
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Competition between reverse water gas shift reaction and methanol synthesis from CO2: influence of copper particle size†
Laura
Barberis
 a,
Amir H.
Hakimioun
b,
Philipp N.
Plessow
b,
Nienke L.
Visser
a,
Joseph A.
Stewart
c,
Bart D.
Vandegehuchte
c,
Felix
Studt
bd and
Petra E.
de Jongh
*a
a,
Amir H.
Hakimioun
b,
Philipp N.
Plessow
b,
Nienke L.
Visser
a,
Joseph A.
Stewart
c,
Bart D.
Vandegehuchte
c,
Felix
Studt
bd and
Petra E.
de Jongh
*a
aMaterials Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: P.E.deJongh@uu.nl
bInstitute of Catalysis Research and Technology, Karlsruhe Institute of Technology, Eggenstein-Leopoldshafen 76344, Germany
cTotalEnergies OneTech Belgium, B-7181 Seneffe, Belgium
dInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology, Karlsruhe 76131, Germany
First published on 3rd August 2022
Abstract
Converting CO2 into value-added chemicals and fuels, such as methanol, is a promising approach to limit the environmental impact of human activities. Conventional methanol synthesis catalysts have shown limited efficiency and poor stability in a CO2/H2 mixture. To design improved catalysts, crucial for the effective utilization of CO2, an in-depth understanding of the active sites and reaction mechanism is desired. The catalytic performance of a series of carbon-supported Cu catalysts, with Cu particle sizes in the range of 5 to 20 nm, was evaluated under industrially relevant temperature and pressure, i.e. 260 °C and 40 bar(g). The CO2 hydrogenation reaction exhibited clear particle size effects up to 13 nm particles, with small nanoparticles having the lower activity, but higher methanol selectivity. MeOH and CO formation showed a different size-dependence. The TOFCO increased from 1.9 × 10−3 s−1 to 9.4 × 10−3 s−1 with Cu size increasing from 5 nm to 20 nm, while the TOFMeOH was size-independent (8.4 × 10−4 s−1 on average). The apparent activation energies for MeOH and CO formation were size-independent with values of 63 ± 7 kJ mol−1 and 118 ± 6 kJ mol−1, respectively. Hence the size dependence was ascribed to a decrease in the fraction of active sites suitable for CO formation with decreasing particle size. Theoretical models and DFT calculations showed that the origin of the particle size effect is most likely related to the differences in formate coverage for different Cu facets whose abundancy depends on particle size. Hence, the CO2 hydrogenation reaction is intrinsically sensitive to the Cu particle size.
Introduction
Global warming is considered one of the most serious environmental problems in recent years. The greenhouse gas contributing the most from human activities is carbon dioxide (CO2).1 A promising approach is the conversion of CO2 into value-added bulk chemicals and fuels, such as oxygenates (alcohols and dimethyl ether) and hydrocarbons (olefins, liquid hydrocarbons, and aromatics). Methanol is a common chemical feedstock in industry and potentially a substitute for fossil fuels.2 Global methanol demand reached 98.3 Mt in 2019, and is expected to surpass 120 Mt by 2025.3 Currently, methanol is almost exclusively produced from either natural gas or coal, mostly due to economic reasons. The industrial process involves the hydrogenation of CO2-enriched syngas over Cu/ZnO/Al2O3 catalysts at 240–260 °C and 40–100 bar.4,5It is generally accepted that the main carbon source in the process is CO2, which is generated by the water–gas shift reaction (CO + H2O → CO2 + H2) and converted into methanol (CO2 + 3H2 → CH3OH + H2O).6 The WGS reaction is beneficial for the catalyst's stability as it consumes the water generated during methanol synthesis. When CO2 is the only carbon source in the feedstock, besides being hydrogenated to methanol, CO2 is also consumed by the reverse water gas shift reaction (CO2 + H2 → CO + H2O). The undesired competitive rWGS reaction has to be avoided to optimize the MeOH selectivity. Moreover, the extra water formed as by-product by rWGS reaction could cause rapid catalyst degradation. These two factors make pure CO2 hydrogenation more challenging than conventional methanol synthesis.7 Another, challenge is the high thermodynamic stability of CO2, which means that a very good catalyst to operate at low temperatures is needed.8
A multitude of new catalytic formulations, based on Pd/Pt monometallic, intermetallic catalysts,9,10 and In2O3,11,12 have been proposed to overcome the challenges posed by a pure CO2 feed. Nevertheless, copper remains the most studied metal mainly thanks to its abundance, low price and highly reactive nature.7,13,14 The commercial Cu–ZnO–Al2O3 catalyst, successfully used for more than 50 years for the syngas to methanol reaction, shows reduced efficiency and faster loss of active surface area.15–19 Additives, such as ZrO2, TiO2, Ga2O3 and CeO2, have been introduced in the catalyst formulation to improve the performance for CO2 hydrogenation.20–23 This has led to a greater complexity of the catalytic systems, linked to their multi-component nature, i.e. simultaneous presence of multiple oxidic components as promoters and supports, which makes fundamental studies on the nature of the copper active sites and reaction mechanisms involved during CO2 hydrogenation more challenging. The use of an inert component, such as a graphitic carbon support, is of importance for reducing the catalyst's complexity by avoiding, e.g., strong metal–support interactions.24–26 These aspects determined the choice of the catalysts considered in this study: a series of size-controlled Cu nanoparticles supported on a rather inert graphitic carbon material.
Copper is believed to reach and remain in the metallic state under reaction conditions, as confirmed by experimental studies involving (in situ) XPS, DRIFT, TEM and XAS.17,26–30 The catalytic properties of the different facets of copper and, consequently, the effect of the size of the nanoparticles is still a matter of debate.31–34 Activity studies of model single crystals such as Cu(100), Cu(111), Cu(110) and Cu(211) have suggested structure sensitivity, with defect-rich stepped surfaces being more active than highly coordinated flat surfaces.19,29,35–42 Campbell et al.29,36 reported that the rate of CO2 hydrogenation was higher on Cu(110) than on Cu(100) and polycrystalline copper foil at a total pressure of 5 bar and a H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2 ratio of 11:1. Behrens et al.19 proposed that the presence of low-coordinated sites, such as on stepped Cu(211) surfaces, could help the adsorption of oxygen-bound intermediates and lower the barrier of their hydrogenation. However, when supported copper catalysts were studied, controversial results have been reported. On the one hand, studies claim structure-insensitivity and a linear relationship between methanol formation and copper surface area.43,44 Karelovic et al.34 studied the MeOH and CO formation rates for Cu/ZnO catalysts at 7 bar, 160–225 °C and an H2/CO2 ratio of 3. MeOH formation rates were reported as independent of copper particle size, while CO formation rates were enhanced over catalysts with smaller copper particles. Large Cu particles (ca. 200 nm) have been reported to favor the MeOH selectivity. On the other hand, other studies report activity increasing or decreasing with Cu particle size.31,32,34,45 Karelovic et al.45 reported a 3-fold increase in the intrinsic formation rate of both MeOH and CO for particles larger than 10 nm compared to 4 nm copper particles over Cu/SiO2 catalysts at 8 bar, 230–270 °C and an H2/CO2 ratio of 3. Arena et al.31 report a linear relationship between specific activity and copper particle size for Cu–Zn/ZrO2, with turn-over frequency (TOF) increasing for nanoparticles from 2 to 32 nm. In contrast, Natesakhawat et al.32 reported, for Zr/Zn/Ga/Y promoted Cu catalysts, an opposite relationship with decreasing activity with increasing particle size. The divergent trends reported could be explained by the different types of support and the presence of one or more promoters which unequivocally influence the catalytic performance and hamper studies on copper particle size effects during CO2 hydrogenation.5
:CO2 ratio of 11:1. Behrens et al.19 proposed that the presence of low-coordinated sites, such as on stepped Cu(211) surfaces, could help the adsorption of oxygen-bound intermediates and lower the barrier of their hydrogenation. However, when supported copper catalysts were studied, controversial results have been reported. On the one hand, studies claim structure-insensitivity and a linear relationship between methanol formation and copper surface area.43,44 Karelovic et al.34 studied the MeOH and CO formation rates for Cu/ZnO catalysts at 7 bar, 160–225 °C and an H2/CO2 ratio of 3. MeOH formation rates were reported as independent of copper particle size, while CO formation rates were enhanced over catalysts with smaller copper particles. Large Cu particles (ca. 200 nm) have been reported to favor the MeOH selectivity. On the other hand, other studies report activity increasing or decreasing with Cu particle size.31,32,34,45 Karelovic et al.45 reported a 3-fold increase in the intrinsic formation rate of both MeOH and CO for particles larger than 10 nm compared to 4 nm copper particles over Cu/SiO2 catalysts at 8 bar, 230–270 °C and an H2/CO2 ratio of 3. Arena et al.31 report a linear relationship between specific activity and copper particle size for Cu–Zn/ZrO2, with turn-over frequency (TOF) increasing for nanoparticles from 2 to 32 nm. In contrast, Natesakhawat et al.32 reported, for Zr/Zn/Ga/Y promoted Cu catalysts, an opposite relationship with decreasing activity with increasing particle size. The divergent trends reported could be explained by the different types of support and the presence of one or more promoters which unequivocally influence the catalytic performance and hamper studies on copper particle size effects during CO2 hydrogenation.5
During the CO2 hydrogenation reaction, methanol and CO are both formed. Two main intermediates, formate (HCOO*) and carboxylic species (COOH*), can be formed over copper catalysts during methanol synthesis.38,46,47 Formate is believed to be the main intermediate for methanol synthesis.27,29,36,48,49 Formate species are formed by the interaction of atomic hydrogen with carbon dioxide to form HCOO* which can be further hydrogenated to formic acid (HCOOH*) and/or dioxymethylene (H2COO*) and, in both cases, further hydrogenation leads to the formation of adsorbed methoxy species (H3CO*) and ultimately to the final product, methanol. The second species reported as reaction intermediate are carboxyl groups.50 Carboxyl species have been proposed as intermediate for methanol synthesis, but the majority of the studies report their involvement in CO formation via an associative path.38,51 The rWGS reaction could occur also via the redox mechanism, which implies the direct dissociation of CO2 without the assistance of adsorbed H atoms.48,52
In this study, carbon-supported copper catalysts were synthetized by impregnation, resulting in particle sizes ranging from 5 to 20 nm. The graphitic carbon material support was used to allow the intrinsic effect of copper particle size to be studied, avoiding any influence of oxidic components. A strong influence of particle size on methanol selectivity was found. DFT calculations were employed to calculate the formation free energy for various reaction intermediates over the different exposed Cu facets and unravel the origin of the copper particle size effect during the CO2 hydrogenation reaction.
Experimental
Catalyst synthesis
Carbon graphene nanoplates (GNP500 from XG Sciences) were used as support. The pristine support had a Brunauer–Emmett–Teller (BET) surface area of 496 m2 g−1 and a total pore volume of 0.91 cm3 g−1. Copper was deposited by incipient wetness impregnation using an aqueous copper nitrate solution (1–3 M), followed by drying and heat treatment in order to obtain the active copper metallic phase. A range of synthesis approaches was used to vary the copper particle size.53,54 Firstly, the metal concentration in the precursor solution was varied, and hence the Cu weight loading (5.2–14.1%). Secondly, the temperature (200–300 °C) and atmosphere (pure N2 followed by 10 vol% H2/N2 or only 20 vol% H2/N2) during the precursor decomposition and reduction of the oxide were varied. Further details on the synthesis procedure and adopted parameters are described in section S1.† Copper-based hydrogenation catalysts often show, during the first hours in a reaction gas feed, slight changes in the catalytic performance mainly due to nanoparticle restructuring.15,53,55 Hence an activation procedure was performed. This procedure consisted of exposing the catalysts to the reaction conditions (260 °C, 40 bar(g) pressure of CO2/H2/He gas mixture) for 20 hours. After the activation procedure, performed in the catalytic setup, the samples were slowly passivated by air exposure at room temperature and subsequently analysed by transmission electron microscopy (TEM) and powder X-ray diffraction (XRD). The catalysts were named X_Cu/C, in which X represents the surface-averaged CuO size in the activated catalyst measured by TEM.Catalyst characterization
Surface area (SBET) and total pore volume (Vp) of the pristine supports were determined from N2 physisorption measurements at −196 °C in a TriStar Plus II gas-volumetric apparatus (Micromeritics), using Carbon Black STSA as a reference. The specific surface area of the support was calculated by fitting with a Brunauer–Emmett–Teller isotherm,56 whereas the total pore volume was derived from the absorbed volume of nitrogen at p/p0 = 0.95.X-ray diffraction (XRD) was performed on a Bruker AXS D8 Advance diffractometer using Co (Kα = 1.79026 Å) radiation. Patterns were collected from 20° to 70° 2θ. Diffractograms were measured in inert Ar atmosphere (silicon wafers with an X-ray transparent airtight dome) for the reduced catalysts and in air for the passivated and activated catalysts. All diffractograms were normalized to the intensity of the graphitic carbon (002) diffraction peak at 30.9° 2θ. The crystallite sizes were calculated by applying the Scherrer equation to the Cu0 (200) diffraction at 59.3 degrees.57
Transmission electron microscopy (TEM) was performed either on a Tecnai 20 (FEI) or on a Talos F200X (FEI), both operated at 200 kV. TEM samples were prepared by directly applying the pre-ground sample powder to copper carbon-coated TEM grids (Agar, 300 mesh). TEM imaging was performed on the passivated catalysts, the activated catalysts and the used catalysts. The size of >400 nanoparticles was determined using ImageJ software. All particle sizes are reported as surface-averaged particle size 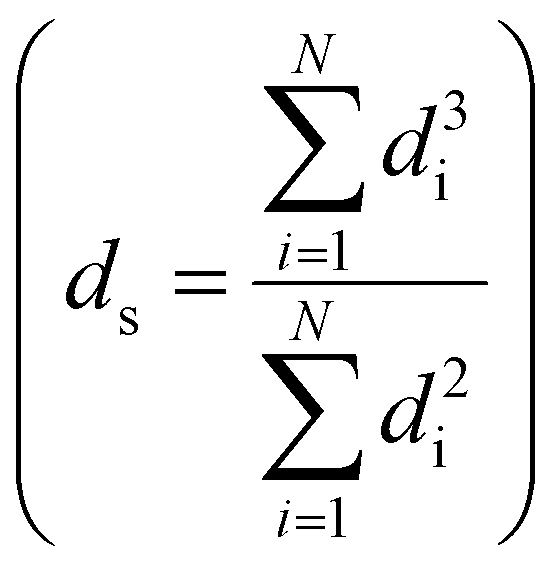 with the corresponding standard error in the particle size
with the corresponding standard error in the particle size 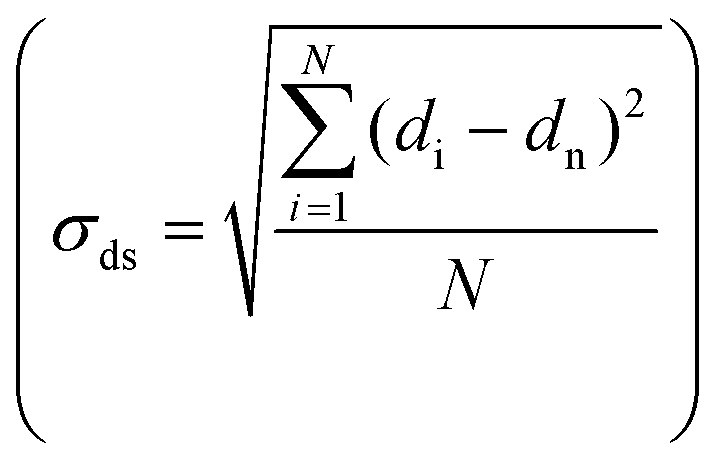 .
.
Catalyst performance
The performance of the catalysts was investigated in a gas-phase fixed-bed 16 parallel reactor system (Flowrence, Avantium). Each stainless steel plug flow reactor of 2.6 mm inner diameter was loaded with 4.2 mg of Cu mass on carbon (sieve fraction 75–150 μm) diluted with SiC (212–425 μm), with an inert fraction between 0.6 and 0.9. Prior to reaction, the catalysts were reduced in situ in 10 vol% H2/N2 flow at 10.9 mL min−1 200 °C for 3.5 hours with 2 °C min−1 under atmospheric pressure. Afterwards, the temperature was lowered to 120 °C and the atmosphere was switched to H2/CO2/He = 67.5/22.5/10 vol% and the reactor was pressurized. Two separate runs were performed to evaluate the long-term stability of the catalysts and the catalytic performances at different temperatures.For the isothermal stability test, after introduction of the H2/CO2/He = 67.5/22.5/10 vol% mixture, the temperature was set to 260 °C and the pressure to 40 bar(g). These conditions were kept constant for over 140 hours. During the performance evaluation test, first a 23 hours stabilizing protocol was performed to ensure the steady state operation and minimize the influence of activation/deactivation behavior. The stabilizing protocol was performed at 260 °C temperature and 40 bar(g) pressure under H2/CO2/He = 67.5/22.5/10 vol% at 2.5 mL min−1 atmosphere. The catalysts characterized after undergoing the stabilizing protocol will be referred to as ‘activated catalysts’. Subsequently, without changes in the gas flow, the reactor temperature was lowered to 200 °C and the temperature was then varied between 200 and 260 °C, with isothermal segments of 20 hours for each temperature.
Reactants and products of the reactions were analyzed online with an Aligent 7890B gas chromatograph at 14 min intervals. Permanent gasses were separated on a HayeSep Q column (0.5 m × 1/8 inch ID) and MolSieve 5A column (2.4 m × 1/8 inch ID) and analysed using a thermal conductivity detector. Hydrocarbons and oxygenates were analysed using two separate flame ionization detectors, in combination with a GS-GasPro column (30 m × 0.32 mm ID) and a HP-Innowax column (30 m × 0.32 mm ID). Details on the calculations of activity and selectivity are given in section S2.†
DFT calculations
DFT calculations were carried out using the Vienna Ab Initio Simulation Package (VASP)58,59 and the Atomic Simulation Environment (ASE)60 employing the Bayesian error estimation functional with van der Waals corrections (BEEF-vdW)61,62 and the projector-augmented wave (PAW) potentials.63 We chose the BEEF-vdW functional as it has a reasonable performance regarding adsorption energies on transition-metal surfaces.64 Importantly, this functional has also been shown to successfully describe processes on copper surfaces relevant for the CO2 hydrogenation to methanol.65 The lattice constant of bulk copper was optimized to 3.664 Å using an energy cutoff of 800 eV and a 16 × 16 × 16 Monkhorst–Pack66k-point sampling. The kinetic energy cutoff for all slab calculations was 450 eV. The surfaces were modelled by 4 layer supercells separated by 18 Å in the z-direction. The different sizes and corresponding k-point sampling used to calculate the various coverages are given in Table S4.† In all calculations the bottom two layers were kept fixed at the bulk position while the top two layers and the adsorbate were allowed to relax until atomic forces were smaller than 0.01 eV Å−1. Adsorption energies were calculated relative to the corresponding gas-phase species (CO2 and ½ H2 in the case of formate and carboxyl). The correction of the well-known gas-phase errors for CO2 (+0.41 eV) and H2 (+0.09 eV) has been taken from the literature.39 Using this correction we obtain an adsorption enthalpy of formate on Cu(100) of 84–91 kJ mol−1 for coverages of 0.12 to 0.5 ML. This compares with 80–100 kJ mol−1 reported for measurements on Cu(100) single crystal surfaces.67 Zero-point energy (ZPE) and entropic contributions were taken from the literature as obtained for the Cu(211) surface calculated with the BEEF-vdW functional39 and are listed in Table S5† and the total energies of all systems reported here in Table S6.†Results and discussion
Catalyst stability and structural properties
Carbon was selected as support material to avoid strong metal–support interactions while studying the relationship between the catalytic performance during CO2 hydrogenation and Cu particle size. However, the use of a support with a relatively weak interaction can result in a rapid loss of accessible metal surface area due to particle growth.55 Hence, we first evaluated the stability of the Cu/C catalysts under isothermal reaction conditions, i.e. at 40 bar(g) and 260 °C for over 140 hours on stream. A typical reaction profile of CO2 conversion and selectivity towards methanol is shown in Fig. 1. After an initial decrease in the first 20–40 hours, the CO2 conversion remained stable around 10%. Under these experimental conditions, equilibrium corresponds to 22% CO2 conversion. MeOH and CO were the two main products detected. MeOH and CO selectivity remained stable around 19% and 81% respectively under these conditions.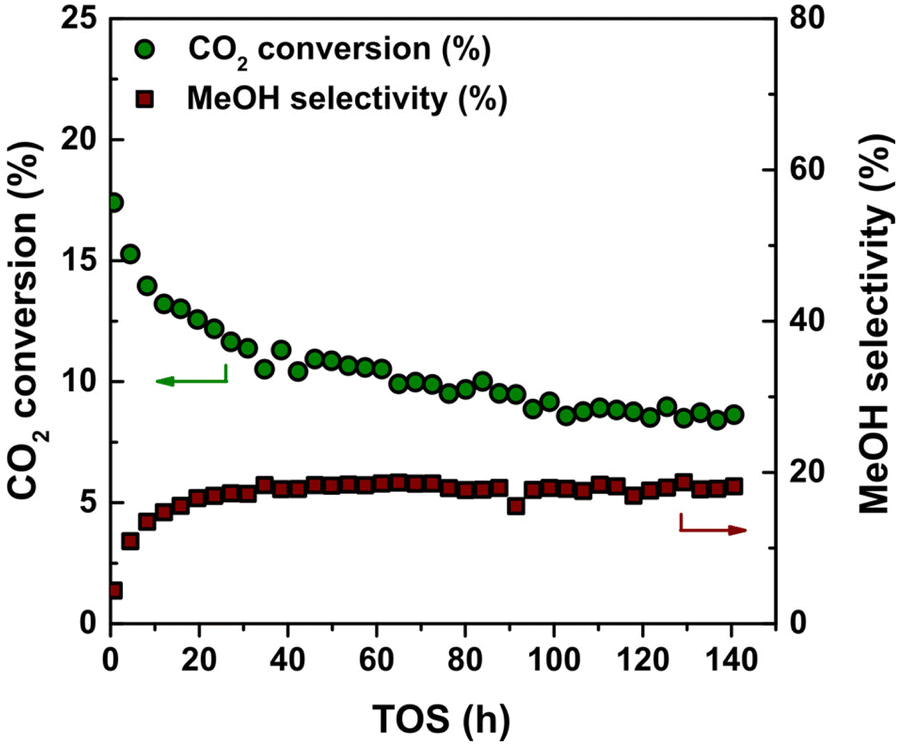 | ||
| Fig. 1 CO2 conversion and methanol selectivity as a function of time on stream (TOS) for the 4.7_Cu/C catalyst with 5.2 wt% Cu. Reaction conditions: 4.2 mg of Cu per reactor, 40 bar(g), 260 °C, 600 mL min−1 gCu−1, H2/CO2/He = 67.5/22.5/10 vol%. | ||
Particle growth during catalysis was evaluated by comparing the Cu particle size before and after catalysis and the surface-normalized turn-over frequencies at the beginning of the catalytic test and after 140 hours on stream. The 4.7_Cu/C catalyst showed an increase in Cu particle size from 3.9 ± 0.9 nm to 4.9 ± 1.2 nm. The complete collection of transmission electron micrographs and particle size distributions of the as-synthetized and used catalysts is depicted in Fig. S1 and Table S1.† The TOF was 3.1 × 10−3 s−1 after 8 hours on stream considering the initial Cu particle size, and 2.4 × 10−3 s−1 after 140 hours on stream considering the final Cu particle size. Hence particle growth was limited, and the loss of active surface area seemed to be the main cause of activity loss.
The most representative image of the catalysts under reaction conditions, considering the initial changes in activity and selectivity during catalysis, is represented by the catalysts after exposure to reaction conditions (260 °C, 40 bar(g) pressure of CO2/H2/He gas mixture) for 20 hours, the so-called activation procedure. Table 1 shows the structural properties of a series of Cu on carbon catalysts that underwent the activation procedure, showing different particle sizes. Fig. 2 (frame a and b) shows transmission electron micrographs and particle size distributions for two representative Cu/C catalysts. The complete collection of micrographs is depicted in Fig. S1,† and properties reported in Table S1.†
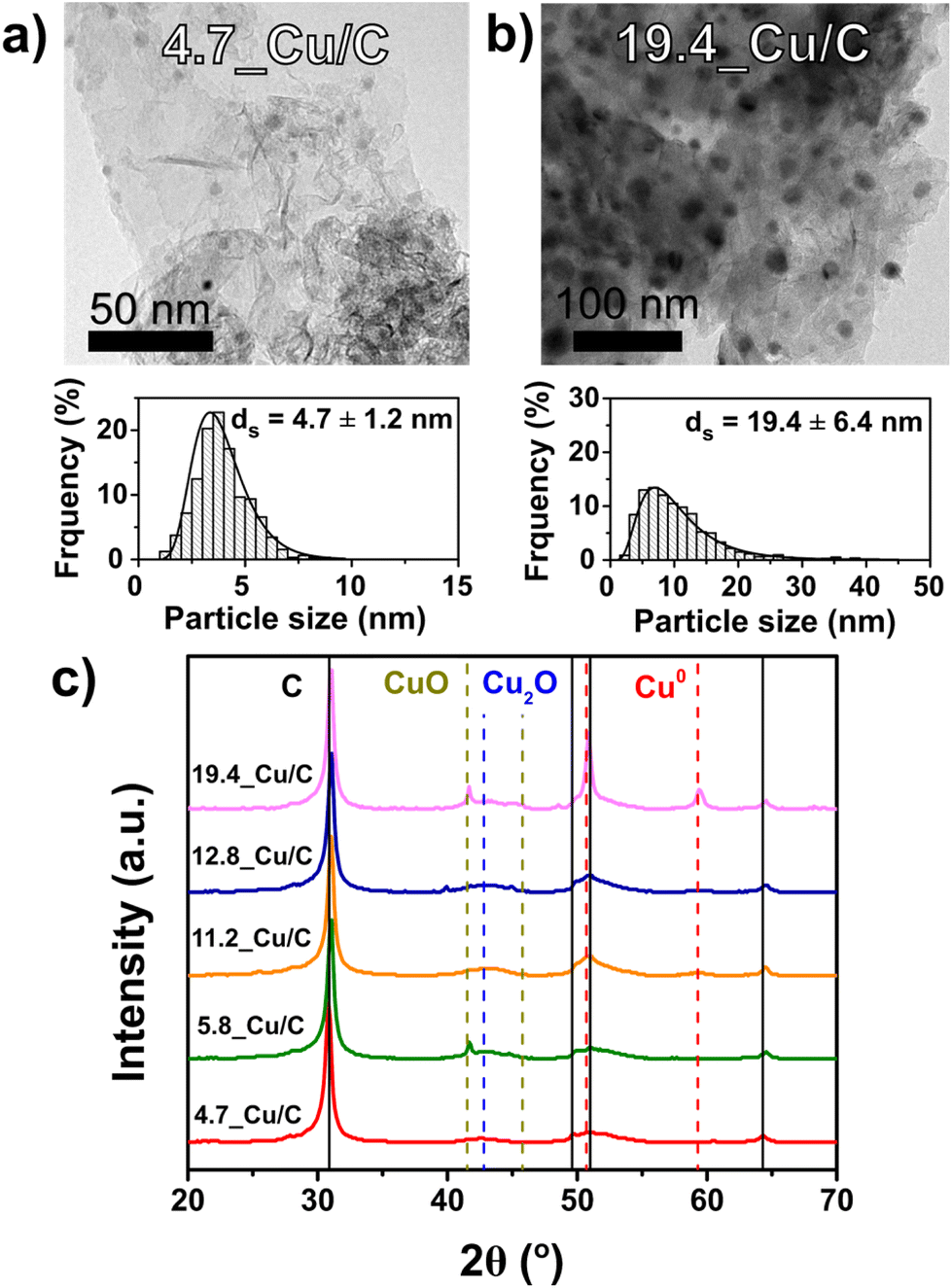 | ||
| Fig. 2 Transmission electron micrographs with corresponding particle size distributions of the 4.7_Cu/C (a) and 19.4_Cu/C (b) catalysts after activation. (c) Powder X-ray diffractograms of the activated Cu/C catalysts, diffractograms normalized to the intensity of the carbon (002) diffraction peak and vertically offset for clarity. | ||
| Name | Cu loading (wt%) | d s ± σdsa (nm) | Cu0 crystallite sizeb (nm) | Cu dispersionc (%) |
|---|---|---|---|---|
| a TEM-derived surface-averaged particle size and relative size distribution. b Cu0 crystallite size as determined by XRD using the Cu0 (200) diffraction peak of the activated catalysts. c The Cu dispersion (fraction of surface Cu atoms at the surface) calculated using the TEM-determined mean diameter (ds) of the activated catalyst. More detail are given in the ESI (section S2†). | ||||
| 4.7_Cu/C | 5.2 | 4.7 ± 1.2 | — | 22.1 |
| 5.8_Cu/C | 9.9 | 5.8 ± 1.6 | — | 17.9 |
| 11.2_Cu/C | 9.9 | 11.2 ± 3.6 | 7.5 | 9.3 |
| 12.8_Cu/C | 9.9 | 12.8 ± 4.1 | 9.6 | 8.1 |
| 19.4_Cu/C | 14.1 | 19.4 ± 6.4 | 12.4 | 5.4 |
TEM analysis showed highly dispersed copper nanoparticle (black dots) on the graphitic carbon sheets for all the catalysts. The diffractograms of the activated catalysts are shown in Fig. 2 (frame c), while the diffractograms of the as-synthetized and used catalysts are shown in Fig. S2.† Next to the graphitic carbon diffraction peaks, diffraction lines are present that can be attributed to CuO, Cu2O and metallic Cu. As expected from the short exposure to air of the samples after activation, different phases of copper oxide were present. The 19.4_Cu/C catalyst exhibited the most intense diffraction peaks of Cu0 (111) and (200) as well as of Cu2+ (002), in accordance with the highest Cu weight loading. TEM and XRD analysis prove that catalysts with particle sizes in the range of 4.7 to 19.4 nm were obtained after activation. By comparing the structural properties and particle sizes of the as-synthetized catalysts, as reported in Table S1,† it is evidenced that the diversity and broad range of sizes, from 3.9 to 18.0 nm in the fresh catalysts, was preserved during the activation procedure. By comparison of the particle sizes of the activated and used catalysts, it is noted that average particle sizes do not vary significantly beyond the activation procedure (Table S1†). The average particle sizes, as determined by TEM, of the 4.7_Cu/C and 19.4_Cu/C catalysts after activation are 4.7 and 19.4 nm, while after 120 hours on stream are, respectively, 4.9 and 21.2 nm. This highlights the importance of the activation procedure to obtain structurally stable catalysts in order to study the influence of the copper particle size during CO2 hydrogenation reaction.
Copper particle size effect on CO2 conversion
The performance of the Cu catalysts in CO2 hydrogenation, with copper particle sizes ranging from 4.7 to 19.4 nm (referred to the activated state), was investigated under a 3:1 ratio of H2/CO2 mixture at 40 bar(g) in the temperature range between 200 and 260 °C. Fig. 3 reports the CO2 conversion and weight-normalized copper time yield (CTY) for selected Cu/C catalysts. A limited loss of activity is observed at 260 °C during the first 20 hours of catalysis, as reported above. After the first 20 hours, the conversion is stable at all the temperatures tested.
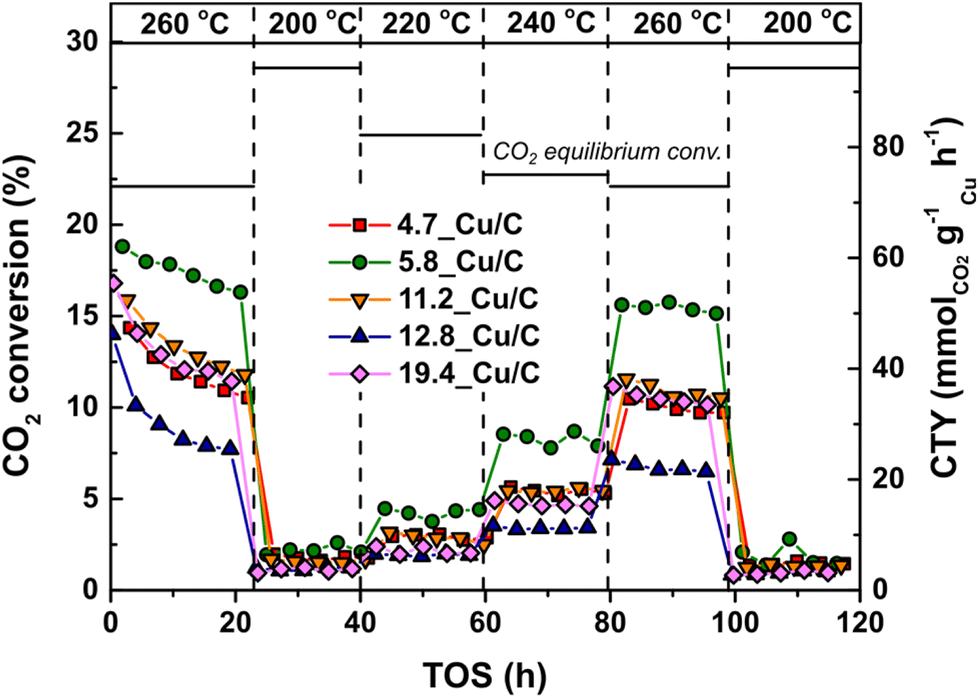 | ||
| Fig. 3 Characteristic profiles for CO2 conversion and weight-normalized copper time yield for Cu/C catalysts as function of TOS (h) at different temperatures. Reaction conditions: 4.2 mg of Cu per reactor, 40 bar(g), 600 mL min−1 gCu−1, H2/CO2/He = 67.5/22.5/10 vol%. | ||
The relationship between TOF and surface-averaged Cu particle size after use is shown in Fig. 4. Approximately a 3-fold increase in TOF was observed with increasing surface-averaged Cu particle size from 5 to 13 nm (at 260 °C the TOF increased from 2.6 × 10−3 s−1 to 7.5 × 10−3 s−1), while no further increase was observed for particles larger than 13 nm. These results confirm that the CO2 hydrogenation reaction is sensitive to the Cu particle size. A similar trend was observed at lower temperatures (Fig. S3†). At 260 °C the CO2 conversions were relatively high (7–15%), but still well below the equilibrium conversion (22%); see Fig. S4† for a graphical comparison.
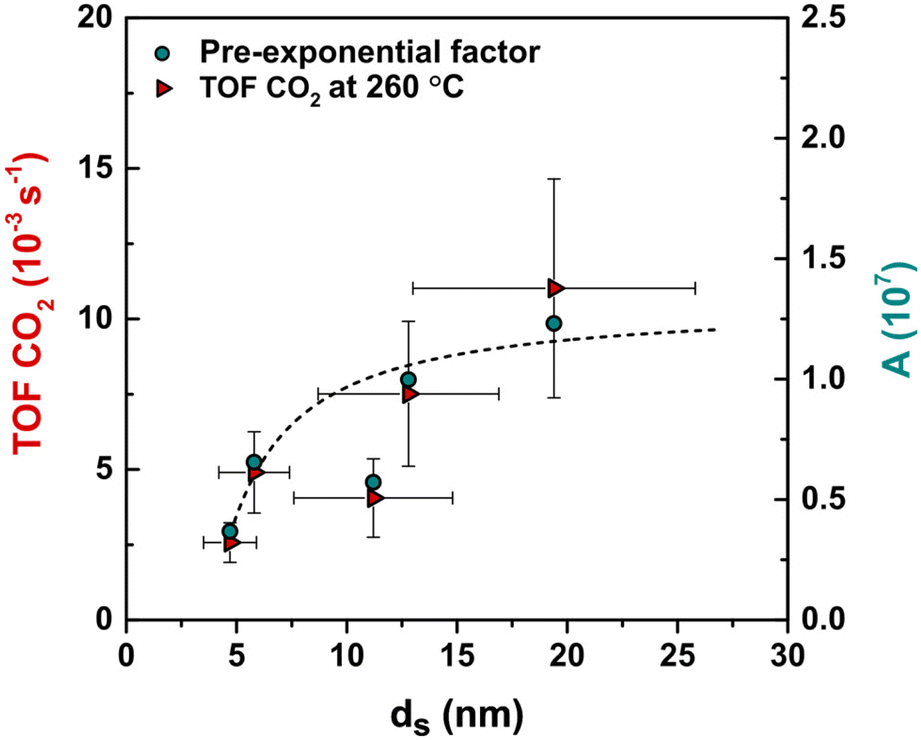 | ||
| Fig. 4 TOF and pre-exponential factor as a function of Cu particle size. Line is drawn to guide the eye. Reaction conditions: 4.2 mg of Cu per reactor, 40 bar(g), 260 °C, 600 mL min−1 gCu−1, H2/CO2/He = 67.5/22.5/10 vol%. | ||
A similar copper particle size effect has been observed for methanol synthesis from CO2 enriched syngas68 as well as for ethyl acetate hydrogenation,25 while for CO2 hydrogenation to methanol divergent results have been reported. Arena et al.31 reported that the TOF of Cu–Zn/ZrO2 catalysts increased linearly with increasing Cu particle size in the range between 2 and 32 nm. On the other hand, Natesakhawat et al.32 showed an opposite trend for Cu catalysts containing Zr/Zn/Ga/Y in different combinations. In both studies, the presence of promoters and the use of oxidic supports might have obscured the intrinsic relationship between the Cu particle size and catalytic performance. An overview of CO2 TOF values observed in this study and values reported in literature is reported in Fig. S5 and Table S2.† CO2 TOF values reported in this study are of the same order of magnitude as those previously reported. However, this is the first time that the effect of the copper particle size supported on rather inert carbon support has been studied at industrially relevant conditions (40 bar(g) pressure and 200–260 °C).
Different phenomena can lead to particle size effects such as size-dependent interactions with the support, electronic particle size effects and structure sensitivity of the reaction itself.69,70 The chemically inert nature of the carbon support makes a strong influence of the support unlikely.71–73 In the size range studied (larger than 3 nm), electronic particle size effects are not expected to play a significant role.69,74,75 The particle size effect observed in these systems is hence likely due to structure sensitivity, which is related to the size dependence of the fraction of different types of copper surface sites.
The apparent activation energies were calculated using the Arrhenius model (Fig. S6† frame a). The obtained energy values for CO2 hydrogenation (Table S3 and Fig. S6† frame b) were, within the error, size-independent with an average Ea of 74 ± 7 kJ mol−1. The pre-exponential factor (A) was calculated using the surface-normalized CTY and an averaged Ea of 74 kJ mol−1. The pre-exponential factor increased from 3.7 × 106 for 4.7 nm particles to 1 × 107 for 12.8 nm and did not increase further for larger nanoparticles. This suggests the presence of well-defined and size-independent active Cu sites responsible for CO2 conversion, for which the relative abundance increases with increasing particle size up to around 13 nm.
Influence of copper particle size on product selectivity
So far we have only considered the effect of copper particle size on the overall conversion of CO2 without considering the products formed. Methanol and carbon monoxide were the main products for all catalysts at all temperatures tested (200–260 °C). Additionally, traces of CH4 (less than 0.6% selectivity) were observed at temperatures above 220 °C, which might be due to minor contamination.24Fig. 5 (frame a) shows the methanol selectivity for different Cu particle sizes as a function of CO2 conversion. Different CO2 conversions were obtained by varying the temperature in the range of 200–260 °C. For all particle sizes, the MeOH selectivity decreased as the conversion increased.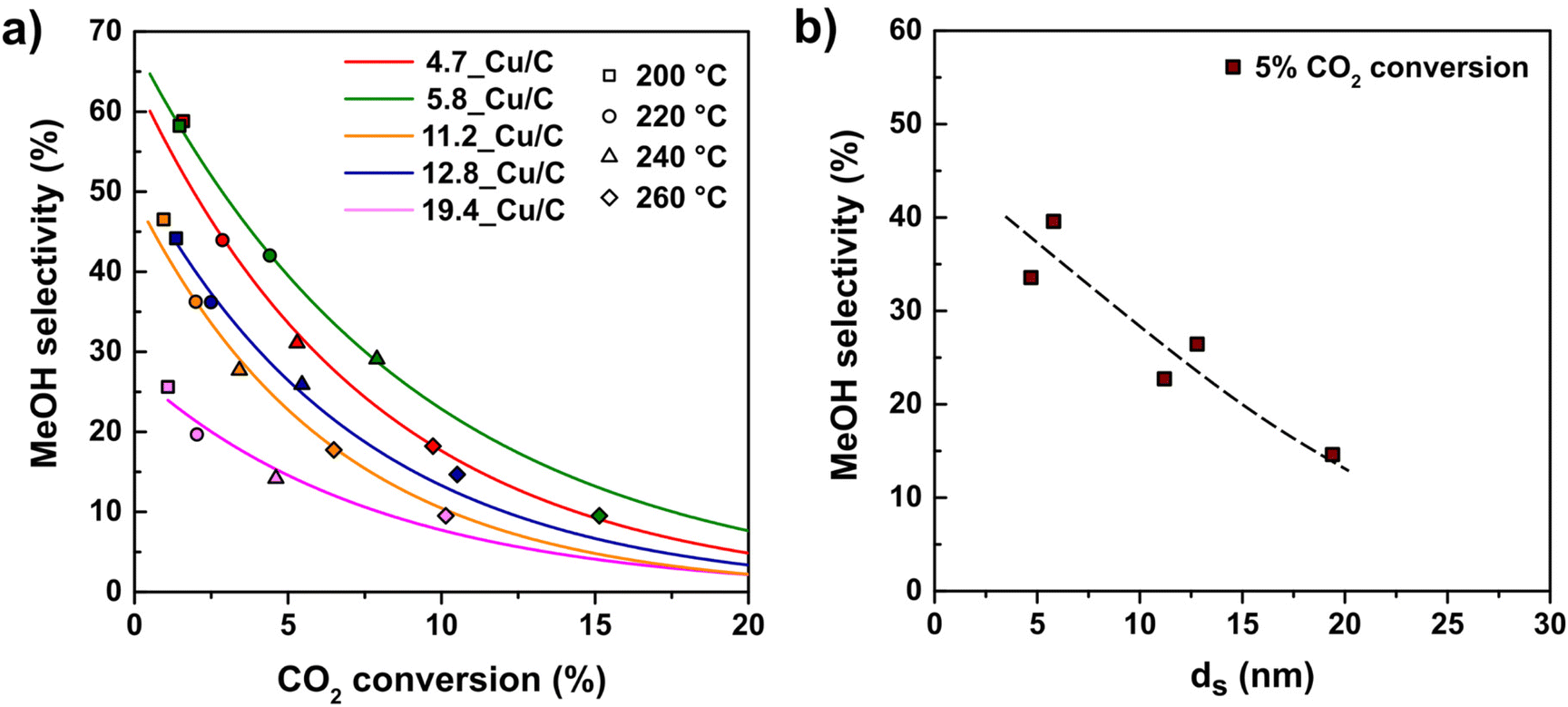 | ||
| Fig. 5 (a) MeOH selectivity as a function of CO2 conversion for catalysts with different Cu particle sizes. (b) Selectivity to MeOH as a function of Cu particle size for a CO2 conversion of ∼5%. The trend line was added to guide the eye. Reaction conditions: 4.2 mg of Cu per reactor, 40 bar(g), 600 mL min−1 gCu−1, H2/CO2/He = 67.5/22.5/10 vol%. | ||
When comparing at constant conversion (Fig. 5 frame b), the influence of particle size on selectivity is even more clear. Small nanoparticles display a greatly enhanced selectivity towards MeOH over CO as a product. For example, at 5% CO2 conversion 4.7_Cu/C shows a MeOH selectivity of 34%, whereas 19.4_Cu/C presents a MeOH selectivity of around 15% only. This influence of the particle size on product selectivity cannot be explained by the impact of conversion effect; it is clearly an intrinsic particle size effect.
To better understand the origin of the observed particle size effect, we considered separately the methanol and carbon monoxide formation rates. MeOH and CO turn-over-frequencies (respectively TOFMeOH and TOFCO) are displayed in Fig. 6. The TOFCO at 260 °C clearly increased from 1.9 × 10−3 s−1 to 9.4 × 10−3 s−1 when the Cu size increased from 5 nm to 20 nm. The data on the formation rate of MeOH suggest a size-independent TOFMeOH of 8.4 × 10−4 s−1 on average, perhaps with the exception of the smallest particles that apparently have a lower TOFMeOH. Consequently, as the CO formation is much less favourable on smaller Cu clusters, they show the highest methanol selectivity.
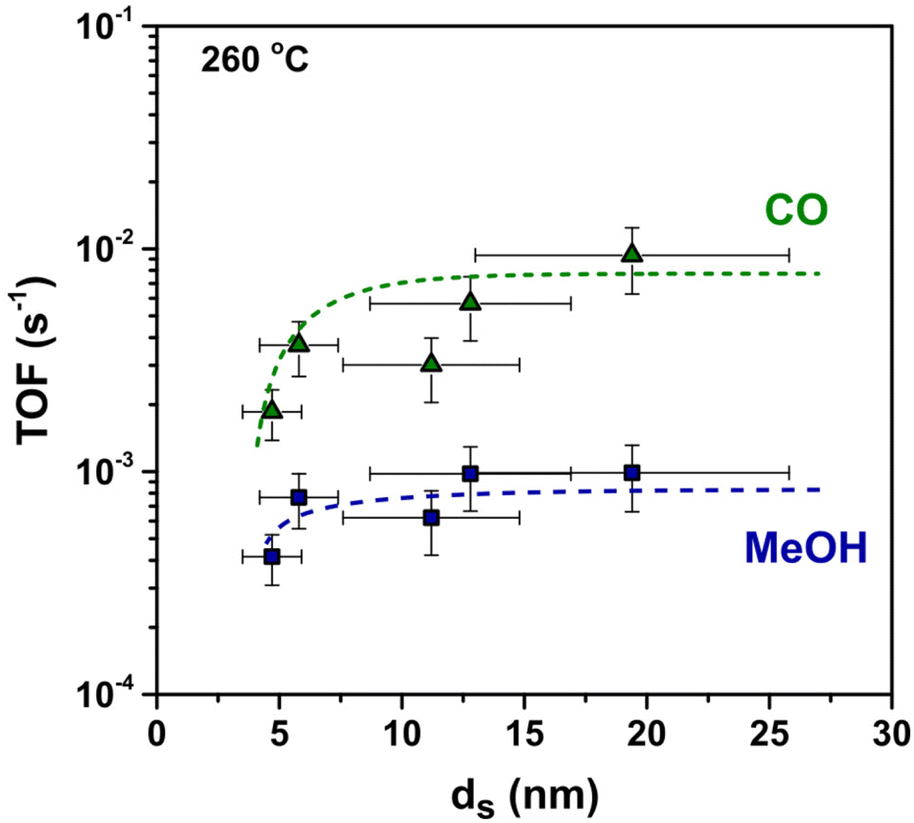 | ||
| Fig. 6 MeOH and CO TOF as a function of the Cu particle size. Trend lines were added to guide the eye. Reaction conditions: 4.2 mg of Cu per reactor, 40 bar(g), 260 °C, 600 mL min−1 gCu−1, H2/CO2/He = 67.5/22.5/10 vol%. | ||
The apparent activation energies for MeOH as well as for CO formation calculated using the Arrhenius model (Fig. S7†) are reported in Table S3.† The apparent activation energies for MeOH formation ranged from 55 to 76 kJ mol−1, with data suggesting a slight increase for larger particle sizes. In literature, apparent activation energies for methanol formation of 73–100 kJ mol−1 have been reported for Cu–SiO2 catalysts and of 77 kJ mol−1 for polycrystalline Cu foil.29,45 Carbon monoxide formation apparent activation energies ranged from 108 to 126 kJ mol−1, with no significant dependence on particle size, while reported literature Ea values for Cu/SiO2 and polycrystalline Cu foil were 100–118 kJ mol−1 and 135 kJ mol−1 respectively.29,45 Therefore, the values obtained in this work are in line with the previously reported values for similar copper-based catalysts. The high MeOH selectivity of the smallest particles is here probably due to a lower fraction of sites that favour CO formation.
The origin of the particle size effect
MeOH and CO formation show two distinctly different and size-independent activation energies suggesting that the nature of the sites where MeOH and CO are formed is the same regardless of the size of the nanoparticles. Moreover, a higher value of apparent activation energy for CO (118 ± 6 kJ mol−1) than MeOH (63 ± 7 kJ mol−1) indicates that MeOH is unlikely to be formed via CO intermediates. TOFCO and TOFMeOH show different particle size-dependence. In particular, CO TOF increases with an increase in particle size, since this dependence cannot be correlated to changes in the nature of the active site, which indicates that the abundance of active sites for rWGS reaction increase with an increase in particle size. In order to understand the particle size effect in terms of surface sites that are likely to be more active for MeOH synthesis and which for CO formation, we calculated the theoretical fraction of different surface sites and the corresponding adsorption free energies for reaction intermediates by DFT calculation.We note that the smallest particles considered in this work are in the range of 3.9 ± 0.9 nm, which is above the limit above where electronic particle size effects have been shown to disappear for copper nanoparticles. This was evident from calculated adsorption energies of atomic oxygen, an important descriptor for CO2 hydrogenation, which reached the bulk limit from 2.5 nm particles onwards.76 We, therefore, turned our attention to another factor that is dependent on the particle size: the ratio of various surface terminations. Thus, we considered how the concentration of these surface sites varies with particle size. This is shown in Fig. 7 (frame a) where the fraction of surface atoms exhibiting a specific generalized coordination number (GCN)77 is given as a function of particle size assuming ideal cuboctahedral copper particles. We stress that this analysis assumes ideal particle shapes and that our aim is to deduct qualitative trends rather than quantitative numbers. Fig. 7 (frame a) shows that the number of edge sites with a GCN of 5.33 decreases from around 20% to less than 5% when going from 3 to 30 nm. On the other hand the fraction of the more close packed (111) and (100) facets increases from 3 and 10% to more than 30 and 50%, respectively. The decrease in rWGS rate with decreasing size seems related to a decrease in concentration of terrace sites (e.g. (100) surfaces).
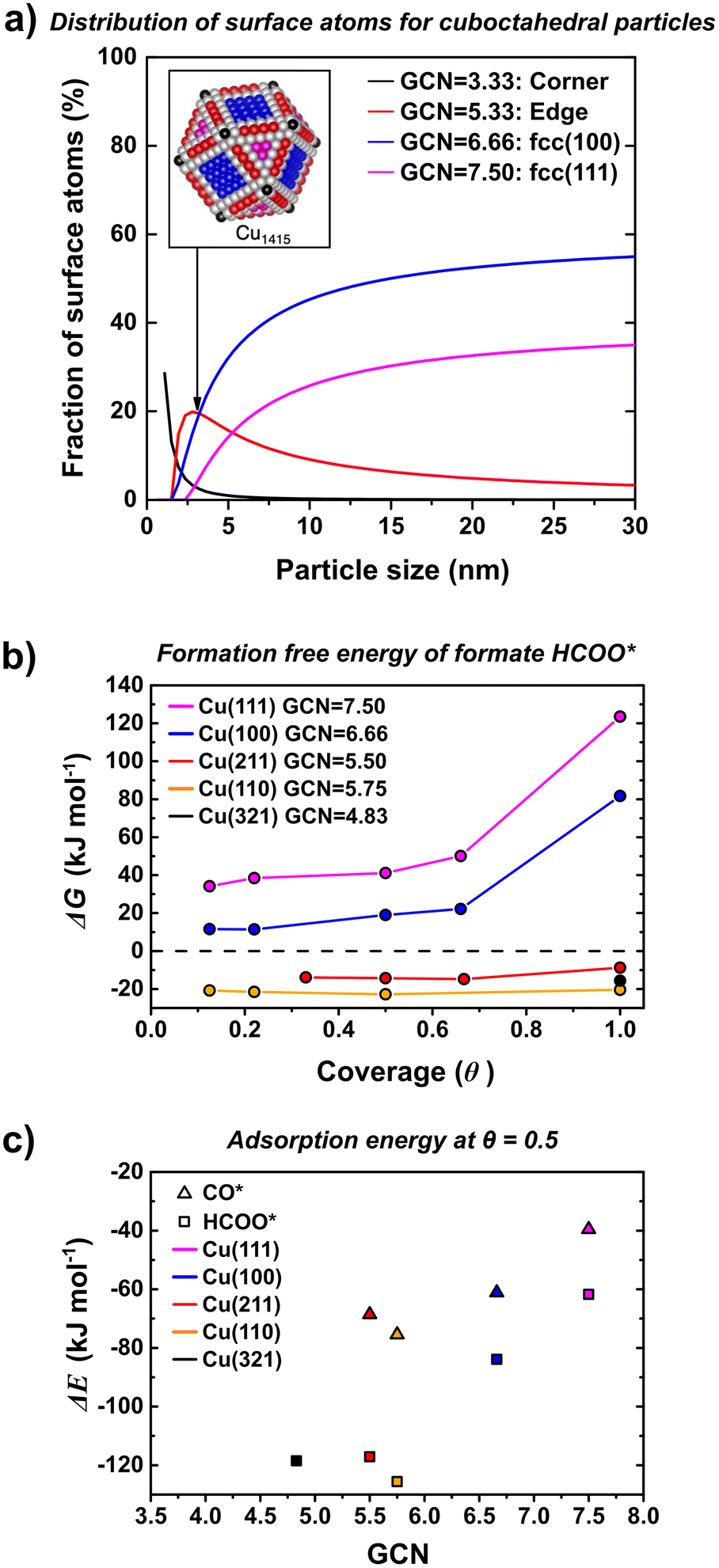 | ||
| Fig. 7 (a) Fraction of surface atoms with a specific GCN as a function of particle diameter for cuboctahedral particles. GCN for the various surface atoms of a Cu1415 particle are shown in the inset. (b) Adsorption free energy of formate as a function of coverage for Cu(111) (pink), Cu(100) (blue), Cu(211) (red), Cu(110) (orange) and Cu(321) (black) surfaces at 500 K and 1 bar of H2 and CO2. The coverage is normalized to 1 ML for one formate per two copper atoms of the corresponding facet (see section S3† for structures). (c) Adsorption energy of formate (squares) and CO (triangles) as a function of GCN. | ||
Both, CO2 hydrogenation to methanol and the rWGS have been shown to be structure sensitive on copper surfaces.19,29,36,38 For methanol synthesis, it has been shown that sites with a low coordination number are more active, e.g. when comparing single crystal measurements on Cu(100) with those on Cu(110).19,29,35,36,78 Surface defects, as e.g. modeled by Cu(211) surfaces have indeed been suggested as active sites for copper-based catalysts.19 Likewise, the rWGS has also been shown to exhibit lower barriers on more undercoordinated surfaces, although kinetic modeling indicates that this does not necessarily translate to higher reaction rates.29,36,48,79,80 An interesting difference between methanol and CO formation is that the former is assumed to proceed via the formate intermediate, while CO formation occurs either via the carboxyl intermediate or through direct splitting of CO2.27,38,46,47,81,82 The higher apparent activation energy for CO than MeOH formation suggests that it is implausible that CO is a main intermediate in the MeOH synthesis. Given that formate is the only intermediate that is found to have significant surface coverages, we calculated the adsorption free energy of formate on a range of different copper surfaces for coverages of up to 1 mono layer (ML) using density functional theory (DFT) (see Fig. 7 frame b). The GCN for our models of edges ((211) and (110) surfaces) and corner atoms ((321) surface) differ slightly from that of the cuboctahedral particle, but they are close enough to be used as a rough guidance. Our calculations reveal that the adsorption free energies decrease in the order Cu(321) ≈ Cu(110) > Cu(211) ≫ Cu(100) ≫ Cu(111). Interestingly, the effect of coverage (here 1 ML is defined as one formate per two copper surface atoms, see section S3† for details) is small for the (211) and (110) surfaces but has a significant influence on the binding energy on Cu(111) and Cu(100). Importantly, at 500 K the adsorption free energy of formate on the (211) and (110) surfaces is negative, but positive on (111) and (100). This indicates that both (211) and (110) are covered by close to 1 ML of formate, while the coverage is significantly smaller on (100) and (111).
Fig. 7 (frame c) shows the adsorption free energies of formate as a function of the GCN at a 1/2 ML coverage for the various surfaces. As can be seen, the adsorption free energies scale linearly with the GCN. This allows to estimate adsorption free energies for the corner and edge sites of cuboctahedral particles (see insert in Fig. 7 frame a). For both facets, the adsorption free energy of formate is exothermic and we thus expect a similar behavior as observed for (211), (110) and (321) facets.
Assuming that formate can only be converted to methanol, this would indicate a strong preference of methanol formation on Cu(211) and Cu(110), which we use here to model the edge sites of the copper nanoparticles. On the other hand, the lower coverage of formate on Cu(111) and Cu(100) might indicate that one or both of these sites are the predominate source for CO formation. Coverages of the carboxyl (COOH*) intermediate as well as CO* reveal similar trends and are given in the section S3 of the ESI.† These findings roughly correlate with the analysis of the fraction of surface sites shown in Fig. 7 (frame a). This would explain why CO formation strongly decreases for the smallest Cu particles and hence overall the smallest particles give the highest MeOH selectively.
Conclusions
The effect of copper particle size was investigated for CO2 hydrogenation reaction over carbon-supported copper catalysts with particle sizes between 5 and 20 nm. We found that CO2 hydrogenation is clearly a structure sensitive reaction. Cu nanoparticles smaller than about 13 nm showed low activity, but high MeOH selectivity. Two distinctly different and particle size-independent activation energy values were found for MeOH and CO formation. DFT calculations revealed that the formation free energy of formate, the main intermediate in methanol formation, is negative on the corner and edge sites, while being positive on terrace sites which are more abundant on larger particles. Therefore, small Cu nanoparticles having a surface dominated by corner and edge sites, have a high ratio of methanol to CO formation, which can be explained by a higher formate coverage. This work is intended to provide new guidelines for the design of novel methanol synthesis catalysts, crucial for the effective utilization of CO2.Author contributions
LB synthesized the catalysts, measured the performance of the catalysts and analyzed the experimental data. LB and PdJ conceptualized the experiments and write-reviewed-edited the manuscript. NV perform the transmission electron microscopy measurements. AH, PP and FS performed the DFT calculations. JS and BV helped in discussing the results. PdJ provided guidance during the project. All authors contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Remco Dalebout and Jan Willem de Rijk for support in catalytic experiments. This project is part of the Consortium on Metal Nanocatalysis funded by TotalEnergies OneTech Belgium, with reference TOTB Contract Ref IPA-5443 between TotalEnergies and Utrecht University and Contract Ref IPA-5441 between TotalEnergies and Karlsruhe Institute of Technology.References
- IPCC, 2014: Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Geneva, Switzerland, 2014 Search PubMed.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond Oil and Gas: The Methanol Economy, 2009 Search PubMed.
- International Renewable Energy Agency (IRENA), Innovation Outlook: Renewable Methanol, 2021 Search PubMed.
- K. C. Waugh, Catal. Today, 1992, 15, 51–75 CrossRef CAS.
- J. B. Hansen and P. E. H. Nielsen, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Weinheim, 2008 Search PubMed.
- G. C. Chinchen, P. J. Denny, D. G. Parker, M. S. Spencer and D. A. Whan, Appl. Catal., 1987, 30, 333–338 CrossRef CAS.
- M. Behrens, Recyclable Catal., 2016, 2, 78–86 Search PubMed.
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- H. Bahruji, M. Bowker, G. Hutchings, N. Dimitratos, P. Wells, E. Gibson, W. Jones, C. Brookes, D. Morgan and G. Lalev, J. Catal., 2016, 343, 133–146 CrossRef CAS.
- A. Ota, E. L. Kunkes, I. Kasatkin, E. Groppo, D. Ferri, B. Poceiro, R. M. Navarro Yerga and M. Behrens, J. Catal., 2012, 293, 27–38 CrossRef CAS.
- J. Ye, C. Liu, D. Mei and Q. Ge, ACS Catal., 2013, 3, 1296–1306 CrossRef CAS.
- M. S. Frei, M. Capdevila-Cortada, R. García-Muelas, C. Mondelli, N. López, J. A. Stewart, D. Curulla-Ferré and J. Pérez-Ramírez, J. Catal., 2018, 361, 313–321 CrossRef CAS.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef PubMed.
- R. van den Berg, G. Prieto, G. Korpershoek, L. I. van der Wal, A. J. van Bunningen, S. Lægsgaard-Jørgensen, P. E. de Jongh and K. P. de Jong, Nat. Commun., 2016, 7, 13057 CrossRef CAS PubMed.
- M. B. Fichtl, D. Schlereth, N. Jacobsen, I. Kasatkin, J. Schumann, M. Behrens, R. Schlögl and O. Hinrichsen, Appl. Catal., A, 2015, 502, 262–270 CrossRef CAS.
- M. Behrens and R. Schlögl, Z. Anorg. Allg. Chem., 2013, 639, 2683–2695 CrossRef CAS.
- F. Amet, C. T. Ke, I. V. Borzenets, J. Wang, K. Watanabe, T. Taniguchi, R. S. Deacon, M. Yamamoto, Y. Bomze, S. Tarucha and G. Finkelstein, Science, 2016, 352, 966–969 CrossRef CAS PubMed.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS PubMed.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Science, 2014, 345, 546–550 CrossRef CAS PubMed.
- C. Li, X. Yuan and K. Fujimoto, Appl. Catal., A, 2014, 469, 306–311 CrossRef CAS.
- F. Arena, G. Mezzatesta, G. Zafarana, G. Trunfio, F. Frusteri and L. Spadaro, Catal. Today, 2013, 210, 39–46 CrossRef CAS.
- B. Ouyang, W. Tan and B. Liu, Catal. Commun., 2017, 95, 36–39 CrossRef CAS.
- R. Dalebout, N. L. Visser, C. E. L. Pompe, K. P. de Jong and P. E. de Jongh, J. Catal., 2020, 392, 150–158 CrossRef CAS.
- R. Beerthuis, J. W. de Rijk, J. M. S. Deeley, G. J. Sunley, K. P. de Jong and P. E. de Jongh, J. Catal., 2020, 388, 30–37 CrossRef CAS.
- R. Dalebout, L. Barberis, G. Totarella, S. J. Turner, C. La Fontaine, F. M. F. de Groot, X. Carrier, A. M. J. van der Eerden, F. Meirer and E. P. de Jongh, ACS Catal., 2022, 12, 6628–6639 CrossRef CAS PubMed.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- A. Beck, M. Zabilskiy, M. A. Newton, O. Safonova, M. G. Willinger and J. A. van Bokhoven, Nat. Catal., 2021, 4, 488–497 CrossRef CAS.
- J. Yoshihara, S. C. Parker, A. Schafer and C. T. Campbell, Catal. Lett., 1995, 31, 313–324 CrossRef CAS.
- J. Schumann, J. Kröhnert, E. Frei, R. Schlögl and A. Trunschke, Top. Catal., 2017, 60, 1735–1743 CrossRef CAS.
- F. Arena, K. Barbera, G. Italiano, G. Bonura, L. Spadaro and F. Frusteri, J. Catal., 2007, 249, 185–194 CrossRef CAS.
- S. Natesakhawat, J. W. Lekse, J. P. Baltrus, P. R. Ohodnicki, B. H. Howard, X. Deng and C. Matranga, ACS Catal., 2012, 2, 1667–1676 CrossRef CAS.
- X. Guo, D. Mao, G. Lu, S. Wang and G. Wu, J. Catal., 2010, 271, 178–185 CrossRef CAS.
- A. Karelovic and P. Ruiz, Catal. Sci. Technol., 2015, 5, 869–881 RSC.
- P. B. Rasmussen, M. Kazuta and I. Chorkendorff, Surf. Sci., 1994, 318, 267–280 CrossRef CAS.
- J. Yoshihara and C. T. Campbell, J. Catal., 1996, 161, 776–782 CrossRef CAS.
- H. Zhenming and R. J. Boyd, J. Chem. Phys., 2000, 112, 9562–9568 CrossRef.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS.
- F. Studt, M. Behrens, E. L. Kunkes, N. Thomas, S. Zander, A. Tarasov, J. Schumann, E. Frei, J. B. Varley, F. Abild-Pedersen, J. K. Nørskov and R. Schlögl, ChemCatChem, 2015, 7, 1105–1111 CrossRef CAS.
- D. Kopač, B. Likozar and M. Huš, Appl. Surf. Sci., 2019, 497, 143783 CrossRef.
- F. Calle-Vallejo, D. Loffreda, M. T. M. Koper and P. Sautet, Nat. Chem., 2015, 7, 403–410 CrossRef CAS PubMed.
- Z. P. Liu and P. Hu, J. Am. Chem. Soc., 2003, 125, 1958–1967 CrossRef CAS PubMed.
- S. I. Fujita, S. Moribe, Y. Kanamori, M. Kakudate and N. Takezawa, Appl. Catal., A, 2001, 207, 121–128 CrossRef CAS.
- G. J. J. Bartley and R. Burch, Appl. Catal., 1988, 43, 141–153 CrossRef CAS.
- A. Karelovic, G. Galdames, J. C. Medina, C. Yévenes, Y. Barra and R. Jiménez, J. Catal., 2019, 369, 415–426 CrossRef CAS.
- Y. Li, S. H. Chan and Q. Sun, Nanoscale, 2015, 7, 8663–8683 RSC.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- T. Fujitani, I. Nakamura, T. Uchijima and J. Nakamura, Surf. Sci., 1997, 383, 285–298 CrossRef CAS.
- S. A. French, A. A. Sokol, S. T. Bromley, C. R. A. Catlow, S. C. Rogers, F. King and P. Sherwood, Angew. Chem., Int. Ed, 2001, 40, 4437 Search PubMed.
- Y. Yang, D. Mei, C. H. F. Peden, C. T. Campbell and C. A. Mims, ACS Catal., 2015, 5, 7328–7337 CrossRef CAS.
- Y. Yang, C. A. Mims, D. H. Mei, C. H. F. Peden and C. T. Campbell, J. Catal., 2013, 298, 10–17 CrossRef CAS.
- L. Dietz, S. Piccinin and M. Maestri, J. Phys. Chem. C, 2015, 119, 4959–4966 CrossRef CAS.
- G. Prieto, J. Zečević, H. Friedrich, K. P. De Jong and P. E. De Jongh, Nat. Mater., 2013, 12, 34–39 CrossRef CAS PubMed.
- P. Munnik, M. Wolters, A. Gabrielsson, S. D. Pollington, G. Headdock, J. H. Bitter, P. E. De Jongh and K. P. De Jong, J. Phys. Chem. C, 2011, 115, 14698–14706 CrossRef CAS.
- H. H. Kung, Catal. Today, 1992, 11, 443–453 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- J. J. Mortensen, K. Kaasbjerg, S. L. Frederiksen, J. K. Nørskov, J. P. Sethna and K. W. Jacobsen, Phys. Rev. Lett., 2005, 95, 1–4 CrossRef PubMed.
- J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Landis, J. K. Nørskov, T. Bligaard and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 32–34 CrossRef.
- D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef.
- J. Wellendorff, T. L. Silbaugh, D. Garcia-Pintos, J. K. Nørskov, T. Bligaard, F. Studt and C. T. Campbell, Surf. Sci., 2015, 640, 36–44 CrossRef CAS.
- F. Studt, F. Abild-Pedersen, J. B. Varley and J. K. Nørskov, Catal. Lett., 2013, 143, 71–73 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- P. A. Taylor, P. B. Rasmussen, C. V. Ovesen, P. Stoltze and I. Chorkendorff, Surf. Sci., 1992, 261, 191–206 CrossRef CAS.
- R. van den Berg, J. Zečević, J. Sehested, S. Helveg, P. E. de Jongh and K. P. de Jong, Catal. Today, 2016, 272, 87–93 CrossRef CAS.
- B. R. Cuenya, Thin Solid Films, 2010, 518, 3127–3150 CrossRef CAS.
- C. Vogt, M. Monai, G. J. Kramer and B. M. Weckhuysen, Nat. Catal., 2019, 2, 188–197 CrossRef CAS.
- B. S. Clausen, J. Schiøtz, L. Gråbæk, C. V. Ovesen, K. W. Jacobsen, J. K. Nørskov and H. Topsøe, Top. Catal., 1994, 1, 367–376 CrossRef CAS.
- C. V. Ovesen, B. S. Clausen, J. Schiøtz, P. Stoltze, H. Topsøe and J. K. Nørskov, J. Catal., 1997, 168, 133–142 CrossRef CAS.
- P. L. Hansen, J. B. Wagner, S. Helveg, J. R. Rostrup-Nielsen, B. S. Clausen and H. Topsøe, Science, 2002, 295, 2053–2055 CrossRef CAS PubMed.
- A. S. Crampton, M. D. Rötzer, C. J. Ridge, F. F. Schweinberger, U. Heiz, B. Yoon and U. Landman, Nat. Commun., 2016, 7, 10389 CrossRef PubMed.
- A. Y. Stakheev and L. M. Kustov, Appl. Catal., A, 1999, 188, 3–35 CrossRef CAS.
- A. H. Hakimioun, E. M. Dietze, B. D. Vandegehuchte, D. Curulla-Ferré, L. Joos, P. N. Plessow and F. Studt, Catal. Lett., 2021, 151, 3165–3169 CrossRef CAS.
- F. Calle-Vallejo, J. I. Martínez, J. M. García-Lastra, P. Sautet and D. Loffreda, Angew. Chem., Int. Ed., 2014, 53, 8316–8319 Search PubMed.
- P. B. Rasmussen, P. M. Holmblad, T. Askgaard, C. V. Ovesen, P. Stoltze, J. K. Nørskov and I. Chorkendorff, Catal. Lett., 1994, 26, 373–381 CrossRef CAS.
- K. H. Ernst, C. T. Campbell and G. Moretti, J. Catal., 1992, 134, 66–74 CrossRef CAS.
- J. Nakamura, J. M. Campbell and C. T. Campbell, J. Chem. Soc., Faraday Trans., 1990, 86, 2725 RSC.
- K. Larmier, W. C. Liao, S. Tada, E. Lam, R. Verel, A. Bansode, A. Urakawa, A. Comas-Vives and C. Copéret, Angew. Chem., Int. Ed., 2017, 56, 2318–2323 Search PubMed.
- G. Wang and J. Nakamura, J. Phys. Chem. Lett., 2010, 1, 3053–3057 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr02612k |
| This journal is © The Royal Society of Chemistry 2022 |