
Achieving ultra-dispersed 1T-Co-MoS2@HMCS via space-confined engineering for highly efficient hydrogen evolution in the universal pH range†
Changle
Yue
a,
Yan
Zhou
 b,
Yang
Liu
a,
Chao
Feng
a,
Wenjing
Bao
a,
Fengyue
Sun
a,
Yongxiao
Tuo
b,
Yuan
Pan
a,
Yunqi
Liu
a and
Yukun
Lu
*a
b,
Yang
Liu
a,
Chao
Feng
a,
Wenjing
Bao
a,
Fengyue
Sun
a,
Yongxiao
Tuo
b,
Yuan
Pan
a,
Yunqi
Liu
a and
Yukun
Lu
*a
aState Key Laboratory of Heavy Oil Processing, College of Chemistry and Chemical Engineering, China University of Petroleum (East China), Qingdao 266580, PR China. E-mail: lyk@upc.edu.cn
bSchool of Materials Science and Engineering, China University of Petroleum (East China), Qingdao 266580, PR China
First published on 12th April 2022
Abstract
The rational design and synthesis of noble-metal-free electrocatalysts for water splitting is always important for the future hydrogen economy. Therefore, it is necessary to design an effective transition metal sulfide down to a molecular level. In this work, a multi-level spatial confinement strategy was developed to fabricate Co-promoted 1T-MoS2 (1T-Co-MoS2) by employing Evans–Showell-type polyoxometalates (POMs) [Co2Mo10O38H4] as molecular precursor. Highly dispersed 1T-Co-MoS2 nanoclusters with few layers (1–3 layers) and ultrasmall size (<5 nm) were synthesized within the hollow mesoporous carbon sphere (HMCS) by in situ vapor phase sulfurization. During the preparation, coordination bonds, organic cations and mesopores provide a triple-confinement environment to limit the growth of 1T-Co-MoS2 from the atomic level, molecular level to mesoscopic scale. The obtained 1T-Co-MoS2@HMCS exhibits remarkable electrocatalytic activity and excellent long-term durability for hydrogen evolution reaction (HER), with overpotentials of 220 and 245 mV to achieve the current density of 200 mA cm−2 in 1 M KOH and 0.5 M H2SO4, respectively. The corresponding theoretical calculations indicate that Co–S edge sites are the most active sites of 1T-Co-MoS2 for HER, reflecting the major significance of Co doping. The superior HER performance could be attributed to the high intrinsic activity from Co-doped 1T-MoS2 sites, abundant exposed active sites from ultra-dispersed nanosheets, and enhanced charge and mass transfer within the HMCS substrate. This work provides a novel design concept via hierarchical multiple-level confinement for the synthesis of high-quality 1T-Co-MoS2 and achieves outstanding performance in electrocatalytic HER.
1. Introduction
Hydrogen energy is one of the effective solutions for the current increasing energy demand and environmental pollution caused by fossil energy.1 In recent years, electrocatalytic water splitting to generate stable and sustainable clean hydrogen fuels has attracted considerable attention.2 Although noble-metal-based hydrogen evolution reaction (HER) catalysts show high activity, their practical applications are limited by their high cost and low abundance.3 Therefore, it is urgent to explore earth-abundant electrocatalysts for electrocatalytic water splitting with high efficiency and stability.4 A large variety of non-noble metal catalysts including metal phosphides,5 carbides,6 and dichalcogenides7,8 have been explored to replace noble-metal-based electrocatalysts. Among them, two-dimensional (2D) molybdenum disulfide (MoS2) is one of the most investigated electrocatalysts for the HER process.7,9 MoS2 has attracted much attention in the field of hydrogen production due to its similar Gibbs free energy (ΔGH*) for hydrogen absorption to that of Pt and its flexible molecular structure.10 The S-edge sites of MoS2 are thermally neutral to hydrogen absorption (ΔGH* = 0.08 eV) and are considered to be the catalytic active centers.11 Among the different crystalline phases of MoS2, 1T-MoS2 is a metallic phase with an electrical conductivity 107 times higher than that of 2H-MoS2, which can greatly improve the charge transfer efficiency in the HER process.12 It was demonstrated that both the basal planes and edges of the 1T phase are active sites for HER, which leads to excellent electrocatalytic activity.13 In particular, 1T-MoS2 shows more efficient HER activity than that of 2H-MoS2 in acidic solutions due to the partially filled Mo 4d and S 3p electronic states around the Fermi level.14 Although 1T-MoS2 offers more active sites and adequate H adsorption, its catalytic performance of HER in alkaline solutions still needs to be improved.15 The slow HER kinetics of 1T-MoS2 in alkaline solutions is due to the rather sluggish initial water splitting step (Volmer step).16Heteroatom doping strategy has been widely applied to MoS2 to further enhance its HER catalytic activity by further tuning its electronic structure.17,18 Grønborg19et al. has demonstrated that the MoS2 cluster can be doped by Co to provide both Mo- and S-terminated edges when the MoS2 cluster size is larger than ∼2 nm. Because the thermodynamic stability of 1T phase is less than that of 2H phase of MoS2, 1T-MoS2 can only be obtained under harsh synthetic strategies.12,20 Although extensive research has been carried out to seek the way of fabricating heteroatom-doped 1T-MoS2, it is rather complicated to obtain single 1T phase MoS2 by a conventional hydrothermal (or solvothermal) method.21–23 Thus, it is difficult to synthesize heteroatom-doped 1T-MoS2 on a large scale. However, it has been found that metastable 1T-MoS2 can be achieved by confining the lateral size and stacking numbers of MoS2 nanosheets.24 Thus, it is reasonable to believe that a confinement approach can lead to successful synthesis of heteroatom-doped 1T-MoS2 when its size can be confined to less than ∼5 nm in diameter and the stacking numbers can be reduced to single or few layers.
Recently, polyoxometalates (POMs) display great advantages as an excellent bimetallic precursor platform.6,7,25–28 It provides more than one type of transition metal element in a single-molecular level. Owing to the sub-nanometer size, well-defined structure, and tunable chemical composition, Mo-containing POMs have been widely studied as molecular-level precursors to fabricate a Mo-based active phase with a relatively uniform size and homogeneous structure.24,29 Our previous investigations also showed that the use of Co5Mo10 as a precursor can successfully obtain ultra-small-sized MoS2 and CoS.25 Single-phase Co-doped 1T-MoS2 was not achieved in the previous investigation because of the rather rigorous sulfidation conditions and the aggregation of the precursors during the sulfidation process. Thankfully, we have discovered a multi-level confinement strategy to achieve ultra-small single-phase Co-doped 1T-MoS2 by ion exchange and vapor phase sulfidation in mesopores with POMs. The cations of POMs can be easily exchanged by cationic surfactants, and thus, they can interconnect the POMs and anchor each POM unit in specific positions up to a supramolecular level. However, supramolecules are more likely to grow into large MoS2 nanosheets or nano-blocks during sulfidation, which is not conducive to the synthesis of small-sized 1T-MoS2.30 Therefore, further spatial confinement, an excellent way to induce the 2D nanosheet with metastable phases, is needed to limit the growth of catalytic materials.23,31 HMCS have been previously reported as typical synthetic nanoreactors, which illustrated their competitive potential in heterogeneous catalysis due to their adjustable microenvironmental effects.32 The mesoporous confinement effect of HMCS proves to be the key to improving catalytic performance, and it is essentially driven by an integrated action of electronic metal–support interaction, reactant enrichment and diffusion, which are mainly ascribed to the peculiar properties of hollow nanoreactors in both electronic and structural aspects, respectively.33,34 Accordingly, multiple confinement strategies at different scales can be integrated to prepare highly dispersed heteroatom-doped 1T-MoS2.
As illustrated in Fig. 1, the bottom-up multi-level spatial confinement strategy is presented for the design and fabrication of ultra-dispersed 1T-Co-MoS2 nanoclusters employing Evans–Showell-type POMs (NH4)6[Co2Mo10O38H4] (denoted as Co2Mo10) as precursors. The multiple confined processes are achieved by rationally utilizing hierarchical interactions from the atomic level to the mesoscopic scale: (1) Chemical bond confinement at the atomic level, in which the Co atoms are anchored via Co–O–Mo coordinative bonding and encapsulated within single POM clusters possessing a stable and specific Co2Mo10 structure, which realizes the close contact of Mo and Co atoms and guarantees enormously the catalytic synergism of binary Co-doped MoS2 active sites for HER. (2) Organic cation confinement at the molecular level, which is achieved by selecting hexamethonium (HM) bromide as the appropriate counter-cation, with single [Co2Mo10]6− polyoxoanion spontaneously separated and integrated into inorganic–organic hybrid networks (Co2Mo10)(HM)3via electrovalent bonding, thereby deriving superior dispersed Co-doped MoS2 nanosheets with few layers. (3) Mesopore confinement at nanoscale, in which the HMCS provides abundant pore structures, with soluble (Co2Mo10)(HM)3 hybrid molecules that were adsorbed and spatially confined within nanosized meso-cages, and then homogeneously distributed throughout conductive H substrates, effectively inhibiting the agglomeration and migration during the following sulfurization, and maintaining the ultrafine Co-MoS2 nanosheets inside the nano-space. As we expected, the obtained 1T-Co-MoS2@HMCS possesses the aforementioned structural superiorities and exhibits remarkable electrocatalytic activities and excellent long-term durability towards HER in both alkaline and acidic electrolytes. Thus, this work provides a novel design concept via hierarchical multi-level confinement for the synthesis of high-quality 1T-Co-MoS2 to enhance the electrocatalytic performance.
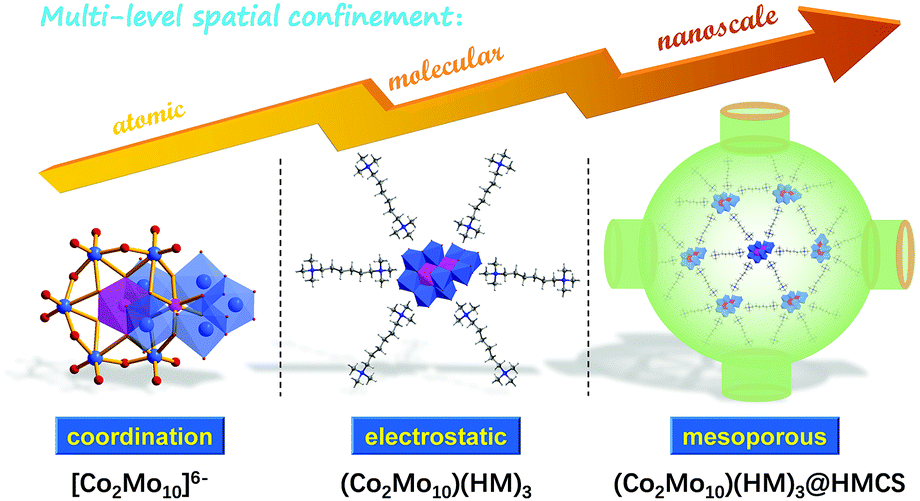 | ||
| Fig. 1 Schematic diagram of the preparation of (Co2Mo10)(HM)3@HMCS precursors by a multi-level confined strategy. | ||
2. Experimental section
All the chemicals used in this study were purchased and used without further purification.2.1 Preparation of HMCS
First, 7.00 mL NH3·H2O was added to 160 mL aqueous solution of ethanol (Vethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Vwater = 7:1) to obtain an alkaline environment. After sufficient magnetic stirring, 7.00 mL TPOS (24.2 mmol) was added slowly to the solution and stirred for 30 min. Then, 1.12 mL CH2O (13.8 mmol) solution and 0.800 g C6H6O2 (7.27 mmol) dissolved in 4.00 mL ethanol by ultrasonication were slowly dropped into the above solution. After stirring for 24 h at room temperature, light yellow SiO2 precipitates were separated from the creamy white solution by centrifugation and dried at 60 °C. The SiO2 powder was then carbonized at 900 °C for 4 h in a N2 (99.999%) atmosphere at a heating rate of 5 °C min−1, followed by natural cooling to produce black SiO2@C spheres. The black HMCS powder could be obtained by SiO2@C spheres stirred in a 13 wt% HF solution for 2 days.
:Vwater = 7:1) to obtain an alkaline environment. After sufficient magnetic stirring, 7.00 mL TPOS (24.2 mmol) was added slowly to the solution and stirred for 30 min. Then, 1.12 mL CH2O (13.8 mmol) solution and 0.800 g C6H6O2 (7.27 mmol) dissolved in 4.00 mL ethanol by ultrasonication were slowly dropped into the above solution. After stirring for 24 h at room temperature, light yellow SiO2 precipitates were separated from the creamy white solution by centrifugation and dried at 60 °C. The SiO2 powder was then carbonized at 900 °C for 4 h in a N2 (99.999%) atmosphere at a heating rate of 5 °C min−1, followed by natural cooling to produce black SiO2@C spheres. The black HMCS powder could be obtained by SiO2@C spheres stirred in a 13 wt% HF solution for 2 days.
2.2 Preparation of (NH4)6[Co2Mo10H4O38]·4H2O (Co2Mo10)
Ammonium salt of decamolybdodicobaltate Co2Mo10 was synthesized according to the published procedure.28 First, 15.0 g (12.1 mmol) (NH4)6Mo7O24·4H2O and 3.10 g (12.4 mmol) Co(CH3COO)2·4H2O were dissolved in 125 mL water, and then 3.00 g active charcoal and 20.0 mL peroxide solution (18%) were added to the solution. After boiling the black solution for 1 h, the active charcoal was separated by filtration. Dark-green Co2Mo10 crystals were obtained by slow evaporation of the solvent at room temperature.2.3 Synthesis of (Co2Mo10)(HM)3@HMCS
Co2Mo10 was used as the source for both Co and Mo. First, 0.248 g (0.133 mmol) Co2Mo10 and 0.144 g (0.399 mmol) hexamethonium (HM) bromide were dissolved in 20.0 mL deionized water. Then, 30.0 mg HMCS was added to the clear green solution and stirred at room temperature for 12 h to obtain (Co2Mo10)(HM)3@HMCS. The black (Co2Mo10)(HM)3@HMCS precipitates were separated by centrifugation, washed several times with deionized water and dried at 60 °C for 24 h.2.4 Synthesis of 1T-Co-MoS2@HMCS
In this work, 1T-Co-MoS2@HMCS was obtained by calcining (Co2Mo10)(HM)3@HMCS under 10 vol% H2S/H2 at 400 °C for 4 h at a heating rate of 5 °C min−1.2.5 Synthesis of Co-MoS2@HMCS
Co-MoS2@HMCS was obtained without HM compared with 1T-Co-MoS2@HMCS. First, 0.248 g (0.133 mmol) Co2Mo10 was dissolved in 20 mL deionized water for 30 min. Then, 30.0 mg HMCS was added to this solution and stirred for 12 h. The as-synthesized black product was separated by centrifugation, washed several times with deionized water and then dried at 60 °C for 24 h. Then, the prepared sample was further calcined under 10 vol% H2S/H2 at 400 °C for 4 h at a heating rate of 5 °C min−1, followed by natural cooling to produce Co-MoS2@HMCS powder.2.6 Synthesis of Co-MoS2
Co-MoS2 was prepared by calcining Co2Mo10 at 400 °C for 4 h at a heating rate of 5 °C min−1 in a 10 vol% H2S/H2 atmosphere.2.7 Synthesis of MoS2@HMCS and CoS@HMCS
First, 0.282 g (0.228 mmol) (NH4)6Mo7O24·4H2O or 0.398 g (1.60 mmol) Co(CH3COO)2·4H2O was dissolved in 20.0 mL deionized water containing 0.144 g (0.399 mmol) hexamethonium (HM) bromide for 30 min. Then, 30.0 mg HMCS was added to this solution and stirred for 12 h. The as-synthesized product was separated by centrifugation, washed several times with deionized water and then dried at 60 °C for 24 h. Then, the prepared sample was further calcined under 10 vol% H2S/H2 at 400 °C for 4 h at a heating rate of 5 °C min−1, followed by natural cooling to produce a MoS2@HMCS or CoS@HMCS powder.2.8 Synthesis of 2H-MoS2
Hydrothermal synthesis in a sealed autoclave system was used for the preparation of 2H-MoS2. First, 1.44 g (10 mmol) MoO3 and 3.04 g (40 mmol) CH4N2S were dispersed in 70 mL deionized water under stirring to form a homogeneous solution. Then, the above solution was transferred into a 100 mL Teflon-lined stainless steel autoclave and kept at 220 °C for 12 h. The final product was washed several times with deionized water and dried at 60 °C.2.9 Structural characterizations
X-ray powder diffraction (XRD) patterns of the products were acquired using an X-ray diffractometer (BRUKER D8 ADVANCE) with Cu Kα radiation (λ = 1.5418 Å, 40 kV, 40 mA) at room temperature. Scanning electron microscopic (SEM) images of the products were acquired using a field-emission SEM (JMS-7900F). High-resolution transmission electron microscopic (HRTEM) images, energy-dispersive X-ray spectroscopy (EDX) spectra and annular dark-field STEM images of the products were recorded using a HRTEM (JEM-2100F). Fourier transform infrared (FT-IR) spectra were recorded using a VERTEX 80 V FT-IR spectrometer. The chemical states of the sample were determined by X-ray photoelectron spectroscopy (XPS) using a Thermo VG ESCALAB250XI surface analysis system. N2 adsorption–desorption isotherms were obtained using a Quantachrome Autosorb-1-MP analyzer at 77 K and the specific surface area was calculated automatically using a multipoint Brunauer–Emmett–Teller (BET) model. Raman spectra of the catalysts were recorded using a Senterra laser Raman spectrometer (λ = 532 nm) equipped with an optical microscope in the confocal mode.2.10 Electrochemical characterizations
All of the electrochemical performance tests in 1.0 M KOH and 0.5 M H2SO4 electrolyte media were performed using a Gamry INTERFACE 1000 E, USA, using a saturated calomel electrode (SCE) electrode, a graphite rod, and as-prepared bimetal sulfide electrodes as the reference electrode, the counter electrode, and the working electrode, respectively. All polarization curves at 2 mV s−1 were iR compensated. The linear sweep voltammetry (LSV) measurements were recorded at a scan rate of 2 mV s−1. The cyclic voltammetry (CV) measurements were carried out with different sweep rates between 40 and 200 mV s−1. The long-term stability tests were performed by CV tests at a scan rate of 100 mV s−1. The electrochemical impedance spectroscopy (EIS) tests were performed by ac impedance spectroscopy in the frequency range of 10° to 1 Hz. According to the Nernst equation ERHE = ESCE + 0.059 pH + 0.254, where ERHE is the potential vs. a reversible hydrogen electrode, ESCE is the potential vs. an SCE electrode, and pH is the pH value of the electrolyte.The surface of the glassy carbon electrode (GCE, 3 mm diameter) was polished with 0.3 μm alumina slurries and sonicated with deionized water and ethanol. Then, the electrode was dried at 25 °C. To prepare modified electrodes, 5.0 mg of electrocatalyst was dispersed into 1.005 mL of Nafion solution [5.0 μL Nafion (5%) dissolved in 1.000 mL of ethanol] to give a homogeneous suspension upon bath sonication. Then, 20.0 μL of the suspension was dropped onto the GCE surface and the electrode was dried at 25 °C.
3. Results and discussion
3.1 Preparation and characterization of 1T-Co-MoS2@HMCS
The detailed synthesis procedures of Co2Mo10 and HMCS were described in the ESI.† Accordingly, 1T-Co-MoS2@HMCS is achieved via three steps, as displayed in Fig. 2. First, SiO2@C was selectively etched to remove the SiO2 template to form a hollow carbon sphere with a mesoporous shell and radially aligned channels. The HMCS can then be served as a nanoreactor to accommodate highly dispersed Co2Mo10 within the cationic surfactants HM2+ to obtain the (Co2Mo10)(HM)3@HMCS precursor. Subsequently, spatially confined 1T-Co-MoS2 in HMCS (1T-Co-MoS2@HMCS) was constructed by vapor phase sulfurization in a tubular furnace at 400 °C for 4 h under a H2S/H2 atmosphere. During this process, the 1T phase Co-MoS2 was facilely derived from Co2Mo10 precursors via in situ O–S exchange and then uniformly dispersed throughout the HMCS frameworks. Thus, the as-prepared 1T-Co-MoS2@HMCS can provide a large number of Co-doped 1T-MoS2 active sites and efficient electric conductivity to achieve high catalytic activity and stability towards HER catalysis.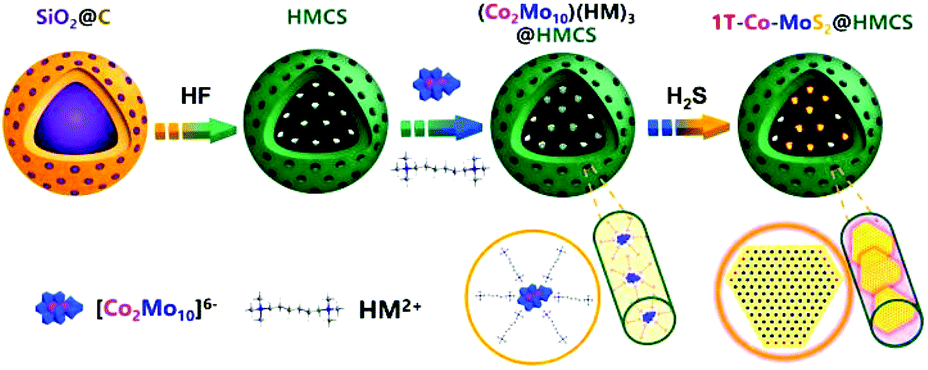 | ||
| Fig. 2 Schematic illustration of the preparation approach of 1T-Co-MoS2@HMCS. | ||
To further elucidate the spatial confinement effects provide by the surfactants and HMCS, XRD characterizations are applied to investigate the crystal structure of Co2Mo10 and (Co2Mo10)(HM)3@HMCS. As shown in Fig. S1a,† the diffraction peaks of (Co2Mo10)(HM)3@HMCS shift to small angles, indicating that the crystal lattice of Co2Mo10 is effectively enlarged by HM2+ organic chains under the action of spatial confined engineering, which provides a basis for ultra-dispersed catalysts. This suggests that the spatial confinement is effective on confining the POMs precursor, and thus, subsequently limits the growth of Co-MoS2. Fig. S1b† represents the FT-IR results of Co2Mo10 and (Co2Mo10)(HM)3@HMCS. The peaks at 615, 679, 863, 907, and 940 cm−1 are assigned to the stretching vibration of the Co–O, Mo–O–Mo, and Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in Co2Mo10 molecules. Besides, the peaks at 1405, 1615, and 3200–3600 cm−1 are ascribed to O–H and N–H modes in Co2Mo10, suggesting the existence of molecular water and NH4+. In comparison, the stretching vibration of Co–O, Mo–O–Mo, and MoO in (Co2Mo10)(HM)3@HMCS shifts toward a lower wave number, which could be attributed to the increased bond length caused by the enlarged lattice space.35 At the same time, the FT-IR spectra of (Co2Mo10)(HM)3@HMCS do not contain peaks associated with O–H and N–H, indicating successful replacement of NH4+ cations on Co2Mo10 by HM2+ cationic surfactants. The successful preparation of host–guest precursors (Co2Mo10)(HM)3@HMCS is attributed to the outstanding structure characteristics and self-assembly ability of [Co2Mo10]6− polyoxoanions.
O in Co2Mo10 molecules. Besides, the peaks at 1405, 1615, and 3200–3600 cm−1 are ascribed to O–H and N–H modes in Co2Mo10, suggesting the existence of molecular water and NH4+. In comparison, the stretching vibration of Co–O, Mo–O–Mo, and MoO in (Co2Mo10)(HM)3@HMCS shifts toward a lower wave number, which could be attributed to the increased bond length caused by the enlarged lattice space.35 At the same time, the FT-IR spectra of (Co2Mo10)(HM)3@HMCS do not contain peaks associated with O–H and N–H, indicating successful replacement of NH4+ cations on Co2Mo10 by HM2+ cationic surfactants. The successful preparation of host–guest precursors (Co2Mo10)(HM)3@HMCS is attributed to the outstanding structure characteristics and self-assembly ability of [Co2Mo10]6− polyoxoanions.
Fig. 3a shows the SEM image of HMCS after the SiO2 template was removed, with an average diameter of 450 ± 50 nm. The spherical structure of HMCS indicates that the removal process of SiO2 did not affect the template morphology. The XRD and FT-IR characterizations shown in Fig. S2† suggest that SiO2 was successfully removed by HF. After loading with (Co2Mo10)(HM)3 and followed by sulfidation, the spherical structure of HMCS is retained (Fig. 3b), suggesting the formation of Co-MoS2 inside the pores of HMCS. The TEM image of broken spheres shown in Fig. 3c indicates the hollow structure of 1T-Co-MoS2@HMCS, with the uniform size and a shell of ∼50 nm thickness. To further understand the compositions within carbon spheres, XRD was applied. As shown in Fig. 3d, the characteristic diffraction peaks of MoS2 are detected in 1T-Co-MoS2@HMCS, and the distinct diffraction angle of the (002) crystal plane of MoS2 significantly shifts to a lower angle (11.8°) compared to 14.4° of MoS2 (PDF# 01–089–3040), due to the increase in MoS2 interlayer spacing caused by the spatial confined effect. No other peaks related to Co species were observed in the XRD patterns, suggesting that after a vapor phase sulfidation process, a single phase of Co-MoS2 was successfully formed in the HMCS. Fig. 3e shows the HRTEM image and the corresponding FFT image (inset) of 1T-Co-MoS2@HMCS, displays that the order lattice spacings are 0.75 and 0.27 nm, corresponding to the (002) and (100) crystal planes of MoS2. This obvious increase in d-spacing in the (002) plane compared to conventional MoS2 (0.62 nm) agrees with the findings from the XRD pattern and the calculated result from Bragg's equation.7 The expanded interplanar spacing of MoS2 can greatly favour the phase transition from 2H to 1T. Meanwhile, compared with Co-MoS2 obtained by direct sulfidation of Co2Mo10, most of the MoO2 phase remains, suggesting that the confined sulfidation process has a higher degree of sulfidation. The complete sulfidation of (Co2Mo10)(HM)3@HMCS is attributed to the higher degree of dispersion of Co2Mo10 due to the confinement effects provided by HMCS and the cationic surfactants. This confirms that the HMCS provides a fine environment for MoS2 to be formed and dispersed inside the carbon spheres due to the spatial confinement effect. In addition, compared to the XRD patterns of 1T-Co-MoS2@HMCS, the materials obtained without addition of surfactant (Co-MoS2@HMCS) show identical diffraction patterns, indicating that the HM2+ surfactant only serves as confinement agents during the sulfidation process and significantly improve the active phase dispersion (Fig. S3†).
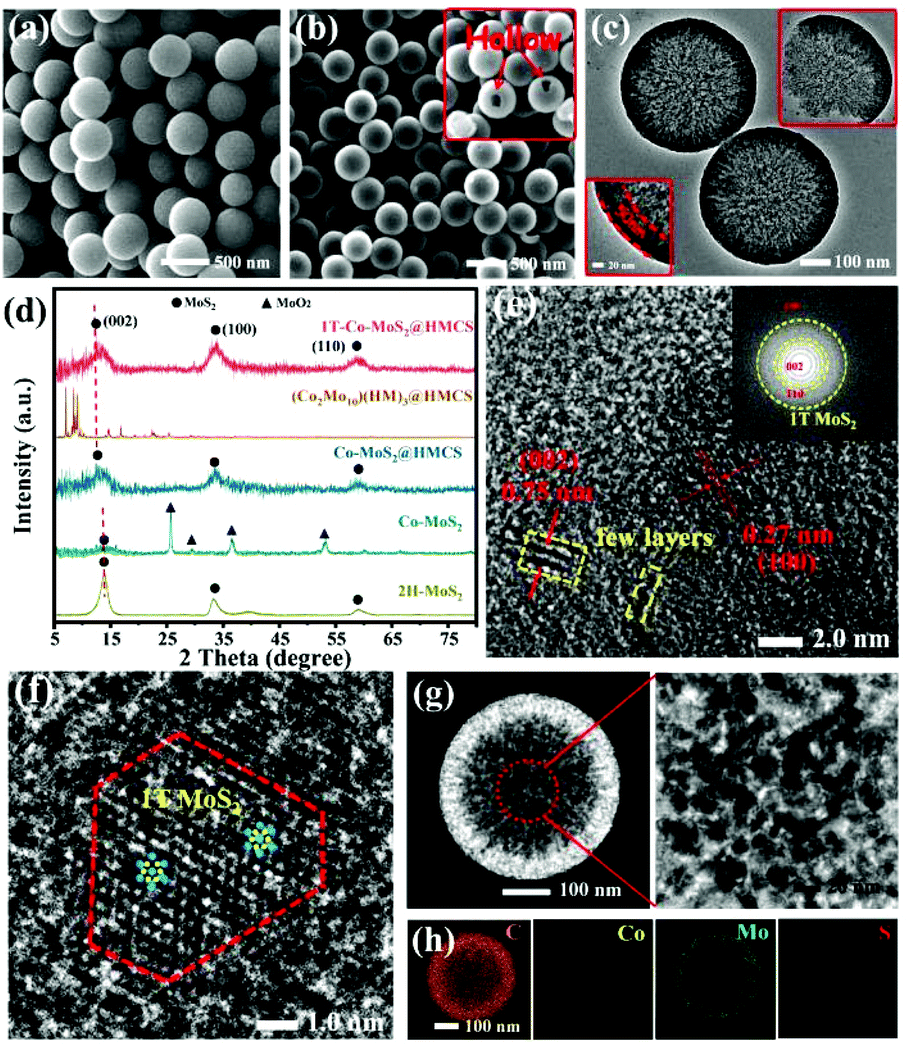 | ||
| Fig. 3 (a and b) SEM images of HMCS and 1T-Co-MoS2@HMCS, (c) TEM images of 1T-Co-MoS2@HMCS, (d) XRD patterns for HMCS, Co-MoS2, Co-MoS2@HMCS, (Co2Mo10)(HM)3@HMCS and 1T-Co-MoS2@HMCS, and (eand f) HRTEM images of 1T-Co-MoS2@HMCS, (g) HAADF-STEM images, and (h) the EDX mapping obtained by TEM for selected area of 1T-Co-MoS2@HMCS. | ||
Fig. 3f and Fig. S4† show the HRTEM images of 1T-Co-MoS2 confined within HMCS. The ultra-small 1T-Co-MoS2 cluster exhibits approximately truncated triangular shape, which is consistent with previous studies of the CoMoS phase.19,36,37 According to the research of Lauritsen et al.,19 this shape can indicate Co doping into a 1T-MoS2 crystal lattice, where Co terminates the growth of 1T-MoS2 along the (001) plane and promotes the formation of the Co-substituted S-edge. In other words, the modification to a partially truncated triangular shape was concluded to be driven by the preferential substitution of Co atoms at the S-edges, which further stabilizes the energy of the S-edge.19 It was also demonstrated in similar research that CoMoS nanoparticles exhibit a truncated hexagonal equilibrium morphology due to Co deposition, exposing both Mo- and S-edges.38 To the best of our knowledge, single-layer Co-MoS2 was prepared with difficultly and observed in addition to the growth on the Au support by chemical vapor deposition. Furthermore, the lateral size of 1T-Co-MoS2 was found to be ∼4 nm, and the stacking number was observed in single or few layers (1–3 layers). The limited pore size and cationic surfactants could avoid the accumulation and growth of MoS2 caused by sulfidation at the basal plane and epitaxial direction, respectively.39 It is speculated that the appearance of Co-doped 1T-MoS2 may originate from the large interlayer spacing with small size regulated by the spatially restricted domain and the electron-modulating effect of Co doping.40,41 Thus, the truncated triangular morphology, few-layer stacking, and reduced lateral size observed in 1T-Co-MoS2@HMCS could be attributed to the synergistically confined growth effect from the atomic level to the mesoscopic scale, which benefits from the hierarchical structural (Co2Mo10)(HM)3@HMCS precursor and in situ vapour-phase sulfurization approach.
The porous structures of HMCS on the shell of hollow nanospheres are further supported by the annular dark-field STEM analysis. As shown in Fig. 3g, the dark areas indicate the enrichment of mesopores in HMCS, and bright areas indicate the dispersion of Co-MoS2 in the HMCS with small sizes. The elemental mappings from EDX show the presence and homogenous distribution of C, Co, Mo, and S (Fig. 3h), indicating that the synthetic method in this study implemented uniform dispersion of small-sized Co-MoS2 in the HMCS nanoreactor and provided more discrete catalytic sites for HER. Meanwhile, it also benefits from the organic cation-confined engineering of Co–O–Mo within the POMs molecule, realizing doping and dispersion of the double transition metals at the molecular level. In addition, the BET analysis shows that the specific surface area of HMCS is about 1377 m2 g−1, and the main pore size is about 7.8 nm in diameter (Fig. S4†). The specific surface area of 1T-Co-MoS2@HMCS is about 189.6 m2 g−1, and the main pore size is about 7.1 nm in diameter, which reveals the successful loading of MoS2 nanosheets inside carbon spheres. These results show that the obtained 1T-Co-MoS2@HMCS maintain the mesoporous structure of HMCS, which can expose the numerous 1T-Co-MoS2 active sites and promote faster charge and mass transfer to enhance the electrocatalytic performance.
Raman spectra further confirm the existence of 1T-MoS2 nanosheets in HMCS (Fig. 4a and S6†). The Raman spectrum of 1T-Co-MoS2@HMCS reveals vibration peaks appearing at 144 (J1), 235 (J2), 283 (Eg), and 333 cm−1(J3), attributed to the highly distorted structure of 1T-MoS2. The broadening of 404 (A1g) and 375 cm−1 (E12g) modes and the suppressed intensity of E12g are also typical for the 1T phase. To further demonstrate the high proportion of 1T-MoS2, the Raman spectrum of pure 2H-MoS2 is compared to 1T-Co-MoS2@HMCS. Two strong fingerprint peaks of 2H-MoS2 appear at 375 and 404 cm−1, assigned to the in-plane and out-of-plane vibration modes of 2H-MoS2, respectively. The comparison results show the high proportion of 1T-MoS2 in 1T-Co-MoS2@HMCS, which also coincides with the results of XPS. In addition, the three peaks between 800 and 1000 cm−1 are ascribed to the molecular structure of Mo3S13,42,43 which locate at the edge sites of MoS2, thus further proving that the ultra-small nanosheets of 1T-Co-MoS2@HMCS can expose a large quantity of under-coordinated Mo–S edge sites. The other two peaks located at 1341 and 1595 cm−1 in 1T-Co-MoS2@HMCS are attributed to the D and G bands of graphitized carbon, which confirms the presence of the HMCS shell. Herein, it is confirmed by Raman spectroscopy that 1T-MoS2 with abundant edge active sites loaded on carbon spheres was indeed prepared. To further verify the chemical composition and valence state of 1T-Co-MoS2@HMCS, XPS analyses were carried out. Fig. S7a† shows the full XPS spectrum of 1T-Co-MoS2@HMCS, indicating the existence of Co, N, C, Mo, and S. The results show a low atomic ratio of 0.65% Co, while the atomic ratio of Mo is 2.83%, which is about 5 times higher than that of Co. This coincides with the atomic ratio of POMs, Co2Mo10. In Fig. 4b, the peaks at around 229.0 and 232.2 eV correspond to the binding energies of Mo 3d3/2 and Mo 3d5/2 for 1T-MoS2.44 Compared to the deconvoluted peaks for 2H-MoS2 (purple peaks), the decrease in the binding energy of 1T-MoS2 is because the Mo–S bond in the unit cell has a shorter bond length than that of the 2H phase. The electron-rich S increases the electron cloud density of Mo due to its strong electronegativity, which increases the shielding effect as a result. The Mo 3d peaks of 1T-Co-MoS2 shift to lower binding energies of approximately 1 eV when compared with the peaks shown in 2H-MoS2, and demonstrate a relaxation energy of 1 eV for the 1T phase derived from the 2H phase. The peaks at about 235.8 and 233.7 eV in the 1T-Co-MoS2@HMCS are attributed to the existence of a small amount of Mo6+. Deconvolution of the high-resolution spectrum in the Mo 3d region shows that the 1T phase content is as high as ∼90%, indicating successful synthesis of 1T phase Co-MoS2. Similarly, for the S 2p spectrum (Fig. 4c), the 2p1/2 and 2p3/2 peaks locate at 163.2 and 161.9 eV, which are approximately 1 eV lower than the corresponding binding energies of 2H-MoS2 (164.3 and 162.6 eV). The changes in these peaks could be ascribed to the formation of 1T-MoS2.45 In addition, the low intensity of S 2s (Fig. 4b) in the 1T-Co-MoS2@HMCS catalysts can be caused by the sulphur vacancy produced during the vapor-phase sulfidation process. The presence of sulphur vacancy leads to the destruction of Mo–S, which allows Mo to combine with oxygen to form MoO3. That is why the peak intensity of Mo6+ of MoO3 is slightly higher than that of Mo4+ of the 2H phase. Of course, leaving the sample in the air for a long time also increases the intensity of the Mo6+ peak.13,46 Fig. S7b† shows the XPS spectrum in the Co 2p region. The spectrum is weaker than that of other elements, indicating relatively low contents of Co in the 1T-Co-MoS2@HMCS, which agrees with the EDX mapping analysis results (Fig. 3h). The deconvoluted peaks at 794.0 and 779.1 eV could be assigned to the binding energy of 2p1/2 and 2p3/2 of Co3+, respectively, whereas the peaks at 798.4 and 782.0 eV could be ascribed to the binding energy of 2p1/2 and 2p3/2 spin orbits of Co2+. The peaks at 802.6 and 787.7 eV are the satellite peaks due to the surface complication.47
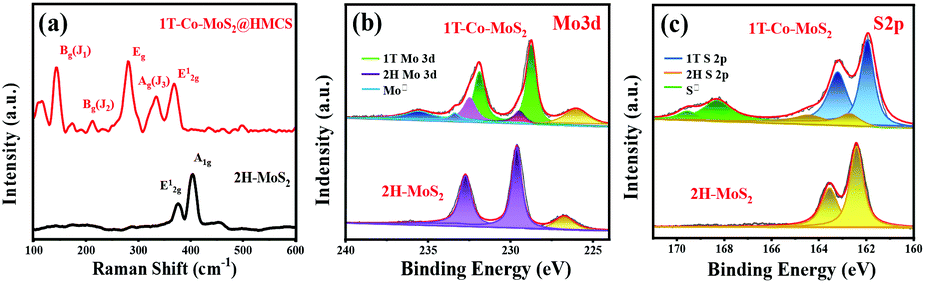 | ||
| Fig. 4 (a) Raman spectra and high-resolution XPS spectra of (b) Mo 3d and (c) S 2p of 1T-Co-MoS2@HMCS catalyst and 2H-MoS2. | ||
In order to exhibit the effectiveness of the multi-level space-confined strategy to obtain 1T-MoS2, the XPS spectra and Raman spectra were further taken to demonstrate the contribution of each level of the strategy to the substable structure. As shown in the Mo 3d and S 2p XPS spectra presented in Fig. S8 and S9,† there is almost no 1T-MoS2 in Co-MoS2, and 1T-MoS2 coexists with 2H-MoS2 in Co-MoS2@HMCS and MoS2@HMCS. These results indicate that at each level, the strategy provides support to enhance the 1T-MoS2 proportion. In particular, HMCS, with mesoporous structures capable of limiting nanosheet growth, play the most critical role, which is consistent with the Raman spectra (Fig. S10†).
Thus, all the above-mentioned results show that the 1T-Co-MoS2@HMCS nanospheres synthesized by the spatial confinement strategy combined with vapor-phase sulfidation have been proven effective to obtain ultra-dispersed nano-sized 1T-Co-MoS2 within the highly disordered mesoporous structure of HMCS. Hence, it is expected to obtain excellent electrocatalytic performance.
3.2 1T-Co-MoS2@HMCS electrocatalytic performance evaluation for HER
To explore the HER electrocatalytic activity of the as-synthesized 1T-Co-MoS2@HMCS, the electrochemical measurements of 1T-Co-MoS2@HMCS were evaluated by linear sweep voltammetry (LSV) at a scan rate of 5 mV s−1 in 1 M KOH and 0.5 M H2SO4 aqueous solutions. As shown in Fig. 5, the HER performance of (Co2Mo10)(HM)3@HMCS, Co-MoS2@HMCS, Co-MoS2, HMCS and commercial 20% Pt/C was also investigated. Fig. 5a exhibits that 1T-Co-MoS2@HMCS delivers smaller overpotentials (74 and 220 mV) compared with (Co2Mo10)(HM)3@HMCS (228 and 365 mV), Co-MoS2@HMCS (123 and 295 mV), Co-MoS2 (168 and 415 mV), and HMCS (358 and 595 mV) at −10 and −200 mA cm−2 in 1 M KOH, respectively. Among them, the HMCS shows the highest overpotential for HER in 1.0 M KOH, suggesting that the HMCS is inactive towards HER catalysis and only serves as a conductive substrate. Similarly, the LSV curve of (Co2Mo10)(HM)3@HMCS precursor also reflects inert hydrogen evolution activity. Compared with spatially confined 1T-Co-MoS2 in HMCS, Co-MoS2 without confinement exhibited low HER catalytic activity, indicating that the confinement of 1T-Co-MoS2 within the mesoporous carbon sphere and the surfactants can significantly enhance the activity of catalytic materials. Besides, Co-MoS2@HMCS shows reasonable HER catalytic activity, suggesting that the presence of HMCS itself without the surfactants can help disperse Co-MoS2 to some extent. Thus, the above discussion for the LSV image results illustrates the positive effect of multi-level confinements. HER kinetics was further evaluated using Tafel plots. The Tafel slopes of 1T-Co-MoS2@HMCS, (Co2Mo10)(HM)3@HMCS, Co-MoS2@HMCS, Co-MoS2, HMCS and commercial 20% Pt/C were calculated as 63, 81, 101, 107, 141 and 29 mV·dec−1 in 1 M KOH, respectively (Fig. 5b). The rather low Tafel slope of 1T-Co-MoS2@HMCS suggests the fast rate of electron transfer kinetics. In addition, electrochemical impedance spectroscopic measurements were performed under acidic and alkaline conditions. As shown in Fig. S11,† the 1T-Co-MoS2@HMCS electrocatalyst exhibits a much lower charge transfer resistance (Rct) than a series of prepared CoMo catalysts, indicating the rapid interfacial reaction rate of the 1T-Co-MoS2 catalyst in the HER process.48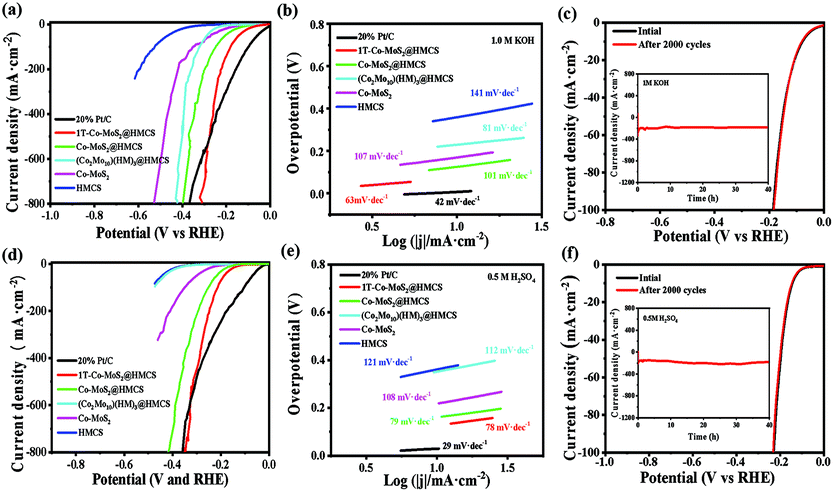 | ||
| Fig. 5 (a) HER polarization curves of different electrodes and electrode materials in 1.0 M KOH. (b) Corresponding Tafel plots obtained from (a). (c) HER durability of 1T-Co-MoS2@HMCS in 1.0 M KOH: polarization curves; the inset is the time dependence of catalytic currents. (d) HER polarization curves of different electrodes and electrode materials in 0.5 M H2SO4. (e) Corresponding Tafel plot obtained from (d). (f) HER durability of 1T-Co-MoS2@HMCS in 0.5 M H2SO4: polarization curves; the inset is the time dependence of catalytic currents. | ||
Generally, the HER activity of catalysts is affected by the electrochemical active surface area (ECSA) and intrinsic activity; therefore, it is necessary to estimate the catalytically active surface area to reveal the effect of spatial confined regulation. The double-layer capacitance (Cdl) was investigated by cyclic voltammetry, which is generally proportional to the ECSA (Fig. S12 and S13†). 1T-Co-MoS2@HMCS catalysts exhibited medium Cdl, suggesting that the improved HER activity of 1T-Co-MoS2@HMCS catalysts originate from the enhanced intrinsic activity of each active site. Among them, Cdl of unconfined Co-MoS2 proves the low electrochemical working area of undispersed Co2Mo10, implying a poor HER performance, which is consistent with the LSV results. Cdl test results show that Co2Mo10 precursors, hexamethonium (HM) bromide surfactants, HMCS substrate, and vapor phase sulfidation all have a decisive contribution to the catalytic performance of the target catalyst. Stability was tested to reflect the durable operation of the electrocatalyst for water electrolysis. As shown in Fig. 5c, LSV curves before and after 20000 cyclic voltammetry (CV) cycles were recorded to examine the stability in an alkaline medium, and the insert figure contains the i-t curve of 40 h at −200 mA cm−2 continuous electrolysis on 1T-Co-MoS2@HMCS, which exhibits almost no deactivation for HER. The above-mentioned electrochemical tests have obtained similar results under acidic conditions. 1T-Co-MoS2@HMCS exhibits superior low overpotential (132 and 245 mV at −10 and −200 mA·cm−2, respectively), low Tafel slope (78 mV·dec−1) and excellent cycle stability compared to other materials in 0.5 M H2SO4 (Fig. 5d–f). Table S1 and S2† list the detailed comparison of the HER performance of 1T-Co-MoS2@HMCS with other reported recent non-precious electrocatalysts, revealing that the 1T-Co-MoS2@HMCS nanosheet is superior to the reported Mo- or Co-based HER electrocatalysts.
In order to prove the benefits of POMs as a high-quality bimetal assembly platform, individual MoS2@HMCS and CoS@HMCS, as well as their mechanically mixed nanostructures, were also prepared based on the related procedures reported in the literature, whose HER performances were evaluated by LSV, as shown in Fig. S14.† The nanospheres MoS2@HMCS and CoS@HMCS both show inferior HER electrocatalytic activities, with potentials of 162 and 446 mV at −10 mA·cm−2 in 1 M KOH, respectively. The similar situation occurred in 0.5 M H2SO4, but MoS2@HMCS had a reduced overpotential compared to that in alkaline solutions, implying a higher intrinsic activity of MoS2 under acidic conditions, in agreement with previous studies.16 In addition, 1T-Co-MoS2@HMCS was also examined in neutral and sea water solutions, and showed reasonable performances (Fig. S15†).
The prominent HER performance of 1T-Co-MoS2@HMCS could be ascribed to the following two reasons: (1) The multilevel confinement effect was shown to be an effective way to synthesize 1T-MoS2 with abundant active sites, providing a structural basis for its high activity. Based on the advantageous platform of POMs, the dispersant improves the dispersion of active sites by inner-space-limited engineering. Meanwhile, the HMCS, as a nanoreactor with a large number of disordered mesoporous structures, was loaded with nanoscale MoS2 sheets by outer-space-limited engineering. The diffusion length of molecules/ions was shortened, resulting in a large amount of H2 production. (2) The bimetallic synergistic effect of 1T-MoS2 was achieved by Co doping at the S-edge, which effectively adjusts the electronic structure and thus improves the absorption and desorption efficiency of H2.
3.3 DFT calculations
We developed a 1T-Co-MoS2 model for DFT calculations and investigated the doping effect on the HER activity of 1T-MoS2 by substituting different Mo sites with Co (Fig. S16†). The adsorption model for acidic and alkaline conditions is shown in Fig. S17 and S18.† As shown in Fig. 6a, we discuss the contribution of different types of S sites to the catalytic activity. Fig. 6b reports the energy barriers of H2O dissociation and H2 desorption in an alkali environment. The H2O dissociation step is the rate-limiting step, and the free energy barrier follows the trend of Mo–S basal sites (2.29 eV) > Co–S basal site (2.21 eV) > Mo–S edge site (2.07 eV) > Co–S edge site (1.94 eV). For the desorption process of H2, ΔGH* on the Co–S edge site (1.14 eV) is much lower than that on the Mo–S basal site of 1T MoS2 (1.60 eV), the Co–S basal site (1.50 eV), and the Mo–S edge site (1.34 eV) of 1T-Co-MoS2, demonstrating the easier desorption process on Co–S edge sites. These calculations are consistent with the experimental finding that the most active site of 1T-Co-MoS2@HMCS is the Co–S edge site, lowering the free energy for H2O dissociation and H desorption, both important steps in alkaline HER, giving rise to enhanced activity towards HER and reflecting the major significance of Co doping.49,50 Moreover, the DFT calculation results exhibited a similar activity trend under acidic solutions (Fig. 6c), indicating the high intrinsic activity of the Co–S edge sites. The calculated results are in agreement with the results of previous studies, which confirmed that the Heyrovsky route for the complete HER pathway is more favourable than the Tafel route.14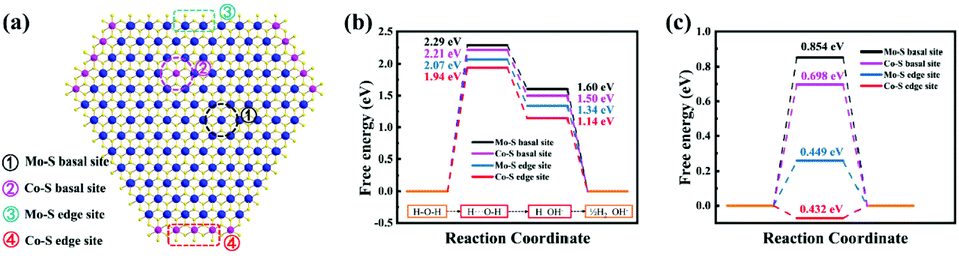 | ||
| Fig. 6 (a) Schematic diagram of the atomic model and sites. Free energies of hydrogen atom adsorption on the basal plane of pristine 1T-MoS2 as well as on the Mo–S edge and Co–S edge of 1T-Co-MoS2@HMCS in alkaline (b) and acidic solutions (c). | ||
The atomic interaction of 1T-MoS2 and Co was further studied based on the electronic structure analysis. Due to the doping effects of Co, the S atoms are more positively charged than pure 1T-MoS2, thus forming a built-in electric field, so that the charges can be quickly transferred between Co and S (Fig. S19†). Compared with the Mo–S basal site of 1T-MoS2 and the Co–S basal site of 1T-Co-MoS2 (cobalt doped inside), the DOS of the Co–S edge site near the Fermi level enhanced (Fig. S20†), indicating that the synergetic effects of Co and S in the boundary significantly increased the charge transfer during water splitting.51 Furthermore, the d-band centre of S atoms in the Co edge site of 1T-Co-MoS2 system is also shifted to a higher energy of −0.752 eV than that in the 1T-MoS2 system (−1.162 eV) and Co basal site of 1T-Co-MoS2 (−1.025 eV) (Fig. S20†). The shift of the d-band centre would further lead to less occupation of the antibonding states between the Co edge of 1T-Co-MoS2 and adsorbed intermediates, thereby enhancing their binding strength. The DOS and PDOS results show the positive roles of the synergetic coupling effects of Co and S in the boundary, confirming that the density of the unoccupied eg orbit state increased in the Co edge of 1T-Co-MoS2 (Fig. S21†). With enhanced conductivity, improved binding strength with intermediates and unoccupied eg orbits, the Co–S edge site of 1T-Co-MoS2 can significantly optimize the HER activity.
3.4 Structure–activity relationship
As shown in Fig. 7, the optimized combination of Co2Mo10 binary POMs, hexamethonium (HM) bromide surfactants, and HMCS substrates shows obvious synergistic effects for the design and synthesis of efficient HER catalysts. Consequently, the well-dispersed (1–3 layers), ultrafine (<5 nm), and metastable Co-doped 1T-MoS2 nanoclusters were in situ fabricated from the POM-based molecular precursor via a multi-level spatial confinement strategy. Based on the aforementioned experimental results, we conclude three advantages for the superior precursors (Co2Mo10)(HM)3@HMCS and the corresponding multi-level spatial confinement strategy, so-called “killing three birds with one stone”, as follows: First, the nanosized Co2Mo10 molecules with a strong and intimate Co–O–Mo interaction facilitated the formation of the Co-promoted MoS2 phase, that is so-called Co-MoS2 active sites. The Mo atom at the S-edge in MoS2 is preferential substituted by Co to generate new Co–S edges, leading to the shape transformation from triangular MoS2 to truncated triangular Co-MoS2 clusters. This result was verified by considering a previously reported study.40 Second, the cationic surfactant hexamethonium (HM) bromide was used for dispersing the Co-MoS2 nanosheets and preventing them from stacking in the [002] direction, resulting in the formation of single- or few-layer Co-MoS2 protected by organic buffer layers. As the previous studies suggested, the expanded interlayer spacing of MoS2 can be beneficial for the formation of 1T-MoS2.52 However, ultra-small Co-MoS2 nanosheets often suffer from severe aggregation and poor thermal stability for their high surface free energy. Third, mesoporous HMCS were therefore adopted to greatly stabilize single-layer nanoclusters and inhibit the epitaxial growth of MoS2, favouring the formation of metastable 1T-MoS2 in the confined spaces.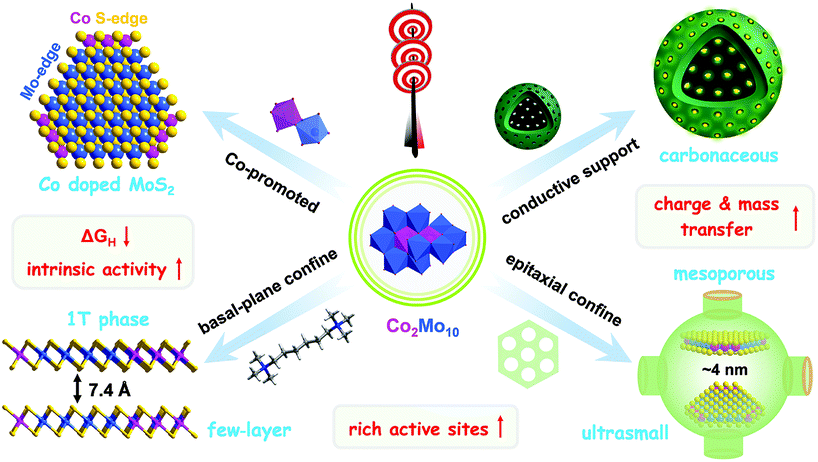 | ||
| Fig. 7 Schematic diagram of the design and fabrication of the 1T-Co-MoS2@HMCS catalyst from the POM-based precursor (Co2Mo10)(HM)3@HMCS via a multi-level spatial confined strategy. | ||
In addition, 1T-Co-MoS2@HMCS not only preserves the mesoporous structure to expose rich Co-MoS2 edge active sites in a network of pores, but also acts as an effective electron conductor, electrolyte transporter and active site protector to enhance charge and mass transfer, which can effectively confine the catalytic HER in hollow sphere structures and make each mesopore configuration as an independent and efficient HER active ecosystem.53 Therefore, the great HER electrocatalytic performance of 1T-Co-MoS2@HMCS should be attributed to its unique structure, high intrinsic activity from Co-doped 1T-MoS2 sites, abundant exposed active sites from ultra-dispersed nanosheets, and enhanced charge and mass transfer within the HMCS substrate.
4. Conclusions
In summary, highly dispersed 1T-Co-MoS2 nanoclusters with few layers and ultra-small size have been successfully fabricated and encapsulated within HMCS conductive substrates by spatially confining engineering. During in situ vapor-phase sulfurization, the optimized precursor combination of Co2Mo10 binary POMs, hexamethonium (HM) bromide surfactants, and HMCS substrates exhibits synergistically confined growth effects from the atomic level, molecular level to mesoscopic scale, thereby achieving the simultaneous regulation of active sites, basal planes, and epitaxial growth of MoS2. When evaluated as an electrocatalyst for the HER process, 1T-Co-MoS2@HMCS shows a prominent activity and excellent long-term durability, and only requires an overpotential of 220 and 245 mV at 200 mA cm−2 current density in 1 M KOH and 0.5 M H2SO4 electrolytes, respectively. DFT calculations further reveal that Co–S edge sites possess highly intrinsic activity in Co-doped 1T-MoS2 for HER, reflecting the major significance of Co doping. Meanwhile, the mesoporous and carbonaceous structures of HMCS can expose the numerous Co-doped 1T-MoS2 active edge sites and promote faster charge and mass transfer to enhance the apparent catalytic activity. Thus, this work provides the first facile preparation of high-purity Co-doped 1T-MoS2 by multi-level spatial confinement and offers an effective way to solve the poor basic activity through Co doping, which offers a reasonable scheme for the design and synthesis of other metastable nanomaterials derived from POM-based hybrid precursors.Conflicts of interest
There are no conflicts to declareAcknowledgements
This work is financially supported by the National Natural Science Foundation of China (21878336, 21805308), the Shandong Provincial Natural Science Foundation, China (ZR2018MB035), the Key Research and Development Project of Shandong Province (2019GSF109075), the Fundamental Research Funds for the Central Universities (20CX02213A), and the China University of Petroleum, Huadong (YCX2021153).References
- M. Li, B. Cai, R. Tian, X. Yu, M. B. H. Breese, X. Chu, Z. Han, S. Li, R. Joshi, A. Vinu, T. Wan, Z. Ao, J. Yi and D. Chu, Vanadium doped 1T MoS2 nanosheets for highly efficient electrocatalytic hydrogen evolution in both acidic and alkaline solutions, Chem. Eng. J, 2021, 409, 128158 Search PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Combining theory and experiment in electrocatalysis: Insights into materials design, Science, 2017, 355, eaad4998 Search PubMed.
- C. C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, Benchmarking hydrogen evolving reaction and oxygen evolving reaction electrocatalysts for solar water splitting devices, J. Am. Chem. Soc., 2015, 137, 4347–4357 Search PubMed.
- J. Yang, J. K. Cooper, F. M. Toma, K. A. Walczak, M. Favaro, J. W. Beeman, L. H. Hess, C. Wang, C. Zhu, S. Gul, J. Yano, C. Kisielowski, A. Schwartzberg and I. D. Sharp, A multifunctional biphasic water splitting catalyst tailored for integration with high-performance semiconductor photoanodes, Nat. Mater., 2017, 16, 335–341 Search PubMed.
- Z. H. Xue, H. Su, Q. Y. Yu, B. Zhang, H. H. Wang, X. H. Li and J. S. Chen, Janus Co/CoP Nanoparticles as Efficient Mott–Schottky Electrocatalysts for Overall Water Splitting in Wide pH Range, Adv. Energy Mater., 2017, 7, 1602355 Search PubMed.
- C. Chen, A. Wu, H. Yan, Y. Xiao, C. Tian and H. Fu, Trapping [PMo12O40]3− clusters into pre-synthesized ZIF-67 toward MoxCoxC particles confined in uniform carbon polyhedrons for efficient overall water splitting, Chem. Sci., 2018, 9, 4746–4755 Search PubMed.
- Y. J. Tang, Y. Wang, X. L. Wang, S. L. Li, W. Huang, L. Z. Dong, C. H. Liu, Y. F. Li and Y. Q. Lan, Molybdenum Disulfide/Nitrogen-Doped Reduced Graphene Oxide Nanocomposite with Enlarged Interlayer Spacing for Electrocatalytic Hydrogen Evolution, Adv. Energy Mater., 2016, 6, 1600116 Search PubMed.
- J. Hou, B. Zhang, Z. Li, S. Cao, Y. Sun, Y. Wu, Z. Gao and L. Sun, Vertically Aligned Oxygenated-CoS2−MoS2 Heteronanosheet Architecture from Polyoxometalate for Efficient and Stable Overall Water Splitting, ACS Catal., 2018, 8, 4612–4621 Search PubMed.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, Catalyzing the Hydrogen Evolution Reaction (HER) with Molybdenum Sulfide Nanomaterials, ACS Catal., 2014, 4, 3957–3971 Search PubMed.
- H. I. Karunadasa, E. Montalvo, Y. Sun, M. Majda, J. R. Long and C. J. Chang, A Molecular MoS2 Edge Site Mimic for Catalytic Hydrogen Generation, Science, 2012, 335, 698–702 Search PubMed.
- J. Zhang, J. Wu, H. Guo, W. Chen, J. Yuan, U. Martinez, G. Gupta, A. Mohite, P. M. Ajayan and J. Lou, Unveiling Active Sites for the Hydrogen Evolution Reaction on Monolayer MoS2, Adv. Mater., 2017, 29, 1701955 Search PubMed.
- Z. Lei, J. Zhan, L. Tang, Y. Zhang and Y. Wang, Recent Development of Metallic (1T) Phase of Molybdenum Disulfide for Energy Conversion and Storage, Adv. Energy Mater., 2018, 8, 1703482 Search PubMed.
- Y. Huang, Y. Sun, X. Zheng, T. Aoki, B. Pattengale, J. Huang, X. He, W. Bian, S. Younan, N. Williams, J. Hu, J. Ge, N. Pu, X. Yan, X. Pan, L. Zhang, Y. Wei and J. Gu, Atomically engineering activation sites onto metallic 1T-MoS2 catalysts for enhanced electrochemical hydrogen evolution, Nat. Commun., 2019, 10, 982 Search PubMed.
- Q. Tang and D. E. Jiang, Stabilization and Band-Gap Tuning of the 1T-MoS2 Monolayer by Covalent Functionalization, Chem. Mater., 2015, 27, 3743–3748 Search PubMed.
- Z. Zhu, H. Yin, C. T. He, M. Al-Mamun, P. Liu, L. Jiang, Y. Zhao, Y. Wang, H. G. Yang, Z. Tang, D. Wang, X. M. Chen and H. Zhao, Ultrathin Transition Metal Dichalcogenide/3d Metal Hydroxide Hybridized Nanosheets to Enhance Hydrogen Evolution Activity, Adv. Mater., 2018, 30, 1801171 Search PubMed.
- D. Cao, K. Ye, O. A. Moses, W. Xu, D. Liu, P. Song, C. Wu, C. Wang, S. Ding, S. Chen, B. Ge, J. Jiang and L. Song, Engineering the In-Plane Structure of Metallic Phase Molybdenum Disulfide via Co and O Dopants toward Efficient Alkaline Hydrogen Evolution, ACS Nano, 2019, 13, 11733–11740 Search PubMed.
- R. Forsythe, C. Cox, M. Wilsey and A. Müller, Pulsed Laser in Liquids Made Nanomaterials for Catalysis, Chem. Rev., 2021, 121, 7568–7637 Search PubMed.
- J. Guo, X. Zhang, Y. Sun, L. Tang and X. Zhang, Self-template Synthesis of Hierarchical CoMoS3 Nanotubes Constructed of Ultrathin Nanosheets for Robust Water Electrolysis, J. Mater. Chem. A, 2017, 5, 11309–11315 Search PubMed.
- S. S. Grønborg, N. Salazar, A. Bruix, J. Rodríguez-Fernández, S. D. Thomsen, B. Hammer and J. V. Lauritsen, Visualizing Hydrogen-induced Reshaping and Edge Activation in MoS2 and Co-promoted MoS2 Catalyst Clusters, Nat. Commun., 2018, 9, 2211 Search PubMed.
- N. H. Attanayake, A. C. Thenuwara, A. Patra, Y. V. Aulin, T. M. Tran, H. Chakraborty, E. Borguet, M. L. Klein, J. P. Perdew and D. R. Strongin, Effect of Intercalated Metals on the Electrocatalytic Activity of 1T-MoS2 for the Hydrogen Evolution Reaction, ACS Energy Lett., 2018, 3, 7–13 Search PubMed.
- J. Strachan, A. F. Masters and T. Maschmeyer, Critical review: hydrothermal synthesis of 1T-MoS2 – an important route to a promising material, J. Mater. Chem. A, 2021, 9, 9451–9461 Search PubMed.
- K. Chang, X. Hai, H. Pang, H. Zhang, L. Shi, G. Liu, H. Liu, G. Zhao, M. Li and J. Ye, Targeted Synthesis of 2H- and 1T-Phase MoS2 Monolayers for Catalytic Hydrogen Evolution, Adv. Mater., 2016, 28, 10033–10041 Search PubMed.
- X. Chen, Z. Wang, Y. Wei, X. Zhang, Q. Zhang, L. Gu, L. Zhang, N. Yang and R. Yu, High Phase-Purity 1T-MoS2 Ultrathin Nanosheets by a Spatially Confined Template, Angew. Chem., Int. Ed., 2019, 58, 17621–17624 Search PubMed.
- Y. Jiao, W. Hong, P. Li, L. Wang and G. Chen, Metal-organic framework derived Ni/NiO micro-particles with subtle lattice distortions for high-performance electrocatalyst and supercapacitor, Appl. Catal., B, 2019, 244, 732–739 Search PubMed.
- Y. K. Lu, C. L. Yue, Y. P. Li, W. J. Bao, X. X. Guo, W. F. Yang, Z. Liu, P. Jiang, W. Yan, S. Liu, Y. Pan and Y. Q. Liu, Atomically dispersed Ni on Mo2C embedded in N, P co-doped carbon derived from polyoxometalate supramolecule for high-efficiency hydrogen evolution electrocatalysis, Appl. Catal., B, 2021, 296, 120336 Search PubMed.
- Y. Y. Ma, C. X. Wu, X. J. Feng, H. Q. Tan, L. K. Yan, Y. Liu, Z. H. Kang, E. B. Wang and Y. G. Li, Highly efficient hydrogen evolution from seawater by a low-cost and stable CoMoP@C electrocatalyst superior to Pt/C, Energy Environ. Sci., 2017, 10, 788–798 Search PubMed.
- B. Huang, D. H. Yang and B. H. Han, Application of polyoxometalate derivatives in rechargeable batteries, J. Mater. Chem. A, 2020, 8, 4593–4628 Search PubMed.
- Y. K. Lu, X. X. Guo, L. Y. Yang, W. F. Yang, W. Sun, Y. Tuo, Y. Zhou, S. Wang, Y. Pan, W. Yan, D. Sun and Y. Liu, Highly efficient CoMoS heterostructure derived from vertically anchored Co5Mo10 polyoxometalate for electrocatalytic overall water splitting, Chem. Eng. J., 2020, 394, 124849 Search PubMed.
- J. S. Li, Y. Wang, C. H. Liu, S. L. Li, Y. G. Wang, L. Z. Dong, Z. H. Dai, Y. F. Li and Y. Q. Lan, Coupled molybdenum carbide and reduced graphene oxide electrocatalysts for efficient hydrogen evolution, Nat. Commun., 2016, 7, 11204 Search PubMed.
- J. A. Toledo-Antonio, M. A. Cortes-Jacome, J. Escobar-Aguilar, C. Angeles-Chavez, J. Navarrete-Bolaños and E. López-Salinas, Upgrading HDS activity of MoS2 catalysts by chelating thioglycolic acid to MoOx supported on alumina, Appl. Catal., B, 2017, 213, 106–117 Search PubMed.
- Z. Li, X. Zhang, H. Cheng, J. Liu, M. Shao, M. Wei, D. G. Evans, H. Zhang and X. Duan, Confined Synthesis of 2D Nanostructured Materials toward Electrocatalysis, Adv. Energy Mater., 2020, 10, 1900486 Search PubMed.
- S. Li, A. Pasc, V. Fierro and A. Celzard, Hollow carbon spheres, synthesis and applications – a review, J. Mater. Chem. A, 2016, 4, 12686–12713 Search PubMed.
- Z. Yu, N. Ji, J. Xiong, X. Li, R. Zhang, L. Zhang and X. Lu, Ruthenium-Nanoparticle-Loaded Hollow Carbon Spheres as Nanoreactors for Hydrogenation of Levulinic Acid: Explicitly Recognizing the Void-Confinement Effect, Angew. Chem., Int. Ed., 2021, 60, 20786–20794 Search PubMed.
- G. Prieto, H. Tüysüz, N. Duyckaerts, J. Knossalla, G. H. Wang and F. Schüth, Hollow Nano- and Microstructures as Catalysts, Chem. Rev., 2016, 116, 14056–14119 Search PubMed.
- A. Misra, K. Kozma, C. Streb and M. Nyman, Beyond Charge Balance: Counter-Cations in Polyoxometalate Chemistry, Angew. Chem., Int. Ed., 2020, 59, 596–612 Search PubMed.
- J. V. Lauritsen, J. Kibsgaard, G. H. Olesen, P. G. Moses, B. Hinnemann, S. Helveg, J. K. Nørskov, B. S. Clausen, H. Topsøe, E. Lægsgaard and F. Besenbacher, Location and coordination of promoter atoms in Co- and Ni-promoted MoS2-based hydrotreating catalysts, J. Catal., 2007, 249, 220–233 Search PubMed.
- J. V. Lauritsen and F. Besenbacher, Atom-resolved scanning tunneling microscopy investigations of molecular adsorption on MoS2 and CoMoS hydrodesulfurization catalysts, J. Catal., 2015, 328, 49–58 Search PubMed.
- A. S. Walton, J. V. Lauritsen, H. Topsøe and F. Besenbacher, J. Catal., 2013, 308, 306–318 Search PubMed.
- N. K. Perkgoz and M. Bay, Investigation of Single-Wall MoS2 Monolayer Flakes Grown by Chemical Vapor Deposition, Nano-Micro Lett., 2016, 8, 70–79 Search PubMed.
- S. Li, S. Wang, T. Xu, H. Zhang, Y. Tang, S. Liu, T. Jiang, S. Zhou and H. Cheng, Growth mechanism and atomic structure of group-IIA compound-promoted CVD-synthesized monolayer transition metal dichalcogenides, Nanoscale, 2021, 13, 13030–13041 Search PubMed.
- P. K. Sahoo, S. Memaran, Y. Xin, L. Balicas and H. R. Gutiérrez, One-pot growth of two-dimensional lateral heterostructures via sequential edge-epitaxy, Nature, 2018, 553, 63–67 Search PubMed.
- J. Staszak-Jirkovský, C. D. Malliakas, P. P. Lopes, N. Danilovic, S. S. Kota, K. C. Chang, B. Genorio, D. Strmcnik, V. R. Stamenkovic, M. G. Kanatzidis and N. M. Markovic, Design of active and stable Co-Mo-Sx chalcogels as pH-universal catalysts for the hydrogen evolution reaction, Nat. Mater., 2016, 15, 197–203 Search PubMed.
- T. An, Y. Wang, J. Tang, W. Wei, X. Cui, A. M. Alenizi, L. Zhang and G. Zheng, Interlaced NiS2-MoS2 nanoflake-nanowires as efficient hydrogen evolution electrocatalysts in basic solutions, J. Mater. Chem. A, 2016, 4, 13439–13443 Search PubMed.
- G. Eda, H. Yamaguchi, D. Voiry, T. Fujita, M. Chen and M. Chhowalla, Photoluminescence from Chemically Exfoliated MoS2, Nano Lett., 2011, 11, 5111–5116 Search PubMed.
- W. Zhou, Z. Yin, Y. Du, X. Huang, Z. Zeng, Z. Fan, H. Liu, J. Wang and H. Zhang, Synthesis of Few-Layer MoS2 Nanosheet-Coated TiO2 Nanobelt Heterostructures for Enhanced Photocatalytic Activities, Small, 2013, 9, 140–147 Search PubMed.
- X. Y. Yu, Y. Feng, Y. Jeon, B. Guan, X. W. Lou and U. Paik, Formation of Ni–Co–MoS2 Nanoboxes with Enhanced Electrocatalytic Activity for Hydrogen Evolution, Adv. Mater., 2016, 28, 9006–9011 Search PubMed.
- Y. Yang, H. Yao, Z. Yu, S. M. Islam, H. He, M. Yuan, Y. Yue, K. Xu, W. Hao, G. Sun, H. Li, S. Ma, P. Zapol and M. G. Kanatzidis, Hierarchical Nanoassembly of MoS2/Co9S8/Ni3S2/Ni as a Highly Efficient Electrocatalyst for Overall Water Splitting in a Wide pH Range, J. Am. Chem. Soc., 2019, 141, 10417–10430 Search PubMed.
- P. Zhou, C. Wang, Y. Liu, Z. Wang, P. Wang, X. Qin, X. Zhang, Y. Dai, M. H. Whangbo and B. Huang, Sulfuration of NiV-layered double hydroxide towards novel supercapacitor electrode with enhanced performance, Chem. Eng. J., 2018, 351, 119–126 Search PubMed.
- J. Liang, C. Ding, J. Liu, T. Chen, W. Peng, Y. Li, F. Zhang and X. Fan, Heterostructure engineering of Co-doped MoS2 coupled with Mo2CTx MXene for enhanced hydrogen evolution in alkaline media, Nanoscale, 2019, 11, 10992–11000 Search PubMed.
- F. Ma, Y. Liang, P. Zhou, F. Tong, Z. Wang, P. Wang, Y. Liu, Y. Dai, Z. Zheng and B. Huang, One-step synthesis of Co-doped 1T-MoS2 nanosheets with efficient and stable HER activity in alkaline solutions, Mater. Chem. Phys., 2020, 244, 122642 Search PubMed.
- Y. Zheng, Y. Jiao, A. Vasileff and S. Z. Qiao, The Hydrogen Evolution Reaction in Alkaline Solution: From Theory, Single Crystal Models, to Practical Electrocatalysts, Angew. Chem., Int. Ed., 2018, 57, 7568–7579 Search PubMed.
- Z. Wang, T. Chen, W. Chen, K. Chang, L. Ma, G. Huang, D. Chen and J. Y. Lee, CTAB-assisted synthesis of single-layer MoS2−graphene composites as anode materials of Li-ion batteries, J. Mater. Chem. A, 2013, 1, 2202–2210 Search PubMed.
- B. Wang, Y. Ye, L. Xu, Y. Quan, W. Wei, W. Zhu, H. Li and J. Xia, Space-Confined Yolk-Shell Construction of Fe3O4 Nanoparticles Inside N-Doped Hollow Mesoporous Carbon Spheres as Bifunctional Electrocatalysts for Long-Term Rechargeable Zinc–Air Batteries, Adv. Funct. Mater., 2020, 30, 2005834 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qi00269h |
| This journal is © the Partner Organisations 2022 |