
Intracellular delivery of bacterial effectors for cancer therapy using biodegradable lipid nanoparticles†
Wenting
Li
ab,
Leihou
Shao
ab,
Ji
Liu
ab,
Jinhan
Sheng
ab,
Qizhen
Zheng
ab and
Ming
Wang
 *ab
*ab
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Analytical Chemistry for Living Biosystems, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 8th March 2023
Abstract
Bacterial effector proteins are virulence factors that are secreted and mediate orthogonal post-translational modifications of proteins that are not found naturally in mammalian systems. They hold great promise for developing biotherapeutics by regulating malignant cell signaling in a specific and targeted manner. However, delivering bacterial effectors into disease cells poses a significant challenge to their therapeutic potential. In this study, we report on the design of a combinatorial library of bioreducible lipid nanoparticles containing disulfide bonds for highly efficient bacterial effector delivery and potential cancer therapy. A leading lipid, PPPDA-O16B, identified from the library, can encapsulate and deliver DNA plasmids into cells. The gene cargo is released in response to the reductive cellular environment that is upregulated in cancer cells, leading to enhanced gene delivery and protein expression efficiency. Furthermore, we demonstrate that PPPDA-O16B can deliver the bacterial effector protein, DUF5, to degrade mutant RAS and inactivate downstream MAPK signaling cascades to suppress cancer cell growth in vitro and in tumor-bearing mouse xenografts. This strategy of delivering bacterial effectors using biodegradable lipid nanoparticles can be expanded for cancer cell signaling regulation and antitumor studies.
Introduction
Bacterial effectors are naturally evolved toxins that hijack host cell signaling to promote bacterial invasion by impairing innate immune responses.1,2 During the evolution of host–pathogen competition, these effectors can specifically interfere with cellular signal components by evolving unique mechanisms to regulate the post-translational modification of protein of host cells.3 Therefore, bacterial effectors hold a great promise for treating genetic diseases,4 including cancers by rewiring the signaling of disease cells.5 For example, Shigella type III effector OspF can dephosphorylate and inactivate extracellular signal-regulated kinase (ERK),6,7 a terminal kinase of the mitogen-activated protein kinase (MAPK) signaling cascade by catalyzing an irreversible β-elimination reaction.8,9 Also, DUF5 (Ras/RAP1-specific endopeptidase, RRSP) can specifically recognize and cleave the conserved domain of RAS protein,10,11 which is frequently mutated in cancer cells. Despite the high efficiency and great potency of bacterial effectors for rewiring disease cell signaling,12 their therapeutic potential is challenged by the difficulty and low efficiency of introducing bacterial effectors to disease cells, particularly in disease cell-selective manner. Previously, it has been reported that fusing bacterial effector protein with protein transduction domain (PTD) or directly delivering genetically engineered bacterial effectors using nanoparticles was effective to regulate cell signaling for cancer therapy.4,13,14 However, the requirement of genetic engineering of bacterial effector protein for intracellular delivery may compromise their activity and therapeutic potency7,15 Therefore, there is a great need for developing new strategies for bacteria effector delivery to advance their therapeutic potential.Lipid nanoparticles (LNPs) are an emerging class of nanocarriers that have been harnessed for intracellular delivery of nucleic acids,16–18 gene editing tools,19–21 and proteins22–24 by making use their high transfection efficiency and biocompatibility. However, the controlled cargo release from LNPs to avoid the degradation of biotherapeutics25 and in response to disease cell26,27 is of great importance for advancing bacteria effector protein into therapeutics. To this end, we and several others have recently designed a series of biodegradable lipid nanoparticles that are responsive to disease cell-associated microenvironment for the delivery of protein,28 mRNA,29,30 and siRNA.31 Their effectiveness to deliver bacterial effector-encoding gene therapeutics, however, remains less explored while highly desired to advance the therapeutic potential of bacterial effector proteins. Herein, we report the design of biodegradable lipid nanoparticles for intracellular delivery of DNA encoding bacterial effector and its use for antitumor study (Scheme 1). To discover effective lipid nanoparticles for DNA delivery, we designed a combinatorial library of biodegradable lipids via the parallel reactions between aliphatic amines and acylates that feature a disulfide bond.32 The incorporation of disulfide bond into lipid nanoparticles can enhance gene delivery efficiency by promoting lipid degradation and intracellular release of therapeutic cargos via the thiol-disulfide exchange reaction in response to the upregulated reduced glutathione (GSH) in tumor cells.33,34 Using a lipid screening approach by delivering DNA encoding green fluorescent protein (GFP), we have identified PPPDA-O16B, synthesized from N-Propyl-1,3-propanediamine (PPPDA) and 2-(dodecyldisulfanyl) ethyl acrylate (O16B) as a leading lipid for DNA delivery from a library containing 16 lipids. Detailed study revealed that PPPDA-O16B can encapsulate DNA plasmid, releasing DNA selectively via lipid degradation catalyzed by GSH. Further, we demonstrate that the intracellular delivery of bacterial effect DUF5-encoding plasmid using PPPDA-O16B effectively cleaves mutant RAS in cancer cells,35 prohibiting tumor cell growth both in vitro and in vivo. The strategy of tumor cell enhanced gene delivery using biodegradable lipid nanoparticles provides an innovative approach to expand the therapeutic potential of bacterial effect proteins.
 | ||
| Scheme 1 . Schematic illustration of bacterial effector delivery using bioreducible lipid nanoparticles (LNPs) for mutant RAS depletion and downstream signaling regulation. | ||
Experimental
General and materials
All chemicals used for lipid synthesis were purchased from Aladdin (Shanghai, China) or Sigma-Aldrich. Lipofectamine 2000 transfection reagent (#11668019, Invitrogen, American), antibodies used in this study were p44/42 MAPK (ERK1/2) (#4695, CST, USA), Phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (#9101, CST, American), RAS (#67648, CST, USA), GAPDH (#5174, CST, USA). The hepatocellular injury of mice was determined by measuring the concentration of albumin, serum aspartate transaminase (AST), alanine aminotransferase (ALT), and total bilirubin (T-BIL) in serum, using quantification kits purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). pEGFP-C1 and DUF5 plasmids were constructed by Beijing Syngentech Co., LTD., pcDNA3.1-OspF-HA and pcDNA3.1-firefly luciferase plasmids were purchased from Miaoling Biotechnology Co., LTD (Wuhan). Fluorescently labelled TAMRA-DNA with 33 bp random sequence was synthesized by Biosyntech (Suzhou, China). All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of National Center for Nanoscience and Technology (NCNST, Beijing, China) and Experiments were approved by the Animal Ethics Committee of NCNST.Analytical flow cytometry was performed on the Beckman Coulter CytoFLEX cytometer. Confocal laser scanning microscopy images were obtained from LSM 700 laser scanning confocal microscope (Zeiss). In vivo bioluminescence imaging and distribution study of PPPDA-O16B/firefly luciferase nanoparticles were performed on IVIS small animal imaging system (PerkinElmer, USA). DLS and Zeta potential measurements were determined on Zetasizer Nano ZS ZEN3600 (Malvern, UK). SEM images were captured by Hitachi S-8020 field-emission scanning electron microscope. TEM images were captured by HT770 transmission electronic microscopy.
Cell culture
HeLa, A375, A549 and HCT116 cells were purchased from National Platform of Experimental Cell Resources for Sci-Tech (Beijing, China). HeLa, A375, A549 cells were maintained in DMEM (Dulbecco's modified Eagle's medium) supplemented with 10% FBS (fetal bovine serum) and 1% penicillin/streptomycin. HCT116 cells were cultured in IMDM medium supplemented with 10% FBS and 1% penicillin/streptomycin. All cells were cultured at 37 °C in the presence of 5% CO2. For the intracellular delivery studies, cells (1 × 105 cell mL−1) were sub-cultured and seeded in 48-well, 96-well plate or 6-well plate 12 h prior to experiment.Lipid synthesis
Lipids were synthesized according to the method we previously reported.28,36 Briefly, O16B (2-(dodecyldisulfanyl) ethyl acrylate) or O16 (hexadecyl acrylate) was mixed and heated with varying amines (as listed in Fig. 1b) at a molar ratio of 3.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in Teflon-lined glass screw-top vials for 48 h at 70 °C. The mixtures were cooled to room temperature and purified using flash chromatography and characterized using 1H-NMR and MS (Fig. S1†).
:1 in Teflon-lined glass screw-top vials for 48 h at 70 °C. The mixtures were cooled to room temperature and purified using flash chromatography and characterized using 1H-NMR and MS (Fig. S1†).
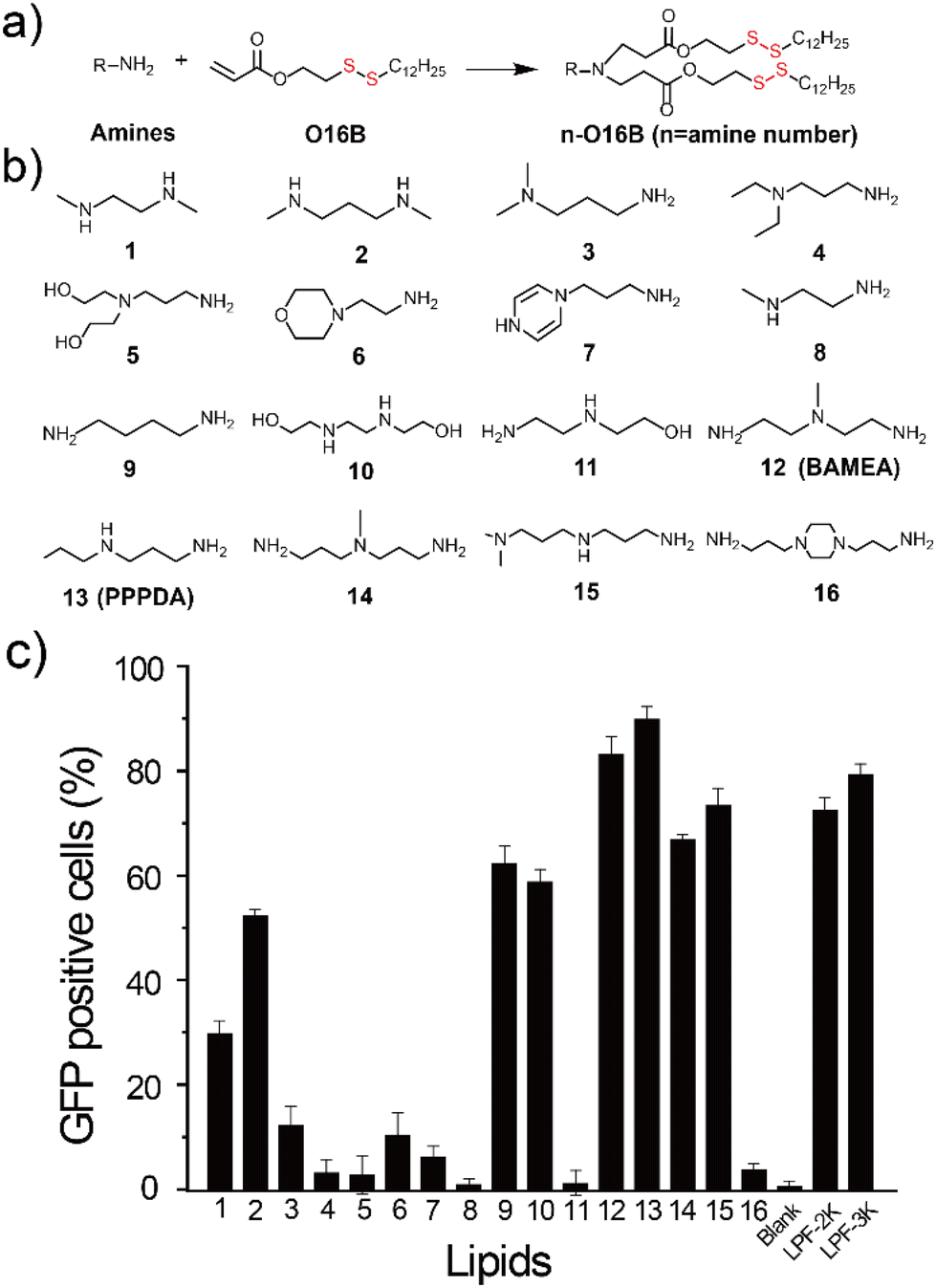 | ||
| Fig. 1 The design of bioreducible lipid nanoparticles for gene delivery. (a) Synthesis route of ionizable bioreducible lipids; (b) chemical structure of head amines used for lipid synthesis; (c) the percentage of GFP-positive HeLa cells after the transfection of pEGFP-C1 (0.66 μg mL−1) using different lipid nanoparticles (4.62 μg mL−1) as indicated. | ||
Formulation of lipid nanoparticles for gene delivery
Different lipids in the library were mixed with cholesterol and DOPE in chloroform at a mass ratio of 16:8:4, the organic solvent was dried overnight to form a lipid film, which was dissolved in ethanol and added dropwise into a sodium acetate buffer (25 mM, pH = 5.2) containing DSPE-PEG2000 at a ratio of lipid to DSPE-PEG2000 of 16:1. The mixed solution was stirred at R. T. for 15 min and diluted to 1 mg mL−1 lipid for intracellular delivery studies.37
Intracellular delivery of pEGFP-C1
HeLa cells were seeded in 48-well plate one day before the experiment. The enhanced green fluorescent protein (EGFP)-encoding plasmid, pEGFP-C1,38 was mixed with different lipid nanoparticles in sodium acetate buffer (25 mM, pH = 5.2) for 15 min at a mass ratio of 1:7. The resulted nanocomplexes were added to cells and incubated for 8 h, then the cell culture medium was replaced. The cells were harvested for flow cytometry analysis to quantify GFP-positive cells 48 h after gene transfection.
GSH-triggered degradation of PPPDA-O16B for enhanced gene cargo release
To study GSH-triggered lipid degradation and DNA release, PPPDA-O16B or PPPDA-O16 (2.1 μg) was mixed with pEGFP-C1 (100 ng) in 25 μL sodium acetate buffered solution (25 mM, pH = 5.2) and incubated at room temperature for 15 min. The resulted lipid/plasmid nanoparticles were further incubated with 0.02–5 mM GSH for 24 h before electrophoresis assay using 1% agarose gel.Intracellular delivery of DUF5 and OspF
To study the therapeutic effect of PPPDA-O16B/DUF5 delivery on prohibiting cancer cell growth, HeLa cells, A549 cells and HCT116 cells were seeded in a 96-well plate at the density of 15000 cells per well one day before the experiment. Cells were treated with PPPDA-O16B/DUF5 nanoparticles and incubated for 48 h, the cell viability was determined using MTT assay.39 Similarly, A375 cells were treated with different concentrations of PPPDA-O16B/OspF nanoparticles to study the therapeutic effect of OspF delivery.
To study RAS degradation or MAPK/ERK inactivation following bacterial effector protein delivery using western blot analysis, HeLa cells were seeded in 6-well plates, the cells were treated with PPPDA-O16B/DUF5 nanoparticles at a final concentration of 0.66 μg mL−1 DUF5 and 4.62 μg mL−1 lipid. After 8 h of incubation, the culture medium was replaced with fresh culture medium, and cells were harvested 36 h for detection RAS and p-ERK using western blot assay. For the delivery of OspF, A375 cells were used the study.
In vivo gene delivery and antitumor study using PPPDA-O16B
To investigate the in vivo gene delivery efficiency of PPPDA-O16B, HCT-116 tumor-bearing xenograft was generated by subcutaneously injecting 1 × 107 HCT116 cells suspended in 100 μL PBS to the left axilla region of 6 weeks-old Nu/Nu female mice (Beijing Vital River Laboratory Animal Technology Co., Ltd, Beijing, China). Tumor-bearing mice with tumor volume around 100 mm3 were intravenously injected with DPBS, firefly luciferase, and PPPDA-O16B/firefly luciferase nanoparticles at a DNA dosage of 0.1 mg kg−1. The major mouse organs, including heart, liver, spleen, lung, and kidney were harvested for bioluminescence imaging using IVIS small animal imaging system (PerkinElmer, USA) 48 h after the injections.For tumor growth suppression study, when the tumor volume reached an average size of 100 mm3, the mice were randomly divided into three groups (6 mice in each group). The mice were intravenously injected with PBS, 8 mg kg−1 PPPDA-O16B encapsulating 0.5 mg kg−1 pEGFP-C1, or 8 mg kg−1 PPPDA-O16B encapsulating 0.5 mg kg−1 DUF5 plasmid every other day for a total of four injections. Tumor size and mouse body weight were measured every two days. The tumor volume was calculated using the following formula:
| Tumor volume = 0.5 × length (mm) × width2 (mm) |
The tumor volume and mouse body weight were normalized to that of mice before nanoparticle injections. At the end of the study, the tumors were excised for western blot assay of RAS degradation and MAPK/ERK inactivation. Mouse blood was collected and centrifuged to isolate serum for liver toxicity assay according to manufacturer's instruction.
Results and discussion
Design of bioreducible lipid nanoparticles for DNA delivery
To enable the delivery of bacterial effector-encoding DNA into tumor cells, we generate a combinatorial library of ionizable lipids via the parallel Michael addition reaction between aliphatic amine and acylates bearing a disulfide bond (Fig. 1a and b). The as-purified lipids were formulated with cholesterol, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and DSPE-PEG2000 for all subsequent cellular delivery studies. To facilitate the screening of lipids for effective gene delivery, enhanced green fluorescent protein-encoding plasmid (pEGFP-C1) was first assembled with different lipids at a weight ratio of 1:7 before transfecting HeLa cells. The percentage of GFP-positive cells was quantified40 and compared to cells treated with pEGFP-C1 alone or pEGFP-C1 complexed with commercial gene transfection reagent, Lipofectamine 2000 (LPF-2K) and Lipofectamine 3000 (LPF-3K). As shown in Fig. 1c, HeLa cells treated with pEGFP-C1 alone did not show detectable EGFP expression, while cells treated with the complexes of plasmid and different lipids showed varied EGFP expression depending on the lipids that have been used for DNA delivery. Among the sixteen lipids we have designed here, lipid nanoparticles assembled from 12-O16B (BAMEA-O16B) and 13-O16B (PPPDA-O16B) showed higher gene transfection efficiency than that of LPF-2K and LPF-3K. Meanwhile, all lipid nanoparticles showed a high biocompatibility for gene delivery, as evidenced by their low cytotoxicity against HeLa cell growth (Fig. S2†). Further optimization of gene delivery using PPPDA-O16B was performed by changing the lipid and DNA ratio to enhance gene delivery and GFP expression efficiency (Fig. S3†). The delivery of 0.66 μg mL−1 pEGFP-C1 using PPPDA-O16B showed EGFP-positive HeLa cells in percentage as high as 90% (Fig. S4†). The as-formulated lipid nanoparticles are stable in biological settings, as evidenced by the slight change of nanoparticle size in a variety of media, suggesting their potency for gene delivery studies. (Table S1†). Moreover, PPPDA-O16B demonstrated generality for gene delivery in various cancer cell lines (Fig. S5†), highlighting the necessity and advantage of high-throughput screening to discover efficient and biocompatible lipid nanoparticles for DNA delivery.
Next, pEGFP-C1-encapsulated PPPDA-O16B nanoparticles were characterized using dynamic light scattering (DLS) analysis and scanning electron microscopy (SEM). DNA encapsulation efficiency determination reveals that the self-assembly of pEGFP-C1 and PPPDA-O16B encapsulated DNA as high as 79% (Table S2†). Meanwhile, it was found that PPPDA-O16B/pEGFP-C1 formed nanoparticles with size around 140 nm in diameter, as revealed by SEM imaging (Fig. S6†) and DLS analysis (Table S3†). Zeta-potential measurement indicated that the nanoparticle surface charge of PPPDA-O16B was decreased after pEGFP-C1 encapsulation, mostly arising from the electrostatic interaction between DNA plasmid and PPPDA-O16B that neutralizes the cationic nature of lipid nanoparticles (Table S3†).
Biodegradation of PPPDA-O16B enhances gene delivery efficiency
To further elaborate whether the integration of disulfide bonds into PPPDA-O16B promotes gene cargo release and enhances gene delivery efficiency in reductive intracellular environment,41 we synthesized a negative control lipid PPPDA-O16 that features the same head amine as PPPDA-O16B, while lacking the disulfide bond in the hydrophobic tails (Fig. 2a). Gel electrophoresis analysis indicated that both lipids can efficiently encapsulate pEGFP-C1 (Fig. 2b). However, only PPPDA-O16B/pEGFP-C1 complex showed effective gene release upon the treatment of reduced glutathione (GSH)42 (5 mM). Meanwhile, compared to PPPDA-O16/pEGFP-C1, the nanoparticle size of PPPDA-O16B/pEGFP-C1 complex was significantly increased after GSH treatment (Table S4†), suggesting the degradation of PPPDA-O16B in the presence of GSH, which is further confirmed by transmission electronic microscopy (TEM) imaging (Fig. S7†). Previous studies have reported that the GSH level in tumor cells (2–10 mM) is higher than in blood and normal cells,43 which results in the increased degradation of PPPDA-O16B, enhancing gene release in cancer cells (Fig. S8†) and, consequently, gene delivery efficiency.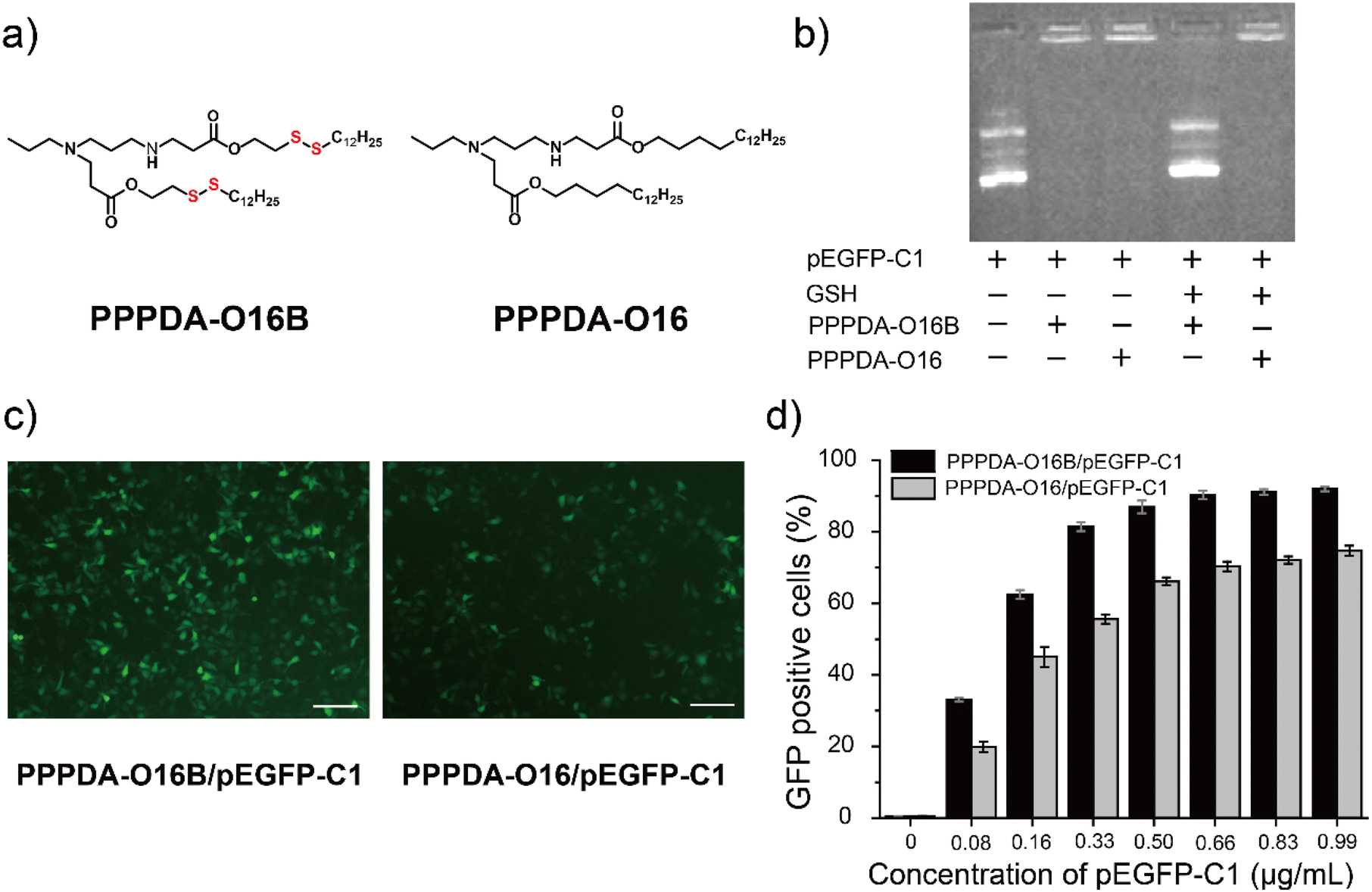 | ||
| Fig. 2 Bioreducible lipid nanoparticles enhance gene delivery efficiency. (a) Chemical structure of bioreducible PPPDA-O16B and non-degradable PPPDA-O16; (b) gel electrophoresis assay of DNA encapsulation by different lipids and GSH-triggered DNA release; (c) fluorescence microscope images of HeLa cells transfected with LNPs composed of pEGFP-C1 (0.33 μg mL−1) and 2.31 μg mL−1 PPPDA-O16B or PPPDA-O16 (scale bar: 20 μm); (d) Flow cytometry analysis of GFP-positive HeLa cells after the delivery of different concentrations of pEGFP-C1 complexed with PPPDA-O16B or PPPDA-O16 as indicated. | ||
pEGFP-C1 delivery and GFP expression efficiency using PPPDA-O16B or PPPDA-O16 was next compared. The GFP expression of HeLa cells treated with PPPDA-O16B/pEGFP-C1 was significantly higher than that treated with PPPDA-O16/pEGFP-C1, as revealed by the fluorescent imaging of cells received different treatments (Fig. 2c). It is noteworthy that the transfection and GFP expression efficiency of PPPDA-O16B/pEGFP-C1 was at least 20% higher than that of PPPDA-O16 under the same experimental condition (Fig. 2d), indicating that the introduction of disulfide bonds into lipids nanoparticles indeed promotes the release of DNA in response to the high concentration of GSH inside cells, which is frequently observed in the progression of tumor, suggesting its potential for delivering bacterial effector DNA for potential cancer therapy.
Bacterial effector delivery using PPPDA-O16B
It was found that the uptake efficiency of PPPDA-O16B complex in HeLa cancer cells was 95% when the concentration was 0.08 μg mL−1 (Fig. S9†), which provided a prerequisite for delivering bacterial-effect factor for tumor therapy. We next investigated the effectiveness of PPPDA-O16B for delivering bacterial effector-encoding DNA for regulating cancer cell signaling and potential cancer therapy. To this end, the delivery of OspF, a phospho-lyase secreted by Shigella spp., that specifically catalyzes the de-phosphorylation of ERK1/2 and inactivates MAPK signaling of host cells using PPPDA-O16B was first studied (Fig. S10†).44,45 A375 melanoma cells were treated with the nanocomplex of OspF and PPPDA-O16B, followed by the study of MAPK/ERK inactivation. An efficient dephosphorylation of ERK was observed when the cells were treated with PPPDA-O16B/OspF nanoparticles (Fig. 3a), while no change of p-ERK level was observed when the cells were treated with PPPDA-O16B/pEGFP-C1 nanoparticles, indicating the specificity of OspF delivery to inactivate MAPK signaling of cancer cells. In addition, PPPDA-O16B/OspF treatment efficiently prohibited A375 cell growth in a DNA concentration-dependent manner, while pEGFP-C1 delivery did not show such a tumor cell growth prohibition effect (Fig. 3b), suggesting the effectiveness and specificity of OspF delivery for potential cancer therapy.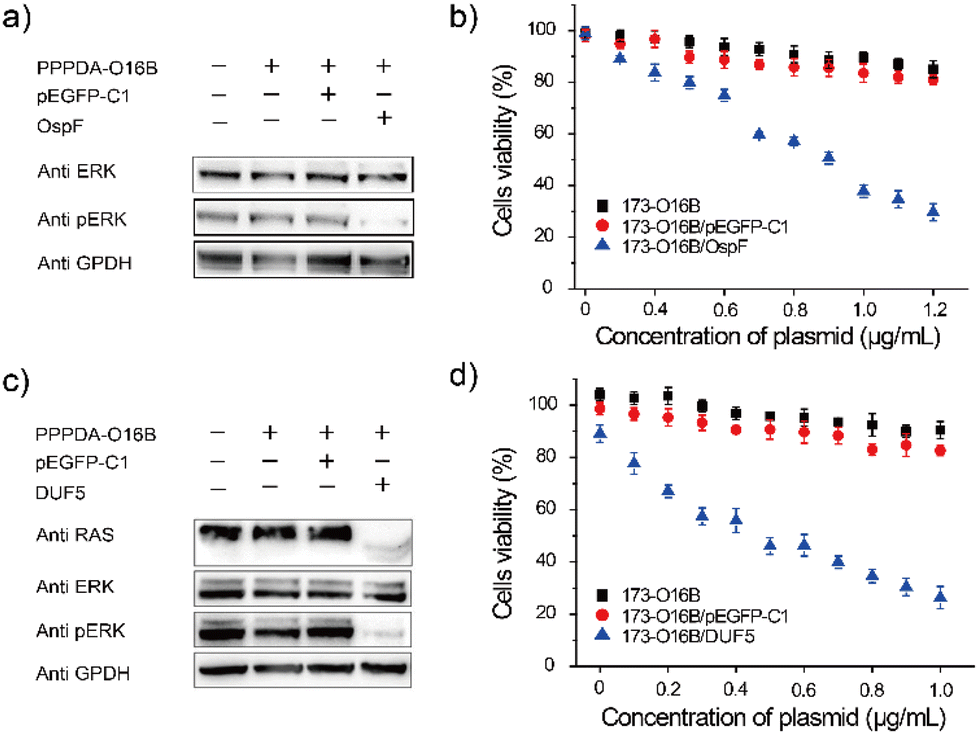 | ||
| Fig. 3 Intracellular delivery of bacterial effector OspF and DUF5 for rewiring tumor cell signaling. (a) Western blot analysis of the dephosphorylation of ERK1/2 in A375 cells transfected with PPPDA-O16B/OspF nanoparticles; (b) cell viability of A375 cells treated with different concentrations of PPPDA-O16B, PPPDA-O16B/pEGFP-C1 or PPPDA-O16B/OspF nanoparticles; (c) western blot analysis of RAS degradation and ERK1/2 dephosphorylation of HeLa cells transfected with PPPDA-O16B/DUF5 nanoparticles; (d) cell viability of HeLa cells received the treatment of different concentrations of PPPDA-O16B, PPPDA-O16B/pEGFP-C1 or PPPDA-O16B/DUF5 nanoparticles. | ||
To further demonstrate the general use of PPPDA-O16B for bacterial effector delivery, DUF5 secreted by Vibrio vulnificus,14,46 was delivered to HeLa cells using PPPDA-O16B. DUF5 can directly and irreversibly inactivate RAS signaling of cancer cells by cleaving the conserved domain of RAS protein,47 the upstream factor of MAPK/ERK signaling cascade (Fig. S11†). It has been previously reported that over one-third of human cancer cells shows RAS mutations with different types, a selective inhibition of RAS signaling therefore holds a great promise for developing new treatments for cancers by targeting RAS mulation.48,49 Indeed, PPPDA-O16B achieved efficient gene release (Fig. S12†), and therefore intracellular delivery of DUF5 to HeLa cells resulted in decreased expression of RAS and subsequent dephosphorylation of ERK (Fig. 3c),50 suggesting the effectiveness of DUF5 delivery to specifically inactivate RAS signaling cascade,51 and its potential for treating RAS mutation-associated cancers. Furthermore, PPPDA-O16B/DUF5 delivery showed significant cell growth prohibition effect on multiple cancer cell lines, including HeLa cells (Fig. 3d), human non-small cell lung cancer A549 cells and HCT116 human colon cancer cells (Fig. S13†), indicating the general use of PPPDA-O16B-mediated gene delivery for rewiring malignant cell signaling for potential cancer therapy.
In vivo delivery of DUF5 using PPPDA-O16B suppressed tumor growth
Encouraged by the high specificity and efficiency of PPPDA-O16B/DUF5 nanoparticles to degrade mutant RAS and inactivate downstream signaling in cultured cells, we next studied its efficacy in vivo. To this end, a HCT116 tumor-bearing mouse xenograft model was generated by subcutaneously injecting HCT116 cells into the armpit of female Nu/Nu nude mouse for studying the in vivo gene delivery efficacy of PPPDA-O16B nanoparticles. The tumor-bearing mice were intravenously injected with firefly luciferase-encoding plasmid formulated using PPPDA-O16B, followed by bioluminescence imaging of mouse tissues52 to study the in vivo distribution and gene delivery efficiency of PPPDA-O16B nanoparticles. As shown in Fig. 4a, the administration of PPPDA-O16B/luciferase nanoparticles resulted in an efficient expression of luciferase in mouse tumors, while a weak luciferase expression was observed in mice injected with free luciferase-encoding plasmid. Furthermore, our study revealed different expression levels of luciferase in normal tissues and tumor. Compared with normal tissues, the high expression of luciferase in tumor implies a higher accumulation of PPPDA-O16B nanoparticles in tumors. We attribute the improved efficiency of gene release in tumor tissues and consider PPPDA-O16B as a potential gene delivery system with tumor targeting potential.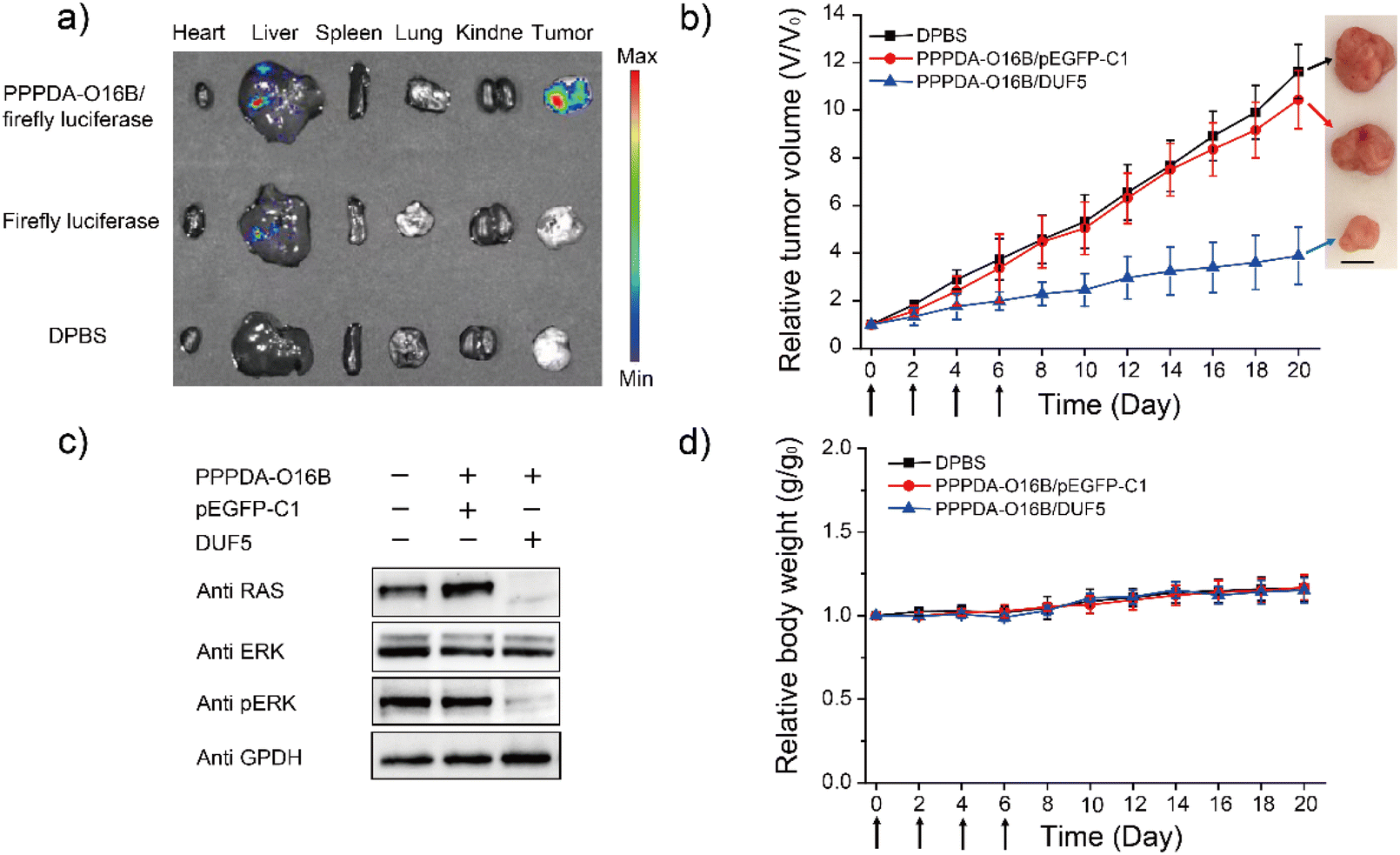 | ||
| Fig. 4 In vivo delivery of DUF5 suppresses tumor growth. (a) Administration of PPPDA-O16B/firefly luciferase nanoparticles resulted in the delivery and expression of luciferase in tumors. HCT116 tumor-bearing mice were injected intravenously with 1 mg kg−1 luciferase-encoding plasmid complexed with PPPDA-O16B; (b) relative tumor volume of HCT116 tumor-bearing mice treated with DPBS, PPPDA-O16B/pEGFP-C1, PPPDA-O16B/DUF5 nanoparticles (scale bar: 1 cm). The results were presented as mean ± SD (n = 6); (c) western blot assay of RAS and downstream signaling of tumors harvested from mice received different treatments as indicated; (d) relative mouse body weight change during the treatment. The results were presented as mean ± SD (n = 6). | ||
The in vivo antitumor efficacy of PPPDA-O16B/DUF5 nanoparticles was subsequently studied by intravenous injection of different nanoparticle formulations to HCT116 tumor-bearing mice, including Dulbecco's Phosphate-Buffered Saline (DPBS), PPPDA-O16B/pEGFP-C1, and PPPDA-O16B/DUF5 at a DNA dosage of 0.5 mg kg−1. We found that the injection of DPBS and PPPDA-O16B/pEGFP-C1 had bare effect on suppressing tumor growth, while PPPDA-O16B/DUF5 injection greatly suppressed tumor growth with tumor volume decreased down to 20% of negative controls (Fig. 4b). Notably, the depletion of RAS and inactivation of MAPK signaling in tumors followed PPPDA-O16B/DUF5 administration were confirmed by the downregulation of RAS and p-ERK levels in tumor tissues (Fig. 4c). In contrast, the delivery of pEGFP-C1 showed a very weak effect on depleting RAS and blocking MAPK signaling. In addition, the mouse body weight change was minimal after different nanoparticle treatments (Fig. 4d). The administration of different PPPDA-O16B nanoparticles did not cause obvious hepatocellular injury, as revealed by the comparable level of albumin, total bilirubin, aspartate aminotransferase (AST) and alanine aminotransferase (ALT) in mouse serum with all treatments (Fig. S14†), suggesting the high biocompatibility of PPPDA-O16B nanoparticles for in vivo delivery of bacterial effector delivery for potential cancer therapy.
Conclusions
In summary, we reported herein the design of bioreducible lipid nanoparticles for intracellular delivery of bacterial effector for rewiring malignant cell signaling for potential cancer therapy. We demonstrated that the bioreducible LNPs enables an effective gene delivery and controlled release of DNA in tumor cells by making use the high concentration of GSH inside tumor cells to promote LNPs degradation. We showed that the delivery of DUF5 using optimized LNPs, PPPDA-O16B, was effective to deplete mutant RAS for suppressing tumor cell growth both in vitro and in vivo. We believe the strategy of delivering bacterial effect protein into tumors using bioreducible lipid nanoparticles could be further expanded to other types of bacterial-derived biotherapeutics for potential cancer therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. Wang acknowledges the financial support from the National Science Foundation of China (22077125 to MW) and Beijing Natural Science Foundation (Z220023).References
- L. Diacovich and J. P. Gorvel, Nat. Rev. Microbiol., 2010, 8, 117–128 CrossRef CAS PubMed.
- E. Lemichez, M. R. Popoff and K. J. F. Satchell, Cell. Microbiol., 2020, 22, e13178 CrossRef CAS PubMed.
- D. Ribet and P. Cossart, Cell, 2010, 143, 694–702 CrossRef CAS PubMed.
- S. J. Yang, Q. Tang, L. Chen, J. Chang, T. Jiang, J. Y. Zhao, M. Wang and P. R. Chen, Angew. Chem., Int. Ed., 2020, 59, 18087–18094 CrossRef CAS PubMed.
- P. Baindara and S. M. Mandal, Biochimie, 2020, 177, 164–189 CrossRef CAS PubMed.
- P. Wei, W. W. Wong, J. S. Park, E. E. Corcoran, S. G. Peisajovich, J. J. Onuffer, A. Weiss and W. A. Lim, Nature, 2012, 488, 384–388 CrossRef CAS PubMed.
- J. Zhao, Y. Liu, F. Lin, W. Wang, S. Yang, Y. Ge and P. R. Chen, ACS Cent. Sci., 2019, 5, 145–152 CrossRef CAS PubMed.
- H. Li, H. Xu, Y. Zhou, J. Zhang, C. Long, S. Li, S. Chen, J. M. Zhou and F. Shao, Science, 2007, 315, 1000–1003 CrossRef CAS PubMed.
- Y. Zhu, H. Li, C. Long, L. Hu, H. Xu, L. Liu, S. Chen, D. C. Wang and F. Shao, Mol. Cell, 2007, 28, 899–913 CrossRef CAS PubMed.
- I. Antic, M. Biancucci, Y. M. Zhu, D. R. Gius and K. J. F. Satchell, Nat. Commun., 2015, 6, 7396 CrossRef CAS PubMed.
- M. Biancucci, A. E. Rabideau, Z. Y. Lu, A. R. Loftis, B. L. Pentelute and K. J. F. Satchell, Biochem., 2017, 56, 2747–2757 CrossRef CAS PubMed.
- J. Downward, Nat. Rev. Cancer, 2003, 3, 11–22 CrossRef CAS PubMed.
- M. Biancucci, G. Minasov, A. Banerjee, A. Herrera, P. J. Woida, M. B. Kieffer, L. Bindu, M. Abreu-Blanco, W. F. Anderson, V. Gaponenko, A. G. Stephen, M. Holderfield and K. J. F. Satchell, Sci. Signaling, 2018, 11, eaat8335 CrossRef PubMed.
- V. Vidimar, G. L. Beilhartz, M. Park, M. Biancucci, M. B. Kieffer, D. R. Gius, R. A. Melnyk and K. J. F. Satchell, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 16938–16948 CrossRef CAS PubMed.
- I. Antic, M. Biancucci and K. J. F. Satchell, Proteins, 2014, 82, 2643–2656 CrossRef CAS PubMed.
- Y. Eygeris, M. Gupta, J. Kim and G. Sahay, Acc. Chem. Res., 2022, 55, 2–12 CrossRef CAS PubMed.
- J. C. Kaczmarek, A. K. Patel, K. J. Kauffman, O. S. Fenton, M. J. Webber, M. W. Heartlein, F. DeRosa and D. G. Anderson, Angew. Chem., Int. Ed., 2016, 55, 13808–13812 CrossRef CAS PubMed.
- E. Alvarez-Benedicto, L. Farbiak, M. Marquez Ramírez, X. Wang, L. T. Johnson, O. Mian, E. D. Guerrero and D. J. Siegwart, Biomater. Sci., 2022, 10, 549–559 RSC.
- J. A. Zuris, D. B. Thompson, Y. Shu, J. P. Guilinger, J. L. Bessen, J. H. Hu, M. L. Maeder, J. K. Joung, Z. Y. Chen and D. R. Liu, Nat. Biotechnol., 2015, 33, 73–80 CrossRef CAS PubMed.
- J. Liu, J. Chang, Y. Jiang, X. D. Meng, T. M. Sun, L. Q. Mao, Q. B. Xu and M. Wang, Adv. Mater., 2019, 31, 1902725 CrossRef PubMed.
- W. Cai, J. Liu, X. Chen, L. Mao and M. Wang, J. Am. Chem. Soc., 2022, 144, 22272–22280 CrossRef CAS PubMed.
- M. Wang, S. Sun, C. I. Neufeld, B. Perez-Ramirez and Q. Xu, Angew. Chem., Int. Ed., 2014, 53, 13444–13448 CrossRef CAS PubMed.
- J. Chang, W. Q. Cai, C. J. Liang, Q. Tang, X. H. Chen, Y. Jiang, L. Q. Mao and M. Wang, J. Am. Chem. Soc., 2019, 141, 18136–18141 CrossRef CAS PubMed.
- W. T. Li, J. Liu, L. H. Shao, L. Q. Mao and M. Wang, ChemBioChem, 2021, 22, 2608–2613 CrossRef CAS PubMed.
- C. J. Liang, Q. Z. zheng, T. L. Luo, W. Q. Cai, L. Q. Mao and M. Wang, CCS Chem., 2022, 4, 3809–3819 CrossRef CAS.
- G. Sahay, W. Querbes, C. Alabi, A. Eltoukhy, S. Sarkar, C. Zurenko, E. Karagiannis, K. Love, D. L. Chen, R. Zoncu, Y. Buganim, A. Schroeder, R. Langer and D. G. Anderson, Nat. Biotechnol., 2013, 31, 653–658 CrossRef CAS PubMed.
- Q. Z. Zheng, W. T. Li, L. Q. Mao and M. Wang, Biomater. Sci., 2021, 9, 7024–7033 RSC.
- J. Chang, X. H. Chen, Z. Glass, F. Gao, L. Q. Mao, M. Wang and Q. B. Xu, Acc. Chem. Res., 2019, 52, 665–675 CrossRef CAS.
- Q. Tang, J. Liu, Y. Jiang, M. Zhang, L. Mao and M. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 46585–46590 CrossRef CAS PubMed.
- W. Q. Cai, T. L. Luo, X. H. Chen, L. Q. Mao and M. Wang, Adv. Funct. Mater., 2022, 32, 2204947 CrossRef CAS.
- C. Liang, J. Chang, Y. Jiang, J. Liu, L. Mao and M. Wang, Chem. Commun., 2019, 55, 8170–8173 RSC.
- M. Wang, K. Alberti, A. Varone, D. Pouli, I. Georgakoudi and Q. B. Xu, Adv. Healthc. Mater., 2014, 3, 1398–1403 CrossRef CAS PubMed.
- Y. M. Li, J. Bolinger, Y. J. Yu, Z. Glass, N. Shi, L. Yang, M. Wang and Q. B. Xu, Biomater. Sci., 2019, 7, 596–606 RSC.
- S. Liu, J. Liu, H. Li, K. Mao, H. Wang, X. Meng, J. Wang, C. Wu, H. Chen, X. Wang, X. Cong, Y. Hou, Y. Wang, M. Wang, Y. G. Yang and T. Sun, Biomaterials, 2022, 287, 121645 CrossRef CAS PubMed.
- M. V. Milburn, L. Tong, A. M. Devos, A. Brunger, Z. Yamaizumi, S. Nishimura and S. H. Kim, Science, 1990, 247, 939–945 CrossRef CAS PubMed.
- S. Sun, M. Wang, S. A. Knupp, Y. Soto-Feliciano, X. Hu, D. L. Kaplan, R. Langer, D. G. Anderson and Q. Xu, Bioconjugate Chem., 2012, 23, 135–140 CrossRef CAS PubMed.
- M. Wang, J. A. Zuris, F. Meng, H. Rees, S. Sun, P. Deng, Y. Han, X. Gao, D. Pouli, Q. Wu, I. Georgakoudi, D. R. Liu and Q. Xu, Proc. Natl. Acad. Sci., 2016, 113, 2868–2873 CrossRef CAS PubMed.
- D. R. Radu, C. Y. Lai, K. Jeftinija, E. W. Rowe, S. Jeftinija and V. S. Lin, J. Am. Chem. Soc., 2004, 126, 13216–13217 CrossRef CAS PubMed.
- Y. Li, J. Bolinger, Y. Yu, Z. Glass, N. Shi, L. Yang, M. Wang and Q. Xu, Biomater. Sci., 2019, 7, 596–606 RSC.
- Y. Li, T. Yang, Y. Yu, N. Shi, L. Yang, Z. Glass, J. Bolinger, I. J. Finkel, W. Li and Q. Xu, Biomaterials, 2018, 178, 652–662 CrossRef CAS PubMed.
- H. F. Abed, W. H. Abuwatfa and G. A. Husseini, Nanomaterials, 2022, 12, 3183 CrossRef CAS PubMed.
- Y. Wang, P. Chen and J. Shen, Biomaterials, 2006, 27, 5292–5298 CrossRef CAS PubMed.
- H. X. Tang, C. Q. Li, Y. Zhang, H. Y. Zheng, Y. Cheng, J. J. Zhu, X. J. Chen, Z. H. Zhu, J. G. Piao and F. Z. Li, Theranostics, 2020, 10, 9865–9887 CrossRef CAS PubMed.
- H. Li, H. Xu, Y. Zhou, J. Zhang, C. Long, S. Li, S. Chen, J. M. Zhou and F. Shao, Science, 2007, 315, 1000–1003 CrossRef CAS PubMed.
- Y. Zhu, H. Li, C. Long, L. Hu, H. Xu, L. Liu, S. Chen, D. C. Wang and F. Shao, Mol. Cell, 2007, 28, 899–913 CrossRef CAS PubMed.
- K. J. F. Satchell, Microbiol. Spectrum, 2015, 3, 3 Search PubMed.
- M. V. Milburn, L. Tong, A. M. deVos, A. Brunger, Z. Yamaizumi, S. Nishimura and S. H. Kim, Science, 1990, 247, 939–945 CrossRef CAS PubMed.
- I. A. Prior, P. D. Lewis and C. Mattos, Cancer Res., 2012, 72, 2457–2467 CrossRef CAS PubMed.
- L. Deng, H. Zhang, Y. Zhang, S. Luo, Z. Du, Q. Lin, Z. Zhang and L. Zhang, Biomater. Sci., 2021, 9, 5599–5611 RSC.
- A. A. Samatar and P. I. Poulikakos, Nat. Rev. Drug Discovery, 2014, 13, 928–942 CrossRef CAS PubMed.
- H. Davies, G. R. Bignell, C. Cox, P. Stephens, S. Edkins, S. Clegg, J. Teague, H. Woffendin, M. J. Garnett, W. Bottomley, N. Davis, E. Dicks, R. Ewing, Y. Floyd, K. Gray, S. Hall, R. Hawes, J. Hughes, V. Kosmidou, A. Menzies, C. Mould, A. Parker, C. Stevens, S. Watt, S. Hooper, R. Wilson, H. Jayatilake, B. A. Gusterson, C. Cooper, J. Shipley, D. Hargrave, K. Pritchard-Jones, N. Maitland, G. Chenevix-Trench, G. J. Riggins, D. D. Bigner, G. Palmieri, A. Cossu, A. Flanagan, A. Nicholson, J. W. Ho, S. Y. Leung, S. T. Yuen, B. L. Weber, H. F. Seigler, T. L. Darrow, H. Paterson, R. Marais, C. J. Marshall, R. Wooster, M. R. Stratton and P. A. Futreal, Nature, 2002, 417, 949–954 CrossRef CAS PubMed.
- M. Manthorpe, F. Cornefert-Jensen, J. Hartikka, J. Felgner, A. Rundell, M. Margalith and V. Dwarki, Hum. Gene Ther., 1993, 4, 419–431 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra, DLS measurements, SEM and fluorescence images, liver function tests. See DOI: https://doi.org/10.1039/d3bm00008g |
| This journal is © The Royal Society of Chemistry 2023 |