
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in micro-/nanostructure array integrated microfluidic devices for efficient separation of circulating tumor cells
Hanyue Kang a,
Yuting Xionga,
Liang Mab,
Tongqing Yanga and
Xiaobin Xu*a
a,
Yuting Xionga,
Liang Mab,
Tongqing Yanga and
Xiaobin Xu*a
aSchool of Materials Science and Engineering, Tongji University, Shanghai, 201804 China. E-mail: xiaobinxu@tongji.edu.cn
bState Key Laboratory of Fluid Power and Mechatronic Systems, Zhejiang University, Hangzhou 310058, China. E-mail: liangma@zju.edu.cn
First published on 6th December 2022
Abstract
Circulating tumor cells (CTCs) released from the primary tumor to peripheral blood are promising targets for liquid biopsies. Their biological information is vital for early cancer detection, efficacy assessment, and prognostic monitoring. Despite the tremendous clinical applications of CTCs, development of effective separation techniques are still demanding. Traditional separation methods usually use batch processing for enrichment, which inevitably destroy cell integrity and affect the complete information acquisition. Considering the rarity and heterogeneity of CTCs, it is urgent to develop effective separation methods. Microfluidic chips with precise fluid control at the micron level are promising devices for CTC separation. Their further combination with micro-/nanostructure arrays adds more biomolecule binding sites and exhibit unique fluid barrier effect, which significantly improve the CTC capture efficiency, purity, and sensitivity. This review summarized the recent advances in micro-/nanostructure array integrated microfluidic devices for CTC separation, including microrods, nanowires, and 3D micro-/nanostructures. The mechanisms by which these structures contribute to improved capture efficiency are discussed. Two major categories of separation methods, based on the physical and biological properties of CTCs, are discussed separately. Physical separation includes the design and preparation of micro-/nanostructure arrays, while chemical separation additionally involves the selection and modification of specific capture probes. These emerging technologies are expected to become powerful tools for disease diagnosis in the future.
1. Introduction
Circulating tumor cells (CTCs), first discovered by Ashworth, are malignant tumor cells shed from solid tumors into circulation.5 In the peripheral blood, they may be free individual cells or multicell clumps that lodge on distal organs and generate metastatic tumors.7,8 About 90% of cancer patients die from tumor metastasis, so early diagnosis and treatment are crucial to reducing cancer mortality.1,10In the early stages of tumor growth, CTCs are present in peripheral blood.11,12 Their concentration is an indicator to evaluate the chemotherapy and patient life expectancy.13,14 The emerging liquid biopsy is a method for diagnosing and monitoring cancer by detecting CTCs or circulating tumor DNA (ctDNA) in blood. Since CTCs inherit some characteristics from the source tumors, which serve for analyzing their origin, location, and nature.18,19 Through systematic sample collection, liquid biopsy can get real-time information about primary tissues, which helps to elucidate metastatic cascade mechanisms and select treatment plans.21–23 The follow-up culture of patients' CTCs also helps personalize anticancer-agent screening. Moreover, the liquid biopsy is facile and non-invasive, prolonging the survival of patients and avoiding the pain from puncture biopsy.25,26 As a result, CTC detection has great potential for early tumor diagnosis, personalized treatment, and patient prognosis.29
In recent years, CTCs have been adjunct to breast cancer screening and prognostic markers for various cancers.31–33 The primary challenge that hinders the clinical application is the lack of practical technologies to separate the low abundance of CTCs.34–36 For patients with early-stage cancer, there might be one CTC mixed with five billion red blood cells (RBCs) and ten million white blood cells (WBCs). Second, the similar size and deformability of CTCs and WBCs set obstacles to separation. Third, separation methods are required to handle very small volumes of samples. With advanced knowledge of CTCs, their applications range from simple counting to cell sequencing, which needs to ensure cell integrity. As a result, high-throughput and high-purity methods for enrichment and separation of intact CTC are desperately needed.
Many techniques have been exploited for CTC separation based on physical properties or affinity.38 Among them, density gradient centrifugation is a simple, rapid, label-free physical separation method. Cells in whole blood are divided into several layers based on density differences.40 The denser WBCs and RBCs precipitate at the bottom of the tube, while CTCs float in the upper layer of the medium. This method retains the viability of CTCs for subsequent culture but tends to leave CTCs in the plasma layer. Filtration is another efficient and straightforward approach regarding cell size.41,42 Simple and high-throughput filtration of CTCs can be achieved according to their phenotypes. Some small CTCs may be filtered out, while larger WBCs are collected by mistake. Other physical strategies such as electrorotation and dielectrophoresis utilize the differences in surface charge and polarization rate between CTCs and WBCs.43 They reduce CTC damage to a large extent but require a long sample processing time. Immunoaffinity is an effective and high-purity chemical separation method based on the specific binding of the surface antigens and antibodies.44 The CellSearch system is the world's first commercially available product approved by U.S. Food and Drug Administration (FDA) for CTC detection and enumeration for malignant disease management. The CTCs in peripheral blood are captured by magnetic nanoparticles bound to specific antibodies. Although non-specific capture of blood cells is avoided, the antigen expression levels vary on the subtypes of tumor cells may cause CTC loss.45 A summary of the above methods is given in Table 1.
| Strategy | Approach/substrates | Separation mechanism | Benefits | Limitations | Ref. |
|---|---|---|---|---|---|
| Conventional approaches | Density gradient centrifugation | Size density | Simple operation | Large sample volume | 51 and 52 |
| Not limited by surface markers | Blood cell contamination | ||||
| Low sensitivity from CTC loss | |||||
| Filtration | Size | Simple operation | WBC contamination | 53 and 54 | |
| Not limited by surface markers | Loss of small CTCs | ||||
| CTC deformation | |||||
| Electrorotation and dielectrophoresis | Dielectric property | Label-free | Low efficiency | 55 and 56 | |
| High purity and viability | |||||
| Immunocapture | Affinity | High specificity, purity and sensitivity | High cost | 57 and 58 | |
| Limited antibody availability | |||||
| Complicated process | |||||
| Microfluidic with micro-/nanostructure (physical approach) | Microrods | Size | Label-free | Complicated nanofabrication | 1 and 4 |
| Low cost | |||||
| DLD | Simple operation | Low specificity and purity | 15 and 59 | ||
| 3D micro-/nanostructures | Adhesion | High throughput | 30 | ||
| Microfluidic with micro-/nanostructure (chemical approach) | DLD-patterned microrods | Size affinity | High capture efficiency, specificity and purity | High cost | 37 and 39 |
| Microrods | Affinity | Limited antibody availability | 20, 28, 60 and 61 | ||
| Nanowires | 3, 9, 17, 27 and 62 | ||||
| 3D micro-/nanostructures | Complicated nanofabrication and modification | 2, 6, 16 and 24 |
Microfluidic chip often called “lab-on-a-chip,” has been used to capture cells or create tumor models due to their advantages, such as high integration, controllable cell manipulation, and convenient viewing.46–49 Compared to conventional methods, the small injection volumes (10−6 to 10−12 L) of microfluidic chips reduce sample and reagent consumption.50 The microchannels' design and the fluid behavior's manipulation allow CTC separation, counting, and release integration on a single chip. The integration avoids loss or contamination from eluting and transferring samples. The surface forces are significantly amplified at the microscale allowing for an effective increase in reaction efficiency, which makes rapid separation and analysis possible. In addition, the portable nature of microfluidic chips has the potential to become point-of-care devices.
Researchers found that microfluidic channels' small surface areas limit their capture capacity. With the development of nanotechnology, the application of micro-/nanostructures to biomedical applications has become a significant trend in today's research. Their small size, high specific surface area, and specially designed structures bring unique physicochemical properties. Compared to flat substrates, the micro-/nanostructure arrays close to cell sizes provide more binding sites, lower cell rolling velocity, and facilitate CTCs-array collision.63,64 The micro-/nanostructures-embedded microfluidic chip often includes a micro-/nanostructured substrate, a manifold to enclose the chip, and a pump for sample injection. Many structures have been designed, such as microrods, microtubes, nanowires, nanofibers, etc. Separation techniques relying on these structures can be mainly classified as physical and chemical. The physical separation method is based on the significant differences in various physical properties (e.g., size, density, and dielectric property) of CTCs and blood cells. In contrast, chemical separation is achieved through various cancer biomarkers on the membranes. Different methods can also be integrated on a single chip for higher performance.
While there have been many reviews on separation CTCs by microfluidic chips, most focus on presenting the differences between physical and chemical separation methods, with little discussion on the role of micro-/nanostructures in improving separation efficiency. With the continuous development of micro and nano fabrication and its deeper integration into the biomedical field, understanding the mechanisms by which different micro and nanostructures promote improved capture efficiency is essential.
This article focuses on the recent advances in micro-/nanostructure integrated microfluidic chips for CTC separation. Three micro-/nanostructures are introduced, including microrods, nanowires, and 3D micro-/nanostructures (Fig. 1). On this basis, physical and chemical separation methods are discussed in subsequent sections. The classic and innovative literature summarized in this review will shed light on improving the efficiency of CTC separation.
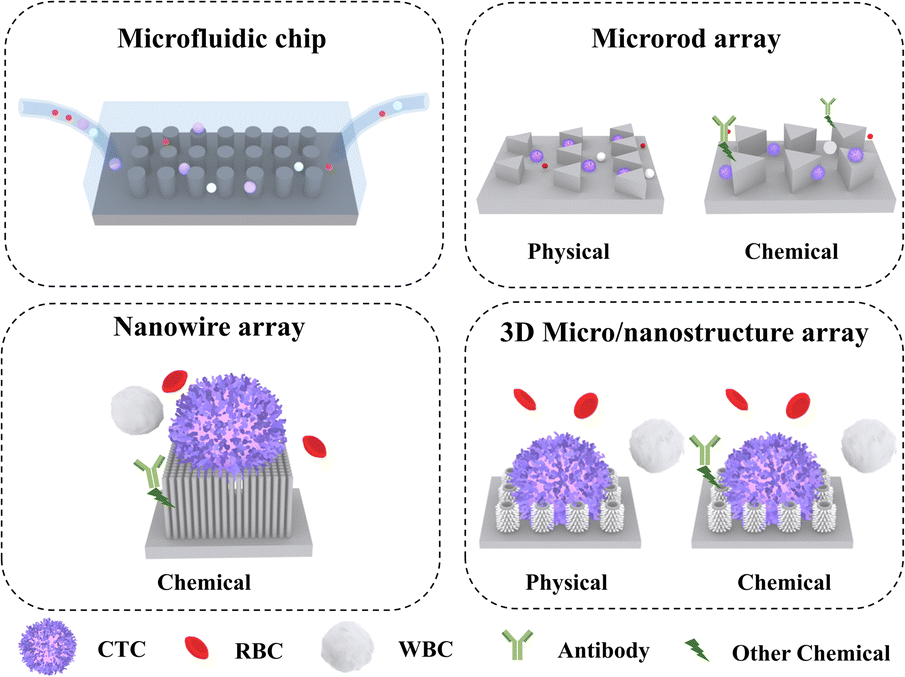 | ||
| Fig. 1 Schematic illustrations of (A) micro-/nanostructure integrated microfluidic device for CTC capture. (B) 1D microrod arrays. (C) 1D nanowire array. (D) 3D hierarchical micro-/nanostructure arrays. | ||
2. Microrod array integrated microfluidic devices
Microrods are one-dimensional (1D) microstructures with a length-to-diameter aspect ratio of 3–5. They are typically fabricated through photolithography and soft lithography on either rigid materials (such as Si,65,66 glass,67 PMMA68,69) or soft materials (such as polydimethylsiloxane, PDMS70,71). The number and topology of microrods can be designed to realize label-free separation based on biophysical properties or specific capture with modified chemical markers.72 When combined with microfluidic devices, the physical interaction of fluid dynamics with the micro-/nanostructure can improve CTC sorting efficiency.73 In the following, we discussed the two major types of CTC separating mechanisms based on microrods, one is physical, and the other is chemical.2.1 CTC physical separation by microrod arrays
Some representative physical separation methods achieved remarkable results, such as membrane or microstructure filtration, hydrodynamic separation (e.g., deterministic lateral displacement and inertial focus), and dielectrophoresis. The physical separation methods are low-cost techniques that rely solely on the structure without biochemical modifications. They preserve the integrity and viability of CTCs, which helps genetic technology obtain accurate cancer information. However, WBCs close in size to CTCs may become interfering factors. This section mainly focuses on microrod arrays for filtration and deterministic lateral displacement.As shown in Fig. 2A, Park et al. developed a matrix of tapered constrictions based on differences in cellular deformability.1 The funnel opening size gradually decreases from the bottom entrance row of 18 μm to the top exit row of 2 μm. The funnel opening size gradually decreases from the bottom entrance row to the top exit row. With the blood sample continuous oscillatory flow through this array, each cell is deformed through a funnel-shaped contraction. When reaching the row of 6 μm constrictions, small RBCs or easily deformed WBCs cross the funnel constriction. At the same time, CTCs are constrained between two rows and wait to be collected by a constant rightward flow. The separation efficiency of the device remains relatively stable at 93–96%. Since oscillatory flow avoids the clogging of high cell density samples, the pre-treatment dilution of the sample is omitted.
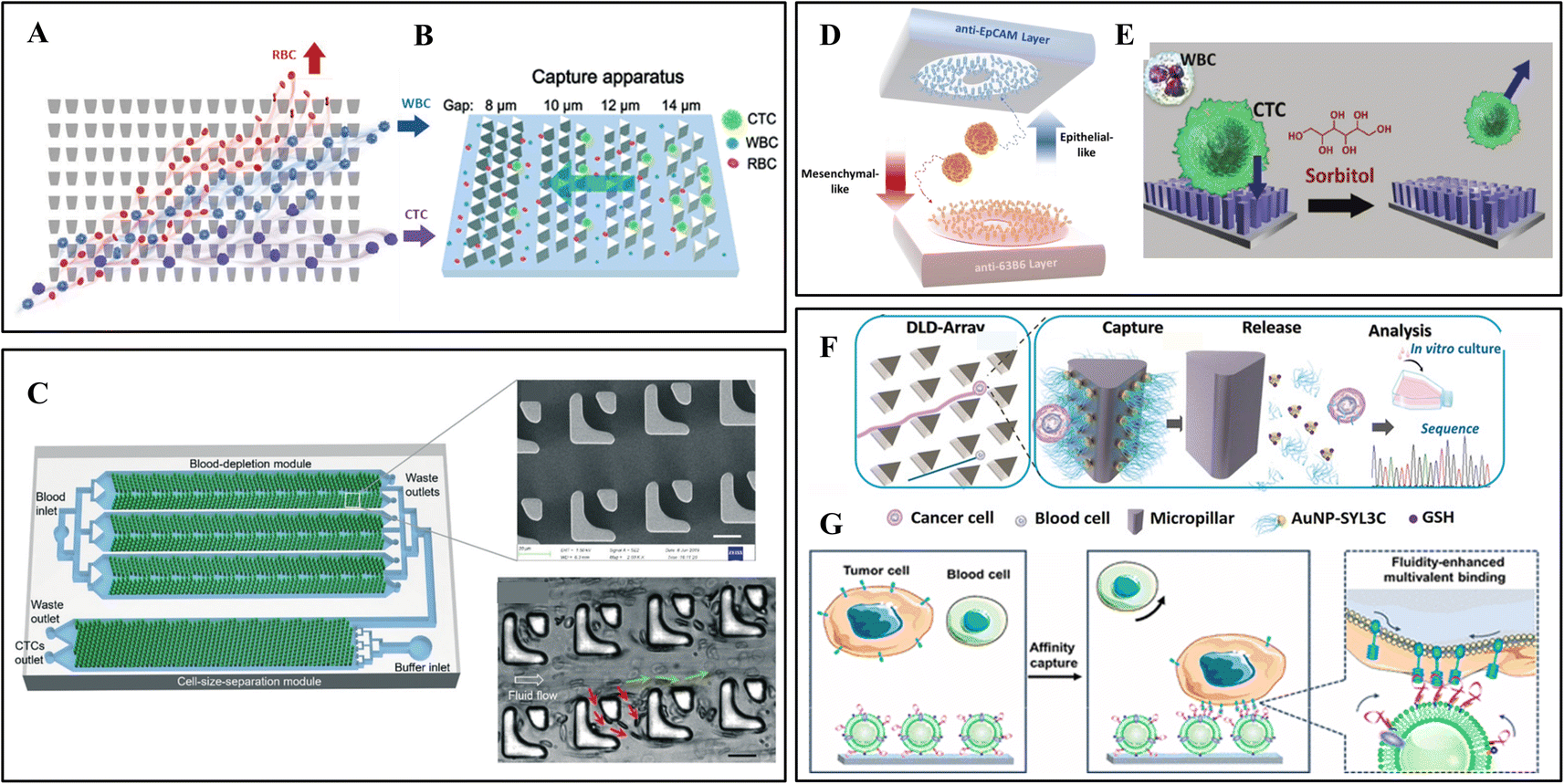 | ||
| Fig. 2 Microrod array integrated microfluidic devices. (A) CTCs filter through a matrix of funnel constrictions. Reprinted with permission from ref. 1. Copyright (2016) John Wiley and Sons. (B) Streamline focus enrichment and triangular prism array for CTC filtration. Reprinted with permission from ref. 4. Copyright (2020) Royal Society of Chemistry. (C) Cascaded filter DLD microchip for size-based CTC separation. Scale bar, 20 μm. Reprinted with permission from ref. 15. Copyright (2020) Royal Society of Chemistry. (D) Dual-immunopatterned (DIP) device modified by anti-EpCAM and anti-63B6. Reprinted with permission from ref. 20. Copyright (2018) Elsevier. (E) CTC capture on poly(3,4-ethylene-dioxythiophene)s (PEDOT) rods array and release by competition with sorbitol. Reprinted with permission from ref. 28. Copyright (2018) John Wiley and Sons. (F) DLD-patterned triangular micropillars modified with multivalent aptamer-functionalized nanospheres for CTC capture and release. Reprinted with permission from ref. 37. Copyright (2019) John Wiley and Sons. (G) DLD-patterned triangular micropillars decorated with aptamer-functionalized leukocyte membrane nanovesicles. Reprinted with permission from ref. 39. Copyright (2020) American Chemical Society. | ||
In addition to the separation according to cell deformability, Lu et al. proposed a size-based device combining a streamline-based focus-separation deceleration design with a triangular prism array (Fig. 2B).4 As the blood flows through the seven focus-separation apparatuses, CTCs are continuously pushed into the main channel. In contrast, some blood cells are removed through the branch channel. This design concentrates CTCs and reduces contamination of blood cells. Subsequently, the fluid flows into the capture apparatus with triangular barrier layer structures of decreasing gaps (14, 12, 10, 8 μm). Different sizes of CTCs were trapped while the remaining blood cells flowed through.
The microrod filtration is a label-free platform that overcomes dependence on specific capture probes. Without complicated modification operations, this method is simple, low cost, and maintains the integrity of CTCs for downstream analysis. However, clogging is inevitable when processing samples with high cell density. In addition, the flow rate must be carefully controlled to avoid cell damage.
Based on this theory, Loutherback et al. designed a triangular-microrod DLD array for continuous-flow capture of CTCs.59 They increased the gap of microrods to 42 μm, corresponding to a Dc of 7 μm, and changed the shape to a triangle, which provides greater throughput and reduces the possibility of clogging. For viscous blood samples, 85% of CTCs were successfully separated at a flow rate of 10 mL min−1. Later, Liu et al. designed a high-throughput cascaded filter-DLD chip, as illustrated in Fig. 2C.15 The filter unit contains two spaced micro-posts. When the sample flows through the filter-DLD array, smaller blood cells are partially removed or move in zigzag mode (red arrows), while CTCs and WBCs move in bumping mode (green arrows). In this chip, the filter-DLD arrays were integrated into two modules. Three sets of symmetric arrays in the blood-depletion module enrich CTCs and a portion of WBCs. Then, large CTCs were collected in the cell-size-separation module with increasing Dc from 8 μm to 22 μm. Compared with conventional DLD structures, filter-DLD shows smaller Dc and a higher depletion rate of smaller cells.
The deterministic lateral displacement also shows the advantages of simple operation, low cost, and minor damage to CTCs. Nevertheless, there are some disadvantages to overcome. The complex blood environment makes cellular interactions challenging to control, and the efficiency will be reduced with high cell concentration.
2.2 CTC separation by chemical modified microrod arrays
In comparison, chemical-based separation relies on binding specific capture probes and cancer biomarkers, which are molecular changes measured between normal and cancerous tissues. The most widely used method immobilizes specific capture probes inside the microchannels. When the sample is injected into the chip, CTCs will be captured while the remaining blood cells flow out. To extend capture capacity, microrod arrays on flat substrates provide larger binding sites and facilitate cell–surface interactions.In 2007, Nagrath et al. prepared an antibody-coated microposts platform to efficiently and selectively isolate CTCs from peripheral blood.60 After modifying anti-EpCAM, this chip identified CTCs for metastatic lung, prostate, pancreatic, breast, and colon cancers with a 99% success rate. Conventional silicon microrod arrays have poor optical transparency and require complex processing. For these reasons, Yan et al. prepared a PDMS microrod array device with high optical transparency using soft lithography and rapid prototyping.61 This device shows high capture efficiency for different cancer cells and allows direct monitoring of the captured cells under the microscope.
However, CTCs may undergo epithelial–mesenchymal transition (EMT), which downregulates the EpCAM expression and leads to false-negative results. Therefore, some researchers explored antibody cocktails to capture the missed CTCs. As shown in Fig. 2D, Kang et al. designed a dual-immunopatterned (DIP) device composed of aligned two layers with rings of polar arrayed microrods.20 The rings are alternately formed from the two layers and maintain the same spacing. The upper and bottom layers were modified with anti-EpCAM for epithelial CTCs and newly developed anti-63B6 for mesenchymal CTCs. The difference in the antigens expressed on the CTC surface can be indicated by comparing their numbers on the two layers. This device achieved good results in separating heterogeneous subtype of CTCs in blood.
Antibody capture avoids non-specific adsorption of blood cells, achieving high efficiency and purity. The release usually requires hydrolytic digestion of the antigen on the cell membrane, hindering subsequent molecular characteristics analysis.80 Shen et al. proposed a method for gentle release of CTC based on competitive binding to other reagents.28 They prepared a phenylboronic acid (PBA)-grafted nanoimprinted poly(3,4-ethylene-dioxythiophene)s (PEDOT) rods array and modified anti-EpCAM via PBA–oligosaccharide bonding (Fig. 2E). The captured CTCs can be released by exposure to sorbitol, which has a stronger affinity to PBA. Through this process, CTCs from blood were substantially purified with well-preserved RNA.
Song et al. from Chaoyong Yang's group developed a multivalent aptamer-modified micropillar array (Fig. 2F).37 Under the principle of DLD, each row of the micropillars has a fixed shift, CTCs larger than the Dc of 13 μm will have constant cell–pillar collisions, while smaller blood cells will migrate in the direction of flow. The triangular micropillars create a smooth hydrodynamic force gradient to slow the flow velocity and achieve maximum CTC attachment. Gold nanoparticles modified on micropillars contain aptamers that bind to cell membranes. The captured CTCs were released nondestructively through the thiol exchange reaction at the AuNP–aptamer interface. However, the non-specifically captured blood cells remained on the surface, which improved the purity. Later, this group modified aptamer-functionalized leukocyte membrane nanovesicles on the triangular micropillars (Fig. 2G).39 Nanovesicles were obtained through disruption and extrusion of biotinylated WBCs and inherited the nature of surface membranes. The blood cell resistance of the membrane reduces the background cells, and the membrane fluidity allows lateral rearrangement of aptamers to achieve high-affinity binding. Moreover, the captured CTCs show excellent CTC viability (97.6%) due to the cushioning of the soft membranes.
Although the aptamers demonstrated excellent capture and release capabilities, they were not as widely used as expected. The primary reason is that their performance is affected by the complex blood environment. In whole blood, they may be rapidly degraded by nucleases or influenced by high-abundance blood cells and proteins.85,86 Therefore, it is necessary to synthesize aptamers that remain effective in the complex blood environment.
3. Nanowire integrated microfluidic devices
Nanostructures in the tissue microenvironment have a significant role in modulating cellular behavior and biochemical support. Nanowires are one-dimensional (1D) microstructures with a high length-to-diameter aspect ratio, usually greater than 1000. Nanowires embedded in substrates precisely mimic the characteristics of cellular microstructures such as extracellular matrix, which in turn are used for cellular behavior studies in biomedical applications. Their diameters are close to the sizes of cellular pseudopods, greatly enhancing cell–substrate adhesion.87–89 By precisely controlling the length, density and arrangement of nanowires, multiple feature size structural substrates can be designed to explore the relationship with CTC capture efficiency.The nanowires are usually obtained by photolithography combined with wet etching techniques, hydrothermal growth, chemical vapor deposition, etc. These techniques are now relatively mature and easy to operate. A typical nanowire microfluidic device for CTC capture is the “NanoVelcro chip” cell affinity substrate concept pioneered by Tseng Lab at UCLA.90 This chip can achieve dynamic and efficient CTC capture by constructing a trapping agent-coated nanostructured substrate. In addition to the common SiNWs, the concept of “NanoVelcro” has been applied to other nanosubstrates, such as conjugated polymer nanofeatures and nanofibers.91,92
The NanoVelcro chip developed by this team has undergone three generations of innovation for different clinical applications. The first generation NanoVelcro chip comprises a patterned SiNW substrate and an overlaid PDMS chaotic mixer, as shown in Fig. 3A.3 The innovative chaotic mixer induced the vertical flow of the sample to enhance CTC–SiNWS contact frequency. The optimized condition achieved more than 85% capture rate in artificial samples. This device was further applied to CTC collection and enumeration to monitor the treatment of cancer patients.93
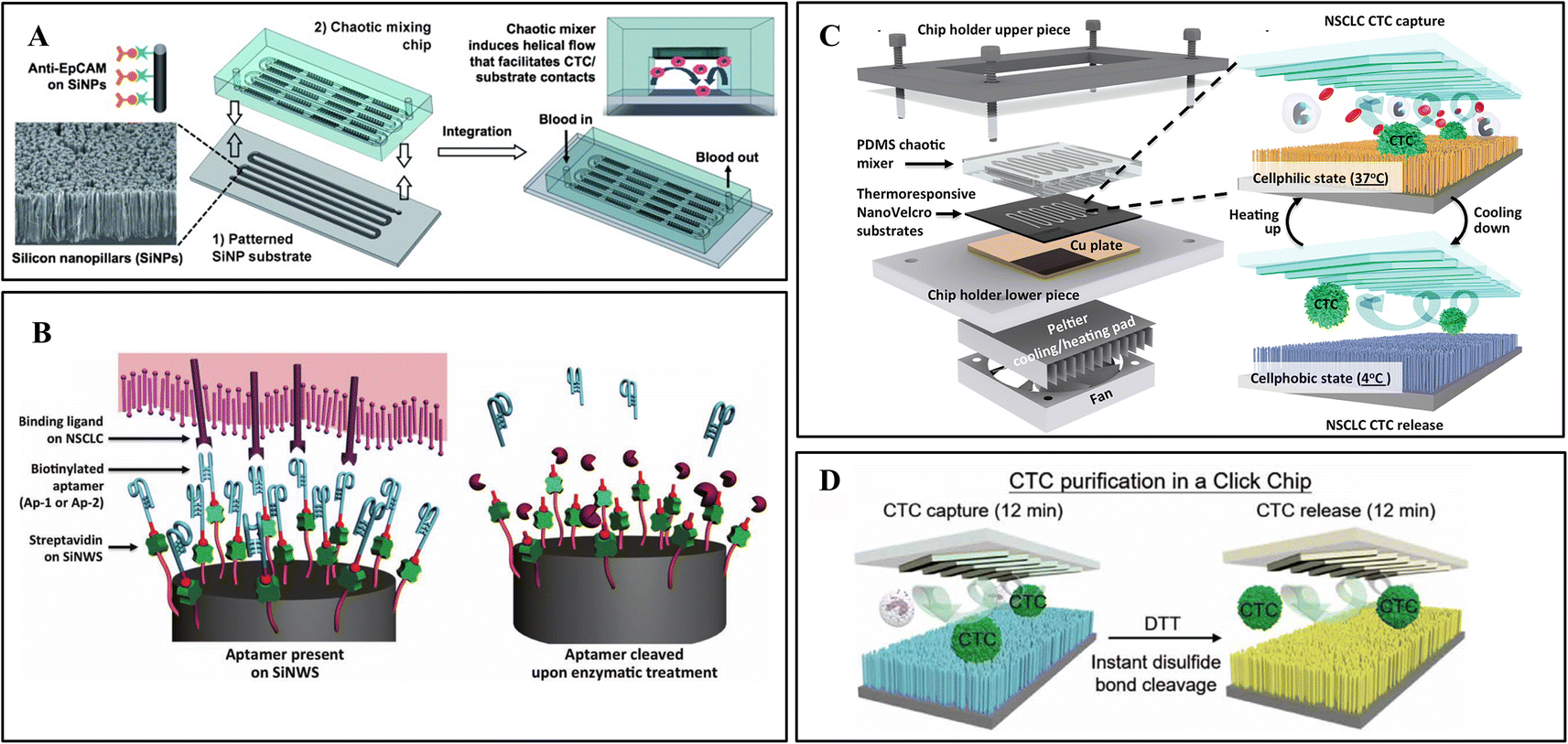 | ||
| Fig. 3 Nanowire integrated microfluidic devices. (A) Antibody modified silicon nanowires with chaotic mixer for immunoaffinity capture. Reprinted with permission from ref. 3. Copyright (2011) John Wiley and Sons. (B) CTC capture and release from aptamer-coated SiNWs. Reprinted with permission from ref. 9. Copyright (2013) John Wiley and Sons. (C) Digitally programmed thermoresponsive NanoVelcro CTC purification system for precisely temperature-dependent capture and release. Reprinted with permission from ref. 17. Copyright (2014) American Chemical Society. (D) The disulfide cleavage-driven release on covalent chemistry-based silicon nanowires. Reprinted with permission from ref. 27. Copyright (2019) American Association for the Advancement of Science. | ||
While NanoVelcro chips allow for efficient and reproducible CTC counting in a clinical setting, empowering NanoVelcro chips with cellular release remains challenging. A diagnostic assay platform capable of efficient capture and specific release of CTCs will pave the way for subsequent downstream applications such as molecular characterization and functional analysis. For release purposes, they introduced an improved first-generation NanoVelcro chip that replaces the antibody-based capture agent (i.e., anti-EpCAM) with two DNA aptamers, Ap-1 and Ap-2 (Fig. 3B).9 Genetically engineered endonucleases were used to specifically degrade the SiNW-grafted DNA aptamers, enabling CTC capture followed by release. Another group proposed a sandwich structured peptide–silicon nanowire (Pe–SiNWS)-based microfluidic device which first employs enzymatic release to collect CTC from blood samples.62 Pe–SiNWS facilitated specific capture, biorelease, and accurate analysis of CTC. This technology can achieve 95.6% CTC capture. After enzymatic digestion, good CTC purity of 28.5% and cell viability of 93.5% can be obtained.
To simplify the procedure and improve cell viability for clinical application, a digitally programmed thermoresponsive-NanoVelcro chip was developed.17 Thermally responsive polymer brushes, covalently-linked biotin group, and antibodies were modified on SiNWs.94 The thermosensitive nanonylon matrix can capture and release CTCs at 37 °C and 4 °C, respectively (Fig. 3C). The release is through internalizing antibodies from the conformational changes in the backbones of polymer brushes. The integrated thermal-electric Peltier cooling/heating system with a thermocouple sensor enables device temperature control. Gentle operating parameters minimize CTC viability and molecular integrity disruption by applying physiologically durable stimuli.
Recently, studies have shown that click chemistry can yield highly pure biomolecules.94 Based on the above research, Tseng lab developed a covalent chemistry-based capture/release platform (“Click Chip”).27 A pair of highly reactive click chemistry motifs (Tz and TCO) were modified on SiNWs and CTCs, leading to instant, high-purity, and irreversible CTC captures under mild conditions. Subsequently, a disulfide cleavage agent was utilized to release CTCs without non-specifically captured WBCs (Fig. 3D). The better-preserved CTC mRNA has been applied to downstream studies for evaluating treatment and monitoring disease progression.
4. 3D micro-/nanostructure array integrated microfluidic devices
Nanostructures are known to provide high specific surface area, thus allowing more immunoaffinity molecules to be immobilized on the surface. With the continuous development of nanotechnology and bionanotechnology, new innovative structures were introduced to improve the performance of biochips. Nowadays, three-dimensional hierarchical nanostructures with optimized performance are a hot topic of current research in enhancing biological cell capture. The 3D micro-/nanostructure here refers to the composition of basic structural units from 1D micro-/nanostructure. For higher capture efficiency, nanowires (e.g., TiO2, ZnO, ITO) are often combined with microrods to form hierarchical topography.90,95 The longitudinally and horizontally combined multi-dimensional forms of micro–nano structures can be generated through various techniques, such as photolithography, atomic layer deposition, multi-step chemical vapor deposition, hydrothermal growth or other combinatorial techniques.96,97 In addition, some researchers have been inspired by nature to prepare 3D micro-/nanostructures using natural structures such as gecko feet, rose petals, and butterfly wings, achieving good results.98,99Recent studies have shown that hierarchical nanostructured substrates exhibit further improved cell capture efficiency and device sensitivity compared to one-dimensional vertical micro–nanostructures (e.g., nanopillars, nanowires) and one-dimensional horizontal nanostructured (e.g., nanofibers) substrates.97 Firstly, the hierarchical nanostructures are better matched in size to the cellular filamentous pseudopods. Secondly, the horizontal branching of the structure can further enhance the interaction between the cellular filamentous pseudopods and the material, improving the contact with the fluid. Besides, the longitudinal structural branches act in concert to further rivet cells, providing additional physical binding sites for cell capture.
For instance, Qiu et al. proposed a 3D micro-/nanostructure from hydrothermally growing TiO2 nanorods on the sidewall of hexagonally patterned silicon microrods (Fig. 4A).2 After antibody modification, this device showed almost twice the capture efficiency of the pure Si nanorods at a range of cell densities. The highest capture efficiency of 76.7% ± 7.1% can be achieved for the artificial whole blood sample. In addition, the excellent biocompatibility of TiO2 nanorods enables the captured CTCs to grow and proliferate well in the microchannel, offering a possible approach for cancer diagnosis and personalized treatment aimed at patients with very low CTC abundance.
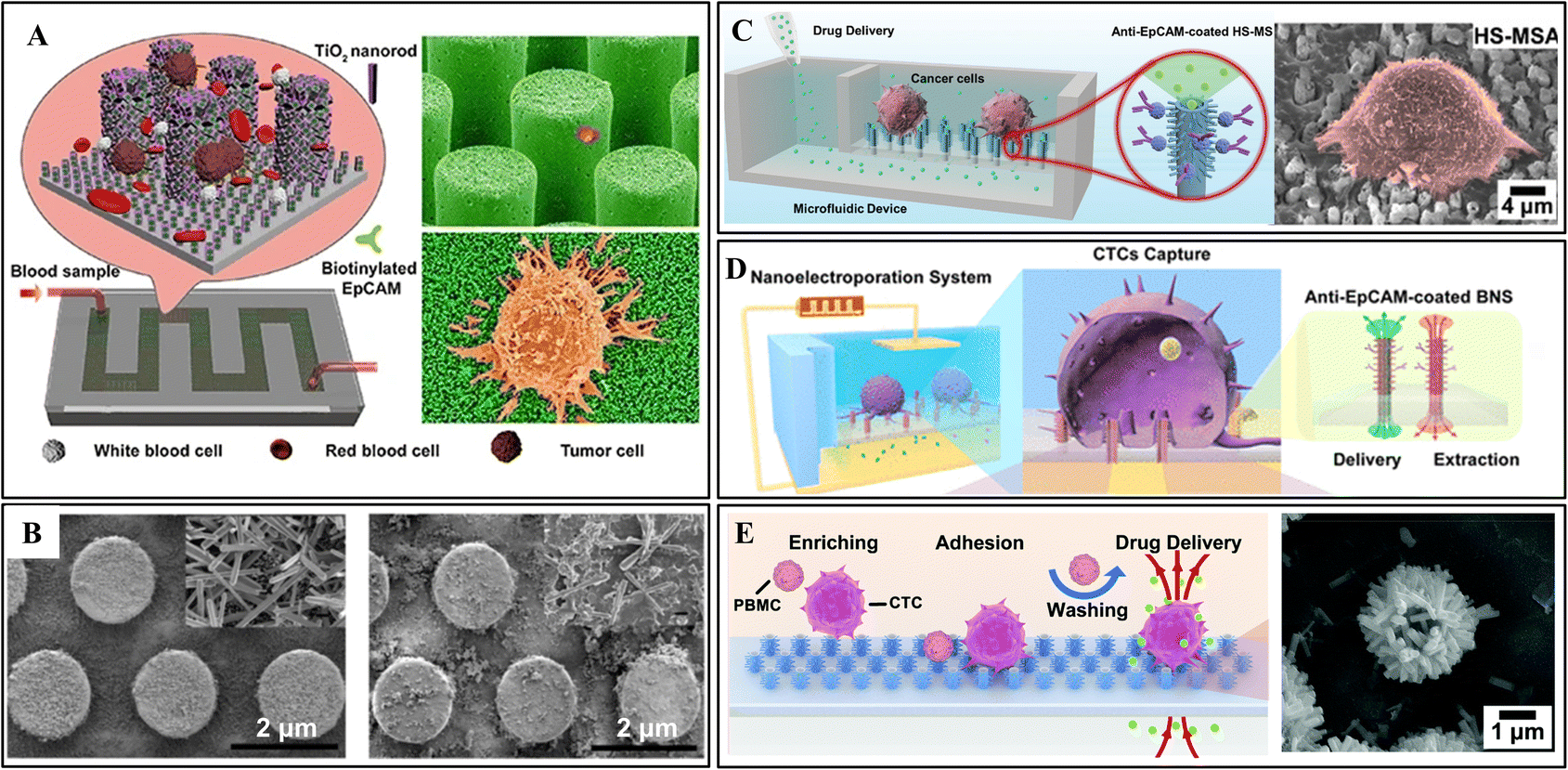 | ||
| Fig. 4 3D micro-/nanostructure integrated microfluidic devices. (A) Vertical growth of TiO2 nanorods on silicon nanopillars for immunoaffinity capture. Reprinted with permission from ref. 2. Copyright (2017) Springer Nature. (B) ZnO coated PDMS microrod array from the first synthesis cycle (left) and the second cycle (right). Reprinted with permission from ref. 6. Copyright (2018) American Chemical Society. (C) Hierarchical spiky microstraw array-integrated microfluidic device for immunoaffinity capture and in situ chemical manipulation. Reprinted with permission from ref. 16. Copyright (2019) John Wiley and Sons. (D) Branched nanostraw-electroporation platform for immunoaffinity capture, downstream regulation and monitoring intracellular activities. Reprinted with permission from ref. 24. Copyright (2019) American Chemical Society. (E) Hybrid TiO2/ZnO branched microtube array for antibody-free separation and in situ regulation. Reprinted with permission from ref. 30. Copyright (2020) Royal Society of Chemistry. | ||
In another study, Hui et al. prepared a self-sterilizing and reproducible ZnO-coated PDMS micrometer column array for home testing.6 Antibody mixture (anti-EpCAM and anti-vimentin) was engrafted to the structural surface to provide more accurate results. The ZnO nanograss enhances the interaction with cells and reduces the adhesion of WBCs. Moreover, ZnO shows robust antibacterial ability making sterile environments unnecessary. Ultimately, the captured CTCs could be released by dissolving a thin layer of ZnO in an acidic solution. As shown in Fig. 4B, the remaining seed layer can be used for the second preparation of ZnO coating. Cui et al. followed the same principle to prepare a ZnO-coated PDMS gear structure array.100 Besides high surface area, the gear structure provides a suitable location for CTC attachment. Due to the increased roughness of the ZnO coating, fully extended pseudopodia have been observed under high magnification SEM images. The captured CTC can also be released by dissolving ZnO in a mild acidic solution, ensuring high viability.
In recent years, cell in situ chemical manipulations has become the exploration target. Nicholas A. Melosh's group at Stanford University demonstrated alumina-coated polycarbonate nanostraws to deliver small molecules and ions into the cytosol.101 To avoid perforating cell membranes, they developed nondestructive electroporation delivery by opening transient pores during perforation and resealing the membrane after voltage removal.102 Subsequently, the nanostraws have been proven effective in delivering signaling ions and extracting proteins and mRNA.103,104
Inspired by their studies, Xi Xie's group fabricated a hierarchical spiky microstraw array (HS-MSA) for high-efficient CTC capture and drug delivery (Fig. 4C).16 The ZnO nanospikes were hydrothermally grown on polycarbonate microstraws. Then, an alumina layer was deposited on the surface to avoid potential cytotoxicity. After anti-EpCAM modification, high capture efficiency (≈84%) was achieved due to the closer contact interfaces provided by nanospikes. The hollow microstraws deliver drugs to local cells through the microfluidic channels, enabling release-free in situ cancer cell drug screening. Following this study, electroporation was also applied to allow intracellular drug delivery and content extraction (Fig. 4D).24
To break through the limitation of antibodies, they explored a hybrid TiO2/ZnO branched microtube array (HBMTA) for antibody-free capture of adherent tumor cells (Fig. 4E).30 After 6 h of incubation, cancer cells preferentially adhered to HBMTA due to the landing sites provided by the rough surface. In contrast, the blood cells freely floated in the sample and were washed off through fluidic shear.
5. Summary and prospect
CTCs have high clinical value as disease markers in cancer diversity research, diagnosis, treatment, and monitoring. In this review, we have summarized the representative works on the micro-/nanostructures array (microrods, nanowires, and 3D hybrid micro-/nanostructures) integrated microfluidic devices for CTC capture during recent years. The large surface area and fluidic perturbation phenomenon of these structures significantly improve CTC capture and screening. The captured CTCs have been applied to explore cancer mechanisms and solve complex biological problems. In recent years, microrod array integrated microfluidic devices using physical and chemical separation methods have enabled small-scale clinical applications. The most common application is CTC enumeration for helping diagnose, detecting cancer progression, and predicting poor prognosis.105–109 The genetic analysis of the captured CTCs provides effective treatment options.110 Besides, CTCs have also been applied to assess sensitivity to chemotherapeutic agents, which helps in the selection of appropriate drugs.111 However, the blockage generated by the physical separation, the loss by low-expression antigens in chemical separation, and the effects of stress forces on CTCs due to fluid perturbation make the large-scale clinical application of these techniques still challenging. In downstream applications, it is also critical to achieving controllable CTC release or single-site individual CTC release to investigate the heterogeneity without cell damage. Also, improve specific selectivity (e.g., missed detection due to low expression in mesenchymal cells) for genomic or protein profiling.112 In addition, a large number of clinical samples are required to ensure the feasibility, reproducibility, and sensitivity of microfluidics before it is introduced into clinical CTC analysis practice. Recently, researchers have strived for 3D micro-/nanostructures by hydrothermal growing or etching nanowires on the surface of microrods, which show better capture performance and high detection repeatability. The innovative bio-inspired 3D structures also open up avenues for CTC capture. The benefits and limitations of micro-/nanostructures mentioned in this paper are summarized in Table 1, and each device is shown in Table 2. In the last few years, 3D printing has emerged for fabricating truly 3D microfluidic devices in one step at a low cost.113,114 With technological change, microfluidic devices will move toward miniaturization and integration, eventually realizing integrated clinical applications for CTC enrichment, separation, and detection, such as single-cell sequencing, metabolic analysis, and drug sensitivity. Portable, miniaturized, and rapid clinical diagnosis and treatment of cancer may emerge in the future.| Structure | Modification | CTC source | Sample | Volume | Flow rate or time | Capture efficiency | Ref. |
|---|---|---|---|---|---|---|---|
| Microrod | None | UM-UC13 | Human blood | 5 mL | 1 mL h−1 | >90% | 1 |
| HeLa, MCF-7 | Rabbit blood | 2 mL | 40 mL h−1 | 95.8% ± 0.9% | 4 | ||
| MDA-MB-231 | 95.7% ± 3.5% | ||||||
| 96.3% ± 1.2% | |||||||
| NCI-H226 | 91.3% ± 3.2% | ||||||
| MCF10A | Human blood | 5 mL | 10 mL min−1 | >85% | 59 | ||
| A549, K562 | Human blood | 10 mL | 60 mL h−1 | >96% | 15 | ||
| Anti-EpCAM | NCI-H1650 | Human blood | n/a | 1 mL h−1 | 65% | 60 | |
| MCF-7 | PBS buffer | 1 mL | 1 mL h−1 | ∼88% | 61 | ||
| NCI-H1650 | Human blood | ||||||
| Anti-EpCAM | MCF-7 | PBS buffer | 1 mL | 1 mL h−1 | 93.81% ± 8.58% | 20 | |
| Anti-63B6 | MDA-MB-231 | Human blood | 95.13% ± 4.28% | ||||
| Anti-EpCAM | LNCap, PC3 | Human blood | 100 μL | 60 min | 72.5% ± 3.0% | 28 | |
| 22Rv1 | 75.2% ± 3.2% | ||||||
| 67.8% ± 1.7% | |||||||
| SYL3C aptamer | SW480 | PBS buffer | n/a | 1 mL h−1 | 86.7–89.4% | 37 | |
| LNCap, K-562 | |||||||
| KATO IIIl | Human blood | ||||||
| SW480, MCF-7 | DMEM | 0.35 mL h−1 | 84.3–91.3% | 39 | |||
| HCT 116 | Human blood | ||||||
| LNCap | |||||||
| Nanowire | Anti-EpCAM | MCF-7 | DMEM | n/a | 1 mL h−1 | >95% | 3 |
| PC3, T24 | Human blood | ||||||
| Ap1, Ap2 aptamer | A549, HCC827 | PBS buffer | 1 mL | 1 mL h−1 | >85% | 9 | |
| Human blood | |||||||
| Peptides CKAAKN | BxPC3 | Artificial sample | 200 μL | 1 mL h−1 | 95.6% | 62 | |
| Anti-EpCAM | H1957, H2228 | Artificial sample | n/a | 0.5 mL h−1 | 90–95% | 17 | |
| A549 | Human blood | ||||||
| HCC78, H2228 | Human blood | 200 μL | 1 mL h−1 | 94 ± 3% | 27 | ||
| HCC827 | |||||||
| H1985 | |||||||
| 3D hybrid micro-/nanostructure | Anti-EpCAM | MCF-7 | Human blood | 1 mL | 0.5 mL h−1 | 76.7 ± 7.1% | 2 |
| Anti-EpCAM | MCF-7 | PBS buffer | 1 mL | 30 min | 85.47 ± 1.88% | 6 | |
| Artificial blood | |||||||
| Anti-vimentin | MDA-MB231 | Human blood | |||||
| Anti-EpCAM | MCF-7 | Human blood | 200 μL | 60 min | 84 ± 6.2% | 16 | |
| 85 ± 2.5% | |||||||
| PC3 | 400 μL | 60 min | 93.1 ± 5.1% | 24 | |||
| 86.0 ± 9.5% | |||||||
| None | MCF-7 | Artificial sample | 1 mL | 6 h | ∼93.1% | 30 | |
| HeLa | ∼79.6% | ||||||
| PC3 | ∼79.2% |
Author contributions
All authors have read and agreed to the published version of the manuscript. HYK and YTX were involved in writing (original draft, and review and editing); LM and TQY were involved in supervision; XBX was involved in supervision and writing (review and editing and visualization).Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is financially support by the National Natural Science Foundation of China (Grant No. 51901159), and the Fundamental Research Funds for the Central Universities.References
- E. S. Park, C. Jin, Q. Guo, R. R. Ang, S. P. Duffy, K. Matthews, A. Azad, H. Abdi, T. Todenhofer, J. Bazov, K. N. Chi, P. C. Black and H. S. Ma, Small, 2016, 12, 1909–1919 CrossRef CAS PubMed.
- J. C. Qiu, K. Zhao, L. L. Li, X. Yu, W. B. Guo, S. Wang, X. D. Zhang, C. F. Pan, Z. L. Wang and H. Liu, Nano Res., 2017, 10, 776–784 CrossRef CAS.
- S. T. Wang, K. Liu, J. A. Liu, Z. T. F. Yu, X. W. Xu, L. B. Zhao, T. Lee, E. K. Lee, J. Reiss, Y. K. Lee, L. W. K. Chung, J. T. Huang, M. Rettig, D. Seligson, K. N. Duraiswamy, C. K. F. Shen and H. R. Tseng, Angew. Chem., Int. Ed., 2011, 50, 3084–3088 CrossRef CAS PubMed.
- C. Y. Lu, J. Xu, J. T. Han, X. Li, N. T. Xue, J. S. Li, W. H. Wu, X. L. Sun, Y. G. Wang, Q. Ouyang, G. Yang and C. X. Luo, Lab Chip, 2020, 20, 4094–4105 RSC.
- Z. Y. Shen, A. G. Wu and X. Y. Chen, Chem. Soc. Rev., 2017, 46, 2038–2056 RSC.
- L. L. Hui, Y. Su, T. T. Ye, Z. Liu, Q. C. Tian, C. J. He, Y. Q. Zhao, P. Chen, X. J. Wang, W. D. Hang, Y. Luo and B. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 207–218 CrossRef CAS.
- D. X. Nguyen, P. D. Bos and J. Massague, Nat. Rev. Cancer, 2009, 9, 274–284 CrossRef CAS.
- A. Gribko, J. Kunzel, D. Wunsch, Q. Lu, S. M. Nagel, S. K. Knauer, R. H. Stauber and G. B. Ding, Int. J. Nanomed., 2019, 14, 4187–4209 CrossRef CAS PubMed.
- Q. L. Shen, L. Xu, L. B. Zhao, D. X. Wu, Y. S. Fan, Y. L. Zhou, W. H. OuYang, X. C. Xu, Z. Zhang, M. Song, T. Lee, M. A. Garcia, B. Xiong, S. Hou, H. R. Tseng and X. H. Fang, Adv. Mater., 2013, 25, 2368–2373 CrossRef CAS PubMed.
- A. W. Lambert, D. R. Pattabiraman and R. A. Weinberg, Cell, 2017, 168, 670–691 CrossRef CAS.
- J. M. Lloyd, C. M. McIver, S. A. Stephenson, P. J. Hewett, N. Rieger and J. E. Hardingham, Clin. Cancer Res., 2006, 12, 417–423 CrossRef CAS PubMed.
- C. A. Klein, Nat. Rev. Cancer, 2009, 9, 302–312 CrossRef CAS.
- H. I. Scher, X. Y. Jia, J. S. de Bono, M. Fleisher, K. J. Pienta, D. Raghavan and G. Heller, Lancet Oncol., 2009, 10, 233–239 CrossRef CAS.
- X. T. Li, B. B. Chen, M. He, H. Wang, G. Y. Xiao, B. Yang and B. Hu, Biosens. Bioelectron., 2017, 90, 343–348 CrossRef CAS.
- Z. B. Liu, Y. Q. Huang, W. L. Liang, J. Bai, H. T. Feng, Z. H. Fang, G. Tian, Y. J. Zhu, H. B. Zhang, Y. X. Wang, A. X. Liu and Y. Chen, Lab Chip, 2021, 21, 2881–2891 RSC.
- G. He, C. D. Yang, J. M. Feng, J. M. Wu, L. F. Zhou, R. Wen, S. Huang, Q. N. Wu, F. M. Liu, H. J. Chen, T. Hang and X. Xie, Adv. Funct. Mater., 2019, 29, 1806484 CrossRef.
- Z. F. Ke, M. Lin, J. F. Chen, J. S. Choi, Y. Zhang, A. Fong, A. J. Liang, S. F. Chen, Q. Y. Li, W. F. Fang, P. S. Zhang, M. A. Garcia, T. Lee, M. Song, H. A. Lin, H. C. Zhao, S. C. Luo, S. Hou, H. H. Yu and H. R. Tseng, ACS Nano, 2015, 9, 62–70 CrossRef CAS.
- G. P. Gupta and J. Massague, Cell, 2006, 127, 679–695 CrossRef CAS PubMed.
- P. S. Steeg, Nat. Med., 2006, 12, 895–904 CrossRef CAS.
- Y. T. Kang, Y. J. Kim, J. Bu, S. Chen, Y. H. Cho, H. M. Lee, C. J. Ryu, Y. Lim and S. W. Han, Sens. Actuators, B, 2018, 260, 320–330 CrossRef CAS.
- J. W. Franses, J. Philipp, P. Missios, I. Bhan, A. Liu, C. Yashaswini, E. Tai, H. L. Zhu, M. Ligorio, B. Nicholson, E. M. Tassoni, N. Desai, A. S. Kulkarni, A. Szabolcs, T. S. Hong, A. S. Liss, C. Fernandez-del Castillo, D. P. Ryan, S. Maheswaran, D. A. Haber, G. Q. Daley and D. T. Ting, Nat. Commun., 2020, 11, 3303 CrossRef CAS.
- R. Klotz, A. Thomas, T. Teng, S. M. Han, O. Iriondo, L. Li, S. Restrepo-Vassalli, A. Wang, N. Izadian, M. MacKay, B. S. Moon, K. J. Liu, S. K. Ganesan, G. Lee, D. S. Kang, C. S. Walmsley, C. Pinto, M. F. Press, W. Lu, J. Lu, D. Juric, A. Bardia, J. Hicks, B. Salhia, F. Attenello, A. D. Smith and M. Yu, Cancer Discovery, 2020, 10, 86–103 CrossRef CAS PubMed.
- L. L. Wu, L. Zhu, M. J. Huang, J. Song, H. M. Zhang, Y. L. Song, W. Wang and C. Y. Yang, Trends Anal. Chem., 2019, 117, 69–77 CrossRef CAS.
- G. He, J. M. Feng, A. H. Zhang, L. F. Zhou, R. Wen, J. M. Wu, C. D. Yang, J. Yang, C. W. Li, D. M. Chen, J. Wang, N. Hu and X. Xie, Nano Lett., 2019, 19, 7201–7209 CrossRef CAS.
- R. Vaidyanathan, R. H. Soon, P. Zhang, K. Jiang and C. T. Lim, Lab Chip, 2019, 19, 11–34 Search PubMed.
- J. Sierra, J. Marrugo-Ramirez, R. Rodriguez-Trujillo, M. Mir and J. Samitier, Sensors, 2020, 20, 1317 CrossRef CAS PubMed.
- J. T. Dong, Y. J. Jan, J. Cheng, R. Y. Zhang, M. Meng, M. Smalley, P. J. Chen, X. H. Tang, P. Tseng, L. R. Bao, T. Y. Huang, D. J. Zhou, Y. P. Liu, X. S. Chai, H. Zhang, A. Q. Zhou, V. G. Agopian, E. M. Posadas, J. J. Shyue, S. J. Jonas, P. S. Weiss, M. Y. Li, G. J. Zheng, H. H. Yu, M. P. Zhao, H. R. Tseng and Y. Z. Zhu, Sci. Adv., 2019, 5, evva9186 CrossRef PubMed.
- M. Y. Shen, J. F. Chen, C. H. Luo, S. Lee, C. H. Li, Y. L. Yang, Y. H. Tsai, B. C. Ho, L. R. Bao, T. J. Lee, Y. J. Jan, Y. Z. Zhu, S. Cheng, F. Y. Feng, P. L. Chen, S. Hou, V. Agopian, Y. S. Hsiao, H. R. Tseng, E. M. Posadas and H. H. Yu, Adv. Healthcare Mater., 2018, 7, 1700701 CrossRef.
- J. S. de Bono, H. I. Scher, R. B. Montgomery, C. Parker, M. C. Miller, H. Tissing, G. V. Doyle, L. Terstappen, K. J. Pienta and D. Raghavan, Clin. Cancer Res., 2008, 14, 6302–6309 CrossRef CAS.
- J. M. Feng, J. S. Mo, A. H. Zhang, D. Liu, L. F. Zhou, T. Hang, C. Yang, Q. N. Wu, D. H. Xia, R. Wen, J. Yang, Y. P. Feng, Y. Huang, N. Hu, G. He and X. Xie, Nanoscale, 2020, 12, 5103–5113 RSC.
- Y. Gao, W. H. Fan, C. H. Duan, W. H. Zhao, J. Zhang and X. X. Kang, Front. Oncol., 2021, 11, 643003 Search PubMed.
- A. Matikas, A. Kotsakis, S. Apostolaki, H. Politaki, M. Perraki, K. Kalbakis, M. Nikolaou, P. Economopoulou, D. Hatzidaki and V. Georgoulias, Br. J. Cancer, 2022, 11, 1563–1569 Search PubMed.
- H. I. Scher, A. J. Armstrong, J. D. Schonhoft, A. Gill, J. L. Zhao, E. Barnett, E. Carbone, J. Lu, E. S. Antonarakis, J. Luo, S. Tagawa, C. H. dos Anjos, Q. Yang, D. George, R. Szmulewitz, D. C. Danila, R. Wenstrup, M. Gonen and S. Halabi, Eur. J. Cancer, 2021, 150, 83–94 CrossRef CAS.
- M. Antfolk, C. Magnusson, P. Augustsson, H. Lija and T. Laurell, Anal. Chem., 2015, 87, 9322–9328 CrossRef CAS PubMed.
- F. Del Ben, M. Turetta, G. Celetti, A. Piruska, M. Bulfoni, D. Cesselli, W. T. S. Huck and G. Scoles, Angew. Chem., Int. Ed., 2016, 55, 8581–8584 CrossRef CAS.
- H. M. Pei, L. Li, Z. J. Han, Y. G. Wang and B. Tang, Lab Chip, 2020, 20, 3854–3875 Search PubMed.
- Y. L. Song, Y. Z. Shi, M. J. Huang, W. Wang, Y. Wang, J. Cheng, Z. C. Lei, Z. Zhu and C. Y. Yang, Angew. Chem., Int. Ed., 2019, 58, 2236–2240 Search PubMed.
- J. Kaiser, Science, 2010, 327, 1072–1074 CrossRef CAS PubMed.
- L. L. Wu, H. M. Ding, X. Qu, X. N. Shi, J. M. Yang, M. J. Huang, J. L. Zhang, H. M. Zhang, J. Song, L. Zhu, Y. L. Song, Y. Q. Ma and C. Y. Yang, J. Am. Chem. Soc., 2020, 142, 4800–4806 Search PubMed.
- P. Banko, S. Y. Lee, V. Nagygyorgy, M. Zrinyi, C. H. Chae, D. H. Cho and A. Telekes, J. Hematol. Oncol., 2019, 12, 48 Search PubMed.
- Y. T. Kang, I. Doh, J. Byun, H. J. Chang and Y. H. Cho, Theranostics, 2017, 7, 3179–3191 CrossRef CAS PubMed.
- S. J. Hao, Y. Wan, Y. Q. Xia, X. Zou and S. Y. Zheng, Adv. Drug Delivery Rev., 2018, 125, 3–20 CrossRef CAS PubMed.
- F. F. Becker, X. B. Wang, Y. Huang, R. Pethig, J. Vykoukal and P. R. C. Gascoyne, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 860–864 Search PubMed.
- V. Murlidhar, L. Rivera-Baez and S. Nagrath, Small, 2016, 12, 4450–4463 CrossRef CAS.
- M. Jamal-Hanjani, E. Thanopoulou, K. S. Peggs, S. A. Quezada and C. Swanton, Curr. Opin. Pharmacol., 2013, 13, 497–503 Search PubMed.
- A. Dalili, E. Samiei and M. Hoorfar, Analyst, 2019, 144, 87–113 Search PubMed.
- T. A. Duncombe, A. M. Tentori and A. E. Herr, Nat. Rev. Mol. Cell Biol., 2015, 16, 554–567 CrossRef CAS PubMed.
- X. Mu, W. F. Zheng, J. S. Sun, W. Zhang and X. Y. Jiang, Small, 2013, 9, 9–21 Search PubMed.
- M. Li, X. E. Song, S. Jin and K. M. Ye, Bio-Des. Manuf., 2021, 4, 526–540 CrossRef.
- H. Okano, T. Konishi, T. Suzuki, T. Suzuki, S. Ariyasu, S. Aoki, R. Abe and M. Hayase, Biomed. Microdevices, 2015, 17, 59 Search PubMed.
- A. G. Oh, D. W. Lee and Y. H. Cho, Sens. Actuators, B, 2012, 166, 24–30 CrossRef.
- J. M. Park, J. Y. Lee, J. G. Lee, H. Jeong, J. M. Oh, Y. J. Kim, D. Park, M. S. Kim, H. J. Lee, J. H. Oh, S. S. Lee, W. Y. Lee and N. Huh, Anal. Chem., 2012, 84, 7400–7407 Search PubMed.
- Y. C. Ma, L. Wang and F. L. Yu, Technol. Cancer Res. Treat., 2013, 12, 295–309 Search PubMed.
- D. L. Adams, P. X. Zhu, O. V. Makarova, S. S. Martin, M. Charpentier, S. Chumsri, S. H. Li, P. Amstutz and C. M. Tang, RSC Adv., 2014, 4, 4334–4342 RSC.
- P. R. C. Gascoyne, J. Noshari, T. J. Anderson and F. F. Becker, Electrophoresis, 2009, 30, 1388–1398 CrossRef CAS PubMed.
- V. Gupta, I. Jafferji, M. Garza, V. O. Melnikova, D. K. Hasegawa, R. Pethig and D. W. Davis, Biomicrofluidics, 2012, 6, 024133 Search PubMed.
- A. L. Allan, S. A. Vantyghem, A. B. Tuck, A. F. Chambers, I. H. Chin-Yee and M. Keeney, Cytometry, Part A, 2005, 65A, 4–14 Search PubMed.
- A. H. Talasaz, A. A. Powell, D. E. Huber, J. G. Berbee, K. H. Roh, W. Yu, W. Z. Xiao, M. M. Davis, R. F. Pease, M. N. Mindrinos, S. S. Jeffrey and R. W. Davis, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3970–3975 CrossRef CAS.
- K. Loutherback, J. D’Silva, L. Y. Liu, A. Wu, R. H. Austin and J. C. Sturm, AIP Adv., 2012, 2, 042107 CrossRef.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 Search PubMed.
- S. Q. Yan, X. Zhang, X. F. Dai, X. J. Feng, W. Du and B. F. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 33457–33463 Search PubMed.
- Q. L. Shen, H. T. Yang, C. X. Peng, H. Zhu, J. Mei, S. Huang, B. Chen, J. Liu, W. B. Wu and S. K. Cao, Int. J. Nanomed., 2019, 14, 205–214 Search PubMed.
- D. Sun, Z. Chen, M. Wu and Y. Zhang, Nanotheranostics, 2017, 1, 389–402 CrossRef PubMed.
- J. T. Dong, J. F. Chen, M. Smalley, M. P. Zhao, Z. F. Ke, Y. Z. Zhu and H. R. Tseng, Adv. Mater., 2020, 32, e1903663 CrossRef PubMed.
- Z. Jahed, R. Zareian, Y. Y. Chau, B. B. Seo, M. West, T. Y. Tsui, W. J. Wen and M. R. K. Mofrad, ACS Appl. Mater. Interfaces, 2016, 8, 23604–23613 CrossRef CAS.
- A. Cutarelli, S. Ghio, J. Zasso, A. Speccher, G. Scarduelli, M. Roccuzzo, M. Crivellari, N. M. Pugno, S. Casarosa, M. Boscardin and L. Conti, Cells, 2020, 9, 88 Search PubMed.
- A. A. Chang, J. N. Patel, C. Cordoba, B. Kaminska and K. Kavanagh, SPIE Conf. Proc., 2014, 8973, 89730J Search PubMed.
- K. Y. Hwang, J. H. Kim, K. Y. Suh, J. S. Ko and N. Huh, Sens. Actuators, B, 2011, 155, 422–429 CrossRef CAS.
- P. T. Wang, J. W. Su, W. Dai, G. Cernigliaro and H. W. Sun, Appl. Phys. Lett., 2014, 104, 043504 Search PubMed.
- B. P. Zhou, W. Xu, A. A. Syed, Y. Y. Chau, L. Q. Chen, B. Chew, O. Yassine, X. X. Wu, Y. B. Gao, J. X. Zhang, X. Xiao, J. Kosel, X. X. Zhang, Z. H. Yao and W. J. Wen, Lab Chip, 2015, 15, 2125–2132 RSC.
- S. Krishnamoorthy, Z. Y. Zhang and C. X. Xu, Bio-Des. Manuf., 2020, 3, 60–70 CrossRef.
- C. Alix-Panabieres and K. Pantel, Lab Chip, 2014, 14, 57–62 Search PubMed.
- Y. T. Xiong, H. Y. Kang, H. Z. Zhou, L. Ma and X. B. Xu, Bio-Des. Manuf., 2022, 5, 607–616 Search PubMed.
- D. Y. Hu, H. Liu, Y. Tian, Z. Li and X. Y. Cui, ACS Comb. Sci., 2020, 22, 701–711 Search PubMed.
- S. Park, R. R. Ang, S. P. Duffy, J. Bazov, K. N. Chi, P. C. Black and H. S. Ma, PLoS One, 2014, 9, e85264 Search PubMed.
- F. Guilak, J. R. Tedrow and R. Burgkart, Biochem. Biophys. Res. Commun., 2000, 269, 781–786 CrossRef CAS.
- L. R. Huang, E. C. Cox, R. H. Austin and J. C. Sturm, Science, 2004, 304, 987–990 CrossRef CAS.
- P. T. Went, A. Lugli, S. Meier, M. Bundi, M. Mirlacher, G. Sauter and S. Dirnhofer, Hum. Pathol., 2004, 35, 122–128 CrossRef CAS.
- J. P. Gleghorn, E. D. Pratt, D. Denning, H. Liu, N. H. Bander, S. T. Tagawa, D. M. Nanus, P. A. Giannakakou and B. J. Kirby, Lab Chip, 2010, 10, 27–29 Search PubMed.
- W. Y. Qian, Y. Zhang and W. Q. Chen, Small, 2015, 11, 3850–3872 Search PubMed.
- E. J. Cho, J. W. Lee and A. D. Ellington, Annu. Rev. Anal. Chem., 2009, 2, 241–264 CrossRef CAS.
- H. T. Ma, J. P. Liu, M. M. Ali, M. A. I. Mahmood, L. Labanieh, M. R. Lu, S. M. Iqbal, Q. Zhang, W. A. Zhao and Y. Wan, Chem. Soc. Rev., 2015, 44, 1240–1256 RSC.
- J. Cao, X. P. Zhao, M. R. Younis, Z. Q. Li, X. H. Xia and C. Wang, Anal. Chem., 2017, 89, 10957–10964 Search PubMed.
- L. K. Liu, K. G. Yang, H. Gao, X. Li, Y. B. Chen, L. H. Zhang, X. J. Peng and Y. K. Zhang, Anal. Chem., 2019, 91, 2591–2594 Search PubMed.
- M. Poudineh, M. Labib, S. Ahmed, L. N. M. Nguyen, L. Kermanshah, R. M. Mohamadi, E. H. Sargent and S. O. Kelley, Angew. Chem., Int. Ed., 2017, 56, 163–168 CrossRef CAS.
- Y. J. Zhao, D. K. Xu and W. H. Tan, Integr. Biol., 2017, 9, 188–205 CrossRef PubMed.
- H. J. Yoon, M. Kozminsky and S. Nagrath, ACS Nano, 2014, 8, 1995–2017 CrossRef CAS PubMed.
- J. X. Meng, P. C. Zhang, F. L. Zhang, H. L. Liu, J. B. Fan, X. L. Liu, G. Yang, L. Jiang and S. T. Wang, ACS Nano, 2015, 9, 9284–9291 Search PubMed.
- H. Y. Yue, S. Huang, J. Chang, C. Heo, F. Yao, S. Adhikari, F. Gunes, L. C. Liu, T. H. Lee, E. S. Oh, B. Li, J. J. Zhang, T. Q. Huy, N. V. Luan and Y. H. Lee, ACS Nano, 2014, 8, 1639–1646 Search PubMed.
- S. T. Wang, H. Wang, J. Jiao, K. J. Chen, G. E. Owens, K. I. Kamei, J. Sun, D. J. Sherman, C. P. Behrenbruch, H. Wu and H. R. Tseng, Angew. Chem., Int. Ed., 2009, 48, 8970–8973 Search PubMed.
- J. Sekine, S. C. Luo, S. Wang, B. Zhu, H. R. Tseng and H. Yu, Adv. Mater., 2011, 23, 4788–4792 Search PubMed.
- N. A. Zhang, Y. L. Deng, Q. D. Tai, B. R. Cheng, L. B. Zhao, Q. L. Shen, R. X. He, L. Y. Hong, W. Liu, S. S. Guo, K. Liu, H. R. Tseng, B. Xiong and X. Z. Zhao, Adv. Mater., 2012, 24, 2756–2760 CrossRef CAS PubMed.
- Y. T. Lu, L. B. Zhao, Q. L. Shen, M. A. Garcia, D. X. Wu, S. Hou, M. Song, X. C. Xu, W. H. OuYang, W. W. L. OuYang, J. Lichterman, Z. Luo, X. Xuan, J. T. Huang, L. W. K. Chung, M. Rettig, H. R. Tseng, C. Shao and E. M. Posadas, Methods, 2013, 64, 144–152 Search PubMed.
- S. Hou, H. C. Zhao, L. B. Zhao, Q. L. Shen, K. S. Wei, D. Y. Suh, A. Nakao, M. A. Garcia, M. Song, T. Lee, B. Xiong, S. C. Luo, H. R. Tseng and H. H. Yu, Adv. Mater., 2013, 25, 1547–1551 CrossRef CAS PubMed.
- M. Lin, J. F. Chen, Y. T. Lu, Y. Zhang, J. Z. Song, S. Hou, Z. F. Ke and H. R. Tseng, Acc. Chem. Res., 2014, 47, 2941–2950 CrossRef CAS PubMed.
- S. Guo, J. Q. Xu, M. Xie, W. Huang, E. F. Yuan, Y. Liu, L. P. Fan, S. B. Cheng, S. M. Liu, F. B. Wang, B. F. Yuan, W. G. Dong, X. L. Zhang, W. H. Huang and X. Zhou, ACS Appl. Mater. Interfaces, 2016, 8, 15917–15925 Search PubMed.
- F. L. Zhang, Y. Jiang, X. L. Liu, J. X. Meng, P. C. Zhang, H. L. Liu, G. Yang, G. N. Li, L. Jiang, L. J. Wan, J. S. Hu and S. T. Wang, Nano Lett., 2016, 16, 766–772 Search PubMed.
- L. Wang, S. Huang, Q. Y. Li, L. Y. Ma, C. Zhang, F. Liu, M. Jiang, X. Yu and L. Xu, Chem. Eng. J., 2022, 435, 134762 Search PubMed.
- J. J. Gu, W. Zhang, H. L. Su, T. X. Fan, S. M. Zhu, Q. L. Liu and D. Zhang, Adv. Mater., 2015, 27, 464–478 Search PubMed.
- H. Cui, Q. Liu, R. Li, X. Y. Wei, Y. Sun, Z. X. Wang, L. L. Zhang, X. Z. Zhao, B. Hua and S. S. Guo, Nanoscale, 2020, 12, 1455–1463 Search PubMed.
- J. J. VanDersarl, A. M. Xu and N. A. Melosh, Nano Lett., 2012, 12, 3881–3886 Search PubMed.
- X. Xie, A. M. Xu, S. Leal-Ortiz, Y. H. Cao, C. C. Garner and N. A. Melosh, ACS Nano, 2013, 7, 4351–4358 Search PubMed.
- Y. H. Cao, M. Hjort, H. D. Chen, F. Birey, S. A. Leal-Ortiz, C. M. Han, J. G. Santiago, S. P. Pasca, J. C. Wu and N. A. Melosh, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E1866–E1874 Search PubMed.
- A. M. Xu, S. A. Kim, D. S. Wang, A. Aalipour and N. A. Melosh, Lab Chip, 2016, 16, 2434–2439 RSC.
- W. L. Gao, H. J. Yuan, F. X. Jing, S. Wu, H. B. Zhou, H. J. Mao, Q. H. Jin, J. L. Zhao, H. Cong and C. P. Jia, Oncotarget, 2017, 8, 12917–12928 Search PubMed.
- K. Yoneda, T. Kuwata, Y. Chikaishi, M. Mori, M. Kanayama, M. Takenaka, S. Oka, A. Hirai, N. Imanishi, K. Kuroda, Y. Ichiki, T. Ohnaga and F. Tanaka, Cancer Sci., 2019, 110, 726–733 Search PubMed.
- X. F. Zhang, X. Lu, W. L. Gao, Y. M. Wang, C. P. Jia and H. Cong, Transl. Oncol., 2021, 14 Search PubMed.
- V. Murlidhar, R. M. Reddy, S. Fouladdel, L. L. Zhao, M. K. Ishikawa, S. Grabauskiene, Z. Zhang, J. Lin, A. C. Chang, P. Carrott, W. R. Lynch, M. B. Orringer, C. Kumar-Sinha, N. Palanisamy, D. G. Beer, M. S. Wicha, N. Ramnath, E. Azizi and S. Nagrath, Cancer Res., 2017, 77, 5194–5206 Search PubMed.
- K. Kure, M. Hosoya, T. Ueyama, M. Fukaya, K. Sugimoto, Y. Tomiki, T. Ohnaga, K. Sakamoto and H. Komiyama, Oncol. Lett., 2020, 19, 2286–2294 CAS.
- M. Kanayama, T. Kuwata, M. Mori, Y. Nemoto, N. Nishizawa, R. Oyama, H. Matsumiya, A. Taira, S. Shinohara, M. Takenaka, K. Yoneda, K. Kuroda, T. Ohnaga and F. Tanaka, Cancer Sci., 2022, 113, 1028–1037 Search PubMed.
- Y. Zhang, X. F. Zhang, J. L. Zhang, S. Sun, L. L. Zheng, J. Li, S. X. Liu, G. D. Sui and Z. F. Yin, Cancer Biol. Ther., 2016, 17, 1177–1187 CrossRef CAS.
- X. Yu, N. Wu, F. Chen, J. Wei and Y. X. Zhao, Trends Anal. Chem., 2019, 117, 27–38 CrossRef CAS.
- Y. Z. Zhu, D. Joralmon, W. T. Shan, Y. Y. Chen, J. H. Rong, H. Y. Zhao, S. Q. Xiao and X. J. Li, Bio-Des. Manuf., 2021, 4, 405–428 Search PubMed.
- V. Mehta and S. N. Rath, Bio-Des. Manuf., 2021, 4, 311–343 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |