
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Opening the pathway towards a scalable electrochemical semi-hydrogenation of alkynols via earth-abundant metal chalcogenides†
Kevinjeorjios
Pellumbi‡
 ab,
Leon
Wickert‡
ab,
Julian T.
Kleinhaus
b,
Jonas
Wolf
ab,
Allison
Leonard
b,
David
Tetzlaff
ab,
Roman
Goy
c,
Jonathan A.
Medlock
c,
Kai
junge Puring
a,
Rui
Cao
d,
Daniel
Siegmund
*ab and
Ulf-Peter
Apfel
*ab
ab,
Leon
Wickert‡
ab,
Julian T.
Kleinhaus
b,
Jonas
Wolf
ab,
Allison
Leonard
b,
David
Tetzlaff
ab,
Roman
Goy
c,
Jonathan A.
Medlock
c,
Kai
junge Puring
a,
Rui
Cao
d,
Daniel
Siegmund
*ab and
Ulf-Peter
Apfel
*ab
aFraunhofer Institute for Environmental, Safety and Energy Technology UMSICHT, Osterfelder Straße 3, D-46047 Oberhausen, Germany. E-mail: ulf.apfel@umsicht.fraunhofer.de; daniel.siegmund@umsicht.fraunhofer.de
bInorganic Chemistry I, Ruhr University Bochum, Universitätsstraße 150, D-44780 Bochum, Germany. E-mail: ulf.apfel@rub.de; daniel.siegmund@rub.de
cDSM Nutritional Products AG, Wurmisweg 576, CH-4303 Kaiseraugst, Switzerland
dKey Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710119, China
First published on 11th October 2022
Abstract
Electrosynthetic methods are crucial for a future sustainable transformation of the chemical industry. Being an integral part of many synthetic pathways, the electrification of hydrogenation reactions gained increasing interest in recent years. However, for the large-scale industrial application of electrochemical hydrogenations, low-resistance zero-gap electrolysers operating at high current densities and high substrate concentrations, ideally applying noble-metal-free catalyst systems, are required. Because of their conductivity, stability, and stoichiometric flexibility, transition metal sulfides of the pentlandite group have been thoroughly investigated as promising electrocatalysts for electrochemical applications but were not investigated for electrochemical hydrogenations of organic materials. An initial screening of a series of first row transition metal pentlandites revealed promising activity for the electrochemical hydrogenation of alkynols in water. The most active catalyst within the series was then incorporated into a zero-gap electrolyser enabling the hydrogenation of alkynols at current densities of up to 240 mA cm−2, Faraday efficiencies of up to 75%, and an alkene selectivity of up to 90%. In this scalable setup we demonstrate high stability of catalyst and electrode for at least 100 h. Altogether, we illustrate the successful integration of a sustainable catalyst into a scalable zero-gap electrolyser establishing electrosynthetic methods in an application-oriented manner.
Introduction
Hydrogenation reactions constitute some of the most important transformations in the chemical industry providing modern society with important synthons for the production of pharmaceuticals, fragrances, vitamins and margarines amongst others.1,2 Despite their large-scale industrial application, the use of molecular hydrogen under elevated pressures and temperatures, as well as the employment of expensive and scarce noble metal catalysts such as Pt or Pd, have made the development of greener alternatives increasingly more important in recent years.3Recently, synthetic pathways via electrochemical means are gaining increasing attention as tuneable and sustainable replacement of thermal processes.4 In addition to their modular and scalable character, electrochemical approaches operate under mild conditions with high atom economy compared to their thermocatalytic counterparts.5 Regarding hydrogenation reactions in particular, electrochemistry can effectively mitigate safety hazards, transport and storage costs as well as carbon dioxide emissions associated with the supply of highly pressurized dihydrogen.6,7 In the case of electrosynthesis instead of gaseous H2, the solvent, ideally water, acts as the proton source, while the possibility to directly use electrons from renewables enables a sustainable implementation.8 By carefully tuning the applied potential, temperature and electrolyte, a variety of functional groups such as ketones,9 aldehydes, esters,10 olefins,7 alkynes,11 nitriles12 and oximes13 have been electrochemically hydrogenated in recent years.
Albeit their promising perspective, electrolysers for the electrochemical hydrogenation (ECH) often still use expensive noble metals such as Pd and Pt, deeming the discovery and implementation of novel and inexpensive alternatives crucial to accelerate the large-scale implementation of this technology.14 Along this line, transition metal chalcogenide-based electrocatalysts have recently gained interest as cheap and stable alternatives to noble metal electrocatalysts in numerous applications.15 In case of ECH reactions, a few monometallic sulphur-depleted transition metal chalcogenides such as CuSx and CoS2−x and Co3S4−x, have already shown elevated activity for the ECH of alkynes,16 unsaturated aldehydes17 and nitroarenes,18 respectively. Another class of transition metal chalcogenides that could show great promise for the field of ECH in terms of both catalytic activity as well as facile and rapid scalability are pentlandite (Pn) materials. Our group has shown that depending on the applied electrochemical conditions pentlandite-based electrocatalysts, M9X8 (M: Fe, Co, Ni; X: S, Se), possess the ability to generate reactive hydride species on the catalytic surface either leading to the generation of hydrogen or promoting the hydrogenation of acetonitrile to ethane.19,20 Regarding scalability, the synthesis of pentlandite materials has also been established by green and scalable mechanochemical methods21 revealing further possible perspectives towards an industrial implementation. Most significantly, a crucial advantage of the pentlandite structure over other transition metal chalcogenides is the ability to fine-tune structural and electronic properties of the employed catalysts via a plethora of metal (Fe/Co/Ni) and chalcogenide (S/Se) variations.22–25 Consequently, utilising this stoichiometric flexibility more active pentlandite catalysts for the hydrogen evolution reaction (HER), CO2 reduction and oxygen evolution (OER) reaction could be developed in recent years.21–23,25,26 Overall, this interesting subgroup of multi-functional electrocatalysts demonstrates a combination of ideal properties towards the application in the field of ECH.
Regarding conceivable substrates, we turned our focus to the substrate class of alkynols, globally playing a major role in the fields of vitamins and perfumes.1 More specifically, in order to investigate the capability of pentlandite catalysts towards ECH, we selected 2-methyl-3-butyn-2-ol (MBY) as a model substrate, which represents the main industrial synthon for vitamin A and E synthesis, reaching annual productions in the scale of thousands of metric tons.1,27 Currently, the industrial hydrogenation of MBY to the highly desired alkene, 2-methyl-3-buten-2-ol (MBE), is mainly performed in batch-reactors under elevated temperatures and pressures suffering from issues of sustainability. Specifically, the lack of green reductants, employment of Pd-based and Pb-containing catalysts, as well as the necessity for additional purification steps, e.g. separation of products from the catalysts, remain key drawbacks of this approach.1 Similarly, despite the ton-scale production of MBE, the minimization of unwanted over-hydrogenation to 2-methyl-3-buten-2-ol (MBA) or possible side reactions, such as dimerization among others, remain major bottlenecks and key topics of current research of MBY hydrogenation.28 Furthermore, whilst there have been some reports of a continuous MBY-hydrogenation there is still the need for further improvements.29 Most notably, despite its industrial importance attempts towards the generation of MBE by the more sustainable electrochemical route remain limited within academic or patent literature, with base metal catalysts being non-existent (Fig. 1).30
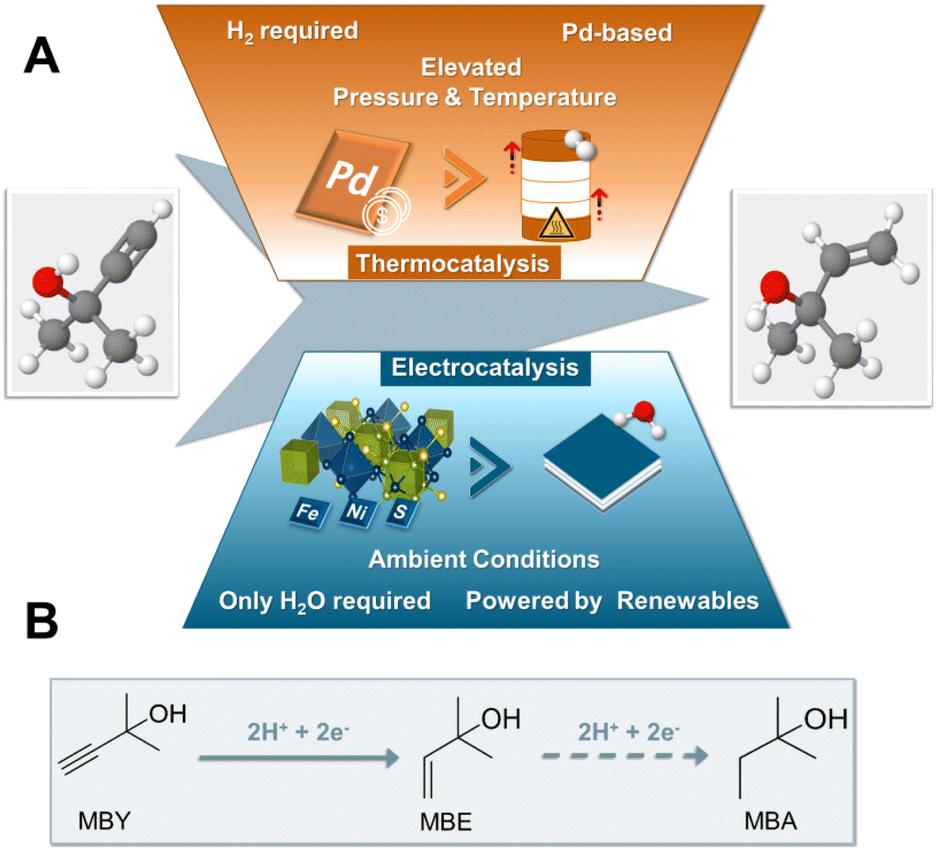 | ||
| Fig. 1 Schematic representation comparing the current state-of-the-art synthesis of MBY to the herein presented work based on pentlandite electrocatalyst (A). Equation depicting the ongoing hydrogenation processes within the ECH electrolyser (B). | ||
Herein, we report for the first time the electrochemical conversion of MBY (2-methyl-3-buty-2-ol) to the respective alkene MBE by a PGM-free catalyst in aqueous solution, without the supply of H2. The conversion of this important vitamin synthon is achieved through a series of robust and cheap transition metal chalcogenides based on the pentlandite structure. Going beyond screening experiments, we integrated the most active composition, Fe3Ni6S8, into industrially relevant zero-gap electrolysers, reaching yields of 70% for MBE at 240 mA cm−2 at a cell voltage of 3.0 V, being on par with current noble metal-based electrolysers, while providing a highly sustainable alternative to the noble-metal-based thermocatalytic state-of-the art.
Results and discussion
Screening of Fe9−xNixS8 –pentlandites for the ECH
Due to our previously gained experiences with this material class, we first focused on already reported materials with variable Ni/Fe ratios (Fe9−xNixS8). This class of catalysts allows us to vary the metal centres without leading to any significant structural changes of the crystal lattice.22 Following reported synthetic procedures for pentlandite materials, we prepared a series of Fe9−xNixS8 (x = 3–6, herein denoted as Nix) catalysts, with different Ni/Fe ratios, via mechanochemical methods (Fig. S1†).21 Subsequently, aiming to investigate the intrinsic activity of the (Fe,Ni)9S8 catalysts, the obtained powders were pressed into pellets and mounted into electrodes with the help of a conductive non-catalytically active carbon glue. The polished pellet electrodes (Ø 3 mm, A = 0.071 cm2) were used as a working electrode in a three-electrode H-type PEEK cell, possessing a compartment volume of 15 mL (Fig. S2†). To directly probe the activity of our pure pentlandite materials while also approaching preparative relevant conditions, electrolytic experiments were performed at 100 mA cm−2 for 2 h. To guarantee sufficient conductivity during testing, our catholyte comprised of 0.3 M KOH and 1 M MBY in water. Notably, the employed reactant as well as the conceivable products are stable under these conditions. All obtained solutions were analysed via headspace GC-MS.Within the investigated series, all tested Pn-materials show good catalytic activity towards ECH in the employed aqueous electrolyte starting from Ni3, possessing a faradaic efficiency for the generation of MBE (FEMBE) of 28% accompanied by a FEMBA of 10% (Fig. 2B). Transitioning to higher Ni-equivalents, the selectivity towards the ECH increases with Ni5 and Ni6 showing similarly elevated FEMBE values of 40% at 100 mA cm−2, with the remaining percentages corresponding to H2 generation as evidenced by gas chromatography. Regarding the observed half-cell potentials, except for Ni4.5 and Ni5, all of the tested materials demonstrate low half-cell potentials of ca. −0.5 V vs. RHE at 100 mA cm−2 (Fig. 2A).
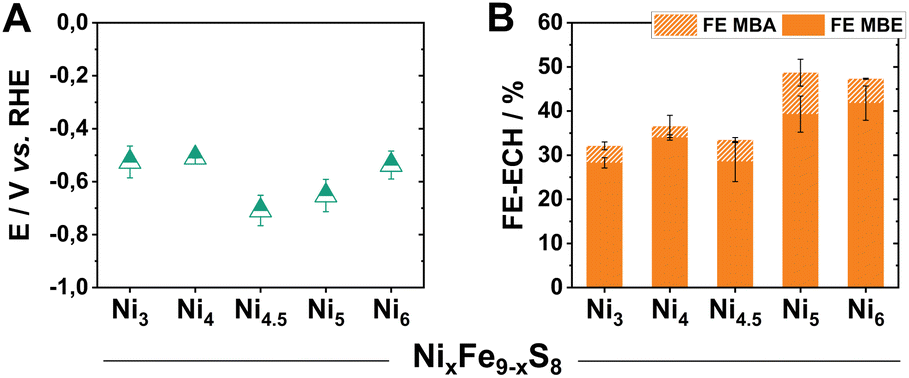 | ||
| Fig. 2 Half-cell potentials of the herein investigated Nix pellet electrodes after 2 h of electrolysis at 100 mA cm−2 (A). The respective faradaic efficiency values via GC-MS quantification (B). | ||
Inspired by these promising results, we extended our screening and elucidated the role of different metal atoms on the ECH in pentlandites. We subsequently also compared the activity of Ni6 with its cobalt analogues, Co9S8 (Co9), FeCo8S8 (FeCo8), and NiCo8S8 (NiCo8), which have been recently synthesized by our group as potential HER-catalysts.31 Interestingly, transitioning from a Fe/Ni-based system to a Co-based one has a major effect on the observed ECH selectivity. Specifically, Co9 shows the lowest activity towards ECH at a FEMBE value of 19%, while the addition of either one equivalent of Ni or Fe leads to an enhancement of the observed ECH activity (Fig. S3†). Specifically, FeCo8 enables the production of MBE with a FE of 50%, while NiCo8 generates a mixture of hydrogenated products consisting of MBE (FEMBE 25%) and MBA (FEMBA 18%). Compared to their Ni/Fe-counterparts, Co-based variants also demonstrate a higher half-cell potential with FeCo8, showing the largest value at −2.7 V vs. RHE. These clear differences not only within the MCo8S8 series but also the different Ni/Fe ratios demonstrate, that small alterations in the composition of our multi-metallic transition metal-rich chalcogenides offer the potential to control the catalytic selectivity and activity for the ECH.
Lastly, to provide a holistic picture within our screening investigation, we compared the activity of our materials to the performance of monometallic sulphides such as FeS and NiS (Fig. S4†). Although these analogues showed some activity for the conversion of MBY, with NiS showing a FEMBE of 62%, they generally suffered from severe instability under the employed electrocatalytic conditions, a behaviour already observed in reports for the hydrogen evolution reaction.20 Consequently, the monometallic sulphides were omitted for this investigation.
Since Ni6 showed the highest selectivity for the ECH, an elevated MBE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MBA ratio as well as a low half-cell potential within our initial screening (Table S1†), we selected it as our catalyst of choice in further comparisons as well as our following implementation into scalable electrolysers.
:MBA ratio as well as a low half-cell potential within our initial screening (Table S1†), we selected it as our catalyst of choice in further comparisons as well as our following implementation into scalable electrolysers.
Implementation of pentlandite materials in zero-gap electrolysers
Having successfully demonstrated that pentlandite-materials efficiently hydrogenate MBY, we shifted our focus towards achieving a link between catalyst development and implementation in scalable cell designs. In recent years, zero-gap electrolysers, have shown great promise in electrolytic applications not only for the H2 generation or the CO2 electroreduction, but also for the ECH.32 Here, an ion-exchange membrane is pressed in between the porous anode and cathode electrodes. This configuration decreases ohmic resistances, allows for a better control of the reaction environment, facilitates reactant transport, and, more importantly, permits the continuous large-scale operation of such organic transformations.33Focusing on the minimization of noble metals, our investigation mainly employs anion-exchange membranes, allowing for the employment of robust and cost-effective anodes such as Ni-foam for O2-formation. The herein investigated membrane electrode assemblies (MEAs) were implemented into an in-house built zero-gap electrolyser, with an active area of 12.57 cm2, consisting of an anion-exchange membrane sandwiched between a Ni-foam anode and carbon supports coated with sulfidic catalysts on the cathode, respectively (Fig. 3A1–A3 and S5†). During electrolysis, 1 M MBY:0.3 M KOH in H2O as well as 2 M KOH were used as catholyte and anolyte respectively, with the electrolyte being continuously recirculated while a current density of 80 mA cm−2 was applied for 2 h. Furthermore, we monitored the substrate and product crossover through the AEM by NMR analysis of both electrolyte compartments (Fig. S6–S9†). The herein discussed results will focus mainly on faradaic efficiencies obtained from the catholyte. The concentration of ECH-products in the anolyte reservoir as well as the respective FE value will only be mentioned when significant crossover was observed.
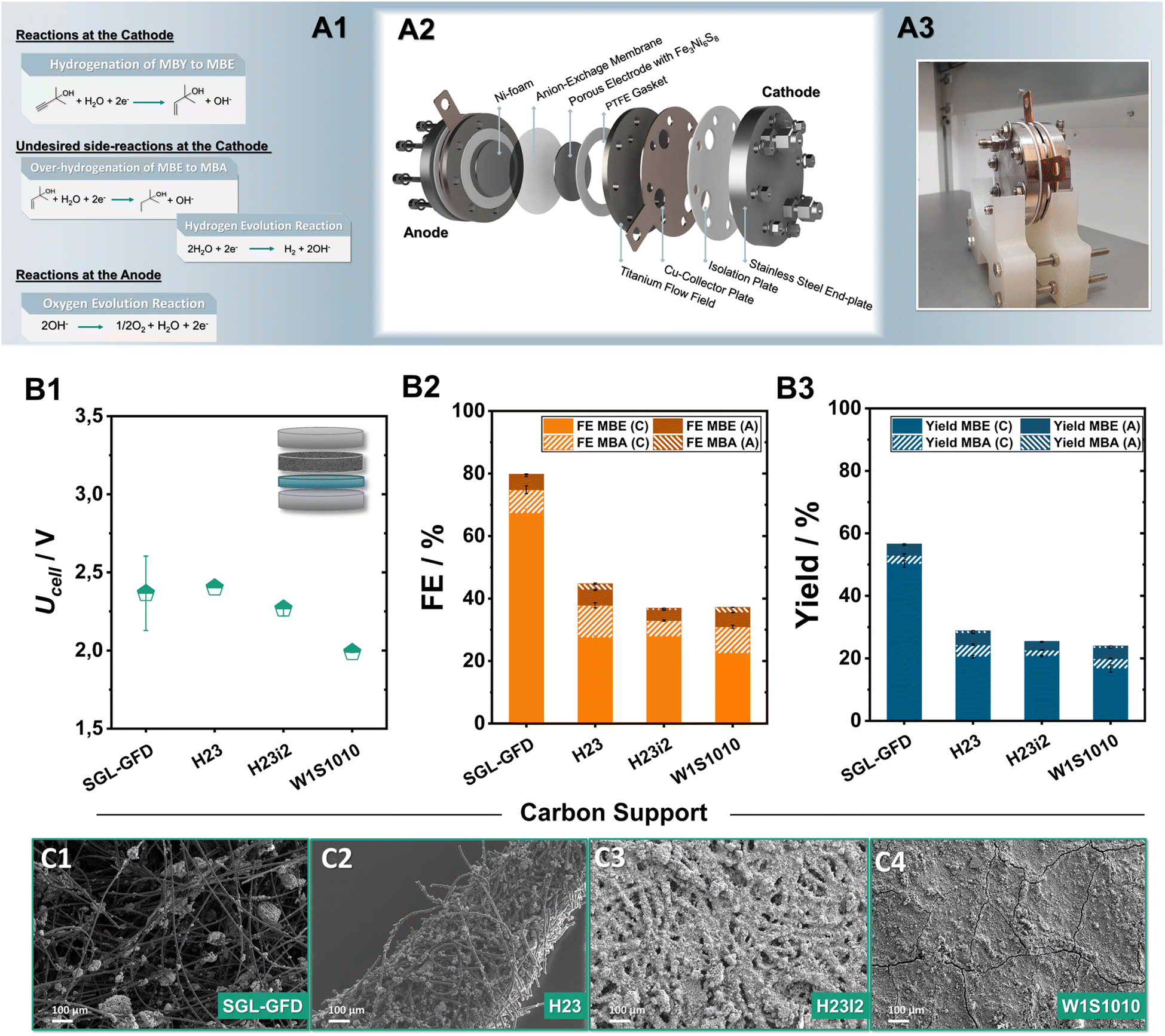 | ||
| Fig. 3 Reaction schemes of the different half-cell reactions during electrolysis (A1). Schematic representation (A2) and picture of the 12.57 cm2 electrolyzer employed in this work (A3). Investigation of the role of the employed carbon-support for the semi-hydrogenation of MBY at 80 mA cm−2 for 2 h of electrolysis. The obtained cell voltage (B1), faradaic efficiency (B2) and yield (B3) through 1H-NMR quantification. Here, FEMBX (C) and FEMBX (A) (X: E, A) correspond to the FE values of ECH products detected in the catholyte and anolyte after electrolysis, respectively. Scanning electron microscopy (SEM) analysis of the resulting catalytic layer on SGL-GFD (C1), H23 (C2), H23i2 (C3) and W1S10101 (C4) at a magnification of 100×. | ||
A key point in our optimization of the ECH in zero-gap electrolysers is the carbon porous transports layer (PTL) employed in the cathode. Although the role of the PTL has been rigorously investigated in the fields of fuel cells as well as electrolysers for the CO2 reduction or hydrogen generation,34 in the case of ECH reactions, it currently constitutes an unexplored parameter. To address this issue and optimize the transport of organic substrates to and of the generated products from the active centres, Ni6 was airbrushed onto a series of commercially available carbon supports; W1S1010, H23, H23i2, and Sigracell GFD 2.5, employing 10 wt% PTFE as a binder. A detailed list of the different PTL properties and vendors can be found in the ESI (Table S2†).
Comparing the two carbon paper substrates H23 and H23i2, it is evident that the hydrophobic treatment plays a minimal role in steering the observed ECH-activity (Fig. 3B2). Both, H23 and its hydrophobically treated counterpart H23i2 demonstrate similar FEMBE values, close to 28% alongside the generation of 4% of MBA. The additional implementation of a microporous layer (MPL) accompanied by a hydrophobic treatment in the case of W1S1010 has a negligible effect on the obtained FEMBE. Notably, transitioning from traditionally employed carbon paper and cloth materials to a porous carbon felt used in redox-flow batteries leads to a significant enhancement of the observed ECH activity.35 Applying a current density of 80 mA cm−2, the use of the Sigracell GFD carbon felt (SGL-GFD) as a conductive support, enables the highest activity for the ECH, reaching FEMBE values of 67% alongside a FEMBA of 8% at a full cell voltage of only 2.4 V, being on par with or outcompeting noble-metal containing MEAs for the ECH.36 The corresponding yields after 2 h of electrolysis were determined to be 53% for MBE and 3% for MBA in the catholyte, confirming appreciable selectivity for alkene formation with a rate of 0.12 mLMBE h−1 cm−2 at 80 mA cm−2 (Fig. 3B3). Regarding crossover of organic products through the membrane by means of either electro-osmosis, or membrane swelling, in the case of all Ni6-based carbon electrodes 5% of the total FEMBE and 2% of the total FEMBA quantified were detected in the anolyte reservoir after 2 h of electrolysis. Moreover, analysis of the observed gaseous products via online GC-MS for the SGL-GFD shows that the remaining FE percentages can be attributed to hydrogen evolution (Fig. S9†).
Analysis of the generated electrodes via SEM-EDX reveals that the different carbon supports result in significantly different morphologies of the catalytic layer (Fig. 3C1–C4). As observed by SEM/EDX-measurements (Fig. S10–S15†), W1S1010 carbon cloth, which shows the lowest FE for the ECH in tested series, shows the most compact catalytic layer, possibly due to the existence of a microporous layer (MPL) on the gas diffusion layer (GDL). Moving towards H23 and H23i2 the Ni6 particles appear to be spread through the surface of the carbon material in the form of spherical agglomerates. Similarly, in case of the SGL-felt, the air-brushed particles are not only located on the upper surface but are also embedded within the 3D structure of the felt forming catalytic clusters on the intersections of the carbon fibres. The formation of these dispersed Ni6-PTFE clusters alongside the significantly larger pores of the SGL-GFD could be responsible for the almost doubled activity through better transport of organic reactants and products to and from the catalytic centres.
In addition to the carbon support, the employed binder also plays a key role in tailoring the local environment around the catalytic particles.37 Therefore, we altered the employed fluoropolymer binder from PTFE to PVDF as well as the added amount, to identify further tuning points. Overall, both PVDF and PTFE bound electrodes on SGL-GFD show FE values for the ECH of over 50%, with PTFE electrodes outcompeting their polymeric counterparts both in terms of activity for the ECH and specifically for the generation of MBE (Fig. 4B1 and B2). Despite the significantly different FE values, the generated electrodes demonstrate similar cell voltages of 2.5 V independently of the employed binder or added amount. In contrast to the employment of PTFE, which leads to the formation of catalytic cluster on the carbon felt fibres, the use of PVDF leads to a significant engulfment of the catalytic particles by the polymeric binder (Fig. 4A1–A6). The thickness of these PVDF-containing layers, as well as the size of the PTFE-formed clusters, appears to increase with the stepwise addition of the fluoropolymer binder. The observed particle agglomeration could also cause the lower FE values for the ECH at higher binder loadings, since a significant proportion of the catalytic material remains encased in these structures not contributing to the ongoing catalytic processes.
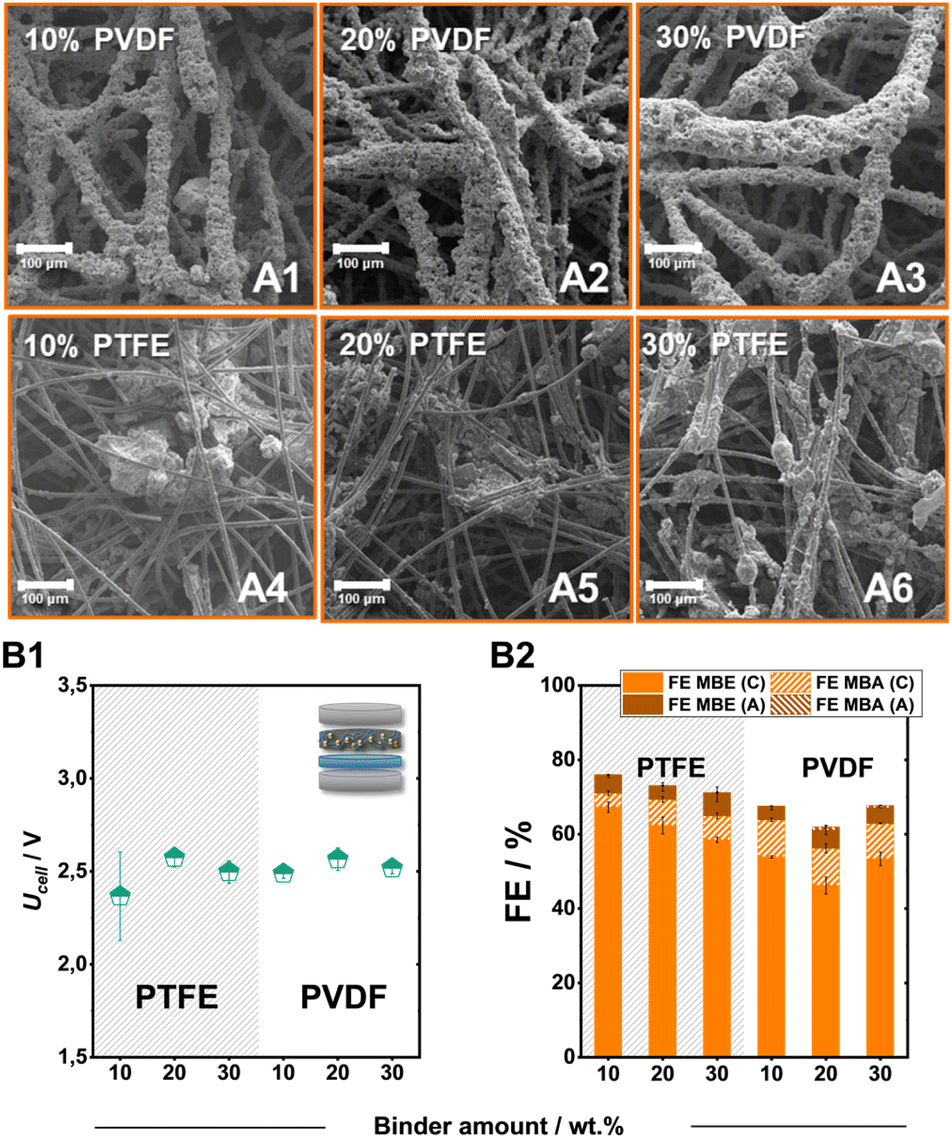 | ||
| Fig. 4 Scanning electron microscopy (SEM) investigation of PVDF (A1–A3) and PTFE bound electrodes (A4–A6). Effect of the binder nature and content on the ECH of MBY at 80 mA cm−2. The obtained cell voltage (B1) and faradaic efficiency (B2). Here, FEMBX (C) and FEMBX (A) (X: E, A) correspond to the FE values of ECH products detected in the catholyte and anolyte after electrolysis, respectively. | ||
Further attempts to increase the energy efficiency of our process by employing elevated temperatures and thinner ion-exchange membranes did not severely alter the obtained values (Fig. S16†).14,38 At a cell temperature of 40 °C, the cell voltage decreases by approximately 0.5 V (2.0 V), resulting in similar FE values compared to ambient conditions. The stepwise increase of the operational temperature leads to a steady decrease of the overall ECH activity. This trend could be associated simultaneously with a limited adsorption of the organic substrate on the catalytic surface as well as improved kinetics for the HER under the elevated temperatures.39 Similarly, employing thinner anion-exchange membranes, of 50 μm and 30 μm thickness (as compared to 130 μm) did not yield any significant improvements in terms of cell voltage, while the total amount of ECH products crossing over to the anolyte increased from 4% (130 μm) to 13% (30 μm), attributed to electroosmosis and membrane swelling.40
Pentlandite electrocatalysts versus the current state-of-the art
Comparing the Ni6-coated electrodes with commonly used ECH catalysts such as Pd particles (0.35–0.8 μm) or Ni-foam under standard conditions further illustrates the high selectivity of pentlandite materials for the targeted semi-hydrogenation reaction (Fig. 5A1–A3). Specifically, identically prepared Pd-based electrodes show a higher tendency towards the hydrogenation of MBY to MBA (FEMBA: 30%) accompanied by a comparatively lower FEMBE at similar MBY conversion. In contrast, the integration of Ni-foam as the cathode material demonstrates a decreased activity for the ECH with a total FE-ECH value of just 15%.8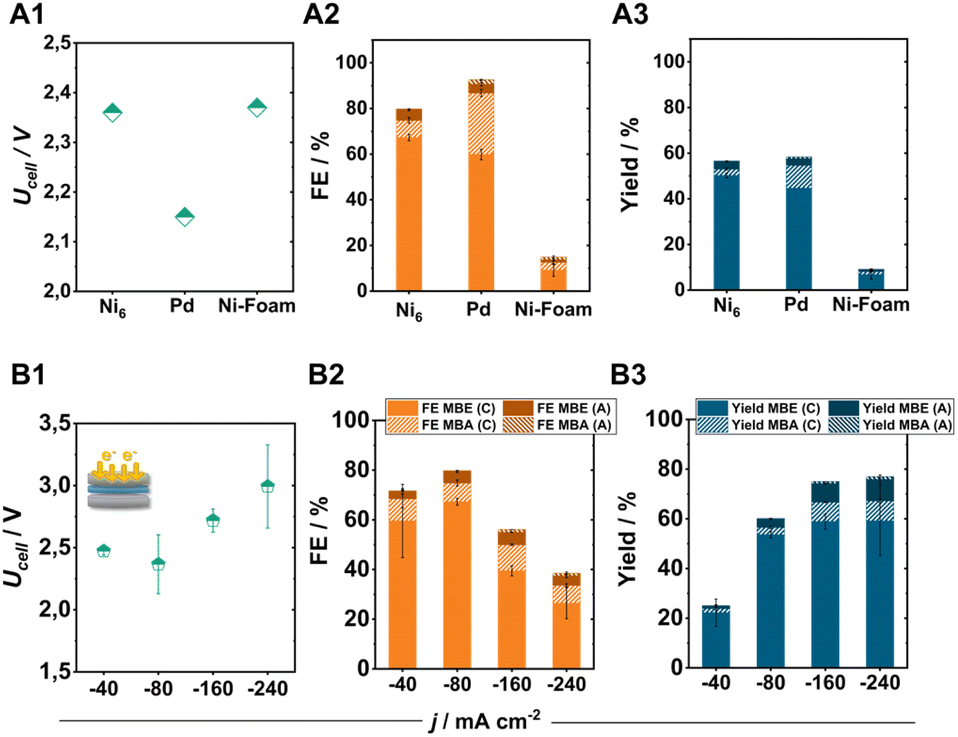 | ||
| Fig. 5 Comparison of Ni6 with the typical electrocatalysts for the ECH, Pd and Ni-foam. The obtained cell voltage (A1), faradaic efficiency (A2), and yield (A3) after 2 h of electrolysis at 80 mA cm−2. Effect of the applied current density on the ECH product distribution for Ni6-based electrodes in a zero-gap electrolyser. The obtained cell voltage (B1) faradaic efficiency (B2) and yield (B3) after 2 h of electrolysis at the applied current density. Here, FEMBX (C) and FEMBX (A) (X: E, A) correspond to the FE values of ECH products detected in the catholyte and anolyte after electrolysis, respectively. | ||
By increasing the applied current density to achieve higher product yields, our optimized electrode was able to perform the ECH at elevated current densities of 240 mA cm−2, with a maintained FEMBE of 30% and a FEMBE:FEMBA ratio of 7:1 (the remaining 70% FE belonging to the formation of H2). Even at these elevated current densities, the crossover of organic products to the anode compartment remains at 5%, allowing us to reach a total MBE yield of 70% in 2 h of electrolysis (Fig. 5B3) when combining the generated amounts of MBE in catholyte and anolyte. Regarding cell efficiency, the obtained cell voltages range from 2.4 to 3.0 V depending on the applied current density, demonstrating the capabilities of electrosynthetic anion-exchange-based MEAs to compete with the often PGM-containing cation-exchange variants.41 Subsequent long-term experiments for 100 h under elevated electrolytic loading, also demonstrate the high stability of pentlandite electrodes (Fig. S17†). Interestingly, although catalyst deactivation is a major issue in thermocatalytic processes, the herein demonstrated elevated stability indicates that ECH reactors could possibly possess a longer lifetime than their counterparts by minimization of by-product formation.
Overall, the observed activity constitutes currently the only example of performing hydrogenation of MBY in aqueous electrolytes without the involvement of any PGM-metals in either cell compartment (Table S3†).
Despite the highly competitive performance of our combined catalyst and electrode optimization in the field of ECH, there is yet still significant room for optimization in direct comparison to thermocatalytic processes. In view of the present results, however, we are confident that the community will be able to establish a green and economically competitive alternative to thermocatalytic processes soon.
Conclusions
In conclusion, we demonstrate the electrochemical reduction of the alkynol 2-methyl-3-butyne-ol (MBY) to its commercially important semi-hydrogenation product 2-methyl-3-butene-ol (MBE) under industrial relevant conditions using a non-noble metal catalyst, without the supply of H2 gas, in aqueous electrolytes. This important milestone was achieved using cheap and robust transition metal chalcogenides based on the mineral pentlandite (M9S8), further demonstrating the multi-functionality of this material class in a range of reactions such as the ECH, HER, CO2RR and OER. Moreover, we herein provide a rare example of the utilization of a PGM-free zero-gap electrolyser in an aqueous-based organic reaction. Via optimization of the carbon support and binder for the ECH, we were able to reach ECH yields of 70% for the conversion of MBY to MBE after 2 h of electrolysis at 240 mA cm−2 at cell voltages as low as 3.0 V. We believe that our catalytic and cell parameter screening provide important stepping-stones towards the development of more financially favourable, active and sustainable electrocatalysts for the ECH of substrates beyond alkynols as well as towards the acceleration of this technology in the industrial scale.Author contributions
Conceptualization: K. P., J. T. K., K. J. P., D. S. and U.-P. A.; methodology: K. P., L. W., J. T. K., K. J. P., D. S. and U.-P. A.; data curation: K. P., L. W., J. W., A. L. and D. T.; formal analysis: K. P., L. W., J. W., A. L. and D. T.; funding acquisition: U.-P. A. and D. S.; supervision: U.-P. A., K. J. P. and D. S.; writing-original draft preparation: K. P., L. W., J. T. K., R. C., J. M., R. G., U.-P. A. and D. S. All authors have read and agreed to the published version of the manuscript.Data availability
Experimental data regarding electrochemical experiments and product analysis are available free of charge in the ESI.†Conflicts of interest
These are not conflicts of interest to declare.Acknowledgements
K. P. acknowledges the Fonds of the Chemical Industry for a PhD Fellowship. The work of J. T. K. and U.-P. A. was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2033 – 390677874 – RESOLV, the Fraunhofer Internal Programs under Grant no. Attract 097-602175 and the Fraunhofer Lighthouse project ShaPID. D. S. is grateful to BMBF for the financial support within the NanoMatFutur Project ‘’H2Organic’’ No. 03XP0421. The authors are also thankful for support by the Mercator Research Center Ruhr (MERCUR.Exzellenz, ‘DIMENSION’ Ex-2021-0034 and ‘KataSign’ Ko-2021-0016).References
- W. Bonrath, J. Medlock, J. Schutz, B. Wustenberg and T. Netscher, in Hydrogenation, ed. I. Karamé, IntechOpen, London, 2012, pp. 69–89 Search PubMed.
- (a) M. Eggersdorfer, D. Laudert, U. Létinois, T. McClymont, J. Medlock, T. Netscher and W. Bonrath, Angew. Chem., Int. Ed., 2012, 51, 12960 CrossRef CAS PubMed; (b) T. Charvillat, P. Bernardelli, M. Daumas, X. Pannecoucke, V. Ferey and T. Besset, Chem. Soc. Rev., 2021, 50, 8178 RSC; (c) X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984 CrossRef CAS PubMed.
- T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484 CAS.
- R. S. Sherbo, A. Kurimoto, C. M. Brown and C. P. Berlinguette, J. Am. Chem. Soc., 2019, 141, 7815 CrossRef CAS PubMed.
- (a) P. Gandeepan, L. H. Finger, T. H. Meyer and L. Ackermann, Chem. Soc. Rev., 2020, 49, 4254 RSC; (b) M. J. Llorente, B. H. Nguyen, C. P. Kubiak and K. D. Moeller, J. Am. Chem. Soc., 2016, 138, 15110 CrossRef CAS PubMed.
- (a) H. B. Goyal, D. Seal and R. C. Saxena, Renewable Sustainable Energy Rev., 2008, 12, 504 CrossRef CAS; (b) S. A. Akhade, N. Singh, O. Y. Gutiérrez, J. Lopez-Ruiz, H. Wang, J. D. Holladay, Y. Liu, A. Karkamkar, R. S. Weber, A. B. Padmaperuma, M.-S. Lee, G. A. Whyatt, M. Elliott, J. E. Holladay, J. L. Male, J. A. Lercher, R. Rousseau and V.-A. Glezakou, Chem. J. Rev., 2020, 120, 11370 CrossRef CAS PubMed.
- J. Yang, H. Qin, K. Yan, X. Cheng and J. Wen, Adv. Synth. Catal., 2021, 363, 5407 CrossRef CAS.
- (a) R. S. Delima, M. D. Stankovic, B. P. MacLeod, A. G. Fink, M. B. Rooney, A. Huang, R. P. Jansonius, D. J. Dvorak and C. P. Berlinguette, Energy Environ. Sci., 2022, 15, 215 RSC; (b) Y. He, L. Deng, Y. Lee, K. Li and J.-M. Lee, ChemSusChem, 2022, e202200232 CAS.
- C. J. Bondue, F. Calle-Vallejo, M. C. Figueiredo and M. T. M. Koper, Nat. Catal., 2019, 2, 243 CrossRef CAS.
- T. Shono, H. Masuda, H. Murase, M. Shinomura and S. Kashiura, J. Org. Chem., 1992, 57, 1061–1063 CrossRef CAS.
- (a) Z. Shi, N. Li, H.-K. Lu, X. Chen, H. Zheng, Y. Yuan and K.-Y. Ye, Curr. Opin. Electrochem., 2021, 28, 100713 CrossRef CAS; (b) B. Li and H. Ge, Sci. Adv., 2019, 5, eaaw2774 CrossRef CAS PubMed.
- (a) R. Mathison and M. A. Modestino, Chem. Catal., 2021, 1, 246 CrossRef; (b) D. Zhang, J. Chen, Z. Hao, L. Jiao, Q. Ge, W.-F. Fu and X.-J. Lv, Chem. Catal., 2021, 1, 393 CrossRef.
- A. J. Fry and J. H. Newberg, J. Am. Chem. Soc., 1967, 89, 6374 CrossRef CAS.
- D. Siegmund, S. Metz, V. Peinecke, T. E. Warner, C. Cremers, A. Grevé, T. Smolinka, D. Segets and U.-P. Apfel, JACS Au, 2021, 1, 527 CrossRef CAS PubMed.
- (a) H. M. A. Amin and U.-P. Apfel, Eur. J. Inorg. Chem., 2020, 2020, 2679 CrossRef CAS; (b) D. Siegmund, N. Blanc, M. Smialkowski, K. Tschulik and U.-P. Apfel, ChemElectroChem, 2020, 7, 1514 CrossRef CAS; (c) M. Nath, H. Singh and A. Saxena, Curr. Opin. Electrochem., 2022, 34, 100993 CrossRef CAS; (d) X. Shao, X. Zhang, Y. Liu, J. Qiao, X.-D. Zhou, N. Xu, J. L. Malcombe, J. Yi and J. Zhang, J. Mater. Chem. A, 2021, 9, 2526 RSC.
- Y. Wu, C. Liu, C. Wang, Y. Yu, Y. Shi and B. Zhang, Nat. Commun., 2021, 12, 3881 CrossRef CAS PubMed.
- S. Han, Y. Shi, C. Wang, C. Liu and B. Zhang, Cell Rep. Phys. Sci., 2021, 2, 100337 CrossRef CAS.
- Y. Zhao, C. Liu, C. Wang, X. Chong and B. Zhang, CCS Chem., 2021, 3, 507 CrossRef CAS.
- S. Piontek, K. Junge Puring, D. Siegmund, M. Smialkowski, I. Sinev, D. Tetzlaff, B. Roldan Cuenya and U.-P. Apfel, Chem. Sci., 2019, 10, 1075 RSC.
- B. Konkena, K. Junge Puring, I. Sinev, S. Piontek, O. Khavryuchenko, J. P. Dürholt, R. Schmid, H. Tüysüz, M. Muhler, W. Schuhmann and U.-P. Apfel, Nat. Commun., 2016, 7, 1 RSC.
- D. Tetzlaff, K. Pellumbi, D. M. Baier, L. Hoof, H. Shastry Barkur, M. Smialkowski, H. M. A. Amin, S. Grätz, D. Siegmund, L. Borchardt and U.-P. Apfel, Chem. Sci., 2020, 11, 12835 RSC.
- D. Tetzlaff, K. Pellumbi, K. j. Puring, D. Siegmund, W. S. K. Polet, M. P. Checinski and U.-P. Apfel, ChemElectroChem, 2021, 8, 3161 CrossRef CAS.
- M. Smialkowski, D. Siegmund, K. Pellumbi, L. Hensgen, H. Antoni, M. Muhler and U.-P. Apfel, Chem. Commun., 2019, 55, 8792 RSC.
- M. Smialkowski, D. Siegmund, K. Stier, L. Hensgen, M. P. Checinski and U.-P. Apfel, ACS Mater. Au, 2022, 2(4), 474–481 CrossRef CAS.
- K. Pellumbi, M. Smialkowski, D. Siegmund and U.-P. Apfel, Chem.–Eur. J., 2020, 26, 9938 CrossRef CAS PubMed.
- H. M. A. Amin, M. Attia, D. Tetzlaff and U.-P. Apfel, ChemElectroChem, 2021, 8, 3863 CrossRef CAS.
- (a) G. L. Parker, L. K. Smith and I. R. Baxendale, Tetrahedron, 2016, 72, 1645 CrossRef CAS; (b) S. Kundu and D. Sarkar, J. Heterocycl. Chem., 2021, 58, 1741 CrossRef CAS.
- (a) N. Semagina and L. Kiwi-Minsker, Catal. Lett., 2009, 127, 334 CrossRef CAS; (b) L. B. Okhlopkova, E. V. Matus, I. P. Prosvirin, M. A. Kerzhentsev and Z. R. Ismagilov, J. Nanopart. Res., 2015, 17, 1 CrossRef CAS; (c) A. González-Fernández, C. Pischetola, L. Kiwi-Minsker and F. Cárdenas-Lizana, J. Phys. Chem. C, 2020, 124, 3681 CrossRef.
- S. Vernuccio, R. Goy, P. Rudolf von Rohr, J. Medlock and W. Bonrath, React. Chem. Eng., 2016, 1, 445 RSC.
- K. Zhu, J. Ma, L. Chen, F. Wu, X. Xu, M. Xu, W. Ye, Y. Wang, P. Gao and Y. Xiong, ACS Catal., 2022, 12, 4840 CrossRef CAS.
- M. Smialkowski, D. Tetzlaff, L. Hensgen, D. Siegmund and U.-P. Apfel, Chin. J. Catal., 2021, 42, 1360 CrossRef CAS.
- (a) K. Nagasawa, Y. Sawaguchi, A. Kato, Y. Nishiki and S. Mitsushima, Electrochemistry (Tokyo, Jpn.), 2018, 86, 339 CrossRef CAS; (b) H. Liu, T.-H. Lee, Y. Chen, E. W. Cochran and W. Li, ChemElectroChem, 2021, 8, 2817 CrossRef CAS; (c) R. I. Masel, Z. Liu, H. Yang, J. J. Kaczur, D. Carrillo, S. Ren, D. Salvatore and C. P. Berlinguette, Nat. Nanotechnol., 2021, 16, 118 CrossRef CAS PubMed; (d) S. Shiva Kumar and V. Himabindu, Mater. Sci. Energy Technol., 2019, 2, 442 Search PubMed; (e) I. Vincent and D. Bessarabov, Renewable Sustainable Energy Rev., 2018, 81, 1690 CrossRef CAS.
- (a) B. Gleede, M. Selt, C. Gütz, A. Stenglein and S. R. Waldvogel, Org. Process Res. Dev., 2020, 24, 1916 CrossRef CAS; (b) C. Gütz, A. Stenglein and S. R. Waldvogel, Org. Process Res. Dev., 2017, 21, 771 CrossRef; (c) M. M. Hielscher, B. Gleede and S. R. Waldvogel, Electrochim. Acta, 2021, 368, 137420 CrossRef CAS; (d) B. Endrődi, E. Kecsenovity, A. Samu, T. Halmágyi, S. Rojas-Carbonell, L. Wang, Y. Yan and C. Janáky, Energy Environ. Sci., 2020, 13, 4098 RSC; (e) D. A. Salvatore, D. M. Weekes, J. He, K. E. Dettelbach, Y. C. Li, T. E. Mallouk and C. P. Berlinguette, ACS Energy Lett., 2018, 3, 149 CrossRef CAS; (f) L. Hoof, N. Thissen, K. Pellumbi, K. Junge Puring, D. Siegmund, A. K. Mechler and U.-P. Apfel, Cell Rep. Phys. Sci., 2022, 3, 100825 CrossRef CAS.
- (a) R. Omrani and B. Shabani, Int. J. Hydrogen, 2017, 42, 28515 CrossRef CAS; (b) S. Park, J.-W. Lee and B. N. Popov, Int. J. Hydrogen, 2012, 37, 5850 CrossRef CAS; (c) R. Küngas, J. Electrochem. Soc., 2020, 167, 44508 CrossRef.
- R. Banerjee, N. Bevilacqua, L. Eifert and R. Zeis, J. Energy Storage, 2019, 21, 163 CrossRef.
- (a) M. Sadakiyo, S. Hata, X. Cui and M. Yamauchi, Sci. Rep., 2017, 7, 17032 CrossRef PubMed; (b) J. Benziger and J. Nehlsen, Ind. Eng. Chem. Res., 2010, 49, 11052 CrossRef CAS.
- (a) K. Junge Puring, D. Siegmund, J. Timm, F. Möllenbruck, S. Schemme, R. Marschall and U.-P. Apfel, Adv. Sustainable Syst., 2021, 5, 2000088 CrossRef CAS; (b) T. H. M. Pham, J. Zhang, M. Li, T.-H. Shen, Y. Ko, V. Tileli, W. Luo and A. Züttel, Adv. Energy Mater., 2022, 12, 2103663 CrossRef CAS.
- R. Krause, D. Reinisch, C. Reller, H. Eckert, D. Hartmann, D. Taroata, K. Wiesner-Fleischer, A. Bulan, A. Lueken and G. Schmid, Chem. Ing. Tech., 2020, 92, 53 CrossRef CAS.
- (a) Y. Shi, E. Xing, J. Zhang, Y. Xie, H. Zhao, Y. Sheng and H. Cao, ACS Sustainable Chem. Eng., 2019, 7, 9464 CrossRef CAS; (b) B. Pierozynski and T. Mikolajczyk, Electrocatalysis, 2015, 6, 51 CrossRef CAS.
- A. Gugliuzza, in Encyclopedia of Membranes, ed. E. Drioli and L. Giorno, Springer, Berlin, Heidelberg, 2019, pp. 1–2 Search PubMed.
- (a) M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, React. Chem. Eng., 2020, 5, 977 RSC; (b) V. Montiel, A. Sáez, E. Expósito, V. García-García and A. Aldaz, Electrochem. Commun., 2010, 12, 118 CrossRef CAS; (c) A. Sáez, V. García-García, J. Solla-Gullón, A. Aldaz and V. Montiel, Electrochim. Acta, 2013, 91, 69 CrossRef; (d) D. Hoormann, C. Kubon, J. Jörissen, L. Kröner and H. Pütter, J. Electroanal. Chem., 2001, 507, 215 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04647d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |