathways by combining machine learning and reaction network approaches. After learning fundamental reactions, the model predicted the products with a top-5 accuracy of 68.6%. In addition, the proposed model could classify several basic reaction rules in the context of organic chemistry and predict the reaction pathway using the acquired chemical knowledge. It was found that the combination of the two simple approaches can provide an explainable learning model for the chemical reaction. Further improvements in the construction of the reaction network and the selection of descriptors are expected for the practical scientific predictor.
This work was supported by the Japan Society for the Promotion of Science KAKENHI Grant Number JP19K05371. We would like to thank Editage (https://www.editage.com) for English language editing. Program codes and all input and output data used in this work are available on GitHub (https://github.com/ida-rnet/RNet/).
Conflicts of interest
There are no conflicts to declare.References
- R. S. Bohacek, C. McMartin and W. C. Guida, Med. Res. Rev., 1996, 16, 3–50 CrossRef CAS PubMed.
- J. Boström, D. G. Brown, R. J. Young and G. M. Keserü, Nat. Rev. Drug Discovery, 2018, 17, 709–727 CrossRef PubMed.
- K. C. Nicolaou and E. J. Sorensen, Classics in Total Synthesis: Targets, Strategies, Methods, Wiley, Hoboken, 1996 Search PubMed.
- S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS PubMed.
- R. Guha, J. Comput. Aid. Mol. Des., 2008, 22, 857–871 CrossRef CAS PubMed.
- T. W. Quadri, L. O. Olasunkanmi, O. E. Fayemi, E. D. Akpan, C. Verma, E.-S. M. Sherif, K. F. Khaled and E. E. Ebenso, Coord. Chem. Rev., 2021, 446, 214101 CrossRef CAS.
- P. Raccuglia, K. C. Elbert, P. D. F. Adler, C. Falk, M. B. Wenny, A. Mollo, M. Zeller, S. A. Friedler, J. Schrier and A. J. Norquist, Nature, 2016, 533, 73–76 CrossRef CAS PubMed.
- C. W. Coley, N. S. Eyke and K. F. Jensen, Angew. Chem., Int. Ed., 2020, 59, 22858–22893 CrossRef CAS PubMed.
- M. A. Kayala and P. Baldi, J. Chem. Inf. Model., 2012, 52, 2526–2540 CrossRef CAS PubMed.
- J. N. Wei, D. Duvenaud and A. Aspuru-Guzik, ACS Cent. Sci., 2016, 2, 725–732 CrossRef CAS PubMed.
- C. W. Coley, R. Barzilay, T. S. Jaakkola, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2017, 3, 434–443 CrossRef CAS PubMed.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, ACS Cent. Sci., 2019, 5, 1572–1583 CrossRef CAS PubMed.
- C. W. Coley, W. Jin, L. Rogers, T. F. Jamison, T. S. Jaakkola, W. H. Green, R. Barzilay and K. F. Jensen, Chem. Sci., 2019, 10, 370–377 RSC.
- A. L. Ferguson, ACS Cent. Sci., 2018, 4, 938–941 CrossRef CAS PubMed.
- E. J. Corey and W. T. Wipke, Science, 1969, 166, 178–192 CrossRef CAS PubMed.
- H. L. Gelernter, A. F. Sanders, D. L. Larsen, K. K. Agarwal, R. H. Boivie, G. A. Spritzer and J. E. Searleman, Science, 1977, 197, 1041–1049 CrossRef CAS PubMed.
- S. Hanessian, J. Franco and B. Larouche, Pure Appl. Chem., 1990, 62, 1887–1910 CrossRef CAS.
- M. H. Todd, Chem. Soc. Rev., 2005, 34, 247–266 RSC.
- A. Cook, A. P. Johnson, J. Law, M. Mirzazadeh, O. Ravitz and A. Simon, WIREs Comput. Mol. Sci., 2012, 2, 79–107 CrossRef CAS.
- O. Engkvist, P.-O. Norrby, N. Selmi, Y.-H. Lam, Z. Peng, E. C. Sherer, W. Amberg, T. Erhard and L. A. Smyth, Drug Discovery Today, 2018, 23, 1203–1218 CrossRef CAS PubMed.
- I. Ugi, J. Bauer, K. Bley, A. Dengler, A. Dietz, E. Fontain, B. Gruber, R. Herges and M. Knauer, Angew. Chem., Int. Ed. Engl., 1993, 32, 201–227 CrossRef.
- B. A. Grzybowski, S. Szymkuć, E. Gajewska, K. Molga, P. Dittwald, A. Wołos and T. Klucznik, Chemistry, 2018, 4, 390–398 CrossRef CAS.
- B. Mikulak-Klucznik, P. Gołębiowska, A. A. Bayly, O. Popik, T. Klucznik, S. Szymkuć, E. P. Gajewska, P. Dittwald, O. Staszewska-Krajewska, W. Beker, T. Badowski, K. A. Scheidt, K. Molga, J. Mlynarski, M. Mrksich and B. A. Grzybowski, Nature, 2020, 588, 83–88 CrossRef CAS PubMed.
- T. Ida, M. Nishida and Y. Hori, J. Phys. Chem. A, 2019, 123, 9579–9586 CrossRef CAS PubMed.
- J. Bradshaw, M. J. Kusner, B. Paige, M. H. S. Segler and J. M. Hernández-Lobato, ICLR, 2019.
- H. Bi, H. Wang, C. Shi, C. Coley, J. Tang and H. Guo, PMLR, 2021, 139, 904–913 Search PubMed.
- M. Yano, Organic Chemistry 1000 Nkocks for Reaction Mechanics, Kagaku-Dojin, Tokyo, 2019 Search PubMed.
- The Pharmaceutical Society of Japan, Essential Organic Reactions, Kagaku-Dojin, Tokyo, 2019 Search PubMed.
- H. Meislich, H. Nechamkin, J. Sharefkin and G. Hademenos, Schaum's Outline of Organic Chemistry, McGraw Hill, New York, 2013 Search PubMed.
- K. P. C. Vollhardt and N. E. Schore, Organic Chemistry: Structure and Function, W. H. Freeman, New York, 2014 Search PubMed.
- D. P. Kingma and J. Ba, arXiv, 2017, preprint, arXiv:1412.6980v9 DOI:10.48550/arXiv.1412.6980.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc03890d |
| This journal is © The Royal Society of Chemistry 2023 |