
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hollow protein microparticles formed through cross-linking by an Au3+ initiated redox reaction†
Laura M. I.
Schijven
 ab,
Thomas D.
Vogelaar
ab,
Simha
Sridharan
a,
Vittorio
Saggiomo
b,
Aldrik H.
Velders
b,
Johannes H.
Bitter
a and
Constantinos V.
Nikiforidis
*a
ab,
Thomas D.
Vogelaar
ab,
Simha
Sridharan
a,
Vittorio
Saggiomo
b,
Aldrik H.
Velders
b,
Johannes H.
Bitter
a and
Constantinos V.
Nikiforidis
*a
aBiobased Chemistry and Technology, Wageningen University and Research, Bornse Weilanden 9, 6708 WG Wageningen, The Netherlands. E-mail: costas.nikiforidis@wur.nl
bBioNanoTechnology, Wageningen University and Research, Bornse Weilanden 9, 6708 WG Wageningen, The Netherlands
First published on 14th June 2022
Abstract
Hollow microparticles (MPs) are of great relevance in the materials industry for a wide range of applications, such as catalysis, coatings, and delivery of theranostics. Here, we report the formation of hollow MPs through the assembly of lipoproteins in CaCO3 templates. Proteins interact in the pores of CaCO3 templates through attractive hydrophobic forces and form dense edges of hollow MPs. To further cross-link the proteins, Au3+ was added to initiate a redox reaction, where proteins were oxidized forming inter- and intramolecular covalent bonds, while Au3+ was reduced and gold nanoparticles (AuNPs) were formed. The obtained protein-based hollow MPs have a diameter of 6 μm and the AuNPs are embedded on their surface. Through this research, we suggest a new route to design biobased Au-protein hollow MPs in simple steps, which can allow new possibilities for carrying functional molecules and bioimaging.
1. Introduction
Microparticles (MPs) offer advantageous structural and functional abilities as a carrier system, which are of interest in a wide range of applications, such as1 encapsulation,2 tissue engineering,3,4 and biosensing.5,6 Whereas the majority of the MPs are fabricated with synthetic polymers,7 it is of great importance to use biobased materials, such as proteins, to expand the range of developing biocompatible MPs with functional properties.For the preparation of protein MPs, proteins are forced to assemble together through the use of liquid or solid templates. The formation of stable MPs includes interactions of reactive amino acid side groups of neighboring protein molecules or the use of cross-linkers, like glutaraldehyde, resulting in a protein gel network.
Besides the formation of MPs with a homogeneous core, hollow MPs are also of high interest because of their ability to encapsulate large quantities of guest molecules in their hollow core.8 Hollow MPs are mostly obtained with the aid of a hard9 or soft10 templates, which have to be removed through thermolysis or extensive acidification, requiring the use of large amounts of chemicals and tedious process steps. Therefore, the use of biobased molecules and environmentally friendly processes to create hollow MPs are highly desired.11
Herein, we demonstrate a facile method to fabricate hollow MPs using egg yolk high-density lipoproteins (HDL), inside sacrificial CaCO3 templates (Scheme 1). HDL spontaneously forms the hollow MPs, due to intra-protein hydrophobic forces, while to increase the rigidity of the MPs, Au3+ ions are used, which has been reported to cross-link HDL through a redox reaction.12 The cross-linking method is based on the oxidation of HDL amino acids and their subsequent covalent bonding, while residual amino acid groups of proteins act as natural reduction sites of Au3+ ions and synthesize gold nanoparticles (AuNPs), which are located on the surface of the MPs. The resulting hollow protein MPs have a well-defined size, morphology and high stability. The ability to produce stabilized hollow protein MPs, using Au3+ ions, with embedded AuNPs, opens avenues for manipulating and navigating microcapsules for controlled release and targeted delivery. The CaCO3 templates can be easily decomposed at acidic pH,13 releasing stable Au-HDL-MPs. The large inner voids of the hollow Au-HDL-MPs could encapsulate and protect molecules of interest. The molecules of interest could be encapsulated through co-precipitation with the proteins and CaCO3,14 or through passive diffusion into the protein MPs.15 The embedded AuNPs make the Au-HDL MPs an attractive platform as a multimodal imaging agent due to their optical,16,17 photothermal,18,19 and high attenuation coefficient properties.20
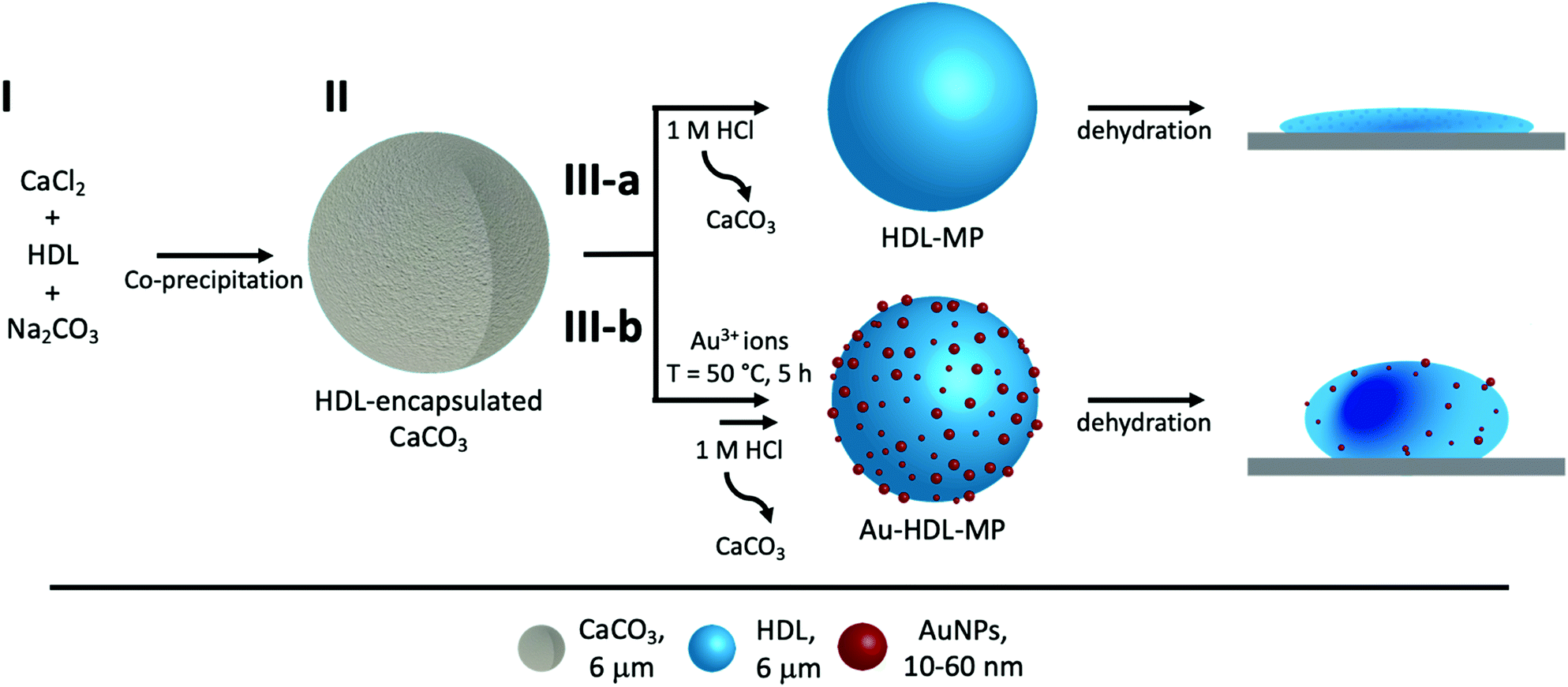 | ||
| Scheme 1 Schematic representation of fabrication of protein hollow MPs. (I) HDL was encapsulated into CaCO3 templates through co-precipitation with CaCl2 and Na2CO3. (II) HDL was encapsulated inside the CaCO3 template (gray). For route (III-a), HDL-MPs (blue sphere) were released from the template by lowering the pH. The HDL-MPs collapsed upon dehydration. For route (III-b), Au3+ ions were added to HDL-encapsulated CaCO3 templates to induce in situ HDL network formation, followed by reduction of Au3+ and synthesis of 10–60 nm AuNPs (red spheres) before template removal. The resulting Au-HDL-MPs formed wrinkled, deflated structures after dehydration. | ||
2. Experimental
2.1 Materials
Fresh hen eggs were purchased from a local organic farm De Hoge Born, Wageningen, the Netherlands. Sodium chloride (NaCl, ≥99.5%), 1 M NaOH and 1 M HCl solutions were purchased from VWR International B.V. Gold(III) chloride hydrate (HAuCl4·xH2O, 99.5%), fluorescein-5-isothiocyanate (FITC, ≥95%), calcium chloride (CaCl2, ≥93%), sodium carbonate (Na2CO3, ≥99.5%) and 76 mm dialysis tubing cellulose membrane (MWCO = 14 kDa) were purchased from Sigma Aldrich. All chemicals were used without further purification and deionized water was used throughout the experiments.2.2 Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 45 min at T = 4 °C, using a Thermo Scientific Sorvall Legend XFR centrifuge. The pellets (granules) were washed with 0.17 M NaCl and centrifuged once more. The granules were suspended in 1.71 M NaCl (10% (w/v)) and the solution's pH was adjusted to 7.25, using 1 M NaOH. The mixtures were collected in dialysis tubings and were dialyzed against deionized water with 3 changes over 24 h. The HDLs were precipitated. The contents of the dialysis tubings were collected and centrifuged. The pellets, rich in HDL, were collected and freeze-dried, using a Salmenkipp alpha 2–4 plus freeze-dryer at a temperature of T = −76 °C and pressure of 0.009 mbar for 72 h.
:1) dissolved in 50 mL of 1.71 M NaCl. The mixture was allowed to react overnight, while covered against light. The mixture was then dialyzed against deionized water with 5 changes over 48 h. The content of the dialysis bag was collected and centrifuged. The supernatant was discarded and the pellet was washed again with deionized water before freeze-drying. The molar ratio of FITC coupled to HDL was 0.8, based on UV-vis absorbance measurements.
480 kDa23), while stirring at 700 rpm using a magnetic stirrer. A white precipitate formed instantly. The stirring speed was subsequently increased to 2500 rpm for 30 s. The sample was then removed from the stirring plate and was left to stand for 15 min to facilitate the formation of CaCO3. The suspended HDL-encapsulated CaCO3 was collected by centrifugation at 3000 × g for 5 min (Thermo Scientific ST 8R centrifuge) and washed 3 times with 10 mL of 1.71 M NaCl to remove non-bound HDL.
000 × g, 10 min, T = 4 °C) and washed 3 times with 10 mL of deionized water, pH 7.
000 × g, 10 min, T = 4 °C) and washed 3 times with 10 mL of deionized water, pH 7.
000 × g for 45 min at T = 4 °C, using a Thermo Scientific Sorvall Legend XFR centrifuge. The pellets (granules) were washed with 0.17 M NaCl and centrifuged once more. The granules were suspended in 1.71 M NaCl (10% (w/v)) and the solution's pH was adjusted to 7.25, using 1 M NaOH. The mixtures were collected in dialysis tubings and were dialyzed against deionized water with 3 changes over 24 h. The HDLs were precipitated. The contents of the dialysis tubings were collected and centrifuged. The pellets, rich in HDL, were collected and freeze-dried, using a Salmenkipp alpha 2–4 plus freeze-dryer at a temperature of T = −76 °C and pressure of 0.009 mbar for 72 h.
:1) dissolved in 50 mL of 1.71 M NaCl. The mixture was allowed to react overnight, while covered against light. The mixture was then dialyzed against deionized water with 5 changes over 48 h. The content of the dialysis bag was collected and centrifuged. The supernatant was discarded and the pellet was washed again with deionized water before freeze-drying. The molar ratio of FITC coupled to HDL was 0.8, based on UV-vis absorbance measurements.
480 kDa23), while stirring at 700 rpm using a magnetic stirrer. A white precipitate formed instantly. The stirring speed was subsequently increased to 2500 rpm for 30 s. The sample was then removed from the stirring plate and was left to stand for 15 min to facilitate the formation of CaCO3. The suspended HDL-encapsulated CaCO3 was collected by centrifugation at 3000 × g for 5 min (Thermo Scientific ST 8R centrifuge) and washed 3 times with 10 mL of 1.71 M NaCl to remove non-bound HDL.
000 × g, 10 min, T = 4 °C) and washed 3 times with 10 mL of deionized water, pH 7.
000 × g, 10 min, T = 4 °C) and washed 3 times with 10 mL of deionized water, pH 7.
2.3 Characterization
For the ultramicrotome sample preparation, the HDL-MPs and Au-HDL-MPs were fixed with 2.5% glutaraldehyde in 0.1 M phosphate/citrate buffer and incubated for 1 h, after which they were washed 3 times with 0.1 M phosphate/citrate buffer. The pellets were re-suspended in 100 μL of 2% gelatin in 0.1 M phosphate and were solidified after 20 min at 4 °C. The pellet was removed from the tube by 15 min incubation in 2.5% glutaraldehyde in 0.1 M phosphate/citrate buffer. The specimens were cut into small pieces (approximately 1 mm3) and were fixed again for 1 h, after which they were washed 6 times with 0.1 M phosphate/citrate buffer. The specimens were fixed again, this time with 1% osmium tetroxide for 1 h, after which they were washed 3 times with MilliQ water. Thereafter dehydration with ethanol was applied, substituting deionized water with 30%, 50%, 70%, 80%, 90%, 96% (5 min for each step) and 2 × 100% ethanol (10 min). Once the specimens were in 100% ethanol, the specimens were infiltrated with Spurr embedding liquid in 3 steps: 2:1, 1:1, and 1:2 (ethanol:Spurr, 30 min per step). Then the specimens were left in 100% Spurr for 1 h and were refreshed once more and incubated overnight. The next day the Spurr was refreshed once more for 1 h, after which the samples were polymerized for 8 h at 70 °C. Once the specimens were hardened, they were sectioned into 50 nm thin coupes using a Leica Ultramicrotome UC7 Rapid. The sections were collected with Formvar film 150 mesh copper TEM grids. The sections were stained by incubation for 10 min in 2% uranyl acetate, after which they were washed 5 times with MilliQ. Then the sections were stained by incubation for 2 min in lead citrate (under a CO2-free environment), after which they were washed 2 times with CO2-free water and 3 times with MilliQ.
3. Results and discussion
Spherical and porous CaCO3 were synthesized by direct mixing of soluble salts of Ca2+ and CO32− to initiate precipitation at supersaturation22 and was used as template for the protein MPs, since it offers a large inner surface area for encapsulation, is biocompatible, economically friendly,25 easy to prepare and sacrificial under mild conditions (e.g. by chelating reagents or under acidic conditions).13 The proteins could be encapsulated through diffusion into pre-synthesized CaCO3 templates or by co-precipitation during CaCO3 synthesis.263.1 HDL encapsulated into the CaCO3 templates
HDL is a relatively hydrophobic and insoluble protein, which forms aggregates in aqueous solutions.27 The aggregates were too large to diffuse into pre-synthesized CaCO3 templates. For this reason, HDL molecules were incorporated into CaCO3 through co-precipitation, which were expected to form a particle at the core of the CaCO3 template.When HDL was mixed with CaCl2 before the addition of Na2CO3, the ions interacted to form the CaCO3 template, and simultaneously encapsulated HDL aggregates, and precipitated (Scheme 1(I-II)). The CaCO3 templates with the encapsulated HDL were collected and initially analyzed using TEM to get information on macroscopic morphology. The obtained HDL/CaCO3 complexes appeared to be spherical with a diameter of around 3.5–5.2 μm and a high electron density, caused by the high mass and crystallinity of the CaCO3 templates (Fig. 1(A)). It has to be noted that some cubic shapes were also observed. However, cubic CaCO3 structures are non-porous,22,26 so it was assumed that those did not contain HDL. Next to TEM, SEM was used for analyzing the surface structures of the HDL/CaCO3 complexes (Fig. 1(B)). In the SEM image, it was observed that the crystal surface was composed of smaller single particles (Fig. 1(B), inset). Spherical, micron-sized CaCO3 is synthesized through the aggregation of primary CaCO3 nuclei, which instantly formed by mixing Ca2+ and CO32−.28 The surface structure is most likely composed of smaller, single CaCO3 nuclei.
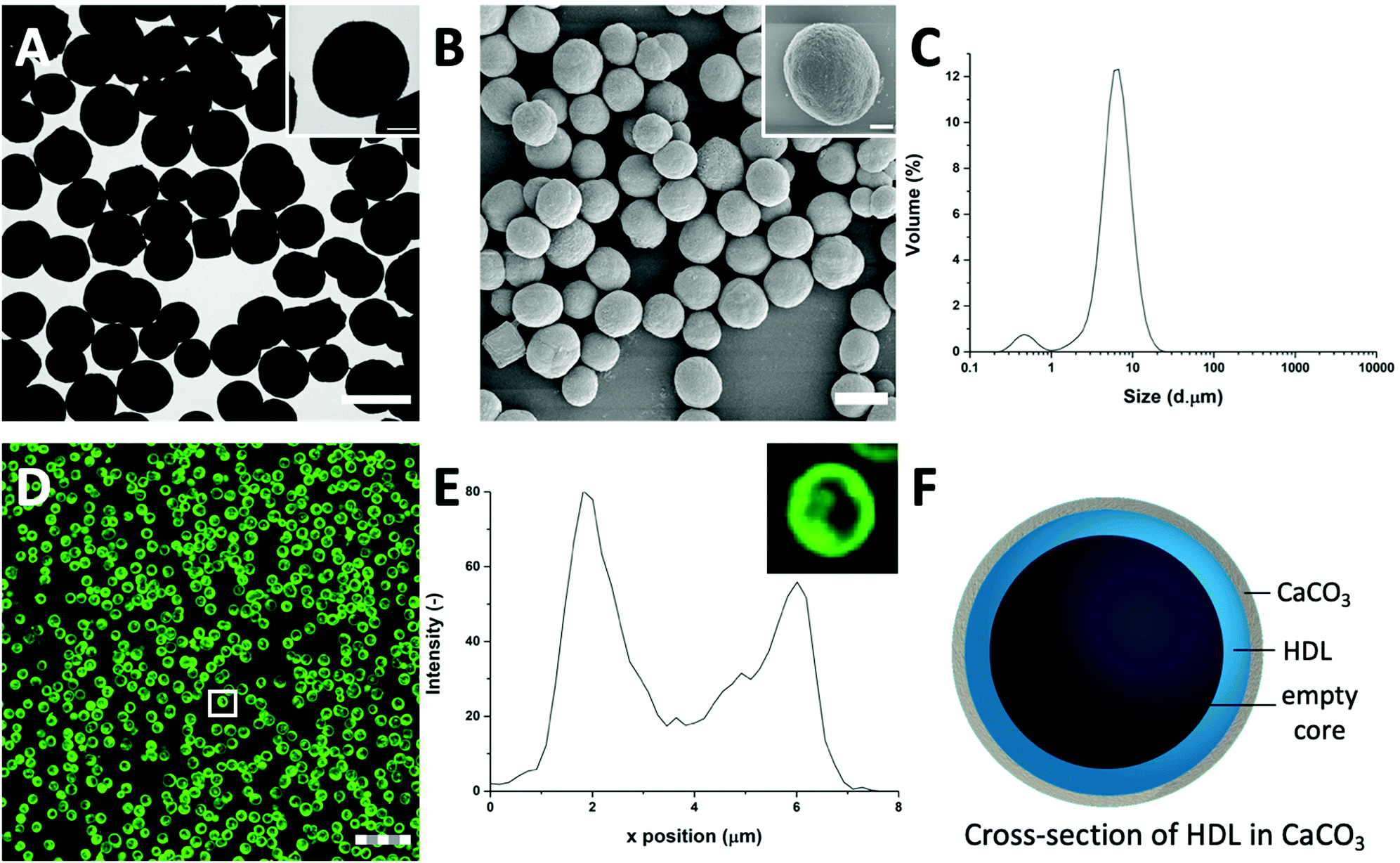 | ||
| Fig. 1 The high mass and crystallinity of the CaCO3 templates provides a high contrast for (A) TEM and (B) SEM (scale bars are 5 μm), inset shows single particles (scale bars are 1 μm). (C) Light diffraction reveals a size distribution of HDL-encapsulated CaCO3 templates with a diameter of 6 μm. (D) FITC-labelled HDL in CLSM reveals that HDL is distributed at the edges of the CaCO3 templates, forming dense rings (scale bar is 25 μm, thickness is 0.6 μm). (E) Corresponding fluorescent profile plot of (inset) a FITC-HDL-encapsulated CaCO3 template, indicated by the white box in (D). (F) Schematic illustration of the cross-section of an HDL-encapsulated CaCO3 template. | ||
To further evaluate the size distribution of the CaCO3/HDL templates, their sizes were measured using light diffraction. As shown in Fig. 1(C), the size distribution was bimodal with peaks at 0.6 μm and 6.7 μm. Previous reports have suggested a size distribution of the CaCO3 templates of around 4–6 μm,22 which agrees with the results we obtained from the TEM analysis. Therefore the smaller peak at 0.6 μm, obtained during the laser diffraction analysis, was attributed to the non-encapsulated HDL aggregates.
To investigate the presence of HDL molecules inside the CaCO3 templates, HDL was covalently labelled with fluorescein isothiocyanate (FITC) before co-precipitation. Using CLSM, the fluorescence of FITC (green color) was detected from the HDL-encapsulated CaCO3 templates, indicating that the excitation wavelength could penetrate the porous templates (Fig. 1(D) and Fig. S1, ESI†).29 As it is shown in the detailed fluorescence profile plot in Fig. 1(E), a higher fluorescent signal was observed at the edges of the templates, while it gradually decreased from the surface to the internal core, showing the formation of shell-like hollow MPs, as shown schematically in Fig. 1(F). The protein border inside the templates had a thickness of 2.0 ± 0.5 μm. The formation of the hollow protein MPs in the CaCO3 template was surprising because it has been reported earlier when proteins such as bovine serum albumins28 and β-lactoglobulins30 were encapsulated in the same template, they formed homogeneous protein MPs. The proposed mechanism of the encapsulation of soluble proteins in the CaCO3 templates was that the proteins adsorb onto the surface of primary CaCO3 nanoparticles and are captured mechanically into the inner pores of CaCO3 during colloidal aggregation of the CaCO3 nanoparticles.28 However, HDL proteins are poorly soluble and form big aggregates in aqueous solutions, and also in the presence of Ca2+ ions. Therefore, it was hypothesized that the HDL aggregates, due to their large size (>100 nm),27 could not adsorb in the pores (20–60 nm)22 of the CaCO3 templates during the formation, but were only deposited on the core of the template at a later stage, making a thin spherical film around it.
3.2 Properties of HDL hollow microparticles
It is possible that strong attractive hydrophobic forces exist between the HDL molecules in the confined environment of the CaCO3 template, which was expected to be the force that can keep the hollow protein MPs intact when the template will be removed (Scheme 1(III-a)). During CaCO3 synthesis, the proteins were under alkaline conditions (pH > 10), which results in the exposure of hydrophobic groups and increases hydrophobic interactions.31 The charge of the HDL molecules was expected to have a minor impact, since it was weak and did not exceed the |20| mV under the storage and CaCO3 decomposition conditions (Fig. S2, ESI†).To investigate the stability of the hollow protein MPs, we initially used CLSM to visualize the particle microstructure (Fig. 2(A) and Fig. S3, ESI†). The fluorescent signal was again predominantly observed at the edges of the HDL-MPs. The fluorescence profile plot showed the highest fluorescence intensity at the edges, but a significant decrease from the surface to the internal core (Fig. 2(B)). This shows the conservation of the protein distribution within the template after the release of the HDL-MPs from the templates. However, the protein border thickness was decreased to 1.6 ± 0.4 μm, which suggests that some proteins were released from the HDL-MPs during template removal. This could be due to the weak hydrophobic interactions, which could not keep all the proteins together. Additionally, the HDL-MPs were aggregated, which could be due to the hydrophobic nature of HDL, leading to extensive hydrophobic interactions between neighboring HDL-MPs.
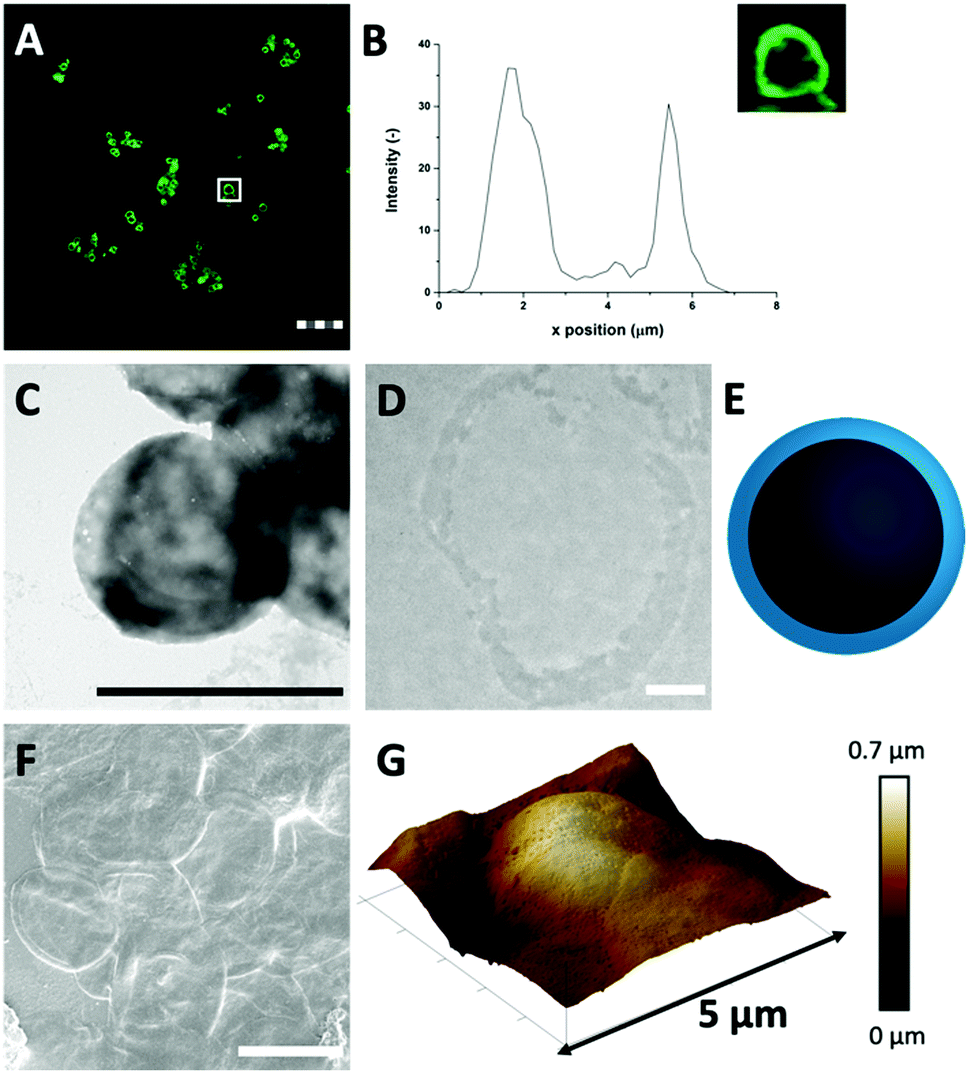 | ||
| Fig. 2 (A) FITC-labelled HDL-MPs (scale bar is 25 μm, thickness is 0.9 μm) in CLSM reveals that the HDL distribution in the HDL-MPs is conserved after template removal. (B) The corresponding fluorescent profile plot of (inset) a single HDL-MP, indicated by the white box in (A). The dense protein network provides contrast for (C) TEM (scale bar is 1 μm) and the (D) thin section (0.05 μm) of the HDL-MP shows that the proteins formed a hollow core–shell morphology (scale bar is 0.5 μm, sample is stained with uranyl acetate and lead citrate). (E) Schematic illustration of the cross-section of an HDL-MP. (F) SEM image of HDL-MPs, coated with tungsten (scale bar is 5 μm). (G) 3-D AFM images of HDL-MPs, with corresponding height scale bar. | ||
Next, TEM imaging was applied to study the HDL-MP structure. The HDL-MPs appeared as slightly loose protein matrices with a low density (Fig. 2(C) and Fig. S4, ESI†) and diameters of 4.7–7.5 μm. The protein–protein interactions in the hollow MPs were constituted by weak hydrophobic forces, resulting in a system with low density. Furthermore, since it was not clear in the TEM image whether the MPs kept their hollow structure, TEM imaging was also performed on an ultramicrotome thin section of an HDL-MP (Fig. 2(D)). In the thin section, a dark-colored border was observed, which consisted of proteins. No proteins were observed in the internal core of the HDL-MP, which confirms that the HDL-MPs formed a hollow structure with a low protein density (Fig. 2(E)).
The surface morphology of the HDL-MPs was further examined by SEM (Fig. 2(F)). After template removal, the HDL-MPs resembled flattened circular discs. During SEM imaging, drying and high vacuum conditions are applied. These conditions could have caused the collapse of the hollow HDL-MPs, which could be due to the absence of proteins in the internal core and weak protein–protein interactions, resulting in low structural integrity of the HDL-MPs in a dried state.32 To further investigate the structural stability of the HDL-MPs, in terms of rigidity and self-standing properties, the particles were dried on a solid substrate under ambient conditions and imaged with AFM. The AFM images show the topography of the HDL-MPs, where the raised (indicated by white-to-light brown color) and lowered (dark brown color) structures are determined. Upon drying, the HDL-MPs have a flattened, collapsed shape with a porous surface with the thickness varying from 0.01 to 0.7 μm (Fig. 2(G)). The collapse of the hollow protein MPs upon drying, indicated that it is necessary to cross-link the proteins, so they will retain their structure.
3.3 Using Au3+ ions to enhance the rigidity of the hollow protein microparticles
Covalent cross-linking of the proteins allows conserving and stabilizing the spherical structure upon template removal. In our previous research, we demonstrated that the addition of (≥150 molar equivalents) Au3+ ions to HDL resulted in gel network formation and subsequent AuNP synthesis.12 The Au3+ ions are known for their strong oxidizing properties on, for instance, thiol- or amine-containing side groups of amino acids, resulting in di-amino acid covalent bonding33,34 and, subsequently, gel network formation. Additionally, several available amino acid side groups (e.g. tryptophan, tyrosine, aspartic acid, and phenylalanine) can act as reduction sites of Au3+ ions and synthesize AuNPs.35–38 In a previous work, FTIR analysis showed the formation of disulfide bonds and fluorescence spectroscopy showed the formation of dityrosine in the gel networks.12 Increasing the amounts of Au3+ ions resulted in a faster formation of the gel networks. However, no changes in morphology were observed.Therefore, 250 molar equivalents of Au3+ ions were added to HDL-encapsulated in CaCO3 templates to induce protein–protein interactions and covalent cross-linking of proteins (Scheme 1(III-b)). During the incubation of the Au3+/HDL/CaCO3 system the suspension gradually changed from white to pink (after 2 h), and then to red, indicating the interactions of Au3+ with the HDL molecules and the subsequent formation of AuNPs (Fig. S5A, ESI†). Increasing the amounts of Au3+ accelerated the color formation of the Au3+/HDL/CaCO3 system. The red color was derived from the characteristic localized surface plasmon resonance (LSPR) absorption of AuNPs, which can be found in the region of λ = 500–600 nm.39 After precipitation of the HDL/CaCO3 complexes, it was observed that only the solid templates appeared red in color, while the solution remained transparent (Fig. S5B, ESI†), showing that both HDL and AuNPs were located at the templates. It is important to mention here that the formation of AuNPs and the subsequent red color was not observed in the absence of HDL, clearly proving that the AuNPs formed after diffusion of Au3+ ions into the templates and their interaction with the HDL molecules that were entrapped. After the removal of the templates, the red color of the Au-HDL-MPs was conserved (Fig. S5C, ESI†). This suggests that HDL not only reduced the Au3+ ions for the formation of AuNPs, but also stabilized the AuNPs through steric stabilization effects, adsorption on the AuNP surface, and by coordinating Au3+ to numerous functional amino acid groups (e.g. amine and carboxylate groups). The steric stabilization could decrease the potential toxicity of the AuNPs, however, since contradictive information on this matter has been published,40 further investigation is needed.
To confirm whether the red color is derived from AuNPs, the Au-HDL MP solutions were measured by UV-vis absorbance at λ = 400–800 nm (Fig. S5D, ESI†). The spectra demonstrated the appearance of an absorption peak centered at λ = 555 nm after 2 h of incubation, which confirmed the presence of the AuNPs.
The Au3+ ions and AuNP formation after template removal may have an impact on the spherical protein structure. For this reason, the microstructure and protein distributions in the protein MPs were studied with CLSM, after labeling HDL using FITC (Fig. 3(A) and Fig. S6, ESI†). The CLSM images showed dense rings, which means that the spherical protein structures were still intact after template removal. The fluorescence profile plot also showed a high fluorescence peak, which corresponds to the observed ring, and a gradual decrease from the surface to the internal core (Fig. 3(B)). The protein border had a thickness of 1.9 ± 0.4 μm, which was similar to the thickness before template removal (Fig. 1(D)). Based on these results, the protein distribution and spherical structure seem to be preserved within the hollow MPs after template removal through the addition of Au3+ ions.
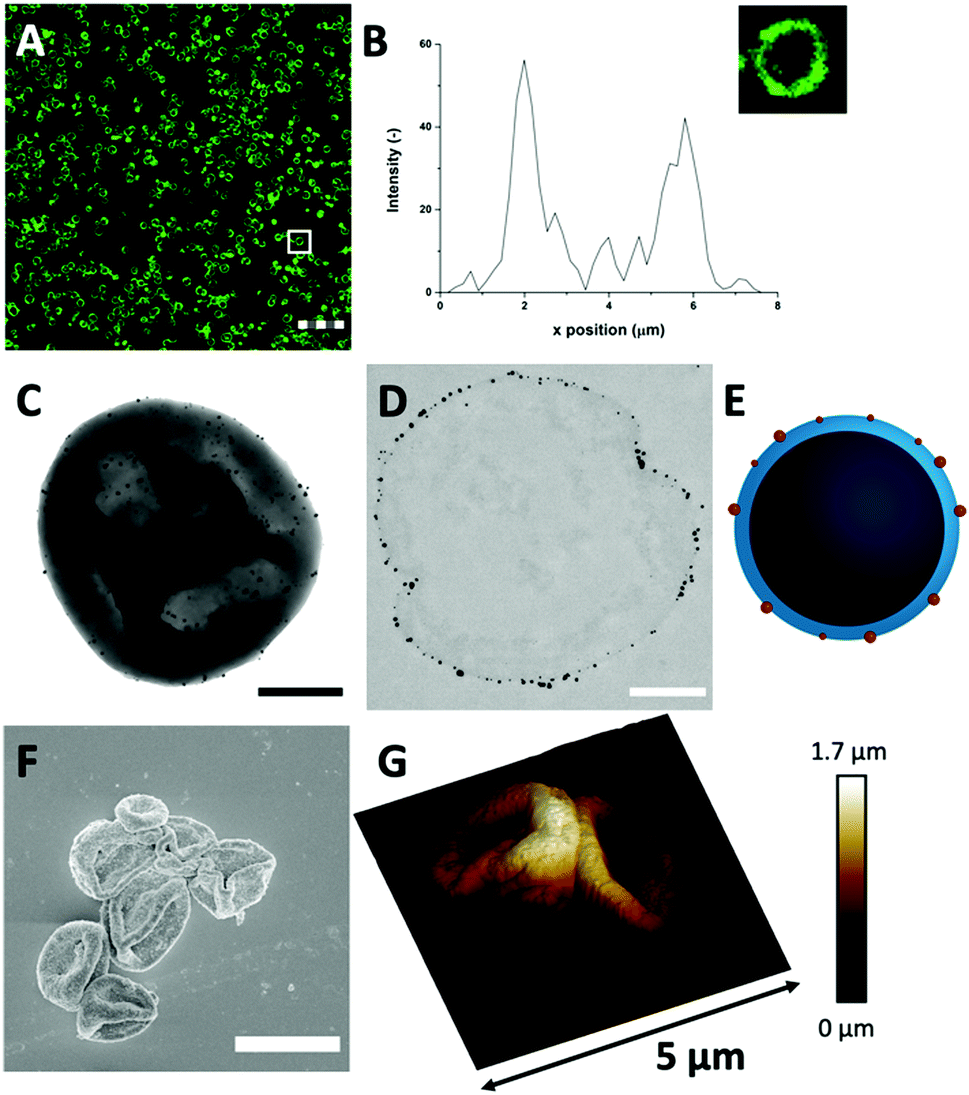 | ||
| Fig. 3 (A) FITC-labelled Au-HDL-MPs (scale bar is 25 μm, thickness is 0.9 μm) in CLSM reveals that the HDL distribution in the Au-HDL-MPs is conserved after cross-linking and template removal. (B) The corresponding fluorescent profile plot of (inset) a single Au-HDL-MP, indicated by the white box in (A). The dense protein network and AuNPs provide a high contrast for (C) TEM (scale bar is 1 μm) and the (D) thin section (0.05 μm) of the Au-HDL-MP shows that the AuNPs are concentrated at the edges of the protein MP surface (scale bar is 0.5 μm, sample is stained with uranyl acetate and lead citrate). (E) Schematic illustration of the cross-section of an Au-HDL-MP with AuNPs on the surface. (F) SEM image of Au-HDL-MPs, coated with tungsten (scale bar is 5 μm). (G) 3-D AFM image of two neighboring Au-HDL-MPs, with corresponding height scale bar. | ||
Next, TEM was used for analyzing the hollow MP structure after the addition of Au3+. In the TEM image, the MPs appeared as dense protein matrices with a size distribution of 1.6–3.1 μm (Fig. 3(C) and Fig. S7, ESI†). Besides the protein MPs, small, dark and spherical spots, with sizes between 10–60 nm, were observed. The AuNPs have a higher electron density than the protein matrix, and this contrast is sufficient to detect the AuNPs in the protein matrix. Based on the contrast difference, the appearance of the red color and the LSPR absorbance, the spherical spots in the TEM image were assigned to AuNPs. However, it could not be clearly observed on the TEM image whether those AuNPs are located on the surface or inside the protein matrix. Therefore, TEM imaging was done on an ultramicrotome thin section (0.05 μm), after agarose embedding (Fig. 3(D)). In the thin section, a dark border was observed, which consisted of stained proteins, and there were some dense electron regions observed in the internal core of the MPs. This confirmed that the MPs formed a stable and rigid hollow structure. The AuNPs were predominantly observed on the outside of the dark borders of proteins. This suggests the ability of Au3+ ions to infiltrate the CaCO3 templates at the protein surface and there they are reduced to AuNPs (Fig. 3(E)). The darker shades that were observed in the TEM image in Fig. 3(C) could be attributed to folds and creases in the structure, which caused a higher local electron density. These folds are likely formed upon dehydration of the hollow-structure morphology of the hollow MPs under drying and high vacuum conditions during TEM imaging.
Additionally, the surface morphology of the Au-HDL-MPs was further examined by SEM (Fig. 3(F)). After template removal, the Au-HDL-MPs appeared flattened with folds and crease structures. The deflated, wrinkled particle structure likely resulted from the hollow-like and cross-linked shell structure of the Au-HDL-MPs, which collapsed upon drying due to the removal of water, but still the protein molecules remained in the border of the MP. AFM was used to study the effect of dehydration on rigidity and self-standing properties of the hollow Au-HDL-MPs (Fig. 3(G)). The drying of Au-HDL-MPs resulted in the formation of a deflated, wrinkled particle shape with the thickness varying from 0.1 to 1.7 μm. The thickness of the Au-HDL-MPs was two times higher than that of the HDL-MPs, indicating that the wall thickness of Au-HDL-MPs was more rigid upon dehydration. This was also observed in the CLSM images, where the wall thickness of the Au-HDL-MPs was higher than that of the HDL-MPs (Fig. 2(A) and 3(A)). Based on that, we hypothesized that the wall thickness is higher due to the covalent cross-linking of the Au-HDL-MPs, which improved the stabilization of the structures upon dehydration.
3.4 Stability of the hollow protein microparticles under different pH conditions
After the preparation of the different particle systems, analytical methods were used to study the stability, in terms of aggregation, of the MPs at different pH values. DLS measurements allowed a comparison of the diameters of the cross-linked (Au-HDL-MPs) and non-cross-linked hollow MPs (HDL-MPs) (Fig. 4(A)). For HDL-MPs, the average size distribution was 0.93 μm at pH 1–3, whereas the sizes increased to 7.7 μm (±19%) at pH 4–10, and decreased to 0.45 μm at pH 11–12. However, it was observed that the sizes of the Au-HDL-MPs were stable at around 6.4 μm (±7%) in the pH range of 1–12. Before the removal of the template, the size distribution of the HDL-encapsulated CaCO3 was 6.7 μm (Fig. 1(C)). The sizes of HDL-MPs were slightly larger at pH 4–10 than before the removal of the template. At extreme low (pH 1–3) and high (11–12) pH values, the sizes were significantly decreased. These results suggest that the HDL-MPs were not stable in size against pH. In contrast, there were no major changes observed in the size of the Au-HDL-MPs after template removal and pH titration. The HDL-MPs and Au-HDL-MP sizes originated from the templates (Fig. 1(C)), which indicates that the MPs are inverse replicates of the CaCO3 templates. Additionally, the pH did not affect the size of the Au-HDL-MPs, indicating that the Au-HDL-MPs are predominantly stabilized by covalent protein–protein interactions. These covalent bonds were derived from oxidized amino acids, like cysteine and tyrosine, due to the addition of Au3+ ions.12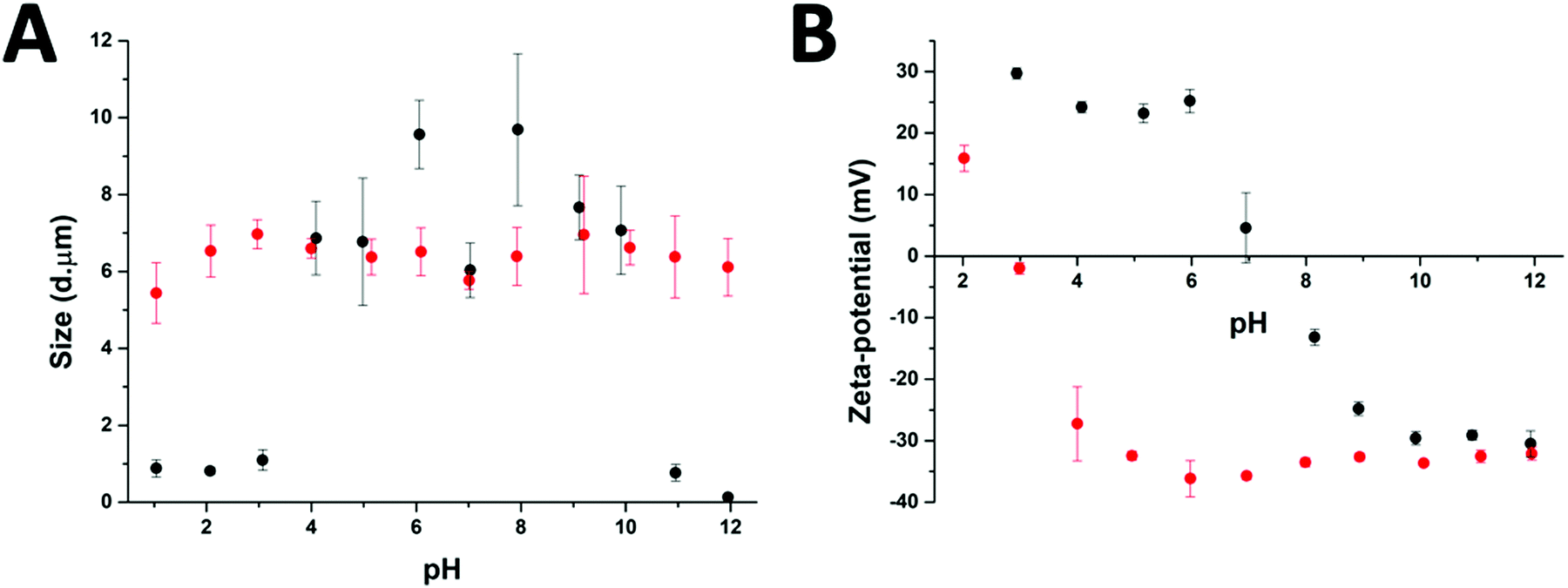 | ||
| Fig. 4 The effect of pH on (A) sizes, as derived from DLS measurements, and (B) zeta-potential of HDL-MPs (black dots) and Au-HDL-MPs (red dots). Separate protein-MP solutions with pH 1–12 were prepared for each individual measurement. | ||
Another important factor in colloidal stability is the surface charge. Zeta-potential measurements allowed a comparison of quantitative values of the overall surface charge and colloidal stability between HDL-MPs and Au-HDL-MPs (Fig. 4(B)). The zeta-potential as a function of pH showed that HDL-MPs have surface charges below |30| mV and a zero surface charge point at pH 7.3. The Au-HDL-MPs have an effective zeta-potential of about −30 mV at pH 4–12, and a zero surface charge point at pH 2.9. When the zeta-potential values are ≥|30| mV, the particles are electrostatically stabilized and cause electrostatic repulsion between the particles and prevent aggregation. However, when the zeta-potential values are <|30| mV, attractive forces exceed electrostatic repulsion, causing the particles to aggregate.30 At pH 4–10, the HDL-MP sizes were increased and the particles bear surface charges of > |20| mV. Based on that, we concluded that electrostatic repulsion caused swelling of the charged shells. However, a size decrease was observed at pH 7, which is close to the surface zero charge point. The decrease in the size of the HDL-MPs could be due to attractive forces, resulting in shrinkage. Additionally, the high zero surface charge point of the HDL-MPs indicates that strong hydrophobic interactions stabilize the HDL-MPs.41 At extreme pH values, the surface charges were |30| mV and the sizes were significantly decreased. This indicates that the high surface charge, and thus high repulsion, at the extreme pH values could lead to more extreme swelling of the HDL-MPs.42 The swollen HDL-MPs were likely sedimented during the size measurements, resulting in a significant size decrease.
The low zero surface charge point of the Au-HDL-MPs suggests that the positively charged groups (e.g. lysine and arginine) of the Au-HDL-MPs are reduced, resulting in a more negative charge. This could be caused by interactions of positively charged groups with the obtained AuNPs,43 or due to the nucleation of the AuNPs,44 or the oxidation of the positively charged groups.33 Since we observed that the sizes of Au-HDL-MPs were stable against pH and have zeta-potential values of −30 mV, we concluded that the covalent cross-linking of the Au-HDL-MPs improved the stabilization of the structures upon template decomposition and could be accelerated by increasing the amounts of Au3+ ions.
4. Conclusions
In this research, we successfully fabricated stable hollow protein MPs with an average size distribution of around 6 μm. For the formation of the hollow MPs, CaCO3 was used as a template. HDL molecules interact in the pores of the template through attractive hydrophobic forces and are predominantly concentrated at the edges of the CaCO3 template. However, it appeared that the hydrophobic forces were not enough to sustain the structure of the MPs. Therefore, Au3+ ions were added to the HDL molecules encapsulated in CaCO3, as it has been shown that Au3+ reacts rapidly with HDL molecules causing protein oxidation and cross-linking. At the same time, Au3+ ions are reduced from the amino acids of the proteins and form AuNPs, which are embedded on the surface of the hollow MPs. The hollow MPs with AuNPs were stable in terms of aggregation and self-standing properties. The design we propose here of stable protein hollow MPs opens new avenues for constructing hollow MPs in simple steps using biobased molecules. Besides that the presence of AuNPs on the surface of the hollow MPs makes the suggested system an attractive platform as a carrier of therapeutics and at the same time a multimodal imaging agent due to their optical, high electron density, and high attenuation coefficient properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research presented in this article was financially supported by the Graduate School VLAG. The authors would like to thank Eleni Ntone for assistance with the CLSM and the Wageningen Electron Microscopy Centre for support with the TEM and SEM measurements, and TEM sample preparation.Notes and references
- M. Lengyel,
et al.
, Sci. Pharm., 2019, 87(3), 20 CrossRef CAS
.
- D. J. McClements, Adv. Colloid Interface Sci., 2018, 253, 1–22 CrossRef CAS PubMed
- S. Shkarina,
et al.
, Sci. Rep., 2018, 8(1), 1–13 Search PubMed
- W. Wijaya,
et al.
, Trends Food Sci. Technol., 2017, 68, 56–69 CrossRef CAS
- Y. H. Roh,
et al.
, BioChip J., 2019, 13(1), 64–81 CrossRef CAS
- F. Bucatariu,
et al.
, ACS Appl. Mater. Interfaces, 2020, 12(33), 37585–37596 CrossRef CAS PubMed
- S. Roberts,
et al.
, Nat. Commun., 2020, 11(1), 1–10 CrossRef PubMed
- J. Han,
et al.
, Chem. Mater., 2007, 19(5), 973–975 CrossRef CAS
- Z. Niu,
et al.
, Adv. Funct. Mater., 2003, 13(12), 949–954 CrossRef CAS
- Z. Wei and M. Wan, Adv. Mater., 2002, 14(18), 1314–1317 CrossRef CAS
- D. Wu,
et al.
, Chem. Rev., 2012, 112(7), 3959–4015 CrossRef CAS PubMed
- L. M. I. Schijven,
et al.
, Soft Matter, 2021, 17, 9682–9688 RSC
- D. V. Volodkin,
et al.
, Angew. Chem., Int. Ed., 2010, 122(48), 9444–9447 CrossRef
- J. Lademann,
et al.
, Acta Biomater., 2016, 30, 388–396 CrossRef CAS PubMed
- R. Elia,
et al.
, J. Coat. Technol. Res., 2015, 12(4), 793–799 CrossRef CAS PubMed
- R. De La Rica and A. H. Velders, Small, 2011, 7(1), 66–69 CrossRef CAS PubMed
- R. de laRica,
et al.
, Angew. Chem., Int. Ed., 2011, 50(25), 5704–5707 CrossRef CAS PubMed
- X. Huang,
et al.
, Lasers Med. Sci., 2008, 23(3), 217–228 CrossRef PubMed
- X. Huang and M. A. El-Sayed, J. Adv. Res., 2010, 1(1), 13–28 CrossRef
- J. Hainfeld,
et al.
, Br. J. Radiol., 2006, 79(939), 248–253 CrossRef CAS PubMed
- O. Castellani,
et al.
, Food Chem., 2004, 85(4), 569–577 CrossRef CAS
- D. V. Volodkin,
et al.
, Biomacromolecules, 2004, 5(5), 1962–1972 CrossRef CAS PubMed
- D. H. Ohlendorf,
et al.
, Nature, 1978, 272(5648), 28–32 CrossRef CAS PubMed
- J. Schindelin,
et al.
, Nat. Methods, 2012, 9(7), 676–682 CrossRef CAS PubMed
- D. V. Volodkin,
et al.
, Langmuir, 2004, 20(8), 3398–3406 CrossRef CAS PubMed
- D. Volodkin, Adv. Colloid Interface Sci., 2014, 207, 306–324 CrossRef CAS PubMed
- M. Anton, J. Sci. Food Agric., 2013, 93(12), 2871–2880 CrossRef CAS PubMed
- A. I. Petrov,
et al.
, Biotechnol. Prog., 2005, 21(3), 918–925 CrossRef CAS
- N. A. Feoktistova,
et al.
, Phys. Chem. Chem. Phys., 2020, 22(17), 9713–9722 RSC
- A. Madadlou,
et al.
, Food Hydrocolloids, 2018, 84, 38–46 CrossRef CAS
- Y. Yang,
et al.
, Food Chem., 2020, 311, 125998 CrossRef CAS PubMed
- S. Schmidt,
et al.
, Adv. Funct. Mater., 2013, 23(1), 116–123 CrossRef CAS
- W. Zhang,
et al.
, Crit. Rev. Food Sci. Nutr., 2013, 53(11), 1191–1201 CrossRef CAS PubMed
- A. Bradshaw,
et al.
, J. Exp. Biol., 2011, 214(10), 1699–1706 CrossRef CAS PubMed
- Y. N. Tan,
et al.
, J. Am. Chem. Soc., 2010, 132(16), 5677–5686 CrossRef CAS PubMed
- J. J. Warren,
et al.
, FEBS Lett., 2012, 586(5), 596–602 CrossRef CAS PubMed
- D. M. Chevrier,
et al.
, J. Nanophotonics, 2012, 6(1), 064504 CrossRef
- J. Xie,
et al.
, J. Am. Chem. Soc., 2009, 131(3), 888–889 CrossRef CAS PubMed
- H. Wei,
et al.
, Nat. Nanotechnol., 2011, 6(2), 93 CrossRef CAS PubMed
- A. Sani,
et al.
, Biochem. Biophys. Rep., 2021, 26, 100991 CAS
- S. Schmidt,
et al.
, Acta Biomater., 2014, 10(3), 1423–1430 CrossRef CAS PubMed
- A. E.-I. Coskun,
et al.
, Food Hydrocolloids, 2015, 45, 291–300 CrossRef
- B. Russell,
et al.
, J. Mater. Chem. B, 2016, 4(42), 6876–6882 RSC
- B. A. Russell,
et al.
, Phys. Chem. Chem. Phys., 2017, 19(10), 7228–7235 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb00823h |
| This journal is © The Royal Society of Chemistry 2022 |