
Ester-functionalized thermally activated delayed fluorescence materials†
Hiroshi
Tasaki
ab,
So
Shikita
ab,
In Seob
Park
a and
Takuma
Yasuda
 *ab
*ab
aINAMORI Frontier Research Center (IFRC), Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: yasuda@ifrc.kyushu-u.ac.jp
bDepartment of Applied Chemistry, Graduate School of Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
First published on 1st November 2021
Abstract
A family of thermally activated delayed fluorescence (TADF) materials functionalized with carboxylate esters is presented herein. Owing to their suppressed concentration-quenching effect in neat films, these ester-functionalized TADF emitters could be applied to the fabrication of non-doped organic light-emitting diodes, demonstrating high external electroluminescence quantum efficiencies of 6.3–18.7%.
Organic thermally activated delayed fluorescence (TADF) materials have attracted significant attention because of their potential to achieve ∼100% internal quantum efficiency (IQE) in organic light-emitting diodes (OLEDs). With these materials, both electrogenerated singlet (S1) and triplet (T1) excitons can be utilized for light emission without using precious metals.1,2 In the electroluminescence (EL) process of TADF-OLEDs, non-radiative dark T1 excitons can be upconverted to emissive bright S1 excitons via reverse intersystem crossing (RISC) driven by ambient thermal energy. Among the vast variety of organic TADF materials ever reported, those containing carbonitrile (CN) group(s) as electron-accepting moieties occupy a prominent position because of their facile synthetic accessibility; high thermal, chemical, and electrochemical stabilities; and excellent TADF capability.1c,3,4 As the first representative, in 2012, Adachi and coworkers reported a series of color-tuneable TADF materials based on phthalonitrile, isophthalonitrile, and terephthalonitrile tethered by multiple carbazoles.1c These CN-based TADF emitters afforded OLEDs with high external EL quantum efficiencies (EQEs) of up to 19.3%, because of the small singlet–triplet energy gap (ΔEST) of less than 0.1 eV. In 2016, by connecting four or five 3,6-di-tert-butylcarbazole (BCz) arms to a benzonitrile core, Duan and coworkers developed 2,3,5,6-tetrakis(3,6-di-tert-butylcarbazol-9-yl)benzonitrile and 2,3,4,5,6-pentakis(3,6-di-tert-butylcarbazol-9-yl)benzonitrile (5BCzBN) as another successful CN-based TADF family.4 The 5BCzBN emitter exhibited a high EQE of 21.2% and an impressive device operational lifetime of over 770 h in TADF-OLEDs. Despite the brilliant success of CN-based TADF materials, thus far, few studies have explored other isosteric electron-withdrawing groups typified by carboxylate ester (CO2R), instead of CN.5 Thus, exploring the applicability of ester functionalization and, thereby, expanding the chemical space for TADF emitters remains a crucial task.
Herein, we report a new family of ester-functionalized TADF emitters 1–4 (Fig. 1) that combine benzoate, isophthalate, and terephthalate esters as electron-acceptor (A) cores with four or five BCz electron-donor (D) units. Compared to the CN-based 5BCzBN,4 the corresponding ester-functionalized emitter (2) exhibited a significantly larger reorganization energy of emission (λS) and, thereby, a broader photoluminescence (PL) spectrum because of the larger structural relaxation of CO2CH3 than that of CN. Additionally, 2 exhibited a considerably high RISC rate (kRISC) of over 106 s−1, originating from an intense spin–orbit coupling (SOC) based on the ester functionalization. Non-doped OLEDs using 1–4 as neat emission layers achieved high maximum EQEs of 18.7%, 18.0%, 12.4%, and 6.3%, respectively, with relatively small efficiency roll-offs.
 | ||
| Fig. 1 Molecular structures of ester-functionalized TADF emitters 1–4. | ||
Compounds 1–4 were synthesized by the sequence of Fischer esterification and nucleophilic aromatic substitution (SNAr) reactions, with commercially available fluorinated benzoic acid, isophthalic acid, and terephthalic acid employed as the starting materials. The detailed synthesis procedures and chemical characterization data are described in ESI.† Peripheral tert-butyl groups of BCz can serve as an insulating shell for suppressing the intermolecular electronic interactions and exciton deactivation arising from the collisional Dexter energy transfer,6 thereby boosting the absolute PL quantum yield (ΦPL) in the condensed solid states, as discussed later.
As revealed by the time-dependent density functional theory (TD-DFT) calculations (ESI†), 1–4 adopt highly distorted D–A conformations between the peripheral BCz and central benzoate (or phthalate) moieties in the optimized geometries. The optimized structure of 2 exhibited large D–A dihedral angles of 60–63° in the ground (S0) state. Accordingly, for 1–4, the highest occupied natural transition orbitals were mainly localized on the multiple BCz units introduced, whereas the lowest unoccupied natural transition orbitals were distributed over the benzoate or phthalate core, including CO2CH3 substituent(s), as in the case of analogous CN-based TADF emitters.1c,4 Such distinct spatial separations of the frontier orbitals for 1–4 resulted in ΔEST values as low as 13–25 meV in the TD-DFT calculations, suggesting the possibility of efficient ISC/RISC, followed by TADF emission.
X-Ray crystallographic analysis revealed that 2 crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . In the crystal, as shown in Fig. 2, 2 was found to exist as an equimolar mixture of the enantiomeric clockwise (C) and counterclockwise (CC) propellers, similar to the case of the propeller chirality (atropisomerism) of hexaarylbenzenes.7 To avoid steric congestion within a molecule, all five BCz propeller blades were tilted in one direction at large D–A dihedral angles of 61–69°, consistent with the foregoing calculation results. This propeller-shaped geometry would be favorable for both lowering the ΔEST and suppressing the concentration quenching.
. In the crystal, as shown in Fig. 2, 2 was found to exist as an equimolar mixture of the enantiomeric clockwise (C) and counterclockwise (CC) propellers, similar to the case of the propeller chirality (atropisomerism) of hexaarylbenzenes.7 To avoid steric congestion within a molecule, all five BCz propeller blades were tilted in one direction at large D–A dihedral angles of 61–69°, consistent with the foregoing calculation results. This propeller-shaped geometry would be favorable for both lowering the ΔEST and suppressing the concentration quenching.
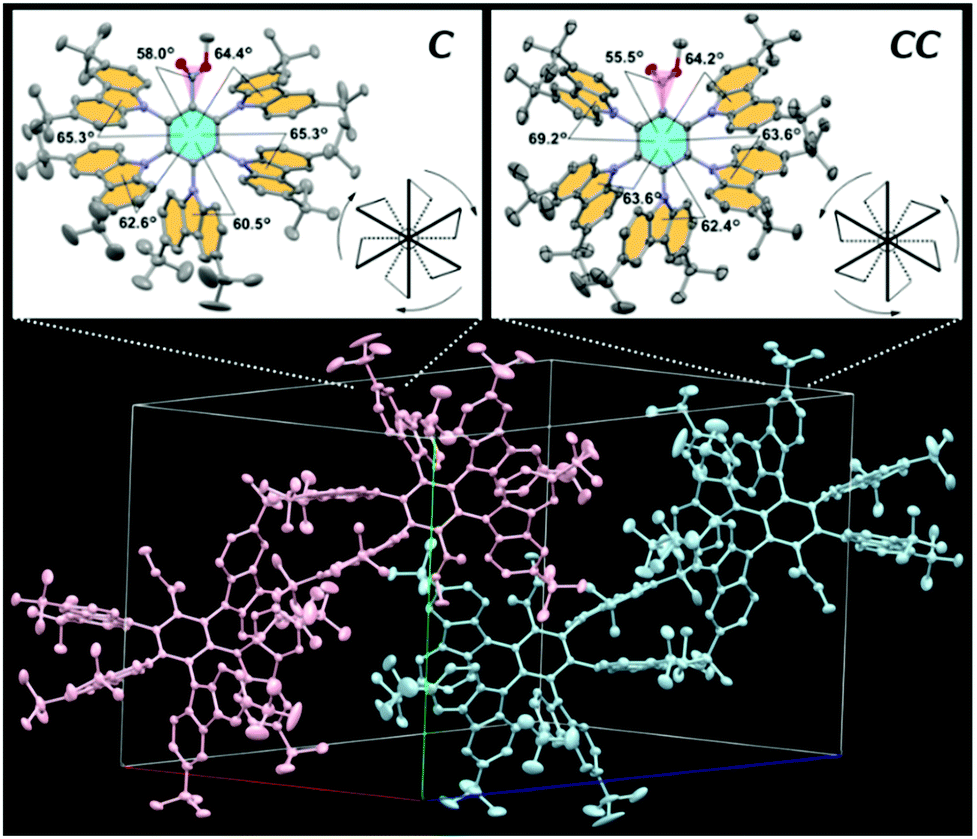 | ||
| Fig. 2 Single-crystal X-ray structure of 2 (CCDC No. 2114087†) with thermal ellipsoids at 50% probability: (top) propeller-shaped enantiomeric clockwise (C) and counterclockwise (CC) isomers, and (bottom) molecular packing diagram containing equimolar C (pink) and CC (blue) isomers. The single crystals of 2 were grown from CHCl3/CH3CN solution. | ||
Fig. 3a depicts the UV-vis absorption and PL spectra of 1–4 in dilute toluene solutions; the relevant photophysical data are compiled in Table 1. In addition to the intense π–π* absorption bands below 390 nm, all these molecules exhibited rather weak, broad absorptions in the range of 390–430 nm, which can be attributed to the intramolecular charge transfer (ICT) originating from their D–A electronic structures. Upon photoexcitation, 1–4 exhibited blue to orange structureless PL with emission peaks (λPL) ranging from 497 to 598 nm (Fig. 3a and b), whose ΦPL values were in the order of 2 (84%) > 1 (50%) > 3 (31%) > 4 (11%) in the deoxygenated toluene solutions. As in the case of the CN-based 5BCzBN, the ester-functionalized emitters showed a positive solvatochromism in PL8 because of the ICT nature of the S1 state (ESI†). Furthermore, 1–4 showed rather broad PL spectra with full width at half-maxima (FWHM) as large as 99–128 nm (0.49–0.52 eV) in comparison with those of their CN-based counterparts.1c,4 Such broadband spectral features can be ascribed to the large structural relaxation upon the ICT (S0 ⇄ S1). As schematically shown in Fig. 3c, the reorganization energies (λS and  ) associated with the emission and excitation processes primarily dominate the resulting spectral FWHM.9 Thus, emitters with larger λS (and
) associated with the emission and excitation processes primarily dominate the resulting spectral FWHM.9 Thus, emitters with larger λS (and  ) should afford a broad ICT emission. Here, we estimated the actual λS values by fitting the PL spectra using the Marcus theory expression (Fig. 3d):9,10
) should afford a broad ICT emission. Here, we estimated the actual λS values by fitting the PL spectra using the Marcus theory expression (Fig. 3d):9,10
 | (1) |
 | ||
| Fig. 3 (a) UV-vis absorption and PL spectra of 1–4 in toluene (1 × 10−5 M). The inset shows a magnified view of the ICT absorption bands. (b) Photograph of the PL emissions from the solutions under UV irradiation. (c) Schematic potential energy diagram of the S0 and S1 states; Eex, Eem, and ECT denote the energies of excitation, emission, and charge transfer, respectively. (d) PL spectra (solid lines) fitted using eqn (1) for the evaluation of λS. | ||
| Emitter | Statea | λ PL [nm] | Φ PL [%] | E FWHM [eV] | λ FWHM [nm] | λ S [eV] | τ p [ns] | τ d [μs] | k r [×106 s−1] | k ISC [×108 s−1] | k RISC [×106 s−1] |
E
ISCa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) j [meV] j [meV] |
E
RISCaj [meV] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Sol = deoxygenated toluene solution (10−5 M) at 300 K; film = pristine neat film at 300 K. b PL emission maximum. c Absolute PL quantum yield evaluated using an integrating sphere. d Full width at half-maxima of the PL spectrum given in energy and wavelength. e Reorganization energy along the S1 → S0 emission estimated from the PL spectrum. f Emission lifetimes of the prompt fluorescence (τp) and delayed fluorescence (τd). g Rate constant of the fluorescence radiative decay (S1 → S0); kr = Φp/τp. h Rate constant of ISC (S1 → T1); kISC = (1 − Φp)/τp. i Rate constant of RISC (T1 → S1); kRISC = Φd/(kIsc·τp·τd·Φp). j The activation energy for ISC and RISC estimated from the Arrhenius plots of kISC and kRISC (Fig. 5d). k See ESI. | |||||||||||||
| 1 | Sol | 497 | 50 | 0.45 | 94 | 0.51 | 5.2 | 3.1 | 10 | 1.8 | 3.0 | — | — |
| Film | 486 | 91 | 0.49 | 99 | 0.64 | 3.0 | 4.6 | 6.8 | 3.2 | 9.5 | 1.1 | 46 | |
| 2 | Sol | 508 | 84 | 0.53 | 113 | 0.79 | 4.7 | 1.9 | 9.4 | 2.0 | 9.8 | — | — |
| Film | 499 | 93 | 0.51 | 105 | 0.75 | 2.2 | 4.2 | 3.4 | 4.6 | 30 | 0.6 | 58 | |
| 3 | Sol | 540 | 31 | 0.51 | 125 | 0.70 | 18 | 0.8 | 9.4 | 0.5 | 1.2 | — | — |
| Film | 517 | 71 | 0.49 | 107 | 0.69 | 8.9 | 1.8 | 11 | 1.0 | 3.8 | 1.6 | 50 | |
| 4 | Sol | 598 | 11 | 0.55 | 163 | 0.99 | 2.3 | 0.06 | 47 | 3.8 | 0.3 | — | — |
| Film | 540 | 41 | 0.52 | 128 | 0.77 | 6.4 | 0.9 | 23 | 1.3 | 2.3 | ≈0 | 64 | |
| 5BCzBN | Sol | 483 | 100 | 0.42 | 83 | 0.38 | 3.7 | 17 | 3.8 | 2.7 | 4.3 | — | — |
| Film | 507 | 100 | 0.44 | 95 | 0.53 | 5.1 | 7.4 | 9.5 | 1.9 | 2.8 | 3.7 | 56 | |
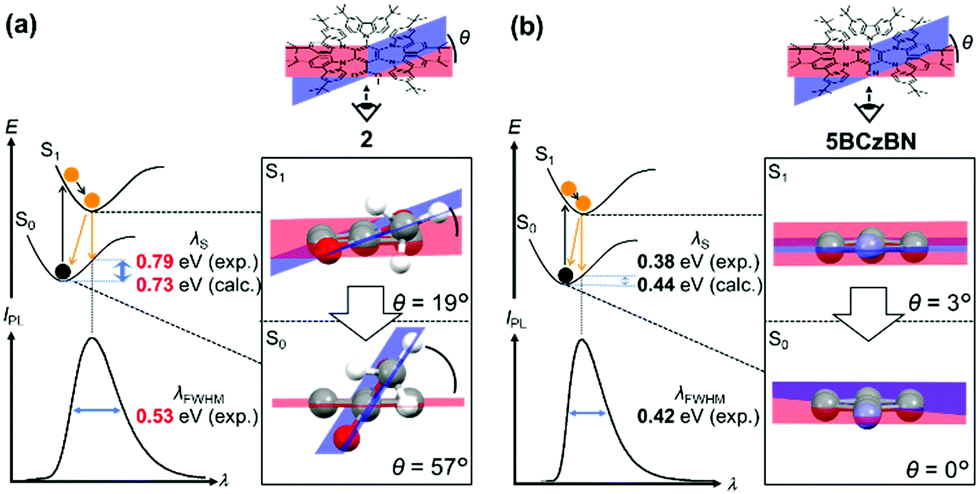 | ||
| Fig. 4 Representation of the structural and spectroscopic differences between (a) 2 and (b) 5BCzBN. Dihedral angle (θ) between the central benzene core and ester (or nitrile) group at the S1 and S0 geometries were simulated using TD-DFT at the PBE0/6-31G(d) level. | ||
Fig. 5 shows the photophysical properties of 1–4 in the pure neat films (see also Table 1 for details). While intense and structureless ICT emissions were observed, the λPL positions blue-shifted by 10–60 nm, compare to those in solutions. Notably, the neat films of 1–4 exhibited rather high ΦPL values of 91%, 93%, 71%, and 41%, respectively (Fig. 5b), indicating suppressed concentration quenching even in the condensed solid states. This result is closely related to the capability of aggregation-induced emission (AIE)11 of these propeller-shaped luminophores (ESI†). To further elucidate the TADF behaviors of 1–4, the transient PL decay characteristics were measured for the neat films (Fig. 5c). Each of the transient PL curves clearly demonstrates two emission components at 300 K and could be fitted by a typical biexponential model. The first component with a lifetime (τp) of ∼10 ns originates from conventional prompt fluorescence (i.e., S1 → S0), and the second, with a lifetime (τd) of ∼10 μs, can be assigned to the TADF involving ISC and RISC (i.e., S1 → T1 → S1 → S0). We further assessed the photophysical rate constants of the fluorescence radiative decay (kr), ISC (kISC), and RISC (kRISC), as listed in Table 1. It is known that lowering the activation energy of RISC (ERISCa) can accelerate spin-flipping RISC, thereby enhancing the TADF emission.9,12 Despite that both 2 and 5BCzBN exhibited comparable ERISCa values of 58 and 56 meV, respectively, the kRISC of 2 (3.0 × 107 s−1) was found to be one order of magnitude higher than that of 5BCzBN (2.8 × 106 s−1) at 300 K. Assuming that the excited state population is described by the classical Boltzmann statistical model, kRISC is given by the following equation:9,13
 | (2) |
 | ||
| Fig. 5 (a) Steady-state PL spectra, (b) PL images and fractional quantum yields for prompt and delayed fluorescence (Φp and Φd), and (c) transient PL decay profiles of 1–4 in the neat films measured at 300 K under N2. (d) Arrhenius plots of kISC and kRISC, measured for the neat films of 1–4, where the solid lines denote the least-squares fittings. | ||
Finally, to evaluate the EL performance of 1–4, we fabricated non-doped TADF-OLEDs with the structure of indium-tin-oxide (ITO, 100 nm)/HAT-CN (10 nm)/TAPC (40 nm)/CCP (10 nm)/1–4 (20 nm)/PPF (10 nm)/B3PyPB (50 nm)/Liq (1 nm)/Al (100 nm). In this device architecture, HAT-CN (2,3,6,7,10,11-hexacyano-1,4,5,8,9,12-hexaazatriphenylene) and TAPC (1,1-bis[4-[N,N-di(p-tolyl)amino]phenyl]cyclohexane) served as the hole-injection layer and hole-transport layer (HTL), respectively, while B3PyPB (1,3-bis[3,5-di(pyridin-3-yl)phenyl]benzene) and Liq (8-hydroxyquinolinato lithium) were adopted as the electron-transport layer (ETL) and electron-injection material, respectively. In addition, the thin layers of CCP (9-phenyl-3,9′-bicarbazole) and 2,8-bis(diphenylphosphinyl)dibenzo[b,d]furan (PPF) with high T1 energies were inserted at the HTL/EML and EML/ETL interfaces to confine the excitons within the neat EML. Fig. 6a–c shows the EL spectra, current density–voltage–luminance (J–V–L) characteristics, and EQE versus L plots of devices A–D using 1–4, and the key EL parameters are summarized in Table 2. Devices A–D were turned on at 3.6–4.0 V, and they emitted sky-blue to yellow EL with emission peaks (λEL) in the range of 494–544 nm. Among the four devices, devices A and B using 1 and 2 exhibited superior EL performance with a maximum EQE (EQEmax) values of 18.7% and 18.0%, respectively. Consequently, the EQEmax values were in the order of device A (18.7%) ≈ B (18.0%) > C (12.4%) > D (6.3%) (Fig. 6c). The reason for the relatively low EQEmax values of devices C and D is the substantially lower ΦPL values of 3 and 4 in the neat films.
 | ||
| Fig. 6 (a) EL spectra, (b) current density–voltage–luminance (J–V–L) characteristics, and (c) EQE vs. L plots of the non-doped TADF-OLEDs based on 1–4 (Devices A–D). | ||
| Device | Emitter | λ EL [nm] | E FWHM [eV] | λ FWHM [nm] | V on [V] | EQEmaxd [%] | EQE100/1000e [%] | CEf [cd A−1] | PEg [lm W−1] | CIEh (x, y) |
|---|---|---|---|---|---|---|---|---|---|---|
| a EL emission maximum. b Full width at half-maxima of the EL spectrum given in energy and wavelength. c Turn-on voltage at a luminance of over 1 cd m−2. d Maximum EQE. e EQE at a luminance of 100 and 1000 cd m−2. f Maximum current efficiency. g Maximum power efficiency. h Commission Internationale de l’Éclairage (CIE) chromaticity coordinates. i See ESI. | ||||||||||
| A | 1 | 494 | 0.49 | 100 | 4.0 | 18.7 | 18.5/13.9 | 46.8 | 37.5 | (0.22, 0.40) |
| B | 2 | 502 | 0.50 | 105 | 3.6 | 18.0 | 18.0/16.5 | 46.9 | 37.5 | (0.24, 0.43) |
| C | 3 | 523 | 0.48 | 108 | 3.6 | 12.4 | 12.4/11.6 | 37.0 | 30.6 | (0.30, 0.52) |
| D | 4 | 544 | 0.48 | 116 | 3.8 | 6.3 | 6.3/6.0 | 19.1 | 14.7 | (0.38, 0.53) |
| Ei | 5BCzBN | 512 | 0.45 | 97 | 3.0 | 15.0 | 15.0/14.2 | 43.9 | 41.0 | (0.26, 0.49) |
In summary, a new family of TADF emitters featuring carboxylate esters was designed and synthesized. Their TADF properties were investigated through steady-state and time-resolved photophysical measurements, together with quantum chemical calculations. Compared to the well-studied CN-based analogs, the ester-functionalized TADF emitters tended to exhibit larger λS and kRISC values. The increase in λS can be ascribed to the structural relaxation of the ester groups upon electronic transitions, resulting in broadband emission spectra. These ester-functionalized TADF materials exhibited rather high ΦPL values even in their neat films, owing to the suppression of aggregation-caused quenching. Consequently, non-doped OLEDs based on the neat emission layers achieved high EQEmax values of up to 18.7%.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by Grants-in-Aid for JSPS KAKENHI (Grant No. JP21H04694), the Murata Science Foundation, and the Mitsubishi Foundation. The authors are grateful for Kyocera Corp. for generous support and helpful discussion, and for the support provided by the cooperative Research Program of the “Network Joint Research Center for Materials and Devices” and computer facilities at the Research Institute for Information Technology, Kyushu University.References
- For the earliest TADF-OLEDs, see: (a) A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef; (b) S. Y. Lee, T. Yasuda, H. Nomura and C. Adachi, Appl. Phys. Lett., 2012, 101, 093306 CrossRef; (c) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS.
- For reviews on TADF, see: (a) Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931 CrossRef CAS PubMed; (b) M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef; (c) Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS; (d) X. Cai and S.-J. Su, Adv. Funct. Mater., 2018, 28, 1802558 CrossRef; (e) X. Liang, Z.-L. Tu and Y.-X. Zheng, Chem. – Eur. J., 2019, 25, 5623 CrossRef CAS PubMed; (f) M. Godumala, S. Choi, M. J. Cho and D. H. Choi, J. Mater. Chem. C, 2019, 7, 2172 RSC; (g) H. Nakanotani, Y. Tsuchiya and C. Adachi, Chem. Lett., 2021, 50, 938 CrossRef CAS.
- For reviews on CN-based TADF, see: (a) X. Cao, D. Zhang, S. Zhang, Y. Tao and W. Huang, J. Mater. Chem. C, 2017, 5, 7699 RSC; (b) Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946 CrossRef CAS; (c) Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915 RSC; (d) J.-M. Teng, Y.-F. Wang and C.-F. Chen, J. Mater. Chem. C, 2020, 8, 11340 RSC.
- D. Zhang, M. Cai, Y. Zhang, D. Zhanga and L. Duan, Mater. Horiz., 2016, 3, 145 RSC.
- G. Kreiza, D. Banevičius, J. Jovaišaitė, K. Maleckaitė, D. Gudeika, D. Volyniuk, J. V. Gražulevičius, S. Juršėnas and K. Kazlauskas, J. Mater. Chem. C, 2019, 7, 11522 RSC.
- (a) J. Lee, N. Aizawa, M. Numata, C. Adachi and T. Yasuda, Adv. Mater., 2017, 29, 1604856 CrossRef; (b) N. Aizawa, S. Shikita and T. Yasuda, Chem. Mater., 2017, 29, 7014 CrossRef CAS.
- (a) J. W. Rakshys. Jr., S. V. Mckinley and H. H. Freedman, J. Am. Chem. Soc., 1970, 92, 3518 CrossRef; (b) T. Kosaka, Y. Inoue and T. Mori, J. Phys. Chem. Lett., 2016, 7, 783 CrossRef CAS; (c) C. Wang, Z.-B. Sun, Q.-W. Xu and C.-H. Zhao, Chem. – Eur. J., 2016, 22, 16750 CrossRef CAS; (d) M. Toyoda, Y. Imai and T. Mori, J. Phys. Chem. Lett., 2017, 8, 42 CrossRef CAS PubMed; (e) T. Kosaka, S. Iwai, Y. Inoue, T. Moriuch and T. Mori, J. Phys. Chem. A, 2018, 122, 7455 CrossRef CAS.
- J. Lee, I. S. Park and T. Yasuda, Bull. Chem. Soc. Jpn., 2017, 90, 231 CrossRef.
- I. S. Park, K. Matsuo, N. Aizawa and T. Yasuda, Adv. Funct. Mater., 2018, 28, 1802031 CrossRef.
- (a) R. A. Marcus, J. Phys. Chem., 1989, 93, 3078 CrossRef CAS; (b) I. R. Gould, D. Noukakis, L. Gomez-Jahn, R. H. Young, J. L. Goodman and S. Faid, Chem. Phys., 1993, 176, 439 CrossRef; (c) K. Vandewal, K. Tvingstedt, A. Gadisa, O. Inganäs and J. V. Manca, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 125204 CrossRef.
- (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (c) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed.
- (a) P. K. Samanta, D. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2017, 139, 4042 CrossRef CAS; (b) H. Noda, X. K. Chen, H. Nakanotani, T. Hosokai, M. Miyajima, N. Notsuka, Y. Kashima, J.-L. Brédas and C. Adachi, Nat. Mater., 2019, 18, 1084 CrossRef CAS PubMed; (c) K. Matsuo and T. Yasuda, Chem. Sci., 2019, 10, 10687 RSC; (d) T. Agou, K. Matsuo, R. Kawano, I. S. Park, T. Hosoya, H. Fukumoto, T. Kubota, Y. Mizuhata, N. Tokitoh and T. Yasuda, ACS Mater. Lett., 2020, 2, 28 CrossRef CAS; (e) H. Min, I. S. Park and T. Yasuda, Angew. Chem., Int. Ed., 2021, 60, 7643 CrossRef CAS PubMed.
- (a) N. Aizawa, A. Matsumoto and T. Yasuda, Sci. Adv., 2021, 7, eabe5769 CrossRef CAS; (b) I. S. Park, H. Min, J. U. Kim and T. Yasuda, Adv. Opt. Mater., 2021, 9, 2101282 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data. CCDC 2114087. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc04933j |
| This journal is © The Royal Society of Chemistry 2022 |