
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombating small molecule environmental contaminants: detection and sequestration using functional nucleic acids
Aimee A.
Sanford
 a,
Brea A.
Manuel
a,
Misael A.
Romero-Reyes
*ac and
Jennifer M.
Heemstra
*ab
a,
Brea A.
Manuel
a,
Misael A.
Romero-Reyes
*ac and
Jennifer M.
Heemstra
*ab
aDepartment of Chemistry, Emory University, Atlanta, Georgia 30322, USA. E-mail: jen.heemstra@emory.edu
bDepartment of Biomedical Engineering, Georgia Institute of Technology, Emory University, Atlanta, GA 30332, USA
cDepartment of Chemistry, Hanover College, Hanover, Indiana 47243, USA. E-mail: romeroreyes@hanover.edu
First published on 6th June 2022
Abstract
Small molecule contaminants pose a significant threat to the environment and human health. While regulations are in place for allowed limits in many countries, detection and remediation of contaminants in more resource-limited settings and everyday environmental sources remains a challenge. Functional nucleic acids, including aptamers and DNA enzymes, have emerged as powerful options for addressing this challenge due to their ability to non-covalently interact with small molecule targets. The goal of this perspective is to outline recent efforts toward the selection of aptamers for small molecules and describe their subsequent implementation for environmental applications. Finally, we provide an outlook that addresses barriers that hinder these technologies from being widely adopted in field friendly settings and propose a path forward toward addressing these challenges.
1. Introduction
Environmental contaminants are potentially hazardous chemicals, microorganisms, or other materials that negatively impact the ecosystem or human health.1,2 Human exposure most often occurs through environmental media such as food, water, surrounding air, or consumer products.2,3 While robust processes exist for detecting and removing large contaminants such as bacteria and fungi,4–9 small molecule contaminants (<1 kDa) are harder to mitigate due to their size, diversity, and limited epitopes. Methods that address these challenges are needed because small molecule contaminants are abundant in environmental media. For example, a 2019 study estimated that 80% of grains are contaminated with naturally occurring mycotoxins.10 This percentage is much higher than previously reported due to improvements in detection limits,10 and this example highlights the need for more accurate monitoring of small molecule contaminants. Further, the number of novel contaminants is expected to drastically increase because of large-scale industry practices that are implemented to meet modern day cultural demands.1 A prominent example is organophosphate pesticides, which revolutionized the agricultural industry, but have led to cases of acute human poisoning and long-term health effects due to their persistence in soil, water, air, and food.11–14 Similarly, factory processes represent the major source of water and soil contamination due to toxic waste dumping practices.15 One example is bisphenol A, which is used in the manufacturing of plastics, and is a commonly reported water contaminant.16,17 Further, increased levels of metals such as lead, mercury, and cadmium from these practices have been shown to persist and bioaccumulate, leading to cases of metal poisoning worldwide.18–20Environmental contaminants cause many human diseases, highlighting the need for rigorous characterization to reduce potential health risks. Global agencies have set guidelines that include parameters such as tolerable daily intake levels to benchmark the maximum amount of an environmental contaminant that is considered safe.21 However, environmental contaminants present in media below established limits can still trigger low dose effects. Recent efforts aimed at designing adaptable methods for direct detection of contaminants in environmental media present a promising new avenue for risk assessment.22,23 Biosensors offer an alternative to traditional detection methods and have gained traction in for a wide range of small molecule detection applications, especially when they obviate the need for expensive equipment such as high-performance liquid chromatography.24,25 To meet field deployable criteria, biosensors must be cost-effective, portable, reproducible, and easy to use.26,27 Additionally, simple field-deployable devices must still have the necessary sensitivity to detect low concentrations of small molecules. Perhaps the most widely known type of biosensor employed for small molecule contaminant monitoring is enzyme-linked immunosorbent assay (ELISA), which utilizes antibodies that bind to the small molecule target and are fused to a reporter enzyme to provide a readable output.28,29 While widely used, ELISA has several disadvantages due to the high batch-to-batch variation in antibody production and cold storage conditions required for stability. Addressing these challenges holds significant promise to advance field friendly molecular recognition-based biosensors.
Beyond biomonitoring, methods are needed to sequester and eliminate environmental contaminants from water sources in order to minimize human exposure. This is especially important given that small molecule environmental contaminants can bioaccumulate in environmental media.16 For instance, many water soluble contaminants end up lakes and streams, where they can further accumulate in fish and other wildlife.30–32 Common decontamination techniques include centrifugation, coagulation, chlorination, photochemical inactivation and the use of membrane systems with varying pore sizes.33 However, these physical or chemical treatments require high amounts of energy, machinery, and complex processes,34 making them poorly suited for the removal of small molecule contaminants in resource limited settings.35 Thus, the development of new field friendly approaches to detection and sequestration of small molecule contaminants have potential to significantly address human and environmental health.
In this perspective, we highlight recent advances in nucleic acids chemistry that could address the aforementioned challenges by enabling new technologies for detection and sequestration of small molecule contaminants (Table 1). Specifically, we will focus on functional DNAs that exhibit activities beyond the canonical role of DNA in storing genetic information, such as recognizing small molecules through non-covalent interactions.36–38 Nucleic acids are inherently field friendly because they are cost-effective to produce, stable to a wide range of conditions, and can be easily functionalized for use in sensors and other platforms. Encouragingly, functional DNAs such as aptamers have already been reported for a variety of natural and synthetic environmental contaminants.24,25 Below, we describe recent efforts to develop and deploy functional DNAs for the detection and sequestration of small molecule contaminants. We highlight key challenges that are encountered and advances in nucleic acid technology that could address these gaps and enable increasingly rapid response to newly emerging environmental threats.
| Nucleic acid | Target | Transduction signal | Reference |
|---|---|---|---|
| Aptamer beacon | Hg2+, Ag+, melamine, and cocaine | Fluorescence | 56–59 |
| Ochratoxin A, aflatoxins, microcystin-LR, BPA, heavy metals | Electrochemical | 118 and 119 | |
| Split aptamer | Cocaine, kanamycin A | Fluorescence | 123 and 124 |
| 17-β-Estradiol, enrofloxacin | Colorimetric | 66 | |
| Structure-switching aptamer | Ochratoxin A, microcystin-LR | Fluorescent | 68, 69, 107, 129 and 130 |
| DNAzyme | Pb2+ | Electrochemical | 134–137, 140 and 141 |
| Pb2+, Cu+, Hg2+ | Colorimetric | 136 and 142–144 | |
| Hg2+, Pb2+ | Fluorescent | 149 |
2. Selection
Detection of small molecule toxins is challenging but critical due to their persistent and evolving effects on the ecosystem. Advantages of functional DNAs such as aptamers and DNAzymes for detection applications include high thermal stability, low cost compared to antibodies, and minimal batch to batch variability. Moreover, functional nucleic acids can be generated using non-natural backbones, which offer very high biostability.39,40 A key component of biosensor function is the affinity and selectivity of the aptamer or DNAzyme. Therefore, having robust methods to generate these functional DNAs is the first, crucial step in biosensor development. Compared to some affinity reagents that must be generated in cells lines or in vivo, functional nucleic acids can be evolved in vitro using Systematic Evolution of Ligands by EXponential Enrichment (SELEX), which involves selection from a diverse pool of nucleic acid sequences to identify candidates that perform a desired function such as binding, conformation change, or catalysis.41,42 The general steps of SELEX (Fig. 1) include the incubation of the nucleic acid library with the target molecule, isolation of sequences having the desired property, and amplification of those sequences to continue to the next round. Once the desired level of enrichment is reached, individual sequences are chosen and characterized based on sequencing data. Isolating active sequences is a critical step that varies based on the target and/or the type of functional nucleic acid desired.41,42 Herein, we will discuss recent efforts in the development and implementation of in vitro selection for functional nucleic acids including aptamers, structure-switching aptamers, and DNAzymes that target small molecule environmental contaminants.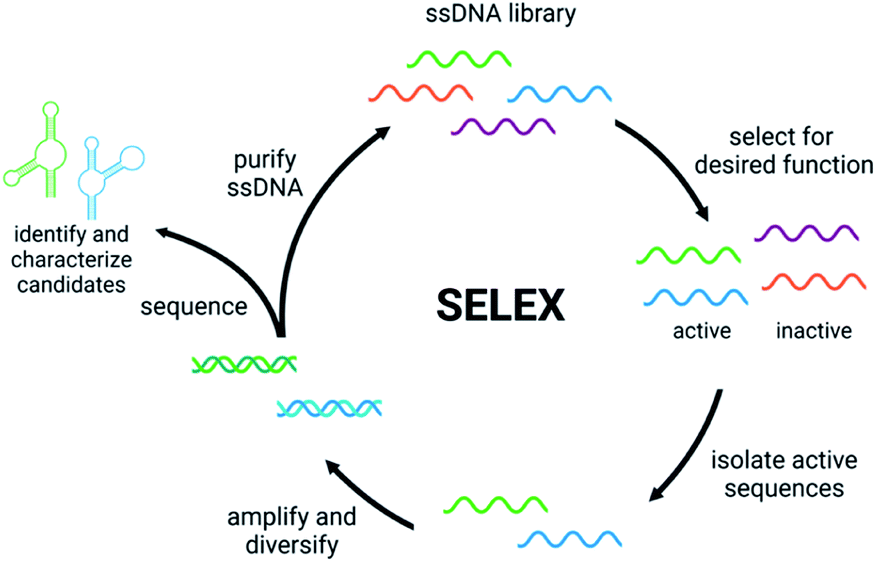 | ||
| Fig. 1 The Systematic Evolution of Ligands by EXponential Enrichment (SELEX). | ||
2.1 Aptamers
DNA aptamers are single stranded oligonucleotides that bind to their cognate target, often with high affinity and specificity.41,42 This activity is selected for in vitro through the isolation of sequences having affinity for a desired target, while removing inactive and non-specifically bound sequences. Aptamer performance vary dramatically depending on the design of this step.43,44 A common strategy is a “panning” approach in which the small molecule is immobilized on a solid support such as magnetic beads or resin to aid in the separation of binding and non-binding sequences. This relies on the ability to conjugate the small molecule to the solid support without perturbing overall structure. This approach has led to successful evolution aptamers to a wide range of small molecules, including common water contaminants such as cyanotoxins, mycotoxins, and pesticides.45–48 However, if a target does not have convenient functional handles for conjugation, this can make the evolution process significantly more time-consuming, and there is a constant risk of generating aptamers that bind to the immobilized target but not to the desired target molecule in solution. In fact, several reported aptamers bind better to immobilized target than free target in solution.49 One way to diminish this problem is to incorporate target-based elution in later rounds of SELEX.44,50Homogeneous isolation methods circumvent this issue altogether because there is no requirement for target modification. Nanomaterials can aid in the partitioning of bound sequences through non-covalent binding interactions. A prominent example is the exploitation of changes in salt-induced aggregation of gold nanoparticles upon small molecule target binding.51,52 The resulting aggregation dependent color change allows for facile tracking of enrichment throughout SELEX rounds. Graphene oxide (GO)-SELEX operates in an analogous manner, where ssDNA participates in π-stacking with graphene oxide when not bound to the target small molecule, enabling separation of functional sequences.53 A major benefit of both designs is the ability to select aptamers having low dissociation constants because the affinity to the target small molecule must be greater than to the partitioning nanomaterial in order for a sequence to advance through the selection. As a result, both nanomaterials have been widely adapted for in vitro selection and downstream detection platforms for small molecule contaminants. However, one limitation is that this does not account for non-specific elution from nanomaterials, which cannot be distinguished from sequences eluting due to target binding.
An alternative to target immobilization involves attaching the library to solid supports, such as seen in Capture-SELEX or Magnetic Cross-Linking Precipitation (MCP)-SELEX.54 These efforts take advantage of complementary strand hybridization to “capture” library members on a complementary strand immobilized on a support, and sequences that bind to the target are eluted off. However, this approach suffers from the same non-specific carryover highlighted in gold-nanoparticle and GO-SELEX. While additional negative selection rounds could mitigate this issue, a more direct approach would be to implement a homogeneous isolation step that is directly related to target binding.
One such homogeneous approach is capillary electrophoresis, where target binding causes a shift in sequence mobility. However, small molecule binding is harder to distinguish via CE because the minimal size change leads to a minimal shift in mobility. This results in poor separation of unbound and bound sequences during the isolation step. Nevertheless, multiple rounds of selection can be used to overcome the loss of active sequences.55
As highlighted above, designing the isolation step in small molecule SELEX is challenging. While many different methods for small molecule aptamer evolution have been reported, they each suffer from at least one major limitation. Moreover, while these approaches can generate sequences having affinity for the target with relatively high reliability, struggles can be encountered when adapting these sequences to function in biosensors.
2.2 Structure-switching aptamers
Functional DNAs that can directly report on small molecule concentration are highly desired for biosensing efforts. Structure-switching aptamers have proven to be particularly useful for these applications, as they are functional nucleic acids that undergo a significant conformational change due to target binding. Structure-switching aptamers can be generated using multiple different motifs, including aptamer beacons, split aptamers, and structure-switching aptamer biosensors (Fig. 2).56 However, in vitro evolution of this class of functional DNAs is particularly challenging because most SELEX methods cannot preferentially isolate sequences having structure-switching activity. While post-SELEX engineering of structure-switching properties is possible, the success rate is often low because not all aptamers can exhibit a significant target induced conformational change upon binding. This likely explains why the majority of aptamer-based sensors reported in literature rely on a small handful of structure switching aptamers, when a much larger number of target-binding aptamers are available. Ellington and coworkers candidly highlighted this challenge for aptamers in general with a section in a review article titled “There Are More Analytes in the World than ATP, Thrombin, Platelet-Derived Growth Factor, and Immunoglobin E.”57 Herein, we detail recent progress and the improvements to the selection process that are needed to address this gap.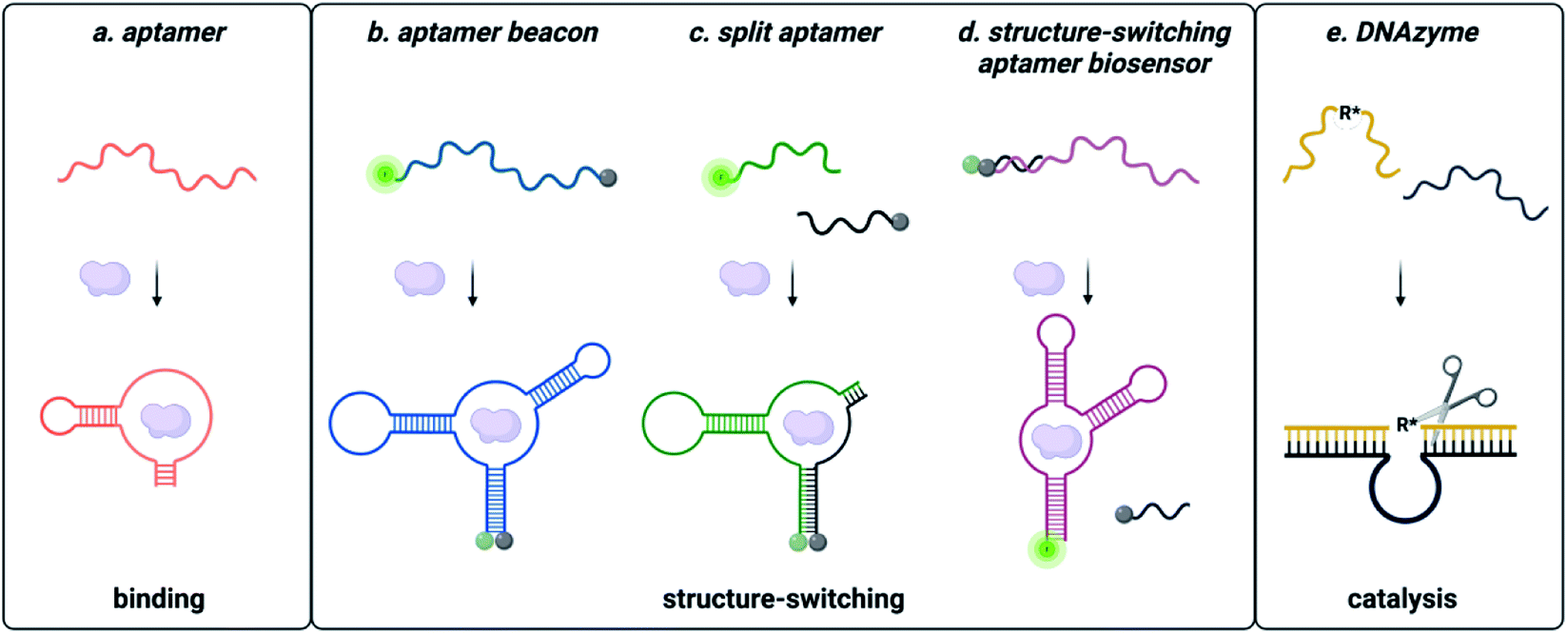 | ||
| Fig. 2 Types of functional nucleic acids. (a) Aptamers bind the small molecule. (b) Molecular beacon structure-switching biosensor. (c) Split aptamer structure-switching biosensor. (d) Structure-switching biosensor. Reprinted (adapted) with permission from B. A. Manuel, S. A. Sterling, A. A. Sanford and J. M. Heemstra, Anal. Chem., 2022, 94(17) 6436–6440, https://doi.org/10.1021/acs.analchem.2c00422. Copyright 2022 American Chemical Society. (e) DNA catalysed reaction. | ||
Aptamer beacons consist of a stem-loop structure with complementary terminal ends.58 In the absence of the target small molecule, the stem is either hybridized with terminal ends in proximity or dehybridized with terminal end separation. Target binding causes a conformational switch where the stem is either opened or closed, respectively. In certain cases, aptamer beacon activity can be rationally engineered based on known target-nucleobase interactions. This approach was successfully applied for heavy metal contaminants such as mercury and silver, based on their characteristic binding between thymine base pairs (T–Hg2+–T) and cytosine base pairs (C–Ag+–C), respectively.59–61
However, most small molecule aptamer beacons rely on manipulation of known aptamer sequences.16,62 One report has outlined in vitro selection of aptamer beacons, wherein a fluorophore labelled ssDNA library is hybridized to a quencher labelled complementary “capture” oligonucleotide that is immobilized on a solid support via a biotin–streptavidin interaction.63 Upon introduction of the target, the capture sequence is dehybridized from the pool, resulting in a fluorescence increase. While this method was validated with an oligonucleotide target that is able to directly interact with the library via Watson–Crick–Franklin binding, it could in principle be extended to small molecule target molecules.63 However, it would still have many of the same limitations as the previously described capture SELEX method.64–66
Split aptamers are functional nucleic acids that, like aptamer beacons, are typically generated by engineering of existing aptamers and are highly dependent on secondary structure. The engineering process involves generating two fragments that do not bind with each other in the absence of the target, but where molecular recognition of the cognate target triggers assembly of the fragments to recapitulate a structure similar to the native aptamer. Generally, aptamers can be split if the structure contains a three-way junction, as this is a privileged architecture.67 Fortuitously, several small molecule aptamers possess structures that were amenable to this process. The most widely cited example is the cocaine split aptamer, which contains a three-way junction structure that was easily split while retaining binding.68 However, many parent aptamers do not inherently have structures that can be easily split and are longer than desired (∼70–90 nt). Moreover, the process of truncations to facilitate splitting can perturb structure as seen for the isocarbophos and 17-β-estradiol aptamers.68,69 To circumvent these challenges, a straightforward method for isolating candidates having three-way junction architectures is of significant utility, and future research in the field would benefit from the development of a method to directly select for sequencing having the split aptamer function of target-dependent assembly.
In contrast to split aptamers, structure-switching aptamer biosensors consist of an aptamer hybridized to a short complementary strand, and target binding causes disassembly of this duplex.70 While this architecture has arguably found the most utility for small molecule biosensing applications, relatively few structure-switching aptamer biosensors have been reported in the literature. Similar to aptamer beacons and split aptamers, most structure-switching biosensors result from post-selection engineering of aptamers that were selected only for target binding. One example is the structure-switching aptamer that recognizes ochratoxin A (OTA). Chen and coworkers constructed a structure-switching aptamer sensor by optimizing DNA concentration, capture strand length, and aptamer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :capture strand ratio.71 Using the optimized conditions, this platform was successfully used to detect OTA in corn samples.71 Due to its success, the OTA structure-switching aptamer is widely used as a model system for optimization of other biosensing platforms.72
:capture strand ratio.71 Using the optimized conditions, this platform was successfully used to detect OTA in corn samples.71 Due to its success, the OTA structure-switching aptamer is widely used as a model system for optimization of other biosensing platforms.72
Given the limitations of post-selection engineering, a preferable approach would be the direct selection of structure-switching architectures. A well cited approach is the capture SELEX workflow outlined above and a few different iterations of this approach exist.64–66,73,74 However, in our hands, non-specific dehybridization has remained a challenge to reliable implementation of these methods and may explain the limited number of structure-switching aptamer biosensors reported using this method. Seeking to overcome these limitations, our lab developed an optimized structure-switching aptamer biosensor evolution method that incorporates a homogeneous isolation step.75 The isolation step takes advantage of the selectivity of restriction enzymes for cleavage at their cognate palindromic double stranded recognition site. This site was incorporated into the capture strand and primer binding site of the library such that sequences for which the capture strand was displaced upon introduction of the target were not digested by the restriction enzyme, but sequences that were inactive and remained duplexed were digested. In the subsequent PCR step, only the undigested sequences were able to undergo amplification. This was demonstrated using kanamycin A, which has become an environmental contaminant due to its overuse in agricultural practices. However, this method is anticipated to be generalizable and thus applicable to multiple small molecule contaminants.
2.3 Modified and xenonucleic acids
Nucleic acids can be modified at the nucleobase, backbone, or both to confer novel activity, resistance to degradation, and increased affinity. We previously reviewed two major types of nucleotide modifications that are incorporated during SELEX.76,77 Herein, we detail a few recent efforts in this area that have significantly impacted small molecule recognition when used either during or after SELEX.The use of modified nucleobases in SELEX has become a widespread approach to incorporate more diverse functional groups into nucleic acids for molecular recognition. Early efforts focused on the use of nucleotide monomers having functional groups appended at sites on the nucleobase that would not significantly impact their ability to be synthesized by polymerases. However, the synthesis of these monomers can be time-consuming and their incorporation via polymerases challenging. A more recent innovative approach utilizes click chemistry to circumvent these issues. Alkynes are incorporated into the DNA library using ethynyl-dU and then copper-catalyzed azide–alkyne cycloaddition used to append diverse functional groups prior to the isolation step. One recent iteration combined this technique with fluorescence activated cell sorting (FACS) to generate a boronic acid modified aptamer having 1 μM affinity for epinephrine. In a separate approach, our lab investigated the use of inosine modification to modulate the binding properties of aptamers post-selection. Through systematic replacement of guanine with insoine at strategic locations in the sequence, we were able to generate cocaine-binding aptamer sequences having a range of affinities, with the best sequence having ∼350-fold improved affinity compared to the parent aptamer.56
Xenonucleic acids (XNAs) are generally considered to be any nucleic acid that is backbone modified, and these benefit from higher biostability, as enzymes generally do not recognize and cleave these molecules as readily as native nucleic acids. As with nucleobase modifications, early efforts used post-SELEX engineering to incorporate XNA monomers into the sequence of aptamers. However, XNA conversion is challenging because any modifications that are incorporated post-selection can significantly impact secondary structure, which subsequently impacts affinity and/or specificity. Alternatively, the generation of novel XNAs using SELEX was envisioned, but required evolution of polymerases capable of transcribing DNA into XNA and reverse transcribing XNA back into DNA. Through significant advances in polymerase engineering, XNA aptamers were generated using a wide range of backbones, but these focused on protein or cellular targets. Generating XNA aptamers for small molecule targets required additional optimization of the SELEX process, and in 2018 our lab reported the first small molecule binding XNA aptamer with a threose nucleic acid (TNA) sequence capable of binding to OTA. These aptamers had similar or better affinity compared to the native DNA aptamers for the same target, but were able to maintain binding in human blood serum over a period of seven days. Since then, scarce additional efforts have been made to generate small molecule binding XNA sequences and progress in this field will require continual improvement of selection processes to accommodate the challenges of working with non-native backbones.
2.4 DNAzymes
DNA enzymes, or DNAzymes are a subset of functional DNAs that act as biocatalysts.78–80 These catalytic DNAs offer similar advantages over protein-based enzymes as described for aptamers relative to protein-based affinity reagents. Through SELEX, DNAzymes have been generated for a variety of reactions, including RNA cleavage,81,82 RNA ligation,83–85 and even carbon–carbon bond forming reactions.86,87 DNAzyme SELEX is particularly challenging because the functional output is unique to every selection. Furthermore, the substrate molecule generally must be immobilized to the DNA library to maintain the genotype–phenotype link during isolation and washing steps.87 However, this can also offer an advantage as target specific affinity handles can then be used to isolate active sequences. For example, a zinc dependent DNAzyme capable of RNA cleavage activity was isolated by appending the target RNA strand with a biotin handle, which allowed for easy isolation using streptavidin-coated magnetic beads.88While DNAzymes have found use in small molecule environmental contaminant detection, it is very rare that they are acting strictly as the biorecognition element. DNAzyme based biosensors instead are selected to rely on a specific heavy metal for an activity such as nucleic acid cleavage, and this metal-dependent activity is then coupled to a fluorescence readout or other amplification and sensing motif.89–93 This format can make DNAzyme biosensors field deployable and cost effective, and in the next section we describe the use of reporter enzymes such as horseradish peroxidase (HRP) to amplify signal. A major limitation, however, is that the catalytic efficiency of DNAzymes remains poor compared to native enzymes. We suggest that this can be addressed by developing homogenous selection methods for DNAzymes, in which the substrate does not need to be tethered to the DNA library during the selection process. This would provide greater control over the stringency of the selection process and enable the direct selection of DNAzymes that function in trans with free substrate molecules. Selection in trans could also lead to improvements in selectivity, as sequences would be sorted based on their ability to produce a specific reaction product rather than a general DNA cleavage or ligation event. A key hurdle to such selection methods is the ability to detect the desired products of the DNAzyme reaction, but we propose that other forms of DNA sensors such as those described below could be leveraged for such applications.
3. Detection
Quantification of small molecule contaminants in environmental samples is crucial for determining potential exposure levels. Biosensors that leverage the molecular recognition capabilities of biomolecules have shown great promise over traditional methods such as HPLC and mass spectrometry due to their high versatility and field deployability.94 There are several types of biosensors in which target binding is transduced into a detectable output. The most promising platforms and implementation strategies using aptamers and DNAzymes for small molecules are summarized below.3.1 Aptamers
Aptasensors are biosensors that utilize the molecular recognition of properties of aptamers. These sensors benefit from the same general advantages of aptamers over antibodies, and these are especially important for small molecule targets. While molecules <1 kDa are challenging targets for antibodies, there are several reports of high affinity aptamers to molecules of this size.95 An added advantage of aptamers over other affinity reagents is that aptamers can be evolved for highly toxic targets that are not compatible with in vivo selection methods, making them well suited for environmental monitoring.50,95A significant challenge faced in aptasensor development is transforming the aptamer-small molecule biorecognition event into a readable output. Many efforts use optical aptasensors because they offer a straightforward readout that is either colorimetric or fluorescent. Small molecule binding is transmitted into an optical output from a chromophore or fluorophore. While promising, there are several hurdles that optical aptasensors must overcome to meet the reproducible, sensitive, specific, and cost-effective criteria. We will emphasize the recent approaches that attempt to address these challenges in colorimetric and fluorescent aptamer-based biosensors for small molecule environmental contaminants.
3.2 DNAzymes
Compared to aptamers, DNAzymes have a significant advantage in biosensing due to their inherent catalytic activity, which can drive signal amplification (Fig. 4).96 As mentioned previously, DNAzymes are typically cation-dependent and can be selected for activity in the presence of specific heavy metals.88–93,97,98 This has, in turn, enabled detection of heavy metals in complex environments including food, water, and soil.99–102 DNAzymes can also be integrated into aptamer architectures to enable small molecule dependent activity, and this strategy can be especially useful for aptasensors that do not meet sensitivity requirements for challenging small molecule targets.101 While most DNAzyme based sensors leverage a catalytic output of nucleic acid cleavage,81,82 it will be interesting to observe whether other catalytic modes can be integrated into sensor architectures in the future.3.3 Colorimetric
Colorimetric aptasensors are a promising class of optical biosensors where the readout can generally be detected using the naked eye, making them well suited for field-deployment. One unique approach to colorimetric sensing involves dye displacement upon target binding, but while this was successful with the cocaine aptamer, it may not be generalizable to all aptamer structures.103 While the above methods have shown utility, the most promising colorimetric sensing approaches incorporate nanomaterials that interact with the aptamers in a target-dependent fashion. Gold nanoparticles are among the most popular materials for these sensors because salt-induced aggregation generates a quantifiable color change from red to blue. In one general sensor motif, the unbound aptamer interacts with the gold nanoparticles, preventing aggregation and causing the nanoparticles to remain red. However, upon binding, the aptamer dissociates from the nanoparticles, resulting in aggregation and a shift to a blue color. This approach has been successfully implemented for mycotoxins, pesticides, heavy metals, and many other small molecule contaminants.51,104–109One major challenge for colorimetric aptasensors is the limited signal generated as a function of binding, as most of the systems above do not include a signal amplification step. Seeking to gain the signal amplification capability of enzymes, Luan and coworkers immobilized horseradish peroxidase and the chloramphenicol aptamer on gold nanoparticles through binding with a magnetic single-stranded binding protein appended on iron oxide nanoparticles. Upon target binding, the gold nanoparticles, aptamer, and HRP are released into the supernatant, while the iron oxide is removed using a magnet. Oxidation of TMB substrate using the HRP then provides an amplified colorimetric signal. A similar approach has also been implemented using binding of aptamers to graphene oxide.110 The aptamers are released upon target binding and this can be coupled to orthogonal enzyme activity that is dependent upon this binding event. Due to the presence of the gold nanoparticles, however, the costs of these particular sensors tend to increase. Another method, however, that has been used to circumvent the use of gold nanoparticles is using a split aptamer. The dramatic change in assembly and folding upon target binding makes elaboration into sensors facile once the split aptamer architecture has been engineered. The majority of reported split aptamer assays have an optical readout, one example is similar in principle to the aptamer-based sensor using gold nanoparticles. Using the split aptamer for 17-β-estradiol, the aptamer fragments bind to the gold nanoparticles and prevent their aggregation. However, upon target introduction, the aptamer fragments instead assemble on the target and leave the nanoparticles free to aggregate, resulting in visible color change.69 Several small molecule food contaminants were also detected using a similar system that harnessed the interaction of gold nanoparticles and magnetic beads, but triggered rolling circle amplification. Taking advantage of the signal amplification provided by the rolling circle step, this sensor provided an impressive limit of detection of 6 pM for enrofloxacin.111 Taking a different approach to signal amplification, our lab developed split-aptamer proximity ligation in which target binding promotes a templated ligation between the split aptamer fragments.112 This was then used to generate an ELISA-like assay in which ligation results in immobilization of a streptavidin–HRP conjugate that generates a colorimetric signal upon oxidation of TMB.113 This format afforded a two-order of magnitude improvement compared to other cocaine split aptamer sensors and allows for translation to field work due to its similarities to currently used ELISA methods. While split aptamer sensors offer easy adaptation into sensors, similar to structure-switching aptamers, the dearth of split aptamer motifs limits their use in sensors to a small number of targets including cocaine, isocarbophos, ATP, or adenosine.68
With colorimetric sensors being attractive for their field deployable nature, DNAzymes are also developed into these sensors, specifically for the detection of heavy metals including copper, lead, and mercury in environmental samples.101,114–116 Colorimetric sensor performance is affected by surface type, immobilization strategy, and catalytic performance.101,114–116 DNAzymes are typically immobilized onto gold nanoparticles, gold nanorods, streptavidin beads, nanotubes, or graphene oxide (Fig. 4b) using the same covalent and non-covalent approaches previously mentioned.101,114–118 Xu and coworkers developed a robust and portable system for detecting copper ions in drinking, lake, and sewage water.101 The presence of copper was revealed in under five minutes and could be discerned by the naked eye, highlighting the potential for DNAzyme colorimetric sensors for rapid field detection. This system is of particular interest because simplicity did not come at the cost of sensitivity. To achieve an LOD of 8 nM, Xu and coworkers utilized HCR with biotinylated target strand and hairpin sequences that allowed for a second significant signal cascade through binding of streptavidin–horseradish peroxidase (SA–HRP) and reaction with TMB.101 The simplicity of this system allowed it to be extended for detection in non-aqueous media as well. As an example, Wang and coworkers demonstrated lead detection in soil with a reported LOD of 50 pM.115 Application of DNAzyme based colorimetric sensors for small molecule detection is possible when an aptamer is inserted as the recognition element and the DNAzyme is used for signal amplification purposes and this approach has been successfully utilized for detection of mycotoxins, cyanotoxins, and antibiotics in a variety of environmental media.115,119–121
3.4 Fluorescent
While colorimetric aptasensors are beneficial from a visibly interpretable output, fluorescent aptasensors are of significant value due to the increased sensitivity they can offer. Most aptamers can be easily functionalized with fluorescent dyes on the terminal ends without significantly impacting affinity or selectivity. In fact, many in vitro evolution methods utilize a 5′ fluorophore to monitor enrichment.122 However, this capability has not been extensively translated into field deployable aptasensors, likely due to the limited change in fluorescence that is produced upon target binding. Rather fluorescent aptasensors can rely on similar approaches to the colorimetric dye-displacement assay described above. Canoura and coworkers developed an exonuclease-based detection system that relies on target binding to increase folding stability of the aptamer and thus prevent exonuclease digestion. Interestingly, exonuclease I digestion is prevented four bases from the binding site. The resulting digestion products can be quantified and visualized using SYBR Gold staining, which produces a fluorescent output.123 Another recent example is a “turn-on” sensor that uses a ssDNA binding fluorophore that shows differential fluorescence upon interaction with different aptamers with and without target due to fluorophore displacement by the formation of stable aptamer–target complexes. Using this motif, cyanotoxins were detected at low nanomolar concentrations.124 Excitingly, in this example the different interactions were translated as a chemical array that was interpreted using a smartphone application. This highlights that while fluorescent sensors were previously considered less field friendly due to the need for more expensive fluorescence detection instrumentation, recent advances in smartphone technology have begun to overcome this limitation.125–127 Like the colorimetric sensors, split aptamers can also be converted into biosensors. The general strategy for utilizing split aptamers in sensors consists of appending a fluorophore and quencher or a FRET pair to the two strands, such that target-dependent association can be tracked through dose-dependent quenching or FRET signal. Using this approach, the cocaine split aptamer has been extensively used to report on cocaine concentrations in the micromolar and millimolar range.128 To increase sensitivity for split aptamer-based detection, a unique approach applied to kanamycin A detection harnesses interaction of CuS-biotin nanoparticles and streptavidin Magnesphere paramagnetic particles. When the target molecule is present and drives assembly, this results in pull-down of the CuS nanoparticles, which can then catalyze formation of a fluorescent signal.129 The reported limit of detection was extremely sensitive with an LOD of 26 pM. However, the number of manipulations needed for this method limit its field deployability.Where our lab has placed much focus, however, is in the area of structure-switching fluorescent biosensing. The structure-switching aptamer for ochratoxin A (OTA) is among the most widely used in biosensors. This aptamer was initially evolved only for binding capability.130 However, it is one of the few aptamers that fortuitously undergo a target-dependent conformational change. Using this scaffold, different sensor motifs can be developed to detect OTA in a variety of matrices. One method uses fluorescence polarization (FP) by hybridizing a short fluorescently labelled complementary oligonucleotide to the aptamer, which is then displaced upon OTA binding.131 This biosensor was optimized by testing the FP response using different complementary oligonucleotides that hybridize to different regions on the aptamer. Once the optimal conditions were identified, the sensor was able to produce a dose-dependent FP signal in response to OTA. Another approach utilizes a similar structure-switching biosensor motif, but where the aptamer is modified with a 5′ fluorophore and the complementary strand is modified with a 3′ quencher.71 After optimizing biosensor concentration, complementary strand length, and complementary strand ratio, successful detection of 2–200 nM OTA was achieved.71 Further, this biosensor was used for quantifying OTA in corn samples with an accuracy of 83–106%.71 Seeking to achieve signal amplification using this biosensor motif, a zinc(II)-protoporphyrin IX probe was utilized that directly interacts with free aptamer and offered an LOD of 0.03 nM.108 Previous work from our lab focused on modifying the OTA structure-switching biosensor by inserting a photocleavable linker between the aptamer and complementary strand, as this imparted temporal control over its function.72 While many sensor motifs have been demonstrated using the OTA structure-switching aptamer, in some cases these sensors have also been developed for or extended to other small molecule targets. Using the capture SELEX method described in Section 2, aptamers having potential biosensor activity were generated for several small molecule environmental contaminants including pesticides/herbicides, antibiotics, and toxic metals.66 In a recent example, this enabled fluorescent detection of small molecules that have been linked to antimalarial resistance using aptamers generated after 15–20 rounds of SELEX.132 Similar to OTA biosensors, this system was optimized for capture strand length and aptamer:capture strand ratio and was able to generate an LOD of 3 nM and 4 nM for piperaquine and mefloquine, respectively. Although structure-switching aptamers to Hg2+ and Pb2+ have been generated using a similar capture approach coupled with particle display technology, the downstream implementation of these aptamers in sensors has not yet been reported.74 Using our RE-SELEX method, we were able to generate a structure-switching sensor for kanamycin A that has a dynamic range of 90 μM to 100 mM, showing the potential utility of our selection methodology to deliver aptamer sequences that are pre-optimized for use as sensors.75
Looking forward, a key need is to increase the sensitivity of these structure-switching sensors. One potential strategy that has been reported is an innovative approach utilizing plasmonic gold nanostars as quenchers of Cy3-labeled complementary strands that are pre-hybridized to microcystin-LR aptamers.133 In the absence of microcystin-LR, the aptamers hybridize to the complementary strand, which prohibits fluorescence quenching by the nanostars. Upon introduction of the target, the aptamer is displaced, and dose-dependent quenching is observed. This approach provides increased sensitivity over previous methods, with a reported LOD of 500 pM and a dynamic range of 100 pM to 50 nM.133 Additional sensor platforms that increase sensitivity without being cost-prohibitive will be critical in building field-deployable sensors for small molecule environmental contaminants.
One approach for improving the signal in these systems is by employing DNAzyme-based fluorescent sensors. Fluorescent DNAzyme-based biosensors also operate similarly to their aptamer counterparts. For instance, the complementary target strand that undergoes metal-dependent cleavage by a DNAzyme can be appended with a fluorophore and quencher on opposite termini such that upon cleavage, a dose-dependent increase in fluorescence signal is generated.99,134 Signal amplifiers including graphene oxide and other nanomaterials can also be included to increase sensitivity for detection of challenging targets.135–137 Alternatively, dyes such as thioflavin T that bind to specific DNA conformations can be used as the reporter in DNAzyme systems. Ravikumar and coworkers immobilized the GR-5 DNAzyme onto a graphene oxide sheet and hybridized it with a GT-rich substrate DNA strand such that in the presence of lead, the substrate strand is cleaved. This results in formation of a G-quadruplex that can bind to thioflavin T and induce fluorescence. Interestingly, in the presence of mercury, the G-quadruplex is unfolded, resulting in loss of thioflavin T fluorescence. This system performed with an LOD of 96 pM for lead and 356 pM for mercury.135
While the sensors above have been deployed in water samples, recent efforts have focused on expanding this capability to other environmental media. Yun and coworkers developed a one-step system that was used to detect mercury in Chinese herbs. In their sensor, a fluorophore-labeled DNA stand is immobilized onto gold nanoparticles along with a DNAzyme having a long thymine repeat segment (E-DNA). In the presence of mercury, the fluorophore-labeled DNA binds to the E-DNA and is cleaved. This releases the fluorescent DNA from the gold nanoparticles, producing signal. Taking advantage of catalytic turnover by the DNAzyme, this sensor provided an LOD of 30 pM of mercury in crops.137
While DNAzyme-based sensors have the benefit of signal amplification and use in multiple output formats, challenges that can be addressed to further increase their utility include increasing the rate of catalysis and improving the ease of adaption for use with small molecule targets. As described in Section 2, we propose that these challenges will most likely be addressed through investment in the development of new selection methods for DNAzymes.
3.5 Electrochemical
Due to predictable structural change, electrochemical readout can be designed. Using the aptamer beacon for aflatoxin B1, immobilization on a gold surface and attachment of a 3′ methylene blue dye results in a signal-on response upon target binding, as the concomitant structure change moves the dye closer to the electrode surface, which increases electric current that is easily measurable using square wave voltammetry (Fig. 3).138 Similar detection systems have also been implemented for diverse of small molecule environmental contaminants including ochratoxin A, aflatoxins, microcystin-LR, bisphenol A, and heavy metals.139,140 These examples utilize covalent attachment of aptamers that is beneficial for long term storage associated with biomonitoring, but the cost of gold for onsite single-use detection is a disadvantage (Fig. 3). One approach to overcome this challenge is utilizing porous gold nanocages with screen-printed carbon electrodes, which are well known for low cost and field deployability.141 While this platform was only tested for aflatoxin B1 detection, the cost effective and facile workflow provides a universal approach for small molecule sensing.141 An alternative technique is through non-covalent attachment of aptamers. The most promising approach uses graphene oxide, to which aptamers bind through π-stacking interactions.142 Graphene oxide can be used as a versatile surface for attachment of additional nanomaterials such as nanoparticles, which are often used for signal amplification. While creation of these electrochemical sensors may require more specialized labor than generating fluorescent sensors, they offer a convenient electrical readout that can be detected with a cell phone, making this a highly promising approach for field-deployable sensing.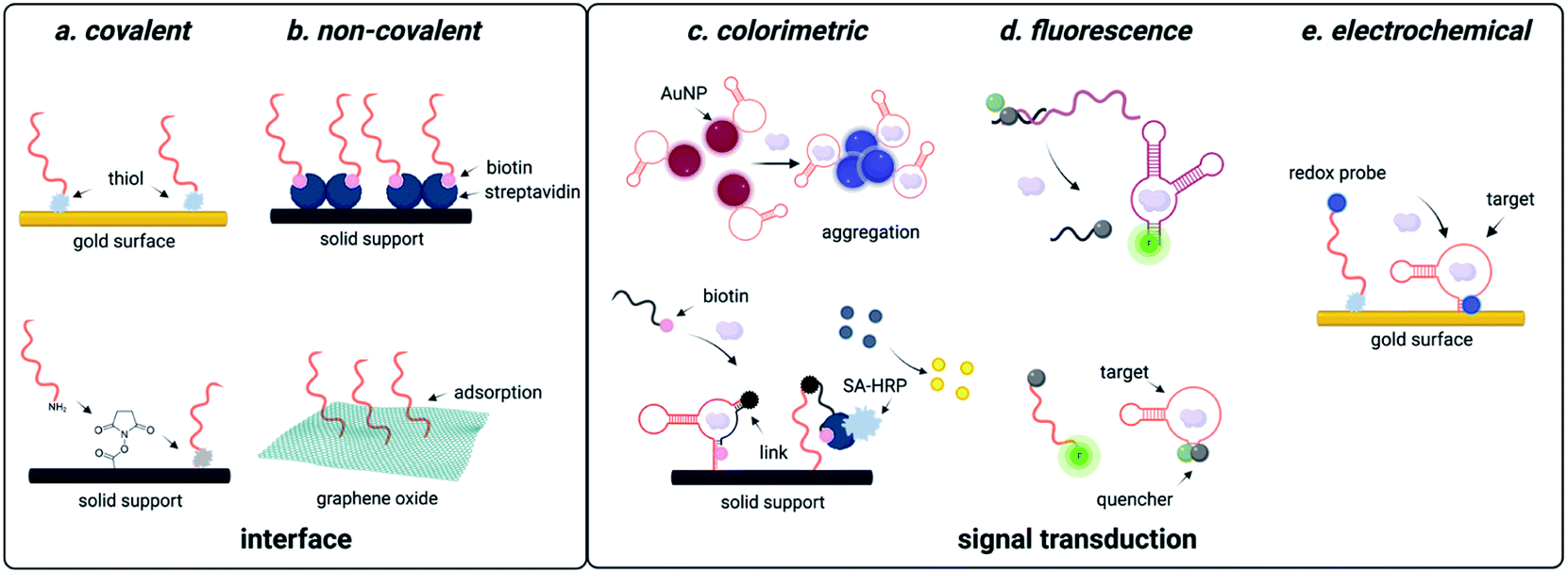 | ||
| Fig. 3 Functional DNA-based detection strategies for small molecule environmental contaminants. (a) Covalent interfaces for aptasensors. (b) Non-covalent interfaces for aptasensors. (c) Colorimetric readouts for aptasensors. (d) Fluorescent readouts for aptasensors. (e) Electrochemical readouts for aptasensors. | ||
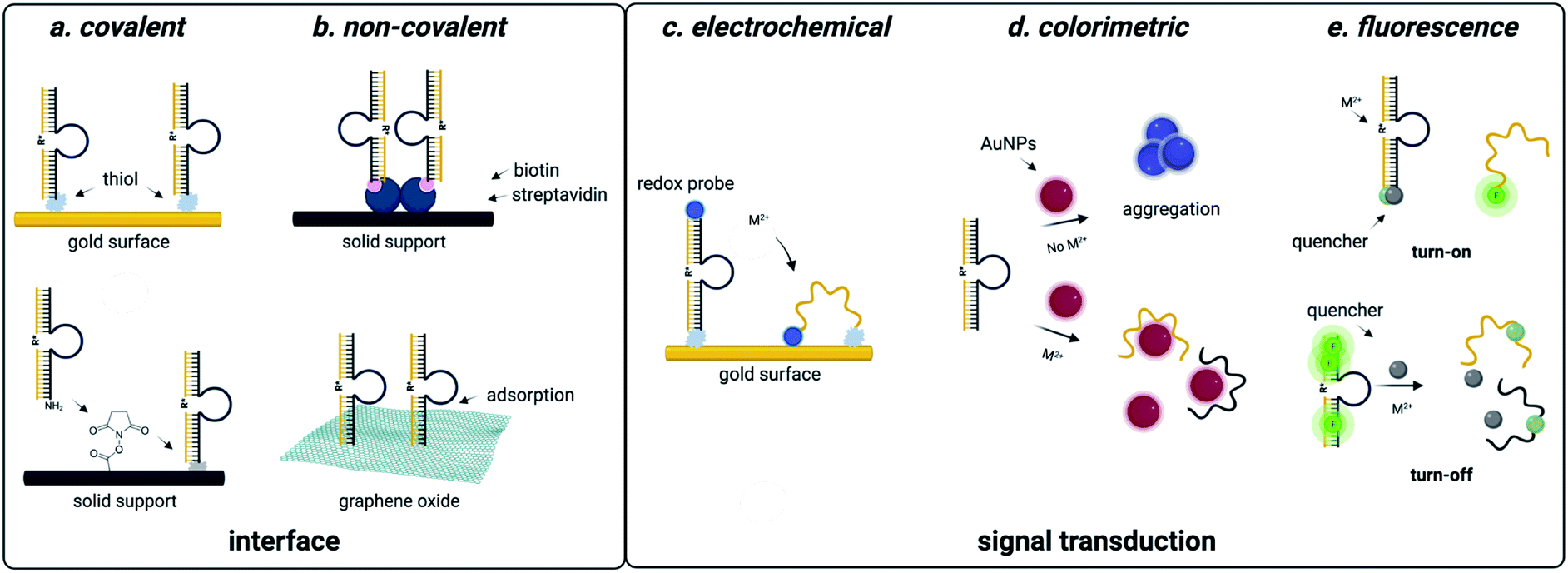 | ||
| Fig. 4 DNAzyme based strategies for detection of heavy metal environmental contaminants. (a) Covalent interfaces for DNAzyme sensors. (b) Non-covalent interfaces for DNAzyme sensors. (c) Electrochemical readouts for DNAzyme sensors. (d) Colorimetric readouts for DNAzyme sensors. Reproduced from ref. 94, https://doi.org/10.3389/fmicb.2018.00179, under the terms of the CC BY 4.0 license https://creativecommons.org/licenses/by/4.0/. (e) Fluorescent readouts for DNAzyme sensors. | ||
Recognizing the need for methods to reliably engineer aptamer beacons, one clever approach that has been identified is inserting aptamer sequences into a G-quadruplex structure that undergoes a significant conformational change upon aptamer-target binding.143 This and related methods hold promise for the elaboration of toxin-binding aptamers into beacons that can be used in optical and electrochemical sensors.
DNAzyme electrochemical sensors function similarly to those described for structure-switching aptamers, as the DNAzyme is functionalized with a redox-active dye such as methylene blue.117 Metal dependent cleavage of the target strand leads to dehybridization, increasing the conformational flexibility of the DNAzyme and allowing the redox active compound to more effectively transfer electrons to the electrode surface.117 The major factors that contribute to the performance of these sensors are DNAzyme immobilization strategy and catalytic efficiency. While catalytic efficiency is inherently dependent on SELEX, there are numerous approaches to DNAzyme immobilization that can impact sensitivity. For sensors using gold surfaces, immobilization strategies include thiol-mediated, ester formation, epoxide opening, and biotin–streptavidin binding. Covalent approaches are favorable because they offer reduced background signal compared to non-covalent attachment strategies, but they can also be more costly due to the need for modified oligonucleotide synthesis.117,118
Electrochemical DNAzyme sensors have proven to be especially useful for heavy metal detection in water. The 8–17 DNAzyme developed by Xiao and coworkers is among the most commonly used, and functionalization with methylene blue provided an LOD of 0.3 μM for lead detection in the electrochemical format. However, this falls short of the sensitivity needed to track environmentally relevant concentrations.144 This highlights a major limitation of DNAzyme-based detection methods in that the output is based on the signal generated by the immobilized methylene blue strand. In this example, methylene blue is tethered to the DNAzyme, resulting in a 1:1 ratio of activity to output.144 Hybridization chain reaction (HCR) can circumvent this challenge by utilizing a hairpin sequence that is partially complimentary to the immobilized DNAzyme. After the initial catalytic event, the liberated DNAzyme can then hybridize the partial compliment, causing the hairpin to open.101,102 This triggers a cascade of hairpin opening events, and subsequent addition of free methylene blue results in binding to the minor groove of the immobilized dsDNA, generating an electrochemical signal.102 This approach has decreased LOD from μM to pM range.99,100,102,145 While electrochemical DNAzyme-based sensors have traditionally been used for metal detection, an exciting potential future direction is their adaptation for small molecule detection. This is, however, difficult due to the lack of small molecule-degrading DNAzymes.
4. Sequestration
While functional nucleic acids selected for toxins have primarily found use in detection applications, the area where their advantages over antibodies truly shine is in sequestration and removal of environmental contaminants. This application space has tremendous potential because DNA is easily functionalized, and thus aptamers can be coupled to diverse scaffolds or solid supports that already exist for environmental decontamination.146 Of the different applications, water decontamination is of utmost importance due to current freshwater scarcity challenges, though aptamer-based sequestration could be applied to a diverse range of media. Below, we highlight how the advances in small molecule aptamer SELEX will allow for significant advances in contaminant removal. Further, we discuss how coupling of aptamers with enzymes holds promise for the creation of self-regenerating purification technologies. There are several factors that impact the ability of a functional nucleic acid to sequester and remove small molecule contaminants from water sources. These include aptamer size, affinity, and method of sequestration95 and the importance of these factors has been investigated using validated purification technologies.1474.1 Dispersed solid supports
Many of the scaffolds that can be used for sequestration act through dispersion in the sample, followed by a collection step. For example, Huang and coworkers developed a method that coupled DNA aptamers against E. coli to TiO2 particles (Fig. 5). Upon irradiation with UV light, TiO2 produces reactive oxygen species, which in turn kills the sequestered bacterial cells.148 While there are no reports of small molecule decontamination using this approach, it is feasible in principle for contaminants that can be effectively neutralized by reactive oxygen. However, a critical challenge that would need to be addressed would be the impact of reactive oxygen species on nucleic acid stability. Aptamers can also be incorporated within the structure of hydrogels and this approach was successful in removing large quantities of BPA from water sources.149 While effective for sequestration, regeneration of the hydrogel material requires complex procedures, limiting scalability. Using a non-covalent approach for incorporation of aptamers, encapsulation in liposomes was implemented for sequestration of oxytetracycline, BPA, and 17-β-estradiol from water.150 This system can leverage multiple small-molecule aptamers for sequestration, but unfortunately optimal function was only observed in buffer, which may limit use in water decontamination applications.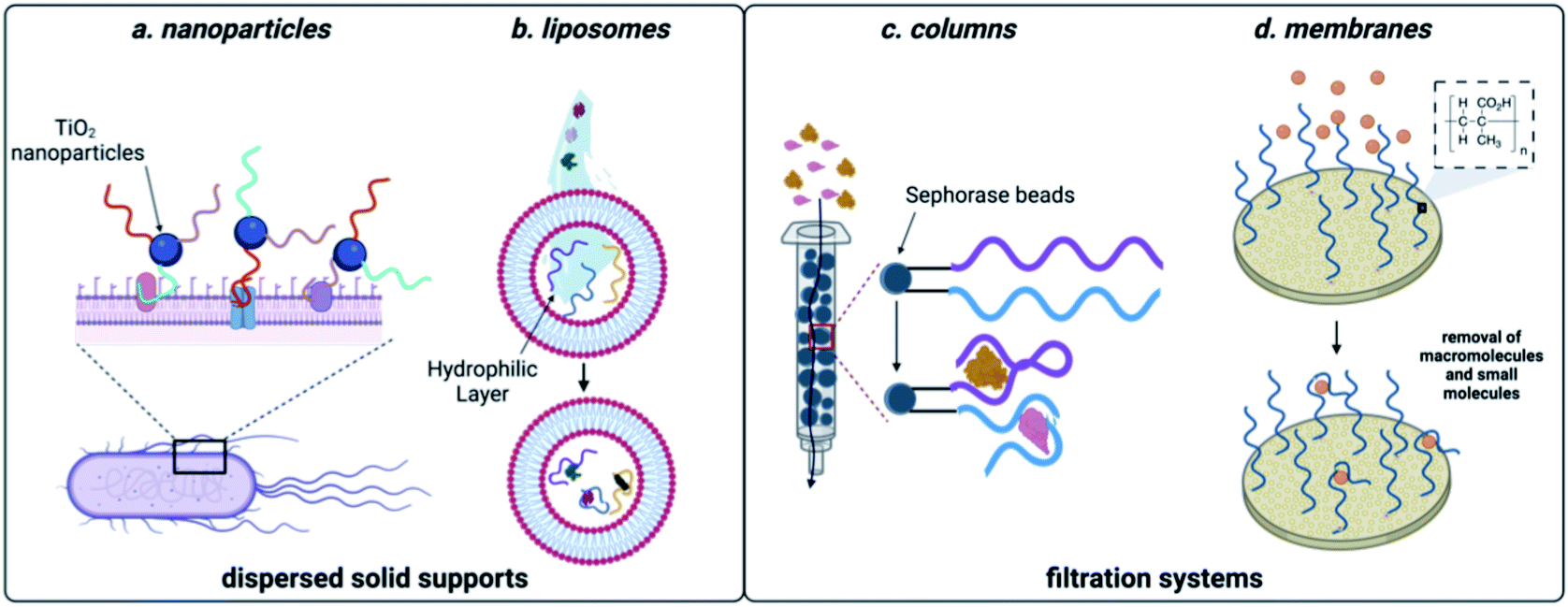 | ||
| Fig. 5 Aptamer-based methods for sequestration of environmental contaminants. (a) Nanoparticle-based aptamer support. (b) Liposome-based aptamer support. (c) Aptamer column filtration.Reproduced from ref. 147, https://doi.org/10.1155/2017/3712070, under the terms of CC BY 4.0 license.(d) Aptamer membrane filtration. | ||
4.2 Filtration systems
While effective, the methods discussed so far can require expensive machinery and most of these materials have not been shown to be capable of regeneration, limiting them to a single use. They do however, demonstrate the power aptamers as versatile affinity reagents for toxin capture. We propose that an ideal aptamer-based decontamination platform will (1) allow for the filtration high volumes of water, (2) be capable of regeneration using minimal technical steps, and (3) have capacity to remove large amounts of contaminants. Immobilization of aptamers on a solid support that is compatible with direct filtration can offers these advantages. In one example, Hu and coworkers attached the cocaine and diclofenac aptamers to Sepharose beads and showed that this enabled contaminant depletion even after a month of storage at 4 °C.151 We recognized that membrane platforms provide a promising alternative to solid-phase beads, owing to their facile preparation, ease of use, and minimal resource consumption.152 Synthetic membranes are broadly classified by pore size and internal structure, and while some membranes can inherently remove small molecule contaminants, the small pore sizes required increase production cost and the energy needed for use. In contrast, ultrafiltration membranes having pore sizes in the high nm to low mm range are widely used for removal of large molecular weight contaminants such as bacteria, parasites, and particulates while still being inexpensive and user friendly.34 We envisioned that attachment of aptamers to ultrafiltration membranes would enable sequestration of small molecule contaminants while maintaining the benefits of ultrafiltration membranes.Using the aptamer for BPA, we attached amine-modified DNA strands to an ultrafiltration membrane having grafted polymethacrylic acid.153 We demonstrated BPA depletion and membrane regeneration using heat to temporarily denature the aptamers. Given that aptamers can be selected for diverse small molecule targets, we recognized that this approach is generalizable and we went on to demonstrate simultaneous removal of pesticides and natural and synthetic toxins.154 To demonstrate scalability, we were able to purify more than 8 L of water in one filtration, making this method highly desirable in settings where regeneration is not feasible.154 Furthermore, we demonstrated simultaneous removal of E. coli and small-molecule toxins using a single ultrafiltration membrane.
We also recognized that enzymes offer complementary activity to aptamers, as they can degrade small molecule organics. However, using enzymes dispersed in solution for water treatment can create downstream purification challenges. Thus, we envisioned a system in which aptamers and enzymes are simultaneously attached to the ultrafiltration membrane. While enzyme alone provides some level of depletion, catalysis is not sufficiently fast to degrade all BPA molecules as they pass through the membrane. However, when combined with BPA-binding aptamers, the result is efficient depletion and autonomous regeneration.155 As research moves forward with identifying new aptamers for contaminants and enzymes capable of degrading these molecules, we envision that this system can be applied to a range of water purification applications.
The use of ultrafiltration membranes combined with aptamers and enzymes has proven to be an excellent starting point for toxin sequestration and degradation as these biomolecules can be evolved for use with a wide range of contaminants. However, a key limitation remains the high cost of DNA relative to the materials used to produce the solid support. We are encouraged by continuing advances in oligonucleotide synthesis, in part spurred on by the COVID-19 pandemic, which are anticipated to make the large-scale synthesis of oligonucleotides increasingly cost effective and practical. Another area for improvement is aptamer stability, which may be addressable through the use of XNA scaffolds, though these can be much most costly than DNA. Despite these limitations, the future of aptamer-based toxin sequestration is promising, and next key steps for the field will include surveying the long-term storage and reusability of these purification systems as well as exploring formats by which they could be scaled up to meet the high demand for clean drinking water.
5. Remaining challenges and future outlook
Functional nucleic acids offer significant utility as affinity reagents and catalysts for sensing and decontamination efforts, owing to their ability to selectively recognize specific ligands and their inherent benefits compared to their protein-based counterparts. However, molecular recognition of small molecule targets presents novel challenges in that they possess few binding epitopes. As a result, there are several aptamers capable of binding to small molecule targets with high specificity, but affinity can be moderate, and this often prevents researchers from achieving the desired level of sensitivity. For instance, in the case of small molecule contaminant biomonitoring, accurate assessment of low-dose effects requires methods that have a limit of detection in the fM to pM range, whereas aptamers typically have affinities in the nM to μM range. We posit that the key hurdle for addressing these challenges lies in aptamer development. Continual improvement of selection methods followed by rigorous characterization and reporting of aptamer candidates is needed.There are many challenges to overcome with small molecule SELEX techniques. Perhaps the most notable is the use of immobilization during the selection. While this allows for facile separation of active from inactive sequences, resulting aptamers often have higher affinity for the immobilized analogue compared to the native target. Furthermore, immobilization introduces difficulty in controlling the ligand concentration during the selection process, which impacts stringency and can prevent effective enrichment of the tightest binding sequences. We propose that to improve small molecule SELEX, methods should aim to utilize homogenous selection steps because they more closely mimic the downstream detection environment and allow for control over ligand concentration. This could not only simplify method development but also increase sensitivity. Additionally, many detection methods rely on structure-switching properties of aptamers, yet sequences selected for target-binding affinity are rarely optimal for such biosensor formats. This leads to the ongoing challenge that the majority of biosensor applications rely on a small number of privileged aptamer structures. We propose that continual effort is needed to develop selection methods that directly enrich for library members having the desired structure-switching or biosensor activity, as this will provide access to the sequences needed to develop biosensors that can address the most pressing needs in toxin detection. Moreover, streamlining and automating these processes would enable the rapid selection of new aptamers and biosensors, equipping researchers to address emerging small molecule contaminant threats.
Improvements to selection methods also hold significant promise to advance the field of DNAzymes. Specifically, the development of an in trans selection method for DNAzymes would not only facilitate application to multiple different toxins, but also likely increase selectivity and catalytic efficiency. Additionally, while enzymes are often used for signal amplification in the case of lower performing aptasensors, DNAzymes could offer a less expensive and more field-deployable alternative if sequences were found that could approach the catalytic efficiency of protein enzymes.
One newly emerging area in which aptamers are being harnessed for environmental applications is that of toxin sequestration. These methods generally rely on utilizing aptamers to capture toxins from water or other matrices, and in particular, membrane filtration appears to hold promise for the efficient removal of toxins and facile regeneration of the sequestration system. Given that the goal of these aptamer-based purification systems is to benefit the environment, it will be critical to focus future efforts on developing platforms that use sustainably produced materials and that offer the greatest potential for reuse or recyclability. While DNA is environmentally benign, many of the reagents used for its synthesis are not, but recent advances in oligonucleotide synthesis demonstrate promise to offer greener routes for the large-scale production of aptamers and other functional DNAs. In the long term, the ideal platforms for toxin sensing and sequestration will both address environmental needs by enabling the detection and removal, and will themselves be environmentally benign.
Author contributions
All authors contributed to research, writing, and editing of the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Defense Threat Reduction Agency [HDTRA118-1-0029 to J. M. H.] and the National Science Foundation [CHE 1904885 and CBET 1818476 to J. M. H.]. Any opinions, findings and conclusions or recommendations expressed in this publication are those of the authors and do not necessarily reflect the views of DTRA.References
- R. Pool and E. Rusch, Identifying and Reducing Environmental Health Risks of Chemicals in Our Society, 2014 Search PubMed.
- S. J. D'Surney and M. D. Smith, Encycl. Toxicol., 2005, pp. 526–530 Search PubMed.
- M. Gavrilescu, K. Demnerová, J. Aamand, S. Agathos and F. Fava, New Biotechnol., 2015, 32, 147–156 CrossRef CAS PubMed.
- Y. Xu, M. M. Hassan, A. S. Sharma, H. Li and Q. Chen, Crit. Rev. Food Sci. Nutr., 2021, 1–19 Search PubMed.
- D. Li, B. Kumari, J. M. Makabenta, A. Gupta and V. Rotello, Nanoscale, 2019, 11, 22172–22181 RSC.
- M. Majdinasab, A. Hayat and J. L. Marty, TrAC, Trends Anal. Chem., 2018, 107, 60–77 CrossRef CAS.
- Y. Hu, Y. Sun, J. Gu, F. Yang, S. Wu, C. Zhang, X. Ji, H. Lv, S. Muyldermans and S. Wang, Food Chem., 2021, 353(1), 129481 CrossRef CAS PubMed.
- X. Guo, F. Wen, N. Zheng, M. Saive, M. L. Fauconnier and J. Wang, Front. Chem., 2020, 8, 195 CrossRef CAS PubMed.
- B. S. Batule, S. U. Kim, H. Mun, C. Choi, W. B. Shim and M. G. Kim, J. Agric. Food Chem., 2018, 66, 3003–3008 CrossRef CAS PubMed.
- M. Eskola, G. Kos, C. T. Elliott, J. Hajšlová, S. Mayar and R. Krska, Crit. Rev. Food Sci. Nutr., 2020, 60, 2773–2789 CrossRef CAS PubMed.
- Y. Zhao, M. Yang, Q. Fu, H. Ouyang, W. Wen, Y. Song, C. Zhu, Y. Lin and D. Du, Anal. Chem., 2018, 90, 7391–7398 CrossRef CAS PubMed.
- S. Lee, R. McLaughlin, M. Harnly, R. Gunier and R. Kreutzer, Environ. Health Perspect., 2002, 110(12), 1175–1184 CrossRef CAS PubMed.
- M. Maroni, C. Colosio, A. Ferioli and A. Fait, Toxicology, 2000, 143, 5–8 CrossRef CAS.
- A. C. Curtis and B. Sattler, J. Am. Assoc. Nurse Pract., 2018, 30, 299–304 CrossRef PubMed.
- U. Epa and O. of Resource Conservation, 2016, 1–478.
- J. L. Wilkinson, P. S. Hooda, J. Swinden, J. Barker and S. Barton, Environ. Pollut., 2018, 234, 864–875 CrossRef CAS PubMed.
- F. Gassara, S. K. Brar, M. Verma and R. D. Tyagi, Chemosphere, 2013, 92, 1356–1360 CrossRef CAS PubMed.
- B. Sharma and P. Shukla, J. Hazard. Mater., 2021, 401, 123285 CrossRef CAS PubMed.
- R. N. Ugbaja, M. A. Enilolobo, A. S. James, T. F. Akinhanmi, A. J. Akamo, D. O. Babayemi and O. Ademuyiwa, Toxicol. Ind. Health, 2020, 36, 863–875 CrossRef CAS PubMed.
- H. Zhang, L. L. Zhang, J. Li, M. Chen and R. D. An, Environ. Sci. Pollut. Res., 2020, 27, 9853–9865 CrossRef CAS PubMed.
- C. Maragoni-Santos, T. Serrano Pinheiro de Souza, J. R. V. Matheus, T. B. de Brito Nogueira, D. Xavier-Santos, R. F. Miyahira, A. E. Costa Antunes and A. E. C. Fai, Crit. Rev. Food Sci. Nutr., 2021, 1–13 CrossRef PubMed.
- M. S. McLachlan, A. Kierkegaard, M. Radke, A. Sobek, A. Malmvärn, T. Alsberg, J. A. Arnot, T. N. Brown, F. Wania, K. Breivik and S. Xu, Environ. Sci. Technol., 2014, 48, 7264–7271 CrossRef CAS PubMed.
- L. Martín-Pozo, B. de Alarcón-Gómez, R. Rodríguez-Gómez, M. T. García-Córcoles, M. Çipa and A. Zafra-Gómez, Talanta, 2019, 192, 508–533 CrossRef PubMed.
- V.-T. T. Nguyen, Y. S. Kwon and M. B. Gu, Curr. Opin. Biotechnol., 2017, 45, 15–23 CrossRef CAS PubMed.
- E. M. McConnell, J. Nguyen and Y. Li, Front. Chem., 2020, 8, 434 CrossRef CAS PubMed.
- W. Thavarajah, A. D. Silverman, M. S. Verosloff, N. Kelley-Loughnane, M. C. Jewett and J. B. Lucks, ACS Synth. Biol., 2020, 9, 10–18 CrossRef CAS PubMed.
- A. J. Agranat, Y. Kabessa, E. Shpigel, B. Shemer, O. Schwartzglass, L. Atamneh, Y. Mizrachi, Y. Uziel, M. Ejzenberg, T. Elad and S. Belkin, SPIE BiOS, 2020, vol. 11258, p. 16.
- M. F. Clark, R. M. Lister and M. Bar-Joseph, Methods Enzymol., 1986, 118, 742–766 CAS.
- E. Engvall and P. Perlmann, J. Immunol., 1972, 109(1), 129–135 CAS.
- K. Wang, H. He, T. C. Zhang, Y. Liang and S. Yuan, ACS Appl. Mater. Interfaces, 2021, 13, 6906–6918 CrossRef CAS PubMed.
- J. M. Galindo-Miranda, C. Guízar-González, E. J. Becerril-Bravo, G. Moeller-Chávez, E. León-Becerril and R. Vallejo-Rodríguez, Water Supply, 2019, 19, 1871–1884 CrossRef.
- L. Schweitzer and J. Noblet, Green Chem. An Incl. Approach, 2018, pp. 261–290 Search PubMed.
- A. J. Whelton, L. K. McMillan, M. Connell, K. M. Kelley, J. P. Gill, K. D. White, R. Gupta, R. Dey and C. Novy, Environ. Sci. Technol., 2015, 49, 813–823 CrossRef CAS PubMed.
- A. Lee, J. W. Elam and S. B. Darling, Environ. Sci.: Water Res. Technol., 2016, 2, 17–42 RSC.
- N. Pandey, S. K. Shukla and N. B. Singh, Nanocomposites, 2017, 3, 47–66 CrossRef CAS.
- S. K. Silverman, in Functional Nucleic Acids for Analytical Applications, 2009, pp. 47–108 Search PubMed.
- R. Micura and C. Höbartner, Chem. Soc. Rev., 2020, 49, 7331–7353 RSC.
- W. Xu, W. He, Z. Du, L. Zhu, K. Huang, Y. Lu and Y. Luo, Angew. Chem., 2021, 133, 6966–6995 CrossRef.
- K. Duffy, S. Arangundy-Franklin and P. Holliger, BMC Biol., 2020, 18 Search PubMed.
- J. C. Chaput and P. Herdewijn, Angew. Chem., Int. Ed., 2019, 58, 11570–11572 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS PubMed.
- M. Roueinfar, K. M. Abraham and K. L. Hong, ACS Omega, 2019, 4, 16201–16208 CrossRef CAS PubMed.
- M. McKeague, E. M. McConnell, J. Cruz-Toledo, E. D. Bernard, A. Pach, E. Mastronardi, X. Zhang, M. Beking, T. Francis, A. Giamberardino, A. Cabecinha, A. Ruscito, R. Aranda-Rodriguez, M. Dumontier and M. C. DeRosa, J. Mol. Evol., 2015, 81, 150–161 CrossRef CAS PubMed.
- X. Ma, W. Wang, X. Chen, Y. Xia, N. Duan, S. Wu and Z. Wang, Food Control, 2015, 47, 545–551 CrossRef CAS.
- M. McKeague, R. Velu, K. Hill, V. Bardóczy, T. Mészáros and M. C. DeRosa, Toxins, 2014, 6, 2435–2452 CrossRef PubMed.
- A. Ng, R. Chinnappan, S. Eissa, H. Liu, C. Tlili and M. Zourob, Environ. Sci. Technol., 2012, 46, 10697–10703 CrossRef CAS PubMed.
- R. M. Williams, C. L. Crihfield, S. Gattu, L. A. Holland and L. J. Sooter, Int. J. Mol. Sci., 2014, 15, 14332–14347 CrossRef CAS PubMed.
- K. Ohsawa, T. Kasamatsu, J. I. Nagashima, K. Hanawa, M. Kuwahara, H. Ozaki and H. Sawai, Anal. Sci., 2008, 24, 167–172 CrossRef CAS PubMed.
- L. Vinet and A. Zhedanov, J. Phys. A: Math. Theor., 2011, 44 Search PubMed.
- W. Li, Y. Luo, T. Gao, L. Yang, J. Wang and R. Pei, J. Mol. Evol., 2019, 87, 231–239 CrossRef CAS PubMed.
- B. Chatterjee, N. Kalyani, A. Anand, E. Khan, S. Das, V. Bansal, A. Kumar and T. K. Sharma, Microchim. Acta, 2020, 187, 1–13 CrossRef PubMed.
- H. Gu, N. Duan, S. Wu, L. Hao, Y. Xia, X. Ma and Z. Wang, Sci. Rep., 2016, 6, 1–9 CrossRef.
- H. Yu, O. Alkhamis, J. Canoura, Y. Liu and Y. Xiao, Angew. Chem., Int. Ed., 2021, 60, 16800–16823 CrossRef CAS PubMed.
- J. Yang and M. T. Bowser, Anal. Chem., 2013, 85, 1525–1530 CrossRef CAS PubMed.
- B. A. Manuel, S. A. Sterling, A. A. Sanford and J. M. Heemstra, Anal. Chem., 2022, 94(17), 6436–6440 CrossRef CAS PubMed.
- E. J. Cho, J.-W. Lee and A. D. Ellington, Annu. Rev. Anal. Chem., 2009, 2, 241–264 CrossRef CAS PubMed.
- E. S. Lee, J. M. Lee, H. J. Kim and Y. P. Kim, Chemosensors, 2021, 9, 54 CrossRef CAS.
- R. Yang, J. Jin, L. Long, Y. Wang, H. Wang and W. Tan, Chem. Commun., 2009, 322–324 RSC.
- Y. Li, J. Yuan and Z. Xu, J. Anal. Methods Chem., 2019, 3712032 Search PubMed.
- G. Wang, Y. Zhu, L. Chen and X. Zhang, Talanta, 2014, 129, 398–403 CrossRef CAS PubMed.
- M. N. Stojanovic, P. de Prada and D. W. Landry, J. Am. Chem. Soc., 2001, 123, 4928–4931 CrossRef CAS PubMed.
- M. Rajendran and A. D. Ellington, Nucleic Acids Res., 2003, 31, 5700 CrossRef CAS PubMed.
- R. Nutiu and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 1061–1065 CrossRef CAS PubMed.
- R. Stoltenburg, N. Nikolaus and B. Strehlitz, J. Anal. Methods Chem., 2012, 1, 14 Search PubMed.
- C. Lyu, I. M. Khan and Z. Wang, Talanta, 2021, 229, 122274 CrossRef CAS PubMed.
- A. D. Kent, N. G. Spiropulos and J. M. Heemstra, Anal. Chem., 2013, 85, 35 CrossRef PubMed.
- X. Qi, X. Yan, Y. Zhao, L. Li and S. Wang, TrAC, Trends Anal. Chem., 2020, 133, 116069 CrossRef CAS.
- J. Liu, W. Bai, S. Niu, C. Zhu, S. Yang and A. Chen, Sci. Rep., 2014, 4, 7571 CrossRef CAS PubMed.
- R. Nutiu and Y. Li, J. Am. Chem. Soc., 2003, 125, 4771–4778 CrossRef CAS PubMed.
- J. Chen, Z. Fang, J. Liu and L. Zeng, Food Control, 2012, 25, 555–560 CrossRef CAS.
- Z. Tan, T. A. Feagin and J. M. Heemstra, J. Am. Chem. Soc., 2016, 138, 6328–6331 CrossRef CAS PubMed.
- N. Qiao, J. Li, X. Wu, D. Diao, J. Zhao, J. Li, X. Ren, X. Ding, D. Shangguan and X. Lou, Anal. Chem., 2019, 91, 13383–13389 CrossRef CAS PubMed.
- H. Qu, A. T. Csordas, J. Wang, S. S. Oh, M. S. Eisenstein and H. T. Soh, ACS Nano, 2016, 10, 7558–7565 CrossRef CAS PubMed.
- A. A. Sanford, A. E. Rangel, T. A. Feagin, R. G. Lowery, H. S. Argueta-Gonzalez and J. M. Heemstra, Chem. Sci., 2021, 12, 11692–11702 RSC.
- K. N. Meek, A. E. Rangel and J. M. Heemstra, Methods, 2016, 106, 29–36 CrossRef CAS PubMed.
- L. K. McKenzie, R. El-Khoury, J. D. Thorpe, M. J. Damha and M. Hollenstein, Chem. Soc. Rev., 2021, 50, 5126–5164 RSC.
- C. Höbartner and S. K. Silverman, Biopolymers, 2007, 87, 279–292 CrossRef PubMed.
- S. K. Silverman, Trends Biochem. Sci., 2016, 41, 595–609 CrossRef CAS PubMed.
- S. K. Silverman, Wiley Encycl. Chem. Biol., 2008 Search PubMed.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1994, 1, 223–229 CrossRef CAS PubMed.
- M. Chandra, A. Sachdeva and S. K. Silverman, Nat. Chem. Biol., 2009, 5, 718–720 CrossRef CAS PubMed.
- K. B. Chapman and J. W. Szostak, Chem. Biol., 1995, 2, 325–333 CrossRef CAS.
- W. E. Purtha, R. L. Coppins, M. K. Smalley and S. K. Silverman, J. Am. Chem. Soc., 2005, 127, 13124–13125 CrossRef CAS.
- Y. Wang and S. K. Silverman, Biochemistry, 2005, 44, 3017–3023 CrossRef CAS PubMed.
- S. K. Silverman, Org. Biomol. Chem., 2004, 2, 2701–2706 RSC.
- S. K. Silverman, Chem. Commun., 2008, 3467–3485 RSC.
- R. Inomata, J. Zhao and M. Miyagishi, Commun. Biol., 2021, 4, 1–8 CrossRef PubMed.
- W. Zhou, Y. Zhang, J. Ding and J. Liu, ACS Sens., 2016, 1, 600–606 CrossRef CAS.
- H. Si, R. Sheng, Q. Li, J. Feng, L. Li and B. Tang, Anal. Chem., 2018, 90, 8785–8792 CrossRef CAS.
- D. Mazumdar, In Vitro, 2008, 8–10 Search PubMed.
- P. J. J. Huang and J. Liu, Anal. Chem., 2016, 88, 3341–3347 CrossRef CAS PubMed.
- Y. Wang, E. Liu, C. H. Lam and D. M. Perrin, Chem. Sci., 2018, 9, 1813–1821 RSC.
- C. K. O'Sullivan, Fresenius' J. Anal. Chem., 2002, 372, 44–48 Search PubMed.
- A. Ruscito and M. C. DeRosa, Front. Chem., 2016, 4, 14 Search PubMed.
- B. Mondal, S. Ramlal, P. S. Lavu, N. Bhavanashri and J. Kingston, Front. Microbiol., 2018, 9, 179 CrossRef PubMed.
- R. Saran and J. Liu, Anal. Chem., 2016, 88, 4014–4020 CrossRef CAS PubMed.
- Y. He, D. Chen, P. J. J. Huang, Y. Zhou, L. Ma, K. Xu, R. Yang and J. Liu, Nucleic Acids Res., 2018, 46, 10262–10271 CrossRef CAS PubMed.
- J. Liu, A. K. Brown, X. Meng, D. M. Cropek, J. D. Istok, D. B. Watson and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2056–2061 CrossRef CAS.
- W. Yun, H. Wu, Z. Yang, R. Wang, C. Wang, L. Yang and Y. Tang, Anal. Chim. Acta, 2019, 1068, 104–110 CrossRef CAS PubMed.
- W. Xu, J. Tian, Y. Luo, L. Zhu and K. Huang, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed.
- J. Zhuang, L. Fu, M. Xu, Q. Zhou, G. Chen and D. Tang, Biosens. Bioelectron., 2013, 45, 52–57 CrossRef CAS PubMed.
- H. Yu, W. Yang, O. Alkhamis, J. Canoura, K.-A. Yang and Y. Xiao, Nucleic Acids Res., 2018, 46, e43 CrossRef PubMed.
- M. Jia, J. Sha, Z. Li, W. Wang and H. Zhang, Food Chem., 2020, 317, 126459 CrossRef CAS PubMed.
- K. M. Song, M. Cho, H. Jo, K. Min, S. H. Jeon, T. Kim, M. S. Han, J. K. Ku and C. Ban, Anal. Biochem., 2011, 415, 175–181 CrossRef CAS PubMed.
- R. Bala, S. Dhingra, M. Kumar, K. Bansal, S. Mittal, R. K. Sharma and N. Wangoo, Chem. Eng. J., 2017, 311, 111–116 CrossRef CAS.
- S. Malhotra, A. K. Pandey, Y. S. Rajput and R. Sharma, J. Mol. Recognit., 2014, 27, 493–500 CrossRef CAS PubMed.
- G. Liu, M. Lu, X. Huang, T. Li and D. Xu, Sensors, 2018, 8(12), 4166 CrossRef PubMed.
- C. Yang, Y. Wang, J. L. Marty and X. Yang, Biosens. Bioelectron., 2011, 26, 2724–2727 CrossRef CAS PubMed.
- Y. Dong, T. Zhang, X. Lin, J. Feng, F. Luo, H. Gao, Y. Wu, R. Deng and Q. He, Microchim. Acta, 2020, 187, 1–18 CrossRef.
- Y. Du, Y. Zhou, Y. Wen, X. Bian, Y. Xie, W. Zhang, G. Liu and J. Yan, Microchim. Acta, 2019, 186, 1–10 CrossRef.
- A. K. Sharma and J. M. Heemstra, J. Am. Chem. Soc., 2011, 133, 12426–12429 CrossRef CAS PubMed.
- A. K. Sharma, A. D. Kent and J. M. Heemstra, Anal. Chem., 2012, 84, 6104–6109 CrossRef CAS PubMed.
- X. Gong, X. Li, T. Qing, P. Zhang and B. Feng, Analyst, 2019, 144, 1948–1954 RSC.
- H. B. Wang, L. H. Ma, B. Y. Fang, Y. Di Zhao and X. Bin Hu, Colloids Surf., B, 2018, 169, 305–312 CrossRef CAS PubMed.
- M. Amouzadeh Tabrizi, J. Ferré-Borrull and L. F. Marsal, Sens. Actuators, B, 2020, 321, 128314 CrossRef CAS.
- C. E. McGhee, K. Y. Loh and Y. Lu, Curr. Opin. Biotechnol., 2017, 45, 191–201 CrossRef CAS PubMed.
- Y. Liu, J. Canoura, O. Alkhamis and Y. Xiao, ACS Appl. Mater. Interfaces, 2021, 13, 9491–9499 CrossRef CAS PubMed.
- L. Wang, F. Zhu, M. Chen, Y. Q. Zhu, J. Xiao, H. Yang and X. Chen, Food Chem., 2019, 271, 581–587 CrossRef CAS PubMed.
- L. Wang, F. Zhu, M. Chen, Y. Q. Zhu, J. Xiao, H. Yang and X. Chen, Food Chem., 2019, 271, 581–587 CrossRef CAS PubMed.
- X. Yu, Y. Lin, X. Wang, L. Xu, Z. Wang and F. F. Fu, Microchim. Acta, 2018, 185, 1–9 CrossRef PubMed.
- R. Stoltenburg, C. Reinemann and B. Strehlitz, Anal. Bioanal. Chem., 2005, 383, 83–91 CrossRef CAS PubMed.
- J. Canoura, Z. Wang, H. Yu, O. Alkhamis, F. Fu and Y. Xiao, J. Am. Chem. Soc., 2018, 140, 9961–9971 CrossRef CAS PubMed.
- M. Kim, S. M. Ko, C. Lee, J. Son, J. Kim, J. M. Kim and J. M. Nam, Anal. Chem., 2019, 91, 10448–10457 CrossRef PubMed.
- Q. Chen, R. Sheng, P. Wang, Q. Ouyang, A. Wang, S. Ali, M. Zareef and M. M. Hassan, Spectrochim. Acta, Part A, 2020, 241, 118654 CrossRef CAS.
- E. H. Lee and A. Son, Chem. Eng. J., 2019, 359, 1493–1501 CrossRef CAS.
- X. Weng and S. Neethirajan, Biosens. Bioelectron., 2016, 85, 649–656 CrossRef CAS PubMed.
- M. N. Stojanovic, P. de Prada and D. W. Landry, J. Am. Chem. Soc., 2000, 122, 11547–11548 CrossRef CAS PubMed.
- A. S. F. Belal, A. Ismail, M. M. Elnaggar and T. S. Belal, Spectrochim. Acta, Part A, 2018, 205, 48–54 CrossRef CAS PubMed.
- J. A. Cruz-Aguado and G. Penner, J. Agric. Food Chem., 2008, 56, 10456–10461 CrossRef CAS PubMed.
- J. A. Cruz-Aguado and G. Penner, Anal. Chem., 2008, 80, 8853–8855 CrossRef CAS PubMed.
- E. S. Coonahan, K. A. Yang, S. Pecic, M. De Vos, T. E. Wellems, M. P. Fay, J. F. Andersen, J. Tarning and C. A. Long, Sci. Transl. Med., 2021, 13, 1535 CrossRef PubMed.
- M. Li, H. Lin, S. K. Paidi, N. Mesyngier, S. Preheim and I. Barman, ACS Sens., 2020, 5, 1419–1426 CrossRef CAS PubMed.
- J. Li and Y. Lu, J. Am. Chem. Soc., 2000, 122, 10466–10467 CrossRef CAS.
- A. Ravikumar, P. Panneerselvam and K. Radhakrishnan, Microchim. Acta, 2017, 185, 1–8 Search PubMed.
- X. Fang, Y. Liu, L. Jimenez, Y. Duan, G. B. Adkins, L. Qiao, B. Liu and W. Zhong, Anal. Chem., 2017, 89, 11758–11764 CrossRef CAS PubMed.
- S. H. Lee, J. H. Choe, C. Kim, S. Bae, J. S. Kim, Q. H. Park and M. Seo, Sens. Actuators, B, 2020, 310, 127841 CrossRef CAS.
- C. Wang, L. Liu and Q. Zhao, ACS Sens., 2020, 5, 3246–3253 CrossRef CAS PubMed.
- E. P. Simão, R. Cao-Milán, G. Costa-Pedro, C. P. De Melo, R. Cao, M. D. L. Oliveira and C. A. S. Andrade, Electroanalysis, 2017, 29, 2268–2275 CrossRef.
- M. Negahdary, Talanta, 2020, 216 Search PubMed.
- K. An, X. Lu, C. Wang, J. Qian, Q. Chen, N. Hao and K. Wang, Anal. Chem., 2019, 91, 8660–8666 CrossRef CAS PubMed.
- S. Eissa, A. Ng, M. Siaj and M. Zourob, Anal. Chem., 2014, 86, 7551–7557 CrossRef CAS PubMed.
- H. Gao, J. Zhao, Y. Huang, X. Cheng, S. Wang, Y. Han, Y. Xiao and X. Lou, Anal. Chem., 2019, 91, 14 Search PubMed.
- Y. Xiao, A. A. Rowe and K. W. Plaxco, J. Am. Chem. Soc., 2007, 129, 262–263 CrossRef CAS PubMed.
- W. Yun, J. Jiang, D. Cai, X. Wang, G. Sang, J. Liao, T. Lu and K. Yan, RSC Adv., 2016, 6, 3960–3966 RSC.
- L. S. Liu, F. Wang, Y. Ge and P. K. Lo, ACS Appl. Mater. Interfaces, 2020, 13, 9329–9358 CrossRef PubMed.
- M. P. Bilibana, M. Citartan, T. S. Yeoh, T. S. Rozhdestvensky and T. H. Tang, J. Nucleic Acids, 2017, 2017 Search PubMed.
- H. Song, J. A. You, C. Chen, H. Zhang, X. Z. Ji, C. Li, Y. Yang, N. Xu and J. Huang, J. Environ. Chem. Eng., 2016, 4, 4653–4660 CrossRef CAS.
- J. Wang, H. Shen, X. Hu, Y. Li, Z. Li, J. Xu, X. Song, H. Zeng, Q. Yuan, J. Wang, H. Shen, X. Hu, Y. Li, Z. Li, J. Xu, Q. Yuan, X. Song and H. Zeng, Adv. Sci., 2016, 3, 1500289 CrossRef PubMed.
- Y. S. Kim, J. H. Niazi, Y. J. Chae, U. R. Ko and M. B. Gu, Macromol. Rapid Commun., 2011, 32, 1169–1173 CrossRef CAS PubMed.
- X. Hu, L. Mu, Q. Zhou, J. Wen and J. Pawliszyn, Environ. Sci. Technol., 2011, 45, 4890–4895 CrossRef CAS PubMed.
- E. O. Ezugbe and S. Rathilal, Membranes, 2020, 10, 89 CrossRef CAS PubMed.
- M. A. Romero-Reyes and J. M. Heemstra, ACS Mater. Lett., 2019, 1, 568–572 CrossRef CAS.
- M. A. Romero-Reyes and J. M. Heemstra, Bioconjugate Chem., 2021, 32, 2043–2051 CrossRef CAS PubMed.
- M. A. Romero-Reyes, K. Patterson and J. Heemstra, ChemRxiv, 2022 DOI:10.26434/chemrxiv-2022-hf3gd.
| This journal is © The Royal Society of Chemistry 2022 |