
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoelectrochemical water oxidation improved by pyridine N-oxide as a mimic of tyrosine-Z in photosystem II†‡
Yong
Zhu
a,
Guoquan
Liu
a,
Ran
Zhao
a,
Hua
Gao
a,
Xiaona
Li
 b,
Licheng
Sun
acd and
Fei
Li
*a
b,
Licheng
Sun
acd and
Fei
Li
*a
aState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116024, China. E-mail: lifei@dlut.edu.cn
bKey Laboratory of Industrial Ecology and Environmental Engineering (MOE), School of Environmental Science and Technology, Dalian University of Technology, Dalian 116024, China
cDepartment of Chemistry, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, Stockholm 10044, Sweden
dCenter of Artificial Photosynthesis for Solar Fuels, School of Science, Westlake University, Hangzhou 310024, China
First published on 1st April 2022
Abstract
Artificial photosynthesis provides a way to store solar energy in chemical bonds with water oxidation as a major challenge for creating highly efficient and robust photoanodes that mimic photosystem II. We report here an easily available pyridine N-oxide (PNO) derivative as an efficient electron transfer relay between an organic light absorber and molecular water oxidation catalyst on a nanoparticle TiO2 photoanode. Spectroscopic and kinetic studies revealed that the PNO/PNO+˙ couple closely mimics the redox behavior of the tyrosine/tyrosyl radical pair in PSII in improving light-driven charge separation via multi-step electron transfer. The integrated photoanode exhibited a 1 sun current density of 3 mA cm−2 in the presence of Na2SO3 and a highly stable photocurrent density of >0.5 mA cm−2 at 0.4 V vs. NHE over a period of 1 h for water oxidation at pH 7. The performance shown here is superior to those of previously reported organic dye-based photoanodes in terms of photocurrent and stability.
Introduction
Photocatalytic splitting of water into hydrogen and oxygen is one of the most attractive routes for solar energy conversion. Among the available methodologies, the photoelectrochemical (PEC) cell based on a semiconductor/liquid junction has been recognized as a promising approach enabling the production of solar fuels at relatively low cost.1–3 In PEC cells, the oxidation of water is a major obstacle of overall water splitting due to the demand for multi-electron and multi-proton transfer. Although a large body of PEC cells has been developed based on light-harvesting inorganic semiconductors, a highly efficient photoanode is yet to be explored. As an alternative approach, dye-sensitized photoelectrochemical cells (DSPECs) have received wide attention considering their ability to be engineered at the molecular level.4 On a TiO2-based DESPEC photoanode, the surface-bound visible dye is integrated with a water oxidation catalyst (WOC). Photoexcitation of the dye leads to electron injection into the conduction band of the TiO2 substrate, followed by cross-surface electron migration from the WOC to the oxidized dye. The latter step is particularly challenging because of the need to compete with back-electron transfer at the oxide interface.5–7In the past decade, the phosphonate-derivatized tris(2,2′-bipyridine)ruthenium(II) dichloride salt (RuP) has been applied as a typical photosensitizer for PEC water oxidation.8 Despite favorable photophysical properties, the employment of a noble metal hindered its practical application in artificial photosynthesis. Aiming at scalable application and economic viability, a DSPEC photoanode should be cost efficient to produce O2 gas. In this regard, organic dyes are attractive candidates in terms of their low cost, earth-abundance and structural tunability by well-established organic synthesis techniques. Previously, a variety of organic dyes including perylene diimide,9,10 porphyrins,11–16 Janus green B17 and triphenylamine-based donor–acceptor compounds18–21 have been integrated in DSPECs for visible-light harvesting. However, organic dyes often suffer from a short excited-state lifetime, and their performance towards solar-driven water oxidation remains poor. The reported photocurrent densities achieved by organic dye-based photoanodes fall in a range of 20 to 200 μA cm−2, the magnitudes of which are far less than that achieved by RuP (up to 1.7 mA cm−2 was reported).22 In addition, compared with noble metal-based photosensitizers, organic dyes are more vulnerable to oxidative degradation, leading to low stability in aqueous environments. In order to overcome these limitations, metal oxides18,23 and polymers such as Nafion24 and poly(methyl methacrylate) (PMMA)25,26 have been deposited on the surface of the TiO2 electrode to stabilize organic dyes and related assemblies. However, the addition of protection layers cannot fully inhibit the decomposition of dyes upon light irradiation. Therefore, a more efficient method to improve the efficiency and stability of organic dyes for photoanodic water oxidation is urgently needed.
In the photosynthetic reaction center of photosystem II (PSII), tyrosine-Z (TyrZ) has been well recognized as a crucial cofactor to shuttle electrons between the CaMn4O4 oxygen evolution complex (OEC) and chromophore P680+˙.27–29 The tyrosine/tyrosyl radical redox couple mediated electron transfer results in quick recovery of the reduced state of the chromophore and thus efficiently protects the chromophore against decomposition.30,31 Another benefit of the redox mediator is that it efficiently retards unproductive back electron transfer by extending the distance between the chromophore and catalyst. In the configurations of DSPECs, synthetic models for the biological electron transfer relay have rarely been investigated. To the best of our knowledge, only two samples have been reported to date, and both are related to RuP sensitized-photoanodes. In one study, a benzoimidazole-phenol moiety was covalently linked with surface-bound IrO2 to accelerate light-induced electron transfer between the IrO2 WOC and RuP.32 Recently, we deposited an ultrathin NiO overlayer on a RuP sensitized SnO2/TiO2 film by atomic layer deposition (ALD), and the NiO layer served as an electron transfer relay between RuP and the exterior Ru-bda (H2bda = 2,2′-bipyridine-6,6′-dicarboxylic acid) catalyst.33 Despite these efforts, preparation of suitable redox mediators that efficiently reproduce the role of tyrosine in PSII remains a challenge.
We report herein that the performance of the organic dye and molecular WOC co-adsorbed TiO2 photoanode could be considerably improved by incorporation with an easily available pyridine N-oxide (PNO) derivative as a molecular electron transfer analog for TyrZ (Fig. 1). During the catalytic cycle, the photogenerated pyridine N-oxide cation radical closely mimics the tyrosyl radical which efficiently extracts electrons from the WOC. With this design, the integrated TiO2|dye|PNO|Cat film (Fig. 2a) exhibited superior photoanodic performance over other organic dye-based photoanodes in terms of photocurrent density and stability.
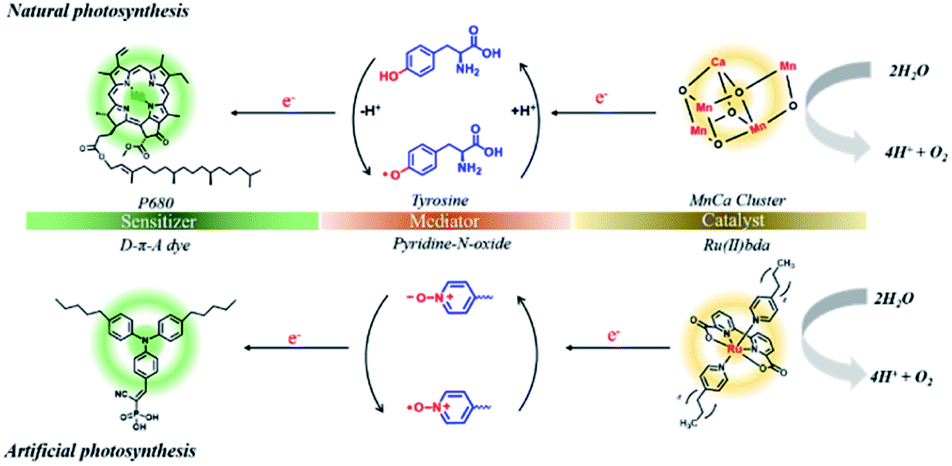 | ||
| Fig. 1 Illustration of the role of the redox mediator in natural (top) and artificial (bottom) photosynthetic systems. | ||
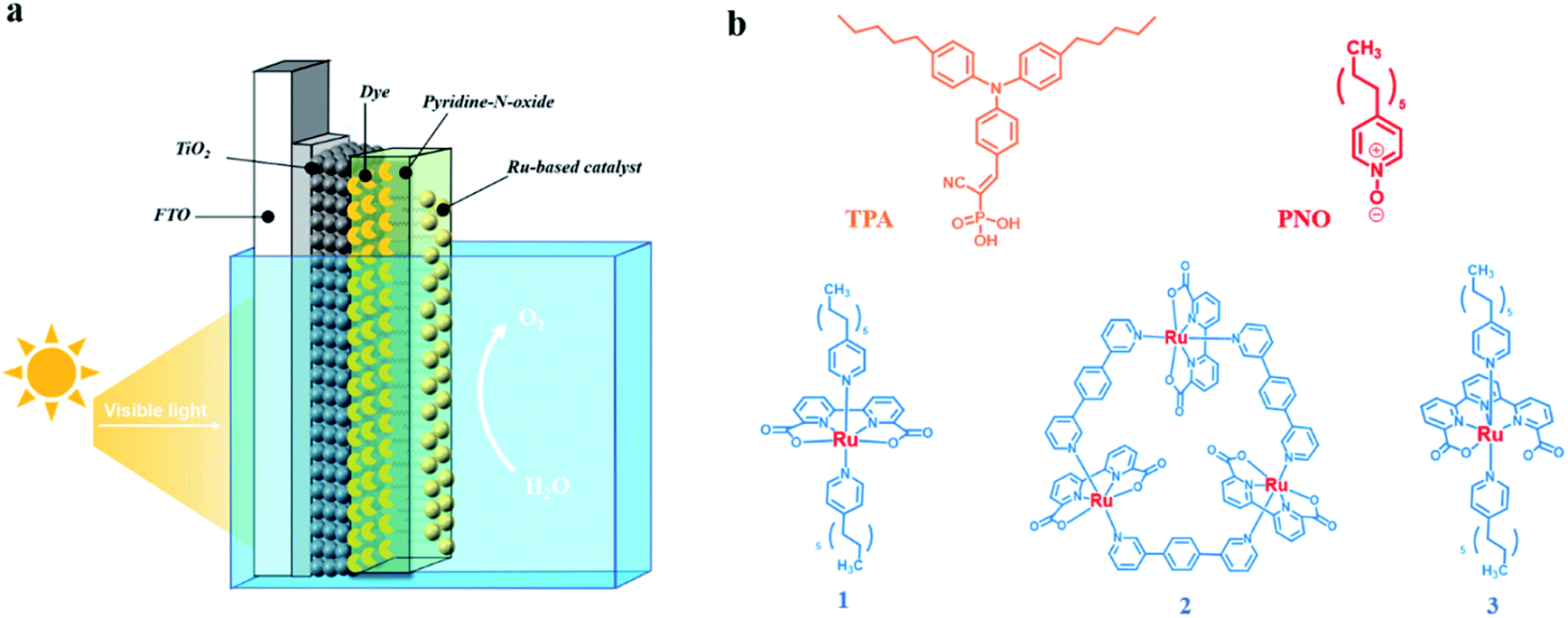 | ||
| Fig. 2 (a) Schematic of the TiO2|dye|PNO|Cat photoanode employed for water oxidation. (b) Molecular structures of TPA dye, PNO and Ru-based molecular water oxidation catalysts 1, 2 and 3 used in the study. | ||
Results and discussion
During the fabrication of the photoanode, a donor–acceptor type triphenylamine (TPA) dye was chosen as the chromophore due to its sufficient driving force for water oxidation. The preparation and characterization of 18 nm nanoparticle TiO2 films on FTO followed a literature procedure.22 As shown in Fig. 2b, the TPA dye was decorated with C5 alkyl chains and a phosphonate anchoring group, which was subsequently sensitized on a metal oxide substrate by immersing the TiO2 thin film in a dye solution for 2 h. Based on the Lambert–Beer law, the coverage of dye on the TiO2 film was 8 × 10−8 mol cm−2, consistent with monolayer adsorption on the electrode surface.34The obtained electrode TiO2|TPA was further decorated with 4-undecyl pyridine N-oxide (PNO) as an electron transfer relay (Fig. 2). Since the DSPEC was operated in aqueous solution, the modification of long alkyl chains on PNO allowed this mediator complex to be loaded as an overlayer via simple drop-casting,35 a method that brings considerable convenience in fabrication of molecular assemblies on metal oxides over the traditional ones via covalent links32 or ALD deposition.33
The SEM image of TiO2|TPA|PNO reveals a rough surface composed of amorphous nanoparticles and the cross-sectional SEM image of TiO2|TPA|PNO shows a clear interface between mesoporous TiO2 and the PNO layer, with an average thickness of 5 μm for the overlayer (Fig. 3). In comparison, a notable difference was observed for the polymer coated TiO2 films. The SEM images of TiO2|TPA|PMMA and TiO2|TPA|Nafion show much flatter and denser membranes over the TiO2 underlayer (Fig. S1‡). Considering that the diameter of a TPA molecule is about 15 Å, the dyes were proposed to be completely buried in the PNO layer.
 | ||
| Fig. 3 SEM images of the TiO2|TPA|PNO film, top view (a) and side view (b). | ||
It has been reported that pyridine N-oxide can be electrochemically or photochemically oxidized in either aqueous or non-aqueous solutions, and the generation of a pyridine N-oxide radical cation was supported by EPR and other techniques.36,37 Agreeing with these findings, cyclic voltammetry (CV) of PNO modified nanoITO in neutral phosphate buffer shows an irreversible anodic wave at 1.1 V vs. NHE (Fig. S2‡), which was ascribed to the oxidation of PNO to the cation radical PNO+˙. The effect of PNO on surface charge transfer was first investigated in the dark with TiO2|TPA coated with different overlayers. The dynamic changes of the absorption spectra for each photoanode were recorded by imposing a constant bias of 1.2 V vs. NHE.18,38 During this process, intense absorption at 700 nm was observed for TiO2|TPA, TiO2|TPA|PMMA and TiO2|TPA|Nafion films due to the oxidation of TPA to TPA+˙ with holes located on the dye (Fig. 4). By contrast, no TPA+˙ radical signal was observed for the electrode TiO2|TPA|PNO during electrolysis. The spectroelectrochemical results obtained here suggest rapid dye regeneration by electron-donating PNO (eqn (1)), which forms the basis for light-driven charge separation at the hybrid interface.
| TiO2 (e−)|TPA+˙|PNO → TiO2 (e−)|TPA|PNO+˙ | (1) |
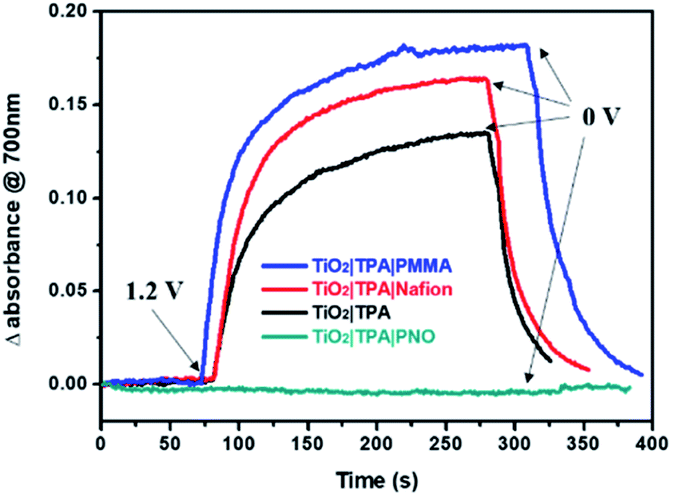 | ||
| Fig. 4 Absorption changes at 700 nm associated with time-dependent oxidation of TiO2|TPA|PMMA (blue), TiO2|TPA|Nafion (red), TiO2|TPA (black) and TiO2|TPA|PNO (green) in pH 7, 0.1 M phosphate buffer containing 0.4 M NaClO4 following a potential step from 1.2 to 0 V vs. NHE. | ||
The photoelectrochemical performance of TiO2|TPA|PNO and its analogues TiO2|TPA, TiO2|TPA|PMMA and TiO2|TPA|Nafion was evaluated in a pH 7 phosphate buffer solution with 0.1 M Na2SO3 as a hole scavenger. The PEC experiments were conducted in a standard three electrode cell under 1 sun illumination (100 mW cm−2). Meanwhile, a 400 nm cutoff filter was used to avoid direct bandgap excitation of the underlying oxide. In the cell, a platinum mesh electrode was used as the counter electrode with an Ag/AgCl reference electrode. In Fig. 5a, the current densities of the four samples under dark/light cycling conditions follow the trend of TiO2|TPA|PNO > TiO2|TPA > TiO2|TPA|PMMA ≈ TiO2|TPA|Nafion. Impressively, a photocurrent of 3 mA cm−2 was achieved by TiO2|TPA|PNO at 0.4 V vs. NHE, which is 2–3 times that for PMMA and Nafion coated films. Since the oxidation of SO32− is kinetically more facile than the oxidation of water, the density of photocurrent reflects the hole transport efficiency of a photoanode. Combined with the electrochemical results, this observation indicates that PNO possesses remarkable hole extraction ability, which is lacking for the polymers (PMMA and Nafion). Another notable feature of comparison is that rapid photocurrent decay occurs for the TiO2|TPA film without a coating layer (Fig. 5b), while the other three samples exhibit stable photocurrents during the test, indicative of simultaneous improvement in hole migration and operation stability by adding PNO.
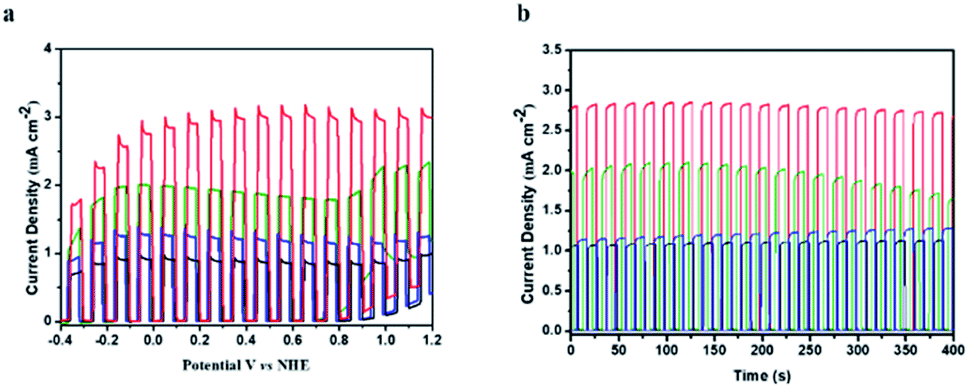 | ||
| Fig. 5 (a) Photocurrent density–voltage (j–v) traces with 5 s light transients for TiO2|TPA|PNO (red), TiO2|TPA (green), TiO2|TPA|Nafion (blue) and TiO2|TPA|PMMA (black) scanned from −0.4 to 1.2 V vs. NHE with a scan rate of 1 mV s−1 under 1 sun illumination in pH 7, 0.1 M phosphate buffer containing 0.1 M Na2SO3. (b) The time dependent photocurrent density (j−t) traces with 10 s light transients at 0.4 V vs. NHE. | ||
In order to realize water splitting, a Ru-bda based water oxidation catalyst (WOC) was chosen to improve the kinetics of oxygen evolution on a photoanode in view of its low overpotential and high activity under neutral conditions. To facilitate catalyst loading on the photoanode, a C11 alkyl chain was modified on the axial pyridyl ligand of Ru-bda (1) (Fig. 2). Catalyst 1 was drop-cast on TiO2|TPA|PNO at a surface loading of Γ = 5 × 10−8 mol cm−2, a value comparable to that of previously reported surface-bound molecular catalysts on mesoporous TiO2 films.22 The SEM image confirmed that the morphology of the PNO underlayer was fully maintained after catalyst loading (Fig. S3‡).
The photoelectrochemical performance for water oxidation was evaluated under ambient conditions in phosphate buffer (pH 7). Linear sweep voltammetry (LSV) of TiO2|TPA|PNO|1 under 1 sun illumination resulted in a prominently enhanced photocurrent with a small onset potential at −0.18 V vs. NHE (Fig. 6a). As a typical behavior for a DSPEC, a saturated current above 500 μA cm−2 is reached after passing a potential of 0.1 V vs. NHE. The effect of a PNO interlayer on photocurrent is notable. For example, TiO2|TPA|1 fabricated by co-loading of dye and catalyst 1 halves the saturated photocurrent (250 μA cm−2). In our control experiment, TiO2|TPA produced negligible photocurrent due to the lack of a catalyst.
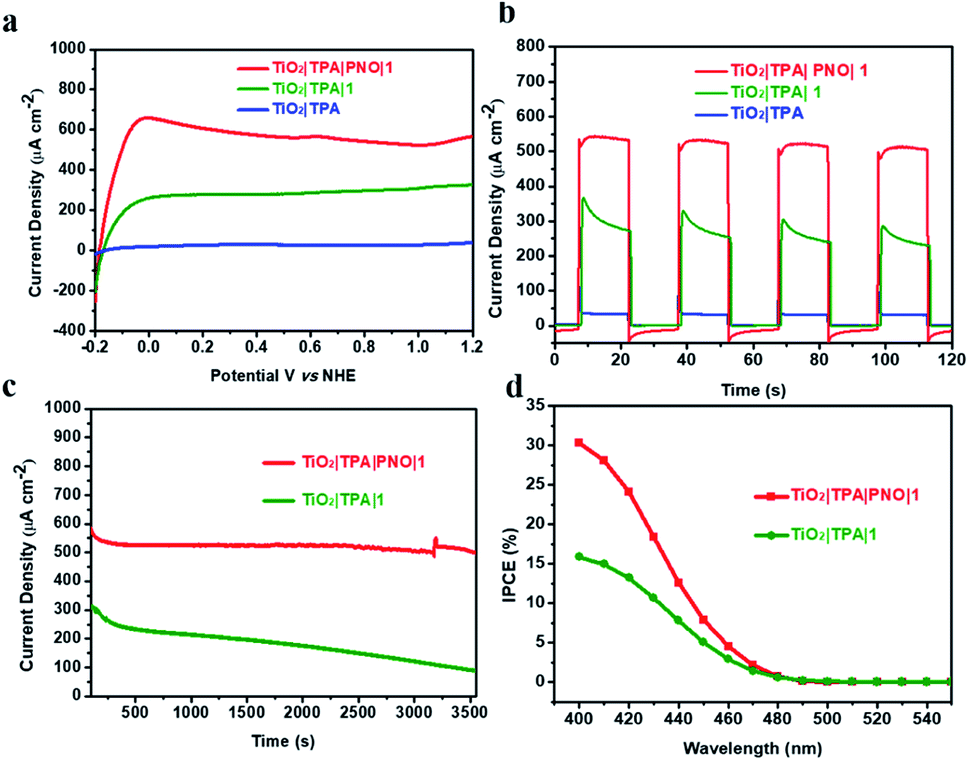 | ||
| Fig. 6 (a) Linear sweep voltammograms for TiO2|TPA|PNO|1 (red), TiO2|TPA|1 (green) and TiO2|TPA (blue) under 1 sun illumination in pH = 7, 0.1 M phosphate buffer containing 0.4 M NaClO4. (b) Photocurrent density–time traces over 15 s dark–light cycles for the three photoanodes at a constant bias of 0.4 V vs. NHE. (c) Photocurrents for TiO2|TPA|PNO|1 (red) and TiO2|TPA|1 (green) at a constant bias of 0.4 V vs. NHE under continuous 1 sun illumination. (d) Incident photon-to-current efficiencies (IPCEs) for TiO2|TPA|PNO|1 (red) and TiO2|TPA|1 (green) at 0.4 V vs. NHE. | ||
The current density–time traces of these photoanodes under chopped illumination and a constant bias of 0.4 V vs. NHE are shown in Fig. 6b. The results again confirmed that the catalyst and light are indispensable factors for water oxidation. The comparison between TiO2|TPA|PNO|1 and TiO2|TPA|1 indicates a two-fold increase in the amplitude of the photocurrent. Another point worthy of note is that the photocurrent exhibited by TiO2|TPA|PNO|1 is more stable than that of TiO2|TPA|1, consistent with the results of the sacrificial experiments. The improved durability as a consequence of insertion of an NPO interlayer between the catalyst and dye was further evidenced in an extended period of photolysis. For TiO2|TPA|PNO|1, a stable anodic photocurrent at 0.4 V vs. NHE, over 500 μA cm−2, was maintained for at least 1 h without obvious decay (<10%), which is in sharp contrast to TiO2|TPA|1, whose photocurrent density was significantly reduced by 70% during the same period due to the degradation of TPA by water molecules and hydroxide ions (Fig. 6c). To ascertain that the observed photocurrents are indeed produced by water splitting, a faradaic efficiency of 82% for O2 production was calculated for long-term photolysis. The loss in faradaic efficiency is consistent with the decomposition and desorption of the catalyst which occur in parallel with water oxidation catalysis.22 The PEC experiment of TiO2|TPA|PNO|1 was also operated in 98% H218O. Isotopically labeled 18O2 was detected by gas chromatography-mass spectrometry (GC-MS) after the PEC reaction, and this result confirms that water was the oxygen source (Fig. S4‡).
During the PEC reaction for 1 h (Fig. 6c), 4 μmol O2 were produced by TiO2|TPA|PNO|1, corresponding to a TON of approximately 80, while 1.2 μmol of O2 was produced by TiO2|TPA|1 with a TON of 13 (see the ESI‡ for experimental details). The higher TON value obtained by TiO2|TPA|PNO|1 is in line with the superior photocurrent response and stability of TiO2|TPA|PNO|1 over TiO2|TPA|1. The differences exhibited by these two photoanodes demonstrate the value of the redox mediator in a multi-step electron transfer reaction. As shown in Table 1, the remarkable PEC performance achieved by TiO2|TPA|PNO|1 is superior to those of other reported photoanodes based on organic dyes in terms of photocurrent and stability. The unique role of PNO was further highlighted by replacing the PNO interlayer with PMMA or Nafion, and the resulting samples (TiO2|TPA|PMMA|1 and TiO2|TPA|Nafion|1) afforded even lower photocurrent densities than that of an interlayer-free electrode, TiO2|TPA|1 (Fig. S5 and S6‡).
| Electrode | Photocurrent densitya (μA cm−2) | Duration | Conditions | Ref. |
|---|---|---|---|---|
| a Data were extracted from current density–time traces in the literature. b JG = Janus green B. | ||||
| SnO2|PMPDI|CoOx | From ∼40 to ∼20 | 300 s | 0.4 V vs. NHE, AM 1.5, pH 7 | 9 |
| TiO2|Al2O3|PMI|Ir–Sil | From ∼75 to ∼20 | 600 s | 0.51 V vs. NHE, 200 mW cm−2, λ > 410 nm, pH 5.8 | 10 |
| TiO2| porphyrin|Cp*–Ir | ∼60 | 60 s | 0.5 V vs. NHE, 200 mW cm−2, λ > 400 nm, pH 7 | 11 |
| TiO2|porphyrins|IrO2 | From ∼120 to ∼30 | 100 s | 0.30 V vs. NHE, AM 1.5, λ > 410 nm, pH 6.8 | 13 |
| TiO2|subporphyrin|Ru(bda) | From ∼150 to ∼70 | 30 s | 0.2 V vs. NHE, 100 mW cm−2, λ > 420 nm, pH 7.3 | 15 |
| TiO2|porphyrin|Ru(bda) | From ∼130 to ∼80 | 60 s | −0.2 V vs. NHE, 35 mW cm−2, λ > 380 nm, pH 7.3 | 16 |
| TiO2|JGb|Prussian blue | From ∼60 to ∼45 | 1 h | 0.8 V vs. NHE, 100 mW cm−2, λ > 420 nm, pH 7 | 17 |
| SnO2/TiO2|TPA|Ru(bda) | From ∼200 to ∼50 | 1 h | 0.4 V vs. NHE, 100 mW cm−2, λ > 400 nm, pH 4.8 | 18 |
| SnO2/TiO2|TPA|Ru(bda) | From ∼200 to ∼100 | 60 s | 0.45 V vs. NHE, 100 mW cm−2, λ > 400 nm, pH 7 | 21 |
| TiO2|TPA|PNO|Ru(bda) | ∼520 | 1 h | 0.4 V vs. NHE, 100 mW cm−2, λ > 400 nm, pH 7 | This work |
The wavelength dependence of the incident photon-to-current conversion efficiency (IPCE) for TiO2|TPA|PNO|1 was evaluated at 0.4 V vs. NHE (Fig. 6d). At the visible absorption maximum of TPA (Fig. S7‡), an IPCE of 30% was achieved, and the value is comparable to the highest value documented for DSPEC photoanodes.18 Based on the above measurements, a 1 sun photocurrent density of 500 μA cm−2 was estimated by integrating the IPCE curves over the AM 1.5G solar spectrum (Fig. S8‡). This result agrees well with the experimental data in Fig. 6a, indicating the reliability of our PEC tests.
The CV of TPA dye in Fig. 7a shows a ground state potential for TPA+˙/0 at 1.2 V vs. NHE. Based on eqn (2), the exited-state potential for TPA+˙/0* on TiO2 was estimated to be −1.9 V vs. NHE in neutral phosphate buffer,39–41 where kB is the Boltzmann constant, F is the Faraday constant and T is the background temperature. The values for E0 and Δṽ0,1/2 were obtained from the spectral fitting described in the ESI (Fig. S9‡).
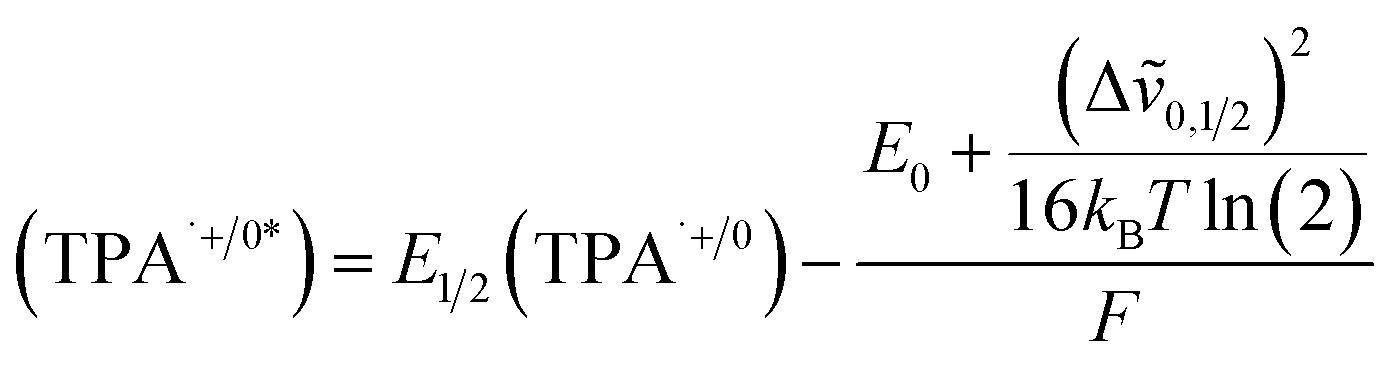 | (2) |
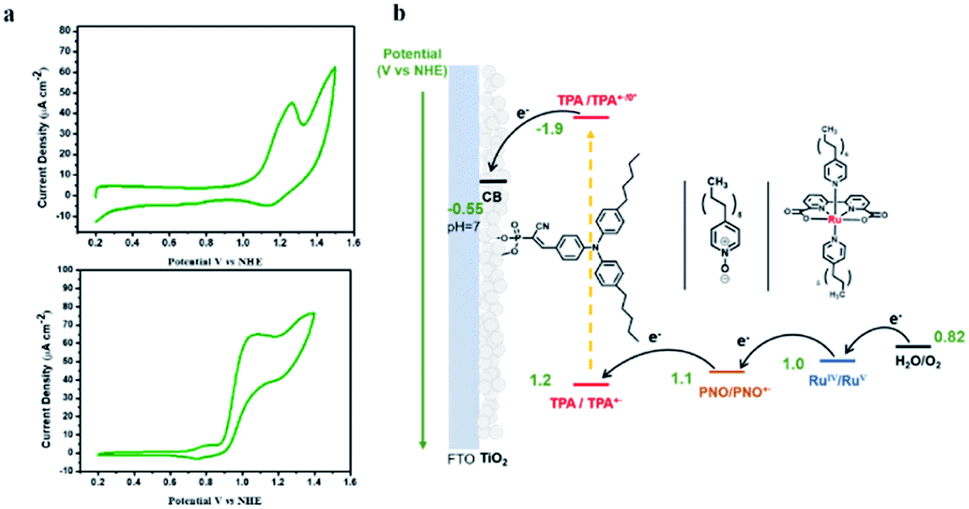 | ||
Fig. 7 (a) Cyclic voltammograms of nanoITO|TPA in pH 7, 0.1 M phosphate buffer (top) and catalyst 1 (1 mM) in mixed CH3CN/H2O (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) solution (bottom) with a scan rate of 20 mV s−1. (b) Schematic energy diagram for TiO2|TPA|PNO|1. :2) solution (bottom) with a scan rate of 20 mV s−1. (b) Schematic energy diagram for TiO2|TPA|PNO|1. | ||
The finding of a more negative potential for the excited-state of TPA dye than that of the TiO2 conduction band (−0.55 V vs. NHE at pH 7) is a direct indication of the ability of excited TPA to inject electrons into the TiO2 underlayer. Since electrochemical water oxidation by 1 on TiO2 or nanoITO is kinetically prohibited by the presence of long alkyl chains (Fig. S10‡), CV of Ru-bda was carried out in homogeneous solution and an onset potential of 0.9 V vs. NHE was determined (Fig. 7a). Bringing these data together, the desired electron flow through spatially arranged redox active components is illustrated in Fig. 7b. The reaction scheme for the activation of the surface assembly and charge separation is shown in eqn (3)–(5). In the assembly, PNO provides a suitable redox potential to establish a free energy gradient allowing electron transfer towards the conducting oxide and hole transfer away from the surface.
| TiO2|TPA*|PNO|1 → TiO2 (e−)|TPA+˙|PNO|1 | (3) |
| TiO2 (e−)|TPA+˙|PNO|1 → TiO2 (e−)|TPA|PNO+˙|1 | (4) |
| TiO2 (e−)|TPA|PNO+˙|1 → TiO2 (e−)|TPA|PNO|1+ | (5) |
| TiO2 (e−)|TPA|PNO|1+ → TiO2|TPA|PNO|1 | (6) |
In order to fully optimise the performance of hybrid photoanodes, a macrocyclic Ru-bda derivative (2)42,43 and a Ru-tda derivative (3) (H2tda = [2,2′:6′,2′′-terpyridine]-6,6′′-dicarboxylic acid)44,45 were selected for catalyst screening (Fig. 2). Both types have shown excellent activity in either electrochemical or photochemical water oxidation. Among them, hydrophobic catalyst 2 was able to be stably immobilized on TiO2|TPA by drop-casting. Akin to catalyst 1, C11 alkyl chains were functionalized on Ru-tda (3) to facilitate its loading on an electrode surface. The performance of TiO2|TPA|PNO|2 and TiO2|TPA|PNO|3 at a catalyst loading of Γ ≈ 5 × 10−8 mol cm−2 was examined under the same experimental conditions used for 1. However, both catalysts showed unsatisfactory catalytic performance (Fig. 8). Particularly, TiO2|TPA|PNO|3 produced a photocurrent lower than its analog TiO2|TPA|3, probably due to the unfavorable thermodynamics (the catalytic onset potential of 3 is more positive than the redox potential of the NPO couple, Fig. S11‡).
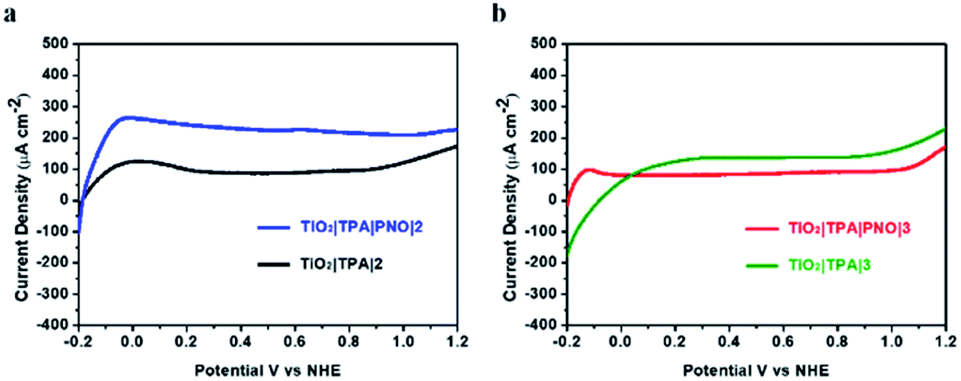 | ||
| Fig. 8 Linear sweep voltammograms for (a) TiO2|TPA|PNO|2 (blue) and TiO2|TPA|2, and (b) TiO2|TPA|PNO|3 (red) and TiO2|TPA|3 (green) under >400 nm illumination at 100 mW cm−2 in pH 7, 0.1 M phosphate buffer containing 0.4 M NaClO4. | ||
The photophysical events that occur upon light absorption are a multi-step electron transfer process, wherein the desired reactions are in kinetic competition with undesired recombination reactions (eqn (6)). To unravel the kinetics of interfacial charge separation and recombination at the photoanode, open-circuit photovoltage decay (OCVAD) was employed as a powerful tool.46 This technique has been used for dye-sensitized solar cells (DSSCs) to monitor the transient of open-circuit photovoltage (Voc) during relaxation from the illuminated quasi-equilibrium state to the dark equilibrium.47 In a DSPEC, excited-state electron injection occurs at the picosecond timescale, this process is followed by cross-surface electron transfer from a WOC to the oxidized dye, and the latter results in the recovery of most of the oxidized dyes.48,49 Under open circuit conditions, the free electron density in TiO2 is roughly determined by electron injection from the photoexcited dye and the back electron transfer of the photogenerated electrons to the oxidized catalysts. Assuming that a majority of the oxidative equivalents are located on the catalyst, the latter step closely resembles the recombination of photogenerated electrons with the oxidized form of the redox couple in DSSCs. Under illumination, more efficient charge separation enables higher free electron density in TiO2 and a larger Voc. When light is stopped, a faster Voc decay is expected due to enhanced charge recombination driven by the high density of free electrons. Based on the research of Zaban and Bisquert et al.,50,51 the lifetime of photogenerated electrons (free electron lifetime τ) in the semiconductor nanostructure and Voc comply with eqn (7), where kB is the Boltzmann constant and T is the temperature.
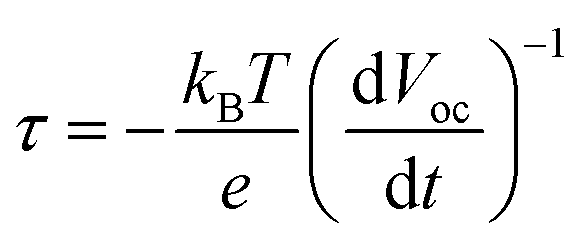 | (7) |
To this end, a free electron lifetime of 8 ms for TiO2|TPA|PNO|1 was quantified at the transient when illumination is stopped. This value was increased by an order of magnitude in the absence of a PNO layer, as a τ value of 90 ms was obtained by TiO2|TPA|1 (Fig. 9). Accordingly, more efficient charge separation under illumination was expected for TiO2|TPA|PNO|1 than TiO2|TPA|1. Longer electron lifetimes were also given by Nafion (150 ms) and PMMA (100 ms) coated photoanodes (Fig. S12‡). The variations in the lifetimes of free electrons are in line with the trend of photocurrent densities achieved by these photoanodes, revealing the crucial role of PNO in facilitating light-induced charge separation at the interface of the photoanode, analogous to tyrosine in PSII.52,53
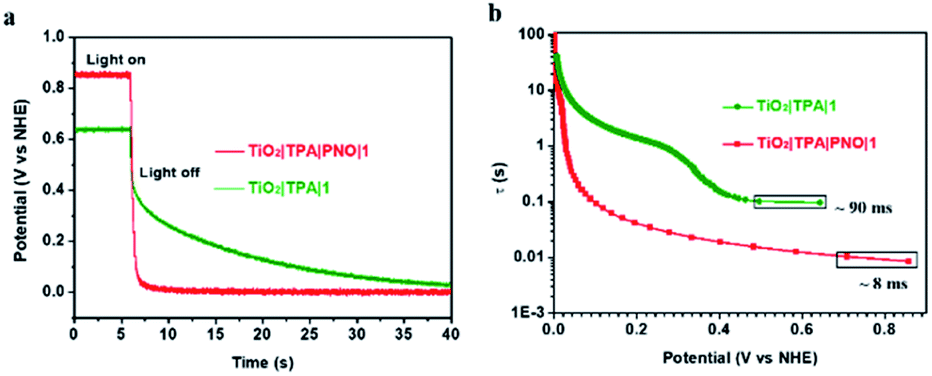 | ||
| Fig. 9 (a) Open-circuit voltage decays for the electrodes TiO2|TPA|PNO|1 (red) and TiO2|TPA|1 electrodes (green). (b) Electron lifetime derived from eqn (7) plotted as a function of Voc at the transient when illumination (>400 nm, 100 mW cm−2) was removed. The results were obtained in 0.1 M phosphate buffer at pH 7. | ||
Conclusions
In summary, we have demonstrated that cheap and easily available pyridine N-oxide can serve as an efficient electron transfer relay in a TiO2 photoanode co-loaded with organic dye and a molecular WOC. As a close mimic of the tyrosine/tyrosyl radical pair in PSII, the PNO interlayer cycles between its reduced form and a radical cation in photolysis and improves light-induced charge separation by forming a suitable electron transfer gradient between the catalyst and semiconductor oxide. The integrated photoanode exhibited a highly stable photocurrent over 500 μA cm−2 that continued for at least 1 hour without obvious decay, which opens a new avenue for rational design of low-cost organic photoanodes. In addition, the hybrid architecture described here was benefited by a facile drop-casting process. The surface assembly organized by hydrophobic interactions overcomes the limitation of a covalent bond between the linker and surface in aqueous solutions and adds an additional advantage to the stability of the photoanode. The observation of an impressive 1 sun photocurrent up to 3 mA cm−2 with a single organic dye under sacrificial conditions implies that further improvement in the efficiency and stability of organic dye-based PEC cells may be accessible with the assistance of faster water oxidation catalysts.Data availability
All relevant experimental procedures and characterization data have been reported above and in the ESI.‡Author contributions
F. L. and L. S. supervised this project; Y. Z., X. L. and F. L. designed the experiments; Y. Z., G. L., R. Z. and H. G. prepared the electrodes, and performed characterization and photoelectrochemical measurements; Y. Z. and F. L. wrote the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22088102, 21872016, and 22172018), the Fundamental Research Funds for the Central Universities of China (DUT20LAB307) and the K&A Wallenberg Foundation.References
- F. Li, H. Yang, W. Li and L. Sun, Joule, 2018, 2, 36–60 CrossRef CAS.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- B. Zhang and L. Sun, J. Am. Chem. Soc., 2019, 141, 5565–5580 CrossRef CAS PubMed.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef CAS PubMed.
- J. Li, Y. Zhu, F. Li, G. Liu, S. Xu and L. Sun, Chin. J. Catal., 2021, 42, 1352–1359 CrossRef CAS.
- J. R. Swierk, N. S. McCool and T. E. Mallouk, J. Phys. Chem. C, 2015, 119, 13858–13867 CrossRef CAS.
- T. J. Meyer, M. V. Sheridan and B. D. Sherman, Chem. Soc. Rev., 2017, 46, 6148–6169 RSC.
- J. T. Kirner and R. G. Finke, J. Mater. Chem. A, 2017, 5, 19560–19592 RSC.
- J. T. Kirner and R. G. Finke, ACS Appl. Mater. Interfaces, 2017, 9, 27625–27637 CrossRef CAS PubMed.
- R. J. Kamire, K. L. Materna, W. L. Hoffeditz, B. T. Phelan, J. M. Thomsen, O. K. Farha, J. T. Hupp, G. W. Brudvig and M. R. Wasielewski, J. Phys. Chem. C, 2017, 121, 3752–3764 CrossRef CAS.
- G. F. Moore, J. D. Blakemore, R. L. Milot, J. F. Hull, H. E. Song, L. Cai, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Energy Environ. Sci., 2011, 4, 2389–2392 RSC.
- A. Nayak, R. R. Knauf, K. Hanson, L. Alibabaei, J. J. Concepcion, D. L. Ashford, J. L. Dempsey and T. J. Meyer, Chem. Sci., 2014, 5, 3115–3119 RSC.
- J. R. Swierk, D. D. Méndez-hernández, N. S. Mccool, P. Liddell, Y. Terazono, I. Pahk, J. J. Tomlin, N. V Oster, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1681–1686 CrossRef CAS PubMed.
- P. K. Poddutoori, J. M. Thomsen, R. L. Milot, S. W. Sheehan, C. F. A. Negre, V. K. R. Garapati, C. A. Schmuttenmaer, V. S. Batista, G. W. Brudvig and A. Van Der Est, J. Mater. Chem. A, 2015, 3, 3868–3879 RSC.
- M. Yamamoto, Y. Nishizawa, P. Chábera, F. Li, T. Pascher, V. Sundström, L. Sun and H. Imahori, Chem. Commun., 2016, 52, 13702–13705 RSC.
- M. Yamamoto, L. Wang, F. Li, T. Fukushima, K. Tanaka, L. Sun and H. Imahori, Chem. Sci., 2016, 7, 1430–1439 RSC.
- T. G. Ulusoy Ghobadi, A. Ghobadi, M. Buyuktemiz, E. A. Yildiz, D. Berna Yildiz, H. G. Yaglioglu, Y. Dede, E. Ozbay and F. Karadas, Angew. Chem., Int. Ed., 2020, 59, 4082–4090 CrossRef CAS PubMed.
- M. S. Eberhart, D. Wang, R. N. Sampaio, S. L. Marquard, B. Shan, M. K. Brennaman, G. J. Meyer, C. Dares and T. J. Meyer, J. Am. Chem. Soc., 2017, 139, 16248–16255 CrossRef CAS PubMed.
- F. Li, K. Fan, B. Xu, E. Gabrielsson, Q. Daniel, L. Li and L. Sun, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef CAS PubMed.
- Y. K. Eom, L. Nhon, G. Leem, B. D. Sherman, D. Wang, L. Troian-Gautier, S. Kim, J. Kim, T. J. Meyer, J. R. Reynolds and K. S. Schanze, ACS Energy Lett., 2018, 3, 2114–2119 CrossRef CAS.
- K. R. Wee, B. D. Sherman, M. K. Brennaman, M. V. Sheridan, A. Nayak, L. Alibabaei and T. J. Meyer, J. Mater. Chem. A, 2016, 4, 2969–2975 RSC.
- Y. Zhu, D. Wang, Q. Huang, J. Du, L. Sun, F. Li and T. J. Meyer, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- K. Hanson, M. D. Losego, B. Kalanyan, D. L. Ashford, G. N. Parsons and T. J. Meyer, Chem. Mater., 2013, 25, 3–5 CrossRef CAS.
- L. Li, L. Duan, Y. Xu, M. Gorlov, A. Hagfeldt and L. Sun, Chem. Commun., 2010, 46, 7307–7309 RSC.
- L. Zhang, Y. Gao and X. Ding, Electrochim. Acta, 2016, 207, 130–134 CrossRef CAS.
- K. R. Wee, M. K. Brennaman, L. Alibabaei, B. H. Farnum, B. Sherman, A. M. Lapides and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 13514–13517 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- J. Z. Zhang and E. Reisner, Nat. Rev. Chem., 2020, 4, 6–21 CrossRef CAS.
- D. Wang, R. N. Sampaio, L. Troian-Gautier, S. L. Marquard, B. H. Farnum, B. D. Sherman, M. V. Sheridan, C. J. Dares, G. J. Meyer and T. J. Meyer, J. Am. Chem. Soc., 2019, 141, 7926–7933 CrossRef CAS PubMed.
- T. Irebo, M. T. Zhang, T. F. Markle, A. M. Scott and L. Hammarström, J. Am. Chem. Soc., 2012, 134, 16247–16254 CrossRef CAS PubMed.
- J. M. Keough, A. N. Zuniga, D. L. Jenson and B. A. Barry, J. Phys. Chem. B, 2013, 117, 1296–1307 CrossRef CAS PubMed.
- Y. Zhao, J. R. Swierk, J. D. Megiatto, B. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612–15616 CrossRef CAS PubMed.
- D. Wang, F. Niu, M. J. Mortelliti, M. V. Sheridan, B. D. Sherman, Y. Zhu, J. R. McBride, J. L. Dempsey, S. Shen, C. J. Dares, F. Li and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12564–12571 CrossRef CAS PubMed.
- L. Zhang, X. Yang, W. Wang, G. G. Gurzadyan, J. Li, X. Li, J. An, Z. Yu, H. Wang, B. Cai, A. Hagfeldt and L. Sun, ACS Energy Lett., 2019, 4, 943–951 CrossRef CAS.
- Y. Wang, F. Li, X. Zhou, F. Yu, J. Du, L. Bai and L. Sun, Angew. Chem., Int. Ed., 2017, 56, 6911–6915 CrossRef CAS PubMed.
- S. I. Kulakovskaya, S. N. Shamaev and V. M. Berdnikov, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1990, 39, 1345–1348 CrossRef.
- H. Li, F. Xie and M. T. Zhang, ACS Catal., 2021, 11, 68–73 CrossRef CAS.
- D. Wang, Z. Xu, M. Sheridan, J. Concepcion, F. Li, T. Lian and T. J. Meyer, Chem. Sci., 2021, 12, 14441–14450 RSC.
- J. P. Claude and T. J. Meyer, J. Phys. Chem., 1995, 99, 51–54 CrossRef CAS.
- A. Ito and T. J. Meyer, Phys. Chem. Chem. Phys., 2012, 14, 13731–13745 RSC.
- E. M. Kober, J. V. Caspar, R. S. Lumpkin and T. J. Meyer, J. Phys. Chem., 1986, 90, 3722–3734 CrossRef CAS.
- M. Schulze, V. Kunz, P. D. Frischmann and F. Würthner, Nat. Chem., 2016, 8, 576–583 CrossRef CAS PubMed.
- V. Kunz, J. O. Lindner, M. Schulze, M. I. S. Röhr, D. Schmidt, R. Mitrić and F. Würthner, Energy Environ. Sci., 2017, 10, 2137–2153 RSC.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786–10795 CrossRef CAS PubMed.
- R. Matheu, M. Z. Ertem, C. Gimbert-Suriñach, J. Benet-Buchholz, X. Sala and A. Llobet, ACS Catal., 2017, 7, 6525–6532 CrossRef CAS.
- M. Zhong, T. Hisatomi, Y. Kuang, J. Zhao, M. Liu, A. Iwase, Q. Jia, H. Nishiyama, T. Minegishi, M. Nakabayashi, N. Shibata, R. Niishiro, C. Katayama, H. Shibano, M. Katayama, A. Kudo, T. Yamada and K. Domen, J. Am. Chem. Soc., 2015, 137, 5053–5060 CrossRef CAS PubMed.
- Y. K. Eom, I. T. Choi, S. H. Kang, J. Lee, J. Kim, M. J. Ju and H. K. Kim, Adv. Energy Mater., 2015, 5, 1–9 Search PubMed.
- J. R. Swierk, N. S. McCool, T. P. Saunders, G. D. Barber, M. E. Strayer, N. M. Vargas-Barbosa and T. E. Mallouk, J. Phys. Chem. C, 2014, 118, 17046–17053 CrossRef CAS.
- P. Xu, C. L. Gray, L. Xiao and T. E. Mallouk, J. Am. Chem. Soc., 2018, 140, 11647–11654 CrossRef CAS PubMed.
- J. Bisquert, A. Zaban, M. Greenshtein and I. Mora-Seró, J. Am. Chem. Soc., 2004, 126, 13550–13559 CrossRef CAS PubMed.
- A. Zaban, M. Greenshtein and J. Bisquert, ChemPhysChem, 2003, 4, 859–864 CrossRef CAS PubMed.
- H. Iwami, M. Okamura, M. Kondo and S. Masaoka, Angew. Chem., 2021, 133, 6030–6034 CrossRef.
- H. Iwami, M. Kondo and S. Masaoka, ChemElectroChem, 2022, 9, 52–58 CrossRef.
Footnotes |
| † This paper is dedicated to Prof. Licheng Sun on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sc00443g |
| This journal is © The Royal Society of Chemistry 2022 |