
Environment tolerant, adaptable and stretchable organohydrogels: preparation, optimization, and applications
Qiongling
Ding
a,
Zixuan
Wu
a,
Kai
Tao
b,
Yaoming
Wei
a,
Weiyan
Wang
a,
Bo-Ru
Yang
a,
Xi
Xie
 a and
Jin
Wu
*a
a and
Jin
Wu
*a
aState Key Laboratory of Optoelectronic Materials and Technologies and the Guangdong Province Key Laboratory of Display Material and Technology, School of Electronics and Information Technology, Sun Yat-sen University, Guangzhou 510275, China. E-mail: wujin8@mail.sysu.edu.cn
bThe Ministry of Education Key Laboratory of Micro and Nano Systems for Aerospace, Northwestern Polytechnical University, Xi'an, 710072, P. R. China
First published on 26th January 2022
Abstract
Multiple stretchable materials have been successively developed and applied to wearable devices, soft robotics, and tissue engineering. Organohydrogels are currently being widely studied and formed by dispersing immiscible hydrophilic/hydrophobic polymer networks or only hydrophilic polymer networks in an organic/water solvent system. In particular, they can not only inherit and carry forward the merits of hydrogels, but also have some unique advantageous features, such as anti-freezing and water retention abilities, solvent resistance, adjustable surface wettability, and shape memory effect, which are conducive to the wide environmental adaptability and intelligent applications. This review first summarizes the structure, preparation strategy, and unique advantages of the reported organohydrogels. Furthermore, organohydrogels can be optimized for electro-mechanical properties or endowed with various functionalities by adding or modifying various functional components owing to their modifiability. Correspondingly, different optimization strategies, mechanisms, and advanced developments are described in detail, mainly involving the mechanical properties, conductivity, adhesion, self-healing properties, and antibacterial properties of organohydrogels. Moreover, the applications of organohydrogels in flexible sensors, energy storage devices, nanogenerators, and biomedicine have been summarized, confirming their unlimited potential in future development. Finally, the existing challenges and future prospects of organohydrogels are provided.
 Qiongling Ding | QiongLing Ding received her BE and MA in micro-electronics from Lanzhou University. She is currently a PhD student in the School of Electronics and Information Technology, Sun Yat-sen University. Her current research interest is stretchable sensing devices. |
 Zixuan Wu | Zixuan Wu received his BE from Sun Yat-Sen University. He is currently pursuing his PhD degree at Sun Yat-Sen University. His research interest includes gas/humidity sensors and stretchable electronics. |
 Yaoming Wei | Yaoming Wei received his BE from Chongqing University in 2019. He is currently pursuing his Master's degree at Sun Yat-Sen University. His research interests focus on gas sensors and stretchable electronics. |
 Jin Wu | Jin Wu received his PhD degree from Nanyang Technological University in 2014. After obtaining his PhD in 2014, he continued to work in SMART program in Nanyang Technological University as a postdoctoral research fellow. Since 2017, he works as an associate professor at the School of Electronics and Information Technology, Sun Yat-sen University. His research interest includes hydrogel-based sensors, flexible and stretchable electronics, etc. |
1. Introduction
Flexible electronics, through the integration of mechanical flexibility and electrical properties in devices, have been well developed in the past decade due to their tremendous application prospects in next-generation portable electronics.1–5 Various stretchable materials, including elastomers, hydrogels, ionogels, and organogels, have been successively developed and used in practice due to their unique properties. Elastomers, such as polydimethylsiloxane (PDMS),6 ecoflex, polymethyl methacrylate (PMMA),7 thermoplastic polyurethanes (TPU),8 and various rubber materials,9 are elastic materials based on high molecular polymer networks and have excellent strength and toughness. However, most elastomers do not possess high strain capacity and conductivity, and are usually used as packaging materials and flexible substrates. Research studies show that dispersing polymer networks in different dispersion media is beneficial to construct gel materials with distinct (desired) characteristics. Hydrogels usually consist of physically and/or chemically crosslinked 3D polymer networks dispersed in a large amount of water. This makes the hydrogels display both solid-like mechanical properties (strength, stretchability, and toughness) and a liquid-like transport capacity (ions and nutrients).10,11 The combination of the unique network structure and composition endows hydrogels with exceptional stretchability, toughness, biocompatibility, and high ionic conductivity,12,13 making them potential candidates in diverse fields, including wearable and implantable electronics, bionic sensors, actuators, soft robotics, tissue engineering, flexible energy storage devices, etc.14–17Nevertheless, the practical applications of hydrogels are still limited by many problems. First, compared with natural load-bearing materials such as tendons, conventional hydrogels have limited mechanical stiffness and low toughness, which are caused by the sparse, homogeneous, and microscale 3D network structure.18,19 Second, there has always been a dilemma of the trade-off between the mechanical performance and ionic conductivity in conventional hydrogels. Unfortunately, higher mechanical properties often require a denser network structure, and this will inhibit ion transport and sacrifice part of the ionic conductivity.20–22 Then, wide environmental adaptability is particularly important for flexible electronic devices for their applications in different environments, even in sub-zero, dry, and underwater environments. However, due to the presence of a large amount of water, the freezing-induced hardening issue at sub-zero temperatures and evaporation-induced structural dehydration will inevitably occur in the hydrogel, which will eventually lead to the loss of its functionality.23,24 Finally, multiple functions, such as self-healing, adhesiveness, transparency, and antibacterial properties, are rarely achieved in conventional hydrogels synchronously.25–27 Due to these inevitable issues, it is of great practical significance to develop anti-freezing and anti-drying hydrogels with excellent mechanical properties and tunable functionalities. Ionogels use ionic liquids as dispersion media and exhibit outstanding stability and mechanical characteristics, and have great potential to be used in flexible electronics, owing to the good ionic conductivity, non-volatility, non-flammable properties, and electrochemical stability of ionic liquids.28–31 However, ionic liquids are relatively expensive and toxic, which are not conducive to applications in biological tissues and large-scale production. Besides, organogels are obtained by mixing polymer chains with organic solution and gelators, which exhibit good viscoelasticity and thermoplasticity.32,33 Generally, various properties of these soft materials can be easily regulated and implemented by changing their composition, structure, concentration, etc.
To date, although remarkable progress has been made in the development of various pristine stretchable materials, their actual applications are always limited by the intrinsic problems of a single system. There is an urgent need to develop gel materials that have excellent mechanical properties, conductivity, and stability in various environments simultaneously. Taking into account both advantages and drawbacks of individual components, the hybrids of these stretchable materials have been widely utilized and have significant advantages in performance optimization by integrating the advantages of each component.34 For example, the incorporation of ionic liquids makes hydrogels not only moderately conductive but also highly stretchable.3,35,36 Alternatively, compared with pristine hydrogels, the binary system of hydrogels and elastomers shows higher mechanical strength and also maintains excellent stretchability.37–40 In addition, due to the entirely opposite physiochemical properties of organogels and hydrogels, it is highly desirable to obtain organogel–hydrogel hybrids with a wealth of unique properties. Correspondingly, Guo et al. formally proposed the concept of organohydrogels by gelling cellulose and poly(vinyl alcohol) (PVA) in a mixed solvent of dimethylformamide (DMF) and water.41 Interestingly, this organohydrogel exhibited relatively high stability in various solvents without dissolution, shrinkage, or swelling, so that the enzyme electrodes based on it could work in various liquid media without being limited by the solubility of the analytes. Over the past few decades, a lot of efforts have been devoted to the preparation, performance optimization, and application expansion of organohydrogels. As a consequence, various types of organohydrogels have been developed, and they are a good alternative to traditional hydrogels through the elimination of the electro-mechanical performance deterioration caused by freezing, dehydration, or swelling of hydrogels under specific harsh conditions, showing good frost resistance, moisture retention, and solvent resistance. Based on the incomparable multi-environmental tolerance of other gel materials, it can be widely used in soft robotics, tissue engineering, and stretchable electronics even in harsh environments, such as cold winters, dry deserts, and liquid environments.15,16,42,43 In addition, the thermoplastic organogel domains and the stable hydrogel elastic network in organohydrogels that feature a binary cooperative phase result in thermally-induced variable mechanical properties, which are expected to serve as promising smart materials to suit a variety of applications.44–47
Beyond this, with the broadened application of flexible materials, new functional requirements for organohydrogels have increased sharply, such as self-adhesiveness, self-healing, antibacterial properties, and transparency. To implement these goals, functionalized organohydrogels can be achieved through the modification of specific functional monomers and delicate composition, and molecular and structural engineering. Nevertheless, it remains a great challenge to develop organohydrogels with all these functions simultaneously, owing to some trade-off dilemma. Herein, a systematic and comprehensive introduction and summary of organohydrogels are presented (Fig. 1). Firstly, the preparation methods and unique characteristics of two different types of organohydrogels are introduced in detail, including heteronetwork and homogeneous organohydrogels. Then, the research significance and strategies for further optimization and additional giving of electro-mechanical properties and various functionalities in organohydrogels reported previously are summarized and discussed, respectively, which will undoubtedly play an indispensable role in the development of multifunctional organohydrogels in the future. Finally, the representative applications of organohydrogels in different fields are described in detail, mainly focusing on sensing devices, energy storage devices, nanogenerators, and biomedicine.
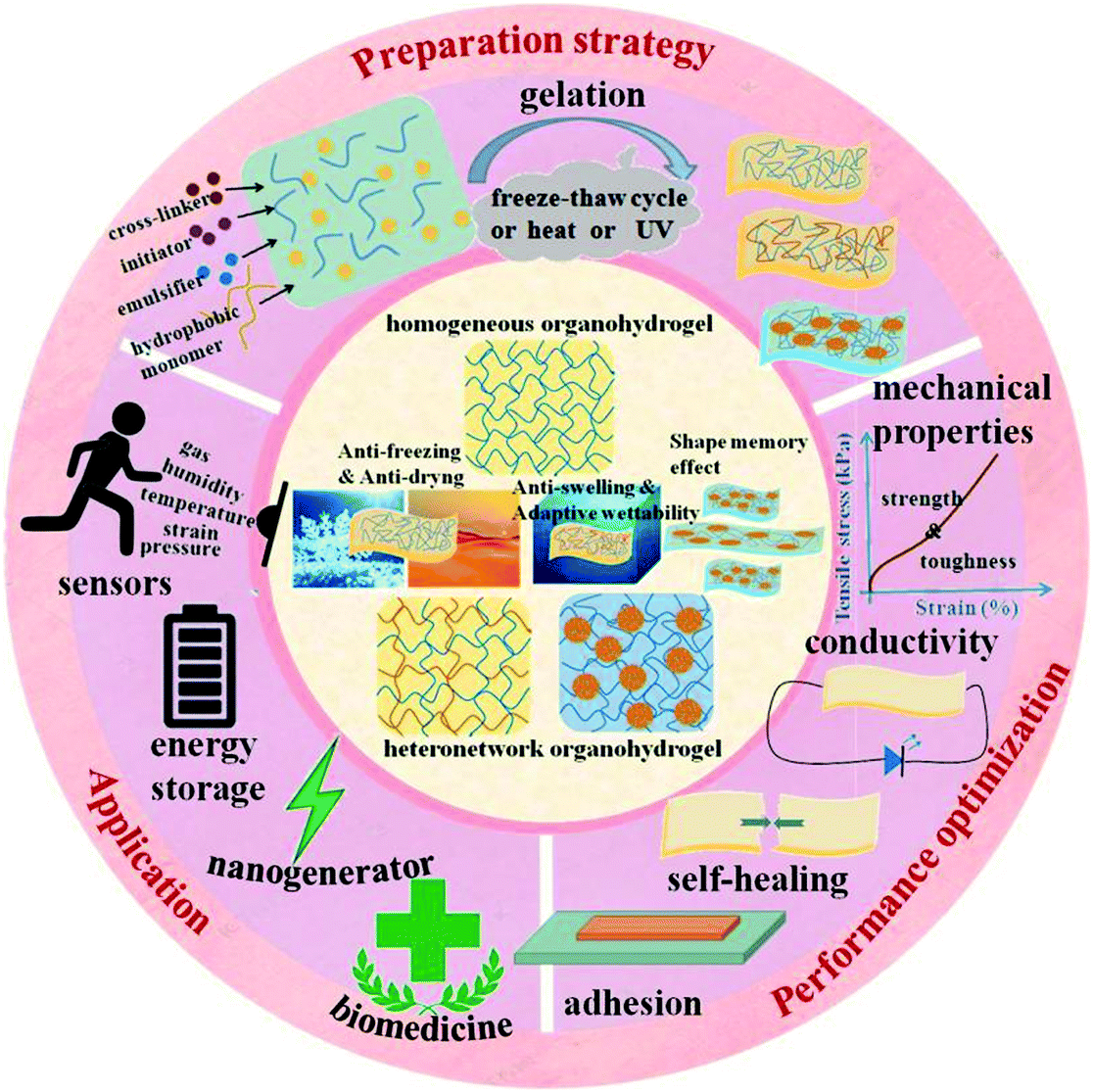 | ||
| Fig. 1 Schematic illustration of the classification, preparation, functionalization, and application of organohydrogels. | ||
2. Configuration design for organohydrogels
2.1. Classification and preparation
Organohydrogel is currently a very typical and common gel material, and its construction generally requires two processes to occur: a polymerization process in which organic monomers are interconnected to form long molecular chains and a cross-linking process in which molecular chains are cross-linked to form a network. During the gelation of the polymer network, two cross-linking mechanisms are mainly involved.48 One is physical cross-linking, and the gel network formed accordingly is mainly cross-linked by non-covalent bonds. To date, the repeated freezing and thawing process is the most common method to obtain physically cross-linked hydrogels through the formation of hydrogen bonds and crystalline domains.24,49 Representatively, PVA-based hydrogels can be directly obtained by dissolving PVA powder in a solvent and then performing several freeze–thaw cycles, and their mechanical properties can be adjusted by the number of freeze–thaw cycles.50,51 As for PVA-based organohydrogels, only one freeze–thaw cycle can form a stable gel.49,52 This is due to the formation of large numbers of hydrogen bonds and the induction of PVA crystallization after the introduction of organic agents, thus increasing the cross-linking points, which greatly improves the production efficiency. Besides, gelatin-based hydrogels can be obtained by heating the gelatin in a solvent to dissolve and dissociate into coil-like chains, and then the gelatin molecules rearrange into triple helix structures through hydrogen bonding and hydrophobic interaction after cooling down, which is also another common method to obtain a physically cross-linked network.53,54 Specifically, the preparation of these polymer networks is completely green and pollution-free as no polymerization reaction occurs.55Another cross-linking mechanism is called chemical cross-linking. In this regard, conventional radical polymerization is deployed to construct a strong covalent cross-linking network as a typical method, requiring a well-designed polymerization step. In the polymer precursor solution, the polymer monomer, initiator, and cross-linker need to be included. In addition, the occurrence of phase transition requires induction from external stimuli, such as heat,56 pH,57 light,58–60 or redox.61,62 Among them, photopolymerization and thermal polymerization are the most common, and the corresponding photoinitiator (2-hydroxy-2-methylpropiophenone63,64 and 2,2’-diethoxyacetophenone (DEAP)65) and thermal initiator (ammonium persulfate (APS)66–68 and 2,2′-azobis(2-methylpropionitrile) (AIBN)69) are used. Chemical cross-linkers such as N,N-methylene bisacrylamide (MBA) are the key to gelation, and the gelation of different polymers often requires different types of cross-linking agents, which is adverse to the expansion of polymer types in gel materials.64,69,70 Apart from these surfactants, solid materials such as nanoclays,71 MXenes,72 and silica nanoparticles73 have also been reported as cross-linkers or to accelerate the gelation process. However, compared to physical cross-linking, the preparation of these chemical reagents is relatively complicated and costly, and the residual reagents after polymerization may cause contamination and damage to biological tissues, which is not conducive to the biocompatibility of the gel materials and their application in biomedicine.63,65,66 Beyond this, partial free radical polymerization generally needs to be carried out under oxygen-free conditions, and the complex polymerization process limits its large-scale production and practical application.74,75
According to the different components in the organohydrogel, there are mainly two different types of organohydrogels that can be constructed: one is a heteronetwork organohydrogel, in which hydrophilic and hydrophobic polymer networks interpenetrate each other or two immiscible phases are independently distributed in the matrix at microscale dimensions.76,77 The other is a homogeneous organohydrogel containing one or more homogeneous hydrophilic polymer networks in a binary-solvent (organic/water) system. So far, various organic solvents for the construction of organohydrogels have been developed, mainly including glycerol (Gly), ethylene glycol (EG), and dimethyl sulfoxide (DMSO).
As for heteronetwork organohydrogels, there are mainly three synthetic methods that have been developed in the past decade (Fig. 2). For example, Gao et al. firstly proposed a diffusion strategy to synthesize heteronetwork organohydrogels in 2017.44 More specifically, an organohydrogel is formed by diffusing an oleophilic polymer dissolved in an amphiphilic solvent into a partially dehydrated hydrogel scaffold and then polymerizing it in situ to swell the organogel in the hydrogel, featuring interpenetrating oleophilic/hydrophilic heteronetworks. Secondly, emulsion polymerization is currently the most commonly used strategy for preparing heteronetwork organohydrogels that feature the binary cooperative phase, which can be applied to various hydrophilic and oleophilic monomers, and was proposed by Zhao et al. in 2017.47 In this strategy, the hydrophilic and oleophilic monomer solutions are vigorously stirred or sonicated to form an oil-in-water emulsion. An emulsifier, including various surfactants or small-sized nanoparticles, should be added to the system to stabilize the emulsion resulting from the Pickering effect. The resulting emulsion shows that a large number of oil emulsion droplets with a diameter of several to tens of μm are uniformly distributed in the continuous water phase. Subsequently, heteronetwork organohydrogels in which micro-organogel domains are uniformly distributed in a continuous hydrogel network can be obtained after polymerization.78–80 Due to the thermoplasticity of the organogels, these organohydrogels show adjustable mechanical properties at different temperatures. Lastly, in some cases, heteronetwork organohydrogels can also be prepared through a one-step copolymerization method in which hydrophilic monomers and hydrophobic monomers are directly added to the binary solvent system and then copolymerized.45,67
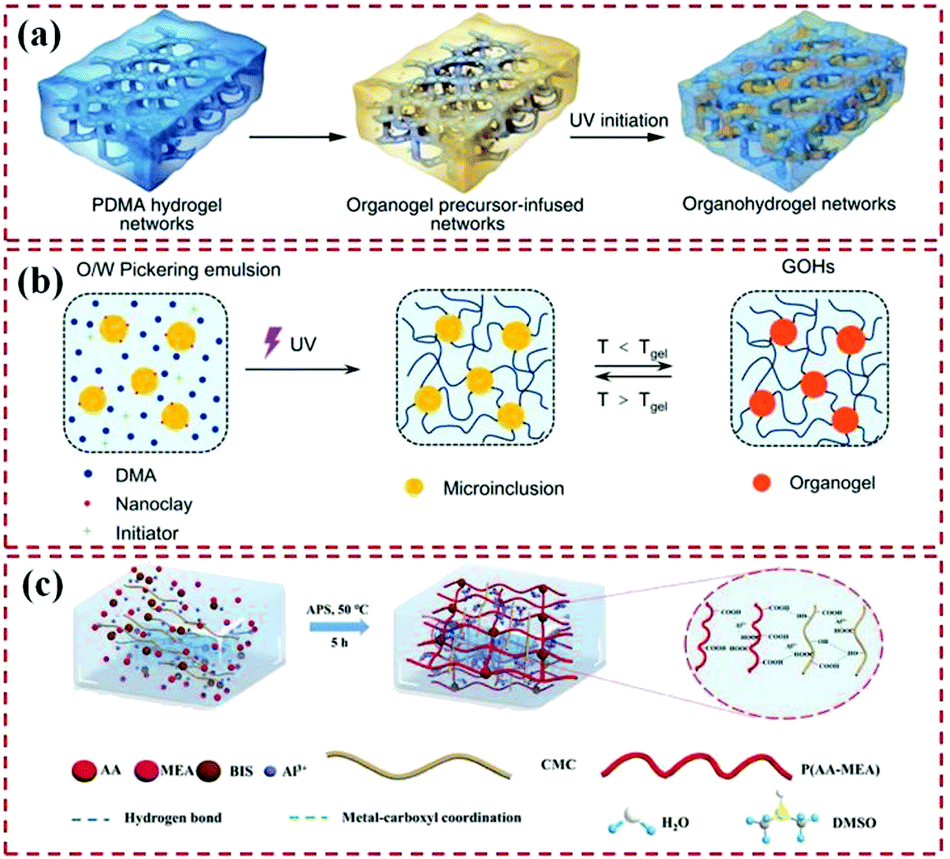 | ||
| Fig. 2 Different preparation strategies of heteronetwork organohydrogels. (a) Preparation of an organohydrogel by diffusing the oleophilic polymer into a hydrogel scaffold to form interpenetrating structures. Reproduced under terms of the CC-BY license.44 Copyright 2017, Springer Nature. (b) Emulsion polymerization. Reproduced with permission.82 Copyright 2021, American Chemical Society. (c) One-step copolymerization of hydrophilic and hydrophobic monomers in a binary solvent. Reproduced with permission.67 Copyright 2021, John Wiley and Sons. | ||
Despite the proven effectiveness, these methods are limited by the complicated polymerization conditions. Given that, other effective preparation methods have also been explored and further developments are needed in the future. Representatively, Li et al. developed a simple “in situ” transformation strategy to prepare organohydrogels by simply soaking poly(pentafluorophenyl acrylate) (pPFPA) gel into the blend solution containing both hydrophilic and oleophilic alkylamines, so that these amines could react with pPFPA and then convert them into the corresponding acrylamide polymers based on the aminolysis of pPFPA.81 Specifically, there are no strict requirements for the solvent in the ammonolysis process, only the selected amine should get dissolved in it and should not react. This strategy can not only efficiently synthesize heteronetwork organohydrogels with outstanding mechanical properties, but also solve the limitation of interfacial copolymerization caused by the immiscibility of water and oil in conventional synthesis methods.
As for homogeneous organohydrogels, there are mainly two methods that have been developed to construct a homogeneous network in a binary water/organic dispersion medium (Fig. 3). Firstly, a facile one-pot sol–gel synthesis method involving in situ gelation of polymers in a binary solvent is proposed.83 In this strategy, all components including polymer monomers, initiators, and cross-linking agents are added to the binary solvent before polymerization takes place. The synthesis process is simple and has been widely used in the preparation of various organohydrogels.15,23,43,70,84 However, the polymerization process is inevitably affected by the solvent environment, and the addition of organic agents may hinder the gelation process. Specific synthesis conditions need to be considered when different monomers are polymerized, which leads to restrictions on the diversity of organohydrogels. Driven by this, Zhou et al. first proposed another strategy for preparing organohydrogels containing a binary-solvent system, which is called solvent replacement.85 More precisely, a pre-prepared hydrogel is immersed in a mixture of organic agents and water, and the organohydrogels are synthesized by replacing part of the free water in the hydrogel with organic molecules owing to the osmotic pressure. The solvent replacement process is very free and does not require additional processing, and can reach equilibrium in a short time. Correspondingly, this strategy avoids the complicated designs of polymerization conditions, and thereby it can be used for various types of organohydrogels.74 Specifically, the performance of the organohydrogels prepared by solvent replacement, such as frost resistance, long-term stability, and mechanical properties, can be easily adjusted by changing the immersion time, concentration, or the type of organic agent.4,86 Based on these advantages, a large number of organohydrogels constructed by the solvent displacement have been reported in the past several years.55,63,66,87,88
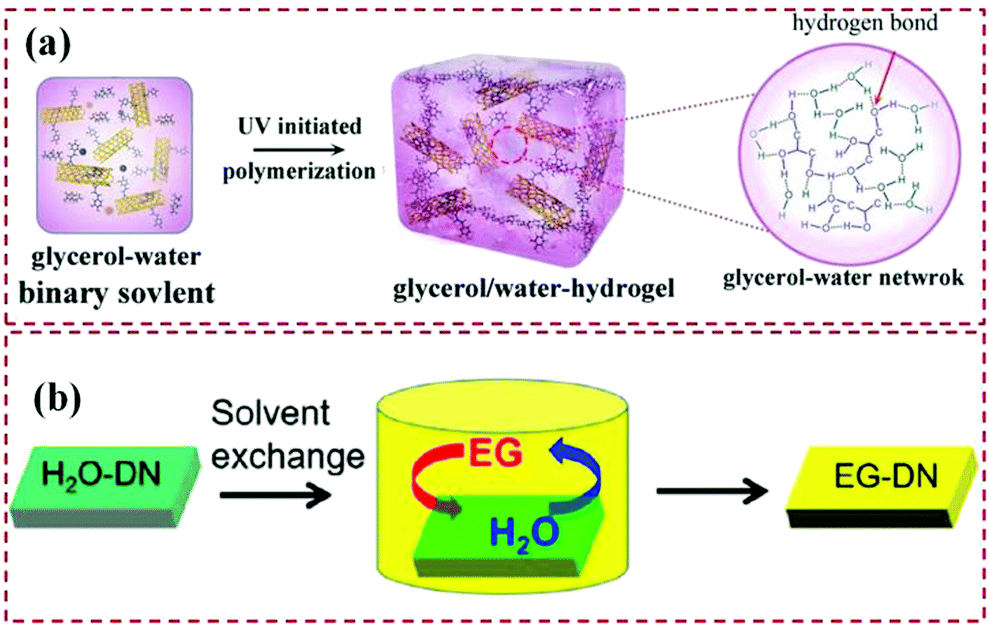 | ||
| Fig. 3 Typical fabrication strategies of homogeneous organohydrogels. (a) A one-pot sol–gel synthesis method. Reproduced with permission.84 Copyright 2017, John Wiley and Sons. (b) The solvent replacement strategy. Reproduced with permission.63 Copyright 2019, Royal Society of Chemistry. | ||
To sum up, we systematically list the advantages and disadvantages of different preparation strategies for organohydrogels in Table 1. Unfortunately, the development of organohydrogels is still in its infancy. Not only do the types of organohydrogels need to be expanded, but it is also necessary to synthesize organohydrogels with different microstructures, such as thin films or fibers, which is conducive to their development in flexible integrated electronics. Driven by this, further development and exploration of more effective, low-cost, and simple preparation methods are still needed, such as three-dimensional (3D) printing technology.
| Types | Methods | Advantages | Disadvantages |
|---|---|---|---|
| Heteronetwork organohydrogel | In situ polymerization of oleophilic polymer diffused into the hydrogel scaffold | Less restricted by polymerization conditions due to separate polymerization process | More preparation steps |
| The resulting organohydrogels both combine the characteristics of hydrophilic and oleophilic polymers and exhibit adaptive surface wettability | The hydrophilic and oleophilic network may not fully interpenetrate due to the intrinsic incompatibility of water and oil phase, resulting in undesirable functionality | ||
| Interfacial copolymerization is suppressed to a certain extent | |||
| Emulsion polymerization | Resulting in the effective combination of hydrogel and organogel at micro/nanoscale dimensions | The use of large amounts of emulsifiers may increase the toxicity of the system | |
| Enabling the organohydrogels to display adjustable mechanical properties | Interfacial copolymerization is restricted | ||
| One-step copolymerization of hydrophilic and hydrophobic monomers in binary solvent | Simple and fast | The polymerization of different polymers under the same conditions is limited, resulting in poor diversity of organohydrogels | |
| Homogeneous organohydrogel | One-pot sol–gel synthesis method | Fast and straightforward | Changes in the polymerization conditions of the polymer limit the successful preparation of various organohydrogels after the introduction of organic solvents |
| Solvent replacement strategy | No restrictions on the polymerization conditions | More steps than one-pot method | |
| Various properties of the final organohydrogels can be easily controlled by the immersion time, concentration or the type of organic agents | The conductivity may be decreased |
2.2. Unique advantages
The manifested macroscopic performance is completely determined by the composition and structure of materials. Due to the combination of binary solvent or complementary phases, organohydrogels hybridized by hydrogels and organogels exhibit not only the unique performances derived from individual gel components but also some synergistic performances, such as anti-freezing and water retention ability, solvent resistance, and shape memory effect. This section describes in detail the generation mechanism, research progress, and application prospects of these unique properties in organohydrogels.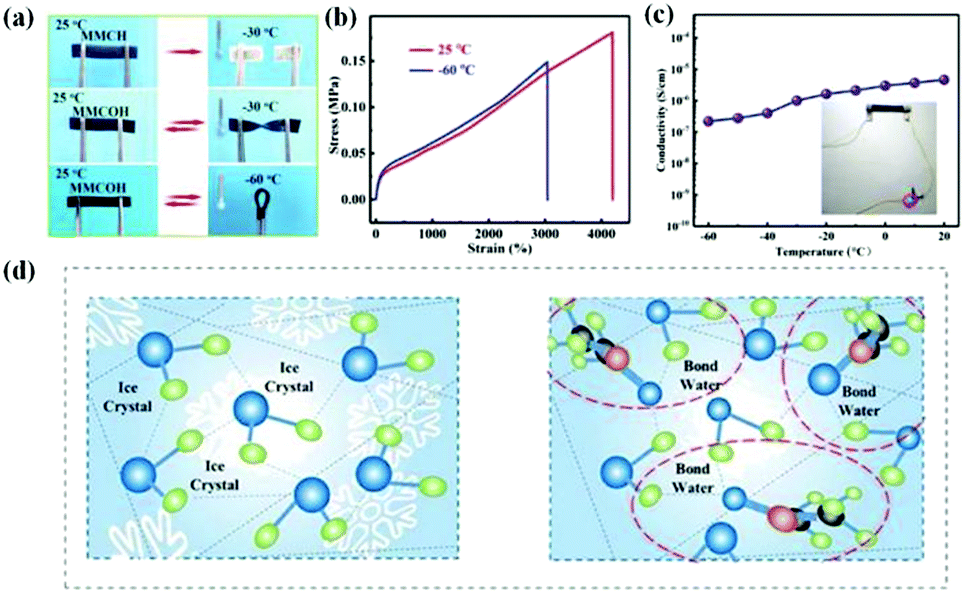 | ||
| Fig. 4 The frost resistance performances and mechanism of organohydrogels. (a) Photographs showing the flexibility; (b) stress–strain curves; and (c) conductivity of MMCH (hydrogel) and MMCOH (organohydrogel) at various temperatures. Organohydrogels maintain excellent mechanical properties and conductivity at sub-zero temperatures, showing an outstanding anti-freezing ability. Reproduced with permission.66 Copyright 2021, John Wiley and Sons. (d) Schematics illustrating the anti-freezing mechanism of organohydrogels. Reproduced with permission.67 Copyright 2021, John Wiley and Sons. | ||
In particular, DMSO is a very common cryoprotectant and has been widely applied for cryopreserving cells.92,93 A water/DMSO binary solvent has also been used for fabricating organohydrogels, and their freezing points are effectively reduced.94 Ye et al. reported a cellulose nanofibril (CNF) based organohydrogel by using the water/DMSO binary solvent, which could remain flexible and well conductive at −70 °C.95 In detail, the freezing point may reach the lowest temperature of −115 °C by adjusting the ratio of DMSO/water to a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 molar ratio. In addition to these, the introduction of other organic agents can also lead to good frost resistance in organohydrogels. Chen et al. reported the utilization of sorbitol to fabricate Ca-alginate/PAM organohydrogels with good mechanical flexibility at −45 °C.85 Gao et al. exploited water and heptanes as the dispersion medium of the hydrophilic/oleophilic heteronetwork organohydrogel, the organohydrogel showed reversible and stable elasticity even at a temperature as low as −78 °C.44 In general, the proportion of binary solvent is vital for the anti-freezing properties of organohydrogels. Previous studies have shown that the freezing point reaches the lowest when the weight percent of Eg and Gly is 70 and 66%, respectively.4,43,84
:3 molar ratio. In addition to these, the introduction of other organic agents can also lead to good frost resistance in organohydrogels. Chen et al. reported the utilization of sorbitol to fabricate Ca-alginate/PAM organohydrogels with good mechanical flexibility at −45 °C.85 Gao et al. exploited water and heptanes as the dispersion medium of the hydrophilic/oleophilic heteronetwork organohydrogel, the organohydrogel showed reversible and stable elasticity even at a temperature as low as −78 °C.44 In general, the proportion of binary solvent is vital for the anti-freezing properties of organohydrogels. Previous studies have shown that the freezing point reaches the lowest when the weight percent of Eg and Gly is 70 and 66%, respectively.4,43,84
In these cases, the ice inhibition effects in organohydrogels strongly put down to the formation of strong hydrogen bonds between the organic agents and the water molecules. There are three different states of water in hydrogels: free water, intermediate water, and strongly bound water.96 The free water can easily freeze at normal freezing points and exchange rapidly with different organic solvents. The intermediate water is weakly bound with polymer chains or other chemicals in the hydrogels, which can freeze at lower temperatures. In particular, the strongly bound water can keep it from freezing even when the temperature is below −100 °C, owing to the formation of more than one hydrogen bond with the polymer and others. In general, water crystallization is the formation of an infinite hydrogen bond network of water molecules.97 The organic solvent in organohydrogels can partially replace free water and form hydrogen bonds with the surrounding water molecules, which is beneficial to reduce the fraction of free water and meanwhile disrupts the hydrogen bonds between water molecules, thereby enhancing frost resistance (Fig. 4d).87 Thus, compared with EG-based organohydrogels, the better freezing resistance in Gly-based organohydrogels can be attributed to the three hydroxyl groups in Gly that can form bonds with the surrounding water molecules.
In addition to water crystallization, water evaporation can also cause detrimental effects on the properties of the hydrogels, including the weakening of mechanical properties and ionic conductivity, shape deformation, or generation of cracks. The excellent water retention ability of hydrogels is necessary to achieve long-term stability even under arid conditions. In previous studies, the simplest and most common strategy to reduce water evaporation is to encapsulate hydrogels with elastomers.98 However, the encapsulation will cause a sophisticated fabrication process or even a dragged down mechanical performance, and the loss of water cannot be completely avoided. Attractively, an excellent drying tolerance can be achieved in organohydrogels. Wu et al. reported a MXene nanocomposite organohydrogel with the water/Gly binary solvent as the dispersion medium, thus leading to the long-lasting moisture retention of these organohydrogels (10 days) (Fig. 5a and b).70 Lu et al. prepared a conductive organohydrogel by using the water/Gly binary solvent, and there was no performance degradation even after a long period of storage (30 days) (Fig. 5c–g).84 Consequently, the construction of organohydrogel seems to be a more effective method to inhibit water evaporation without losing mechanical properties.
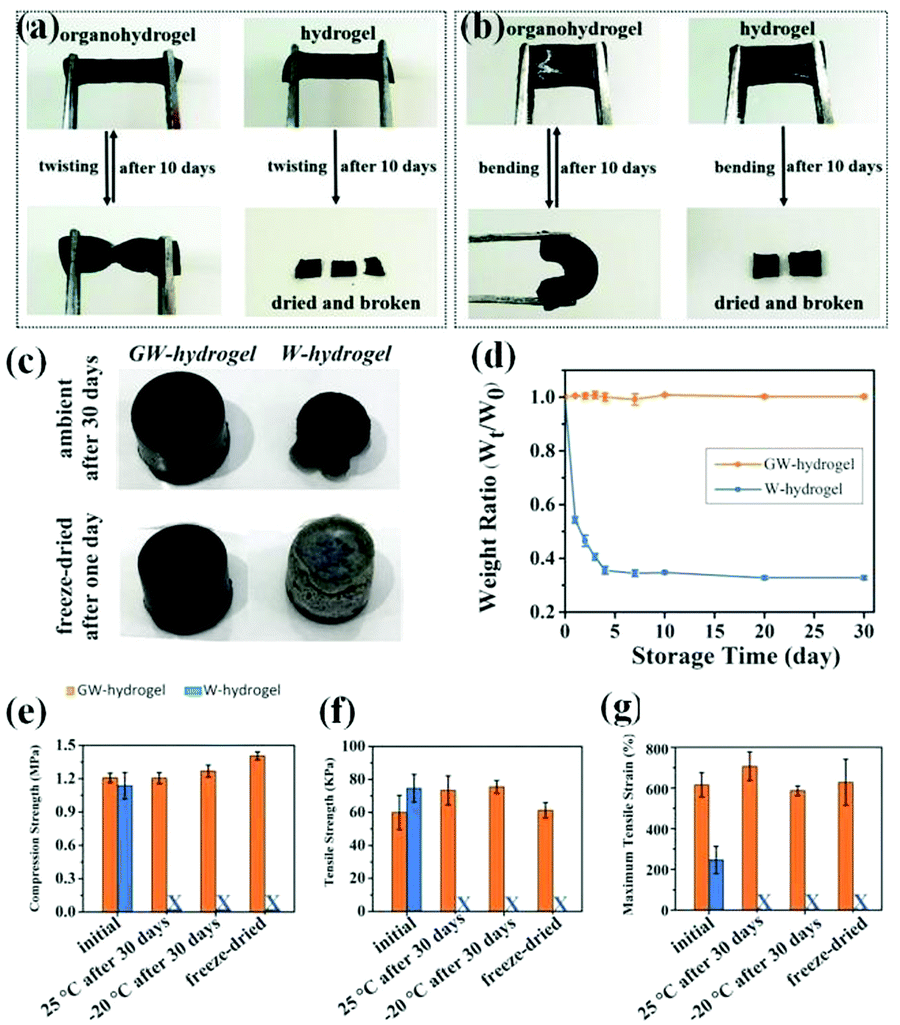 | ||
| Fig. 5 The water retention ability of organohydrogels. Comparison of flexibility between a MXene nanocomposite organohydrogel and a hydrogel through (a) twisting and (b) bending after storage for 10 days. Reproduced with permission.70 Copyright 2020, Royal Society of Chemistry. (c) Photographs of the GW-hydrogel (organohydrogels) and the W-hydrogel (hydrogels) after being placed in air for 30 days or after 1 day of freeze drying; (d) The change curve of the weight of the GW-hydrogel and the W-hydrogel with storage time; Comparison of (e) compression strength, (f) tensile strength and (g) maximum tensile strain of the GW-hydrogel and the W-hydrogel in the initial state or after storage for 30 days at 25 and −20 °C or after freeze-drying, reflecting the excellent stability of organohydrogels. Reproduced with permission.84 Copyright 2017, John Wiley and Sons. | ||
In general, the evaporation of water is caused by the lower vapor pressure of the surrounding environment than that of the hydrogels. Organic agents generally have a higher boiling point and can reduce the vapor pressure after mixing with water. Typically, Gly is a well-known, nontoxic humectant with excellent hygroscopicity. Compared with similar hygroscopic EG, Gly has a better water retention ability due to its higher viscosity, higher boiling point, and lower vapor pressure.63 Consequently, the outstanding water retention ability in the organohydrogels can be attributed to the closer vapor pressure between the materials and surroundings. Additionally, the interaction between organic molecules and the network also plays an important role in inhibiting water evaporation such as the ample and strong hydrogen bonds formed with water molecules.43 Considering the relatively high dissociation energy of these bonds, they can even exist stably at a higher temperature, resulting in a lower drying speed and increased environmental tolerance of the resulting organohydrogels.
The representative developments of the anti-freezing and water retention capabilities of organohydrogels are summarized in Table 2, and obviously their working temperature range and service life are effectively extended. Generally, at a sub-zero temperature, in addition to inhibiting the freezing of water molecules in the bulk phase, the freezing of water molecules adsorbed on the surface of the organohydrogels also needs to be prevented. However, the latter is rarely involved. Additionally, although the loss of water is greatly alleviated in the organohydrogel, dehydration will inevitably occur under extremely arid environments or after long-term use. Ideally, we urgently hope to develop a self-regeneration organohydrogel, which can self-regenerate to its original state for reuse. Recently, a LiCl-based hydrogel with strong water retention and self-regeneration abilities was reported. After vacuum drying, the dehydrated hydrogel could recover to its original state by spontaneously absorbing water molecules from the surrounding environments because of its lower vapor pressure.68 As water absorption from moisture has also been discovered in organohydrogels,64,85 we believe that self-regeneration of organohydrogels can also be achieved by regulating the proportion of organic agents and the humidity of the storage environment. Moreover, there are other feasible methods to fabricate anti-freezing and anti-drying hydrogels, such as the incorporation of inorganic salts (e.g. NaCl, CaCl2, and ZnCl2) and zwitterionic osmolytes (e.g. betaine or proline), which can also be used synergistically to further broaden the working environment of organohydrogels.4,23,68
| Organohydrogel | Solvent composition | Preparation method | Freezing resistance | Dry resistance | Published year |
|---|---|---|---|---|---|
| PVA/PEDOT:PSS organohydrogel49 | Water/EG | One-pot sol–gel synthesis method | −55 °C | — | 2017 |
| Hydrophilic/oleophilic heteronetwork organohydrogel44 | Water/n-decane | Diffusion strategy | −15 °C | — | 2017 |
| Water/heptanes | −78 °C | ||||
| PDA-CNTs/PAM/PAA organohydrogel84 | Water/Gly | One-step copolymerization | −20 °C | 90% weight retention after 7 days (60 °C) | 2018 |
| Ca-Alginate/PAM organohydrogel85 | Water/Gly | Solvent displacement | −50 °C | 88%, 45%, 75% weight retention after freeze-drying | 2018 |
| Water/EG | −70 °C | ||||
| Water/sorbitol | −45 °C | ||||
| Gelatin supramolecular organohydrogel55 | Citrate (Cit)/water/Gly | Solvent displacement | −80 °C | 90% weight retention after 7 days (20 °C, 50% RH) | 2019 |
| PVA/CNF organohydrogel95 | Water/DMSO | One-pot sol–gel synthesis method | −70 °C | 60% weight retention after 24 h (40 °C, 15% RH) | 2020 |
| TA@CNF/MXene/PAM organohydrogel23 | Water/Gly | One-step copolymerization | −80 °C | 80% weight retention after 7 days (20 °C, 35% RH) | 2020 |
| Alginate/(poly(ethylene glycol) diacrylate (PEGDA) organohydrogel fibers74 | Water/Gly/CaCl2/KCl | Solvent displacement | −100 °C | 90% weight retention after 10 days (20 °C, 45% RH) | 2020 |
| PAM/montmorillonite/CNT organohydrogel66 | Water/Gly | Solvent displacement | −60 °C | 90% weight retention after 3 days (25 °C) | 2021 |
| 77.5% weight retention after 72 h (60 °C) | |||||
| Carboxymethyl cellulose sodium salt (CMC)/P(AA-MEA) organohydrogel67 | Water/DMSO | One-step copolymerization | −90 °C | 76% weight retention after 15 days (normal temperature) | 2021 |
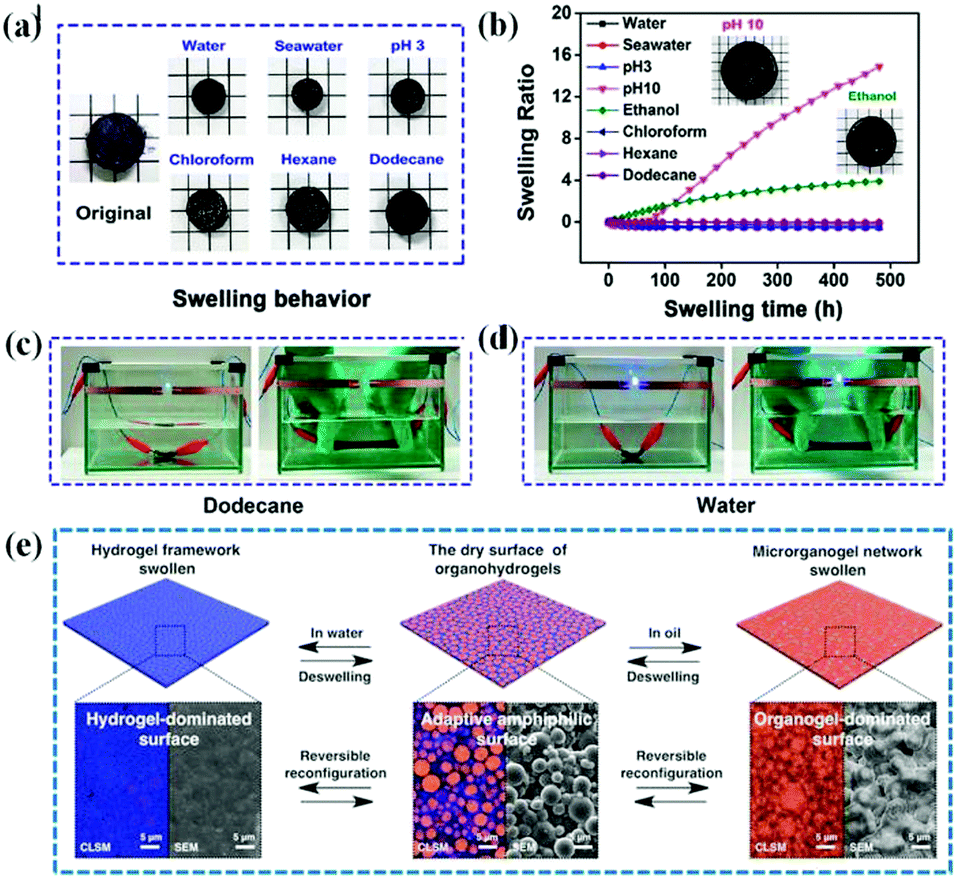 | ||
| Fig. 6 The anti-swelling and adaptive wettability of heteronetwork organohydrogels. (a) Photographs of the swelling behavior of organohydrogels after swelling 20 days in various solvents and the corresponding (b) swelling ratio change of organohydrogels as a function of swelling time; stable conductivity of organohydrogels in (c) dodecane and (d) water for strain sensing in various liquid media. Reproduced with permission.45 Copyright 2020, American Chemical Society. (e) Schematic diagram of reversible switching between the hydrogel- and organogel-dominated surface states of the organohydrogel as the solvent changes. Reproduced with permission.46 Copyright 2019, John Wiley and Sons. | ||
In particular, owing to the intrinsic immiscibility and the opposite solvent affinities of the hydrophilic and hydrophobic networks, the heteronetwork organohydrogels can controllably and reversibly rearrange their surface structure according to the different surrounding media, and realize adaptive superhydrophilic and superoleophilic transitions.46 In this case, the organohydrogel exhibits amphiphilic surface properties in dry air. When the organohydrogel is immersed in water, the hydrophilic network can absorb water and swell while the hydrophobic network is squeezed inward, resulting in a hydrogel-dominated surface reconfiguration and subsequent superhydrophilic surface properties. In contrast, organohydrogels exhibit superoleophilic surface properties in oil due to the occurrence of organogel-dominated surface reconfiguration (Fig. 6e). To this end, an organohydrogel with adaptive macroscopic wetting properties was synthesized by polymerizing an oleophilic monomer (lauryl methacrylate and n-butyl methacrylate) in a three-dimensional crosslinked hydrophilic matrix, which has potential application in smart gating systems, such as on-demand oil/water separation.44
Furthermore, by taking advantage of the hydrophilic/hydrophobic interactions that exist simultaneously in the system, Wan et al. reported a surface patterning technique that uses pre-patterned substrates with different wettability regions to induce patterning of hydrophilic and hydrophobic monomers at the solid–liquid interface through hydrophilic/hydrophobic interactions, and then in situ polymerization to fix the patterns on the organohydrogel surface.65 The surface-patterned organohydrogels can be well maintained in diverse solvents, owing to their excellent solvent resistance. Additionally, focusing on the obvious transparent change of the heteronetwork organohydrogel resulting from the microphase separation in poor solvents and low-swelling in suitable solvents, Liu et al. proposed a dynamic information memory device for recording and encrypting information based on the variable and adjustable optical properties.67 Similarly, this organohydrogel still exhibits anti-swelling properties owing to the introduction of the hydrophobic and hydrophilic components. Therefore, in combination with such commendable solvent resistance, heterogeneous organohydrogels not only exhibit an excellent environmental tolerance but also can work normally in a variety of water or oil-based liquid phase environments like water, n-dodecane, chloroform, and hexane; hence, they have shown great potential for applications in various fields such as underwater robots, underwater sensing, implantable bioelectronics, and oil–water separation.
Given these limitations, a high-strain and tough shape memory organohydrogel with a binary cooperative phase was prepared, and its shape memory effect was closely related to the phase transition of the paraffin composition in the micro-organogel domains by thermal stimuli.47 As illustrated in Fig. 7a and b, at room temperature, this organohydrogel exhibited excellent strength and toughness due to the reinforcement of micro-organogel domains, and the micro-organogel domains exist in spherical shapes to maintain the lowest energy of the system. When the organohydrogel is heated above the melting point (Tm) of paraffin, the micro-organogel domains will soften accordingly. Meanwhile, the sample can be more easily stretched into a specific shape, and the micro-organogel domains change from spherical to ellipsoidal shapes along the stretching direction. By reducing the temperature from over Tm to below Tm, the correspondingly frozen paraffin will keep the micro-organogel domains in the deformed shape and prevent the elastic deformation of the hydrogel network through covalent interconnection at the heterophase interfaces, resulting in a fixed temporary shape of the organohydrogel at the macroscale and the energy storage in the system. When the temperature increases above Tm again, the elastic potential energy in the hydrogel network and the interfacial tension of the micro-organogel domains drive the organohydrogel to release energy so as to completely return to its original shape with the lowest energy. In this case, the organohydrogel is accompanied by energy storage and release during the shape memory process, which is conducive to the recovery of elastic deformation.
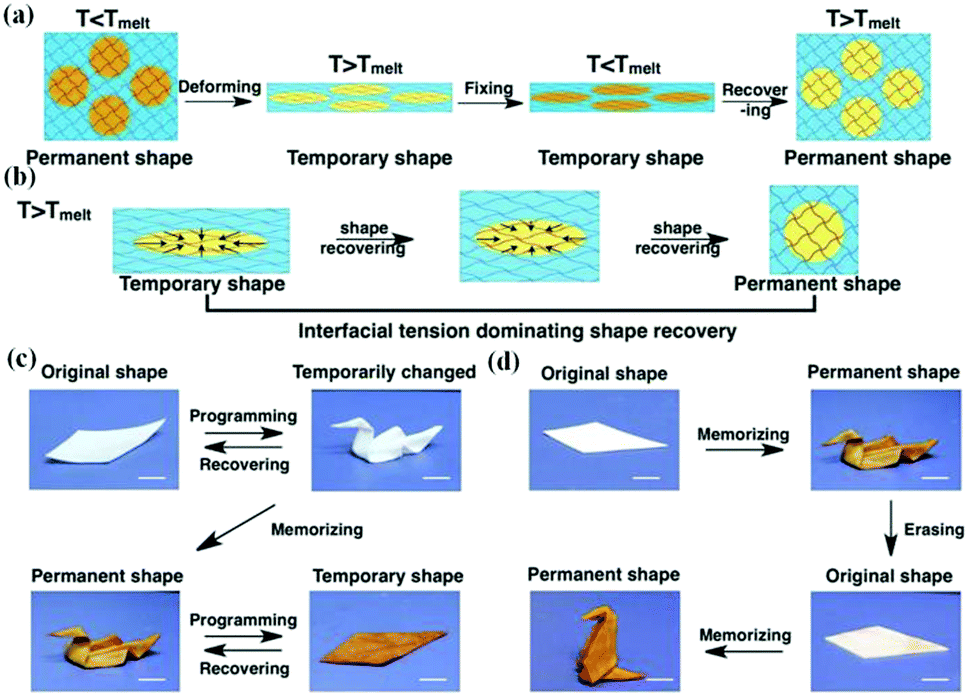 | ||
| Fig. 7 Shape memory effect of the heteronetwork organohydrogels. (a and b) Schematic illustration of the shape memory process of the organohydrogel based on the phase-transition of micro-organogels. Reproduced with permission.47 Copyright 2017, John Wiley and Sons. (c and d) Hierarchical shape morphing process. Reproduced with permission.117 Copyright 2018, John Wiley and Sons. | ||
According to this mechanism, it is anticipated that multiple shape memory effects can be achieved by combining multiple noneutectic phase transition components inside microrganogel inclusions. Subsequently, Zhuo et al. prepared a complex multiphase organohydrogel by incorporating microrganogel inclusions containing multiple oleophilic components with different phase transition temperatures in continuous elastic hydrogel networks.80 For example, assembling C16H34 and C28H58 with significantly different alkyl chain lengths and poly(stearyl methacrylate) (PSMA) within the microrganogel inclusions provided the organohydrogel with precisely controllable thermo-induced triple-switching mechanical properties due to the triple-phase transitions of the microrganogel inclusions from a fully crystalline state to a fully melting state as the temperature varies.
Considering the dual-phase heteronetworks in an organohydrogel, Zhao et al. developed a dual-programmable shape-morphing organohydrogel containing micro-organogel inclusions and the reversible metallo-supramolecular hydrogel framework, which can independently respond to different stimuli.117 On the one hand, the reversible metal–ligand association/dissociation of the hydrogel framework realizes the permanent shape memorization process via depressing the storage energy of the previous temporary state. On the other hand, the micro-organogel inclusions with thermoswitchable phase-transition ability dominate the shape memory process. This hierarchical shape morphing behavior enables versatile step-wise memorization and programming of geometrically complex structures (Fig. 7c and d). Specifically, note that this adaptive smart organohydrogel could adjust the surface topographies and properties simultaneously, a smart microfluidic device was developed accordingly to enable on-demand remote programming of unidirectional liquid transport by the integration of photothermal Fe3O4 nanoparticles.46 Nevertheless, although the huge application potential of these smart organohydrogels has been proven, it is still a long way from the laboratory to practical applications.
3. Optimal design of organohydrogels
In addition to these inherent excellent properties derived from the unique components and structure of organohydrogels, there are still some other properties that need to be included or further optimized through additional means to meet the practical applications of organohydrogels in various fields, such as mechanical properties, conductivity, self-healing, adhesion, antibacterial properties, etc. Herein, this section summarizes the reported optimization strategies for the various properties of organohydrogels in detail.3.1. Mechanical properties
Flexible electronic devices usually need to exhibit good mechanical properties, including strong strength, high stretchability, and good fatigue resistance, and this is mainly determined by the cross-linking method and degree between various components of the gel network.5 In general, chemical cross-linking is mainly based on strong covalent bonds, which can significantly improve the mechanical strength, but the breaking of these covalent chemical bonds under large deformation will cause irreversible damage to the structure, thus leading to poor deformation tolerance and fatigue resistance.2,87 Conversely, a purely physical cross-linking network based on the reversible non-covalent interactions, such as hydrogen bonds, π-stacking interactions, and van der Waals interactions, can easily recover and rebuild under deformation.35 As for traditional hydrogels, they generally exhibit low mechanical strength due to high water content and loose cross-linking, and are prone to breakage during mechanical deformations, which is not conducive to practical applications.1 To solve this problem, multiple reversible non-covalent interactions are introduced into the strong chemical cross-linking network, which can dissipate energy as sacrificial bonds during the stretching process and reassociate after the stress is removed, making the hydrogel exhibit both good mechanical strength and high toughness.4,55Organohydrogels generally exhibit better mechanical properties than hydrogels composed of the same polymer network due to the following two points. First of all, the organic agents in the organohydrogels will form a large number of hydrogen bonds with water and polymers in the network, which can increase the cross-linking density of the polymer network and facilitate the bridging of polymer chains,84,85,87 resulting in enhanced mechanical properties. For instance, Fang et al. prepared a gelatin/ferric-ion-cross-linked polyacrylic acid (PAA) dual dynamic supramolecular network, which can be toughened by incorporating Gly into the gel networks because of the significantly increased physical cross-linking of gelatin and reduced phase separation.88 However, when the organic solvent is excessive, it will cause rapidly increased cross-link density of hydrogen bonds and even volume shrinkage, which is not conducive to the movement of polymer molecular chains, making the resulting organohydrogels extremely fragile.85 In general, the mechanical performance of the organohydrogels largely depends on the proportion of organic solvent in the mixture.
Secondly, organohydrogels exhibit enhanced mechanical strength without sacrificing much flexibility because the organic agents in them can induce polymer crystallization.49,95 For instance, different from other conventional PVA hydrogels with low mechanical strength, high-strength organohydrogels combined with the water/EG binary solvent were reported. This strength enhancement is mainly derived from the crystallization of PVA after the introduction of EG, and the strength continues to increase with the increasing EG concentration due to the formation of more induced PVA crystalline domains.49 Furthermore, a highly stretchable, strong, and tough PVA-based organohydrogel was also fabricated by using the water/DMSO binary solvent; the excellent mechanical strength was also ascribed to the crystallization of PVA chains, which was formed because of the strong intermolecular interaction between DMSO and water and thus the insufficient solvation of PVA chains.95 In these cases, the formed crystalline domains not only served as rigid and high-functionality crosslinkers to strengthen the network, but also toughened the organohydrogels through the crack pinning effect,18,49 thereby providing better mechanical properties compared to hydrogels.
In fact, it is necessary to further improve the mechanical properties of existing organohydrogels. In a single polymer network, the low density or irregular distribution of cross-linking points results in different polymer chain lengths, and this intrinsic structural inhomogeneity easily causes initial cracks. Moreover, organohydrogels are prone to rupture due to the lack of sufficient energy dissipation mechanism to slow down the propagation of cracks. To address this problem, tremendous improvements have been made to fabricate strong and tough organohydrogels by introducing energy dissipation mechanisms, such as forming a double network (DN),43,63,87,118 adding nanofillers,16,84,95,119 introducing other dynamic interactions,86,120,121 and creating nano-crystalline domains.88
Above all, the incorporation of a second polymer network is an effective strategy to strengthen and toughen organohydrogels and keep other functions from being lost.122,123 Generally, the obtained DN hydrogel is composed of two complementary polymer networks with different physical natures, either physically (PVA,43 agar,123 alginate,122 carrageenan,124 gelatin88) or chemically cross-linked (PAM,43,70,87 PAA43,64). During the stretching process of the materials, the physically crosslinked polymer network is broken first to dissociate and dissipate energy, resulting in an increase in toughness. Meanwhile, the chemically cross-linked network remains intact and stabilizes the deformation to provide the materials with good mechanical strength. In addition, the interactions between these two polymer networks also dissipate mechanical energy so as to continue to improve the mechanical properties of the materials. As a consequence, this incorporation ultimately results in the combination of relatively high stiffness and high toughness of the material, which has important advantages over traditional single-network organohydrogels with weak and brittle nature.
Secondly, adding nanofillers to the polymer network can also effectively reinforce the organohydrogels, which have been widely used in previous studies. So far, many nano-filled materials have been reported to be incorporated into organohydrogels, such as CNTs,16,66,84 silk nanofibers (SNFs),119 CNFs,23,95 graphitic carbon nitride (g-C3N4),119 and silica nanoparticles.73 It is worth noting that the mechanical properties of the composite organohydrogels are largely determined by the chemical nature and dispersivity of the fillers.125,126 The weak interfacial interactions between the filler and the polymer matrix are not conducive to an effective load transfer between them.127,128 In order to effectively improve the mechanical properties of the organohydrogel, the filler added generally has a large number of functional groups, and the reinforcing effect can be attributed to the formation of dynamic hydrogen bonds.129 Very recently, Bao et al. reported the utilization of soft SNFs and hard g-C3N4 nanosheets to co-reinforce a PVA organohydrogel.119 The functional groups on the surface of g-C3N4 nanosheets and the molecular chain of silk nanofibers, such as amide groups, carboxyl groups, and amino groups, could form reversible hydrogen bonds with the PVA molecular chains, and thus greatly improve the tensile strength and toughness of the organohydrogel (Fig. 8a and b). Driven by this, the chemical nature of the fillers can be effectively changed by modifying the functional groups to strengthen interfacial interactions with polymer networks, which is an effective approach for hydrogel reinforcement.130 For example, Jaiswal et al. proposed a mechanically stiff nanocomposite hydrogel by incorporating nanoparticles functionalized with nitro-dopamine anchored PEG diacid on the surface, which resulted in a more than 10-fold increase in mechanical stiffness and a 20-fold increase in toughness due to the enhanced nanoparticle–polymer interactions.131
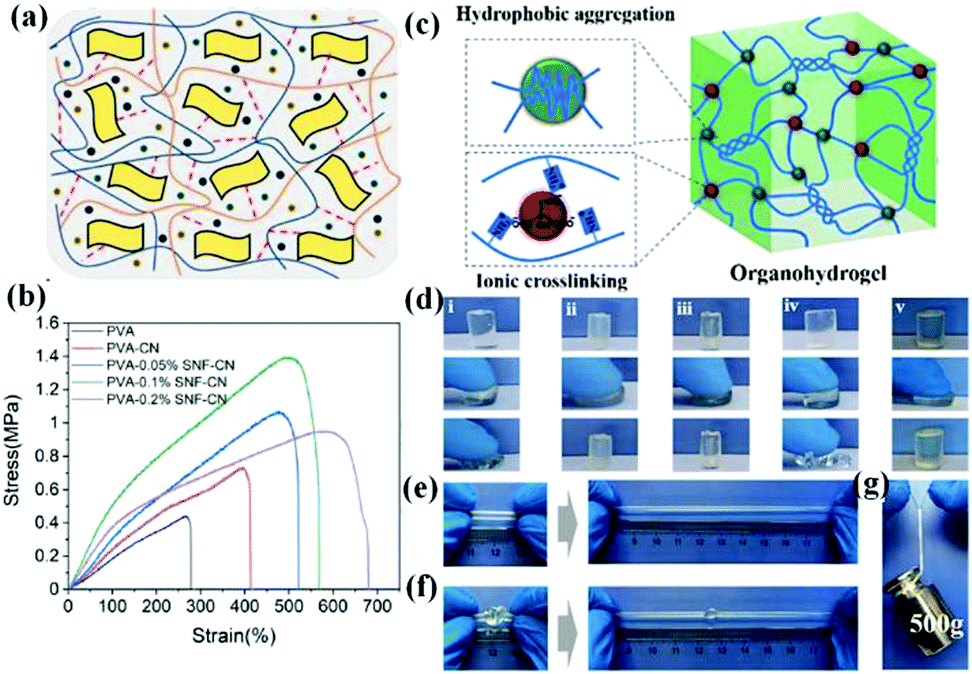 | ||
| Fig. 8 Structural design and mechanical performance characterization of tough organohydrogels. (a) Schematic diagram of SNF and g-C3N4 nanosheet incorporated organohydrogel. (b) Comparison of the stress–strain curves between a nanocomposite organohydrogel and a pure organohydrogel. Reproduced with permission.119 Copyright 2021, Elsevier. (c) Schematic diagram of a supramolecular organohydrogel prepared in a citrate water/glycerol mixture solution. (d) Compression performances of various gelatin gels ((i) hydrogel, (ii) supramolecular hydrogel 1, (iii) supramolecular organohydrogel, (iv) organohydrogel, and (v) supramolecular hydrogel 2), showing the improvement in the mechanical properties by supramolecular interaction; mechanical performance of a supramolecular organohydrogel under (e) stretching, (f) knotting and (g) 500 g load. Reproduced with permission.55 Copyright 2019, American Chemical Society. | ||
In this strategy, it cannot be ignored that these fillers, especially for CNTs, are prone to aggregation in the gel due to their inherent surface hydrophobicity and strong van der Waals force, which in turn lead to a decrease in the mechanical properties of the organohydrogels due to the uneven distribution of the structure. According to previous reports, this can be properly resolved by adding stabilizers or dispersants to the aqueous solution of CNTs, such as polydopamine (PDA),84 tannic acid (TA),16 and montmorillonite (MMT).66,126 The good dispersibility achieved is caused by the repulsion between the functional groups of the agents attached to CNTs or between dispersants and CNTs. In particular, the mechanical properties of these organohydrogels can be further improved due to the increased interaction between dispersants and the polymer network.
Thirdly, the addition of ions enables the strengthening of the organohydrogel by forming ionic interactions with polymer chains and thereby increasing the cross-linking density.55,67,121,132 Particularly, apart from the formed coordination bonds, the introduced ions can also adjust the mechanical properties by influencing the aggregation/crystallization of polymer chains, that is, the Hofmeister effect or ion-specific effect.18 Generally, some ions can combine with the surrounding hydration water molecules of the polymer chains and make them polarized, resulting in the instability of the hydrogen bonds between the polymer chains and their hydration water molecules, and finally the water molecules can be discharged from between the polymer chains (salting-out effect). Meanwhile, the polymer chains aggregate through the formation of hydrogen bonds between the hydrophilic groups of the polymer chains, exhibiting a strong reinforcement effect.133 For example, a tough gelatin supramolecular organohydrogel was prepared by immersing the pre-hydrogel in concentrated sodium citrate water/Gly solution.55 Its excellent flexibility and mechanical strength can be attributed to the formation of multiple non-covalent supramolecular cross-linking, such as the hydrophobic aggregation induced by the salting-out effect, ionic interactions between the –NH3+ of gelatin and Cit3− anions, and quadruple hydrogen bonding (Fig. 8c–g). Specifically, studies have shown that when ions are introduced, the improvement of mechanical properties caused by the formation of induced crystalline domains is more effective than the traditional strengthening achieved by ionic cross-linking.134,135 Additionally, some ions can directly bond with the polymer chain and dissociate the hydrogen bonds between the polymer chains, leading to the increase of polymer solubility and the softening of the network (salting-in effect). For instance, Fe(NO3)3 was found to have a strong salting-in effect on PVA hydrogels, which can make the tough hydrogels soften quickly. Inspired by this, a hydrogel that can switch in situ between the stiff and soft states was proposed, which hopefully can be used as a neuron probe.133 However, organohydrogels with adjustable mechanical properties have not yet been involved, which is quite necessary for their future developments.
Even so, these methods mentioned above mainly focus on composition or molecular engineering, which rarely involve structural changes. In a single and homogeneous covalent polymer network, high crack resistance and high elastic modulus cannot be realized simultaneously.136 In contrast to homogeneous tough polymer networks made by molecular engineering methods, creating anisotropic micro/nanostructures in hydrogel can well resist the growth of a crack, achieving both high toughness and fatigue resistance. General structural engineering methods include ice-templating,137 mechanical stretching,138,139 and compositing.136,140 The ice-templating method can lead to the formation of micro-alignment structures in the material. And mechanical stretching can also effectively create anisotropic micro/nanostructures. The formation of these micro/nanostructures in a hydrogel will lead to an increase in material density to strengthen the material, and the increased toughness is attributed to the increased energy dissipation during fracture. The most common strategy for compositing methods is to add alien micro/nanoscale fibre reinforcements to the hydrogel. To better improve the fatigue resistance of materials, this compositing method needs to follow the principles that the fibre reinforcements should be stiffer than the matrix and the interface adhesion should be strong.136 The soft matrix facilitates the stress transfer in the network. Ideally, the initial crack will not cause serious stress concentration in the fibers but spreads to the entire network, and the crack propagation is also prohibited by the fibers, showing a crack-blunting ability (Fig. 9a). Consequently, this composite hydrogel requires more energy to grow the crack and shows a gradual failure mode featuring stepwise fracture and pull-out of fibres.141–143 Based on this, Ge et al. developed a highly stretchable PAA organohydrogel by incorporating polyaniline nanofibers (PANI NFs) into the network, taking advantage of the fiber-reinforced microstructures.132
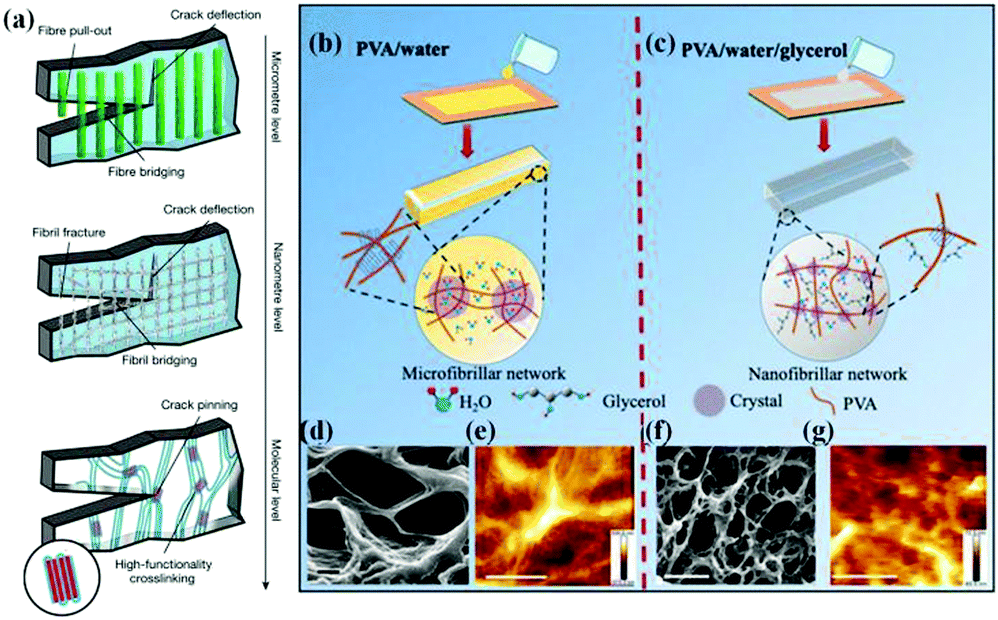 | ||
| Fig. 9 Structural engineering in organohydrogels. (a) Toughening mechanisms at each length scale. Reproduced with permission.18 Copyright 2021, Springer Nature. Schematic design of (b) PVA hydrogels with a microfibrillar network and (c) PVA organohydrogels with a nanofibrillar network. SEM images of (d) a PVA hydrogel and (f) a PVA organohydrogel (scale bar: 1 μm). Atomic force microscopy (AFM) height images of (e) a PVA hydrogel and (g) a PVA organohydrogel (scale bar: 500 nm). Reproduced with permission.52 Copyright 2020, American Chemical Society. | ||
At present, the effective combination of molecular and structural engineering strategies is an important way to construct gel materials with better mechanical properties.144 Recently, Hua et al. presented a freezing-assisted salting-out treatment to produce a hierarchically anisotropic hydrogel, which in turn formed a mesh-like stretchable nanofibril network on the surface of the micrometre-scale aligned pore walls, showing a high toughness value that even surpass natural tendons and spider webs.18 Concretely, directional freeze-casting causes the hydrogel to form aligned micropore walls and correspondingly leads to an increase in material density. The subsequent salting out strongly induces the aggregation and crystalline domains in the nanofibrils by phase separation, which can effectively delay the fracture of individual fibrils by crack-pinning. Similarly, a nanofibrillar PVA organohydrogel cross-linked by numerous PVA nanocrystallites was formed by freeze–thaw and solvent replacement (Fig. 9b–g).52 Thanks to the generation of nanoscale PVA crystalline domains induced by Gly, excellent mechanical properties are achieved accordingly. However, the PVA hydrogel sample exhibits microfibrillar morphology in the absence of Gly, and shows a very low mechanical strength and relatively low elongation at break.
3.2. Conductivity
A good conductivity in organohydrogels is generally required in most application areas, such as sensors, nanogenerators, and storage devices. As for hydrogels, the large amount of water contained facilitates ion transporting within the network and makes them capable of transmitting electronic signals.145,146 Although the conductivity is reduced after the replacement of free water, the anti-freezing and water retention ability achieved in organohydrogels can also stabilize their conductivity even under harsh conditions. Various methods have been developed to improve the conductivity of organohydrogels. At present, the simplest and most effective method is to directly introduce ions into the organohydrogel, which is achieved by using polyelectrolytes or adding inorganic salts. The polyelectrolyte network can release large numbers of free-moving ions into the solvent water, thereby achieving a good ionic conductivity.43,94 Yang et al. prepared a conductive organohydrogel with sodium methacrylate (MAANa) and [2-(methacryloyloxy)-ethyl] trimethyl ammonium chloride (DMC) as monomers, which exhibited a good conductivity even in low-temperature environments.15 Moreover, the conductivity of this organohydrogel was not affected by current frequency and had the potential to be used as signal transmission materials.Nowadays, various inorganic salts including KCl,63,87,147 NaCl,88,95 LiCl,42,148 and AlCl3,64,67,119 have been introduced into organohydrogels to increase their ionic conductivity. The addition of salt can be achieved by directly mixing the salt in the precursor solution or immersing the prepared gel in the salt solution for the ions to diffuse into the network. Accordingly, the conductivity will increase with the salt concentration due to the increased dissociated ion concentration, suggesting the contribution of the added ions to the conductivity. However, this increase in conductivity has an upper limit. Ye et al. proposed that excess salt in the organohydrogels will form ion pairs or clusters rather than existing as dissociated ions, resulting in no longer increased conductance.95 In particular, as mentioned before, the introduction of ionic polymers or salts into organohydrogels is also beneficial to the antifreeze ability of the materials, which can effectively compensate for the contradiction between conductivity and frost resistance.15
The conductivity of organohydrogels is not only related to ion concentration, but also largely depends on the mobility of ions. Generally, a polymer network with a high degree of cross-linking is not conducive to ion migration;149 that is, there is a dilemma between strength and ionic conductivity in hydrogels. Another typical strategy is to introduce a secondary conductive network into the traditional organohydrogel, such as conductive polymers,49,132 CNFs,95 MXene nanosheets,23,70 and CNTs,16,84 which may contribute to the increase in mechanical strength and conductivity simultaneously. The conductive polymer is a kind of functional polymer with highly conjugated polymer chains; it has active π-bonded electrons and can provide channels for carrier transfer and transition, and has intrinsic electronic conductivity.150–152 However, conductive hydrogels based on conductive polymer matrices, such as PANI, polypyrrole, and PEDOT, tend to have lower strength and flexibility, and thus they are usually selected to be incorporated into another polymer matrix.84,153 For example, Rong et al. reported a PVA organohydrogel with adjustable conductivity by adding PEDOT: PSS.49 As for CNFs, the negative surface carboxylate groups on them can attract counter ions and provide more hopping sites for their migration,95,154–156 thus leading to an increase in the conductivity. MXene nanosheets possess superior electrical conductivity, rich surface functional groups, good hydrophilicity, and correspondingly good dispersibility, which will undoubtedly show tremendous potential in the fabrication of organohydrogels with stable electrical properties.157–159 For example, Wei et al. developed a nanocomposite organohydrogel by incorporating conductive MXene nanosheets and TA@CNFs into the polymer networks, which demonstrated outstanding flexibility and electrical conductivity within a wide temperature range.23 Herein, MXene is connected to the organohydrogel matrix to form a conductive skeleton, and thus endows the organohydrogel with high electrical conductivity. Similarly, the introduction of CNTs with high conductivity and specific surface area can also benefit the improvement of conductivity,16 but their aggregation problem needs to be solved first.
3.3. Self-healing ability
As a wearable electronic device, organohydrogels will suffer unavoidable damage during the repeated stretching and bending, including cracks, crazing, puncture, and delamination, which will lead to a sharp degradation in the mechanical and electrical properties of the device. Therefore, self-healing organohydrogels that can self-repair some unexpected damages have a wider application field and longer service life, reducing resource waste. Mechanical damage of the gel network will cause the polymer chains to break and slip with each other, and leave active groups on the broken interfaces.5 In general, as seen in Fig. 10, the intrinsic self-healing process is realized by the diffusion and rearrangement of molecules and inherently reversible chemical bonding between molecular chains160,161 and controlled by thermodynamics or reaction kinetics.162 When a break occurs, the entropy increase of the system and the gradient of the molecular chain network density will drive the chain to spread.163 Specifically, the flexibility of the polymer chain will strongly affect chain diffusion across a fracture surface interface, and often the polymer chains with a low molecular weight and a low glass transition temperature show relatively high flexibility and correspondingly high molecular diffusion speed.164 When the driving force of the system is not enough, the healing process of the gel needs external energy to proceed, such as high temperature165,166 and ultraviolet irradiation.167,168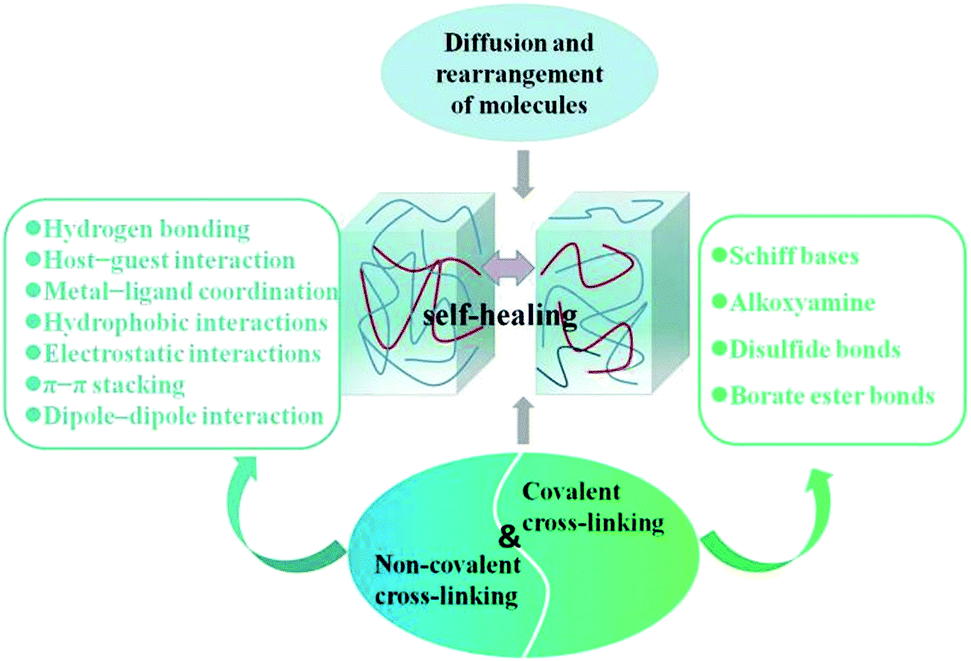 | ||
| Fig. 10 Schematic illustration of the self-healing mechanisms. | ||
The subsequent self-healing process is mainly realized by dynamic covalent cross-linking and non-covalent cross-linking.69 Non-covalent cross-links used to achieve self-healing properties mainly include hydrogen bonding,15,169,170 host–guest interaction,171–173 and metal–ligand coordination.164,174,175 Hydrogen bond is a common reversible weak bond in polymer networks, which can automatically re-bond to repair damage even at room temperature. Hydrogen bond-based self-healing was first proposed in rubber. It can restore damage with the help of multiple hydrogen bonds in molecular chains and does not require any external assistance.176 Therefore, compared with hydrogels, the abundant hydrogen bonds formed between organic molecules and polymer networks make organohydrogels show better self-healing ability.49,87 The host–guest supramolecular interactions originate from the complementary guest and host moieties in polymer networks, such as cyclodextrins and ferrocene molecules171 or cyclodextrin and azobenzene molecules,172 which can form host–guest inclusion complex due to the high affinity between them. When damage occurs, the damaged surface will expose the dangling host and guest molecules, which can autonomously recognize each other and recombine again to repair the damaged surface through the dynamic host–guest interactions. In particular, since the affinity between host and guest molecules can be controlled by external stimuli, these host–guest supramolecular hydrogels can be used in switch soft materials.171 Metal–ligand bonds possess moderate bond energy, which can dynamically associate or dissociate at room temperature and thus endow the hydrogel with self-healing properties. Besides these, other non-covalent supramolecular interactions such as hydrophobic interactions,177,178 electrostatic interactions,179,180 π–π stacking181–183 and dipole–dipole interaction,184 which can be introduced by modifying hydrophobic blocks, charged groups, or surfactant molecules in polymer networks, have also been reported to be used in the preparation of self-healing materials. These reversible breakable bonds can provide an energy dissipation mechanism to prevent the breaking of the molecular backbone and realize subsequent self-healing of damaged materials.185
As for the self-healing mechanism based on covalent cross-linking, various reversible covalent bondings such as Schiff bases,186,187 alkoxyamine,188 disulfide bonds,189,190 and borate ester bonds23,43 have been studied for achieving self-healing properties. Synergistically, high healing efficiency and speed can be achieved by combining these two mechanisms. For example, Wu et al. reported a MXene nanocomposite organohydrogel modified with dopamine (DA) and phenylboronic acid (PBA), and its good self-healing capability is mainly attributed to the existence of abundant dynamic hydrogen bonds and covalent ester bonds.70 The organohydrogel can re-ignite the LED when the two cut parts are brought together, and the stretch strain can be restored to 88.75% after self-healing for 12 h (Fig. 11). For most self-healing organohydrogels, their self-healing process can mostly be carried out at room temperature, and the recovery speed of electrical and mechanical properties is quite different. Generally, their conductivity can be restored within a few seconds because the physical dynamic cross-linking at the fracture interface can quickly reform at room temperature, and the self-healing of mechanical properties is relatively slow due to the rearrangement of the polymer matrix, which can be further accelerated by increasing the temperature. In particular, the healing process of some organohydrogels requires a heating and cooling cycle. For example, for a carrageenan-based organohydrogel, the self-healing of its mechanical properties is largely derived from the dissociation of the double-helical structure of carrageenan at high temperatures and the reformation at low temperatures.63,87 Alternatively, when there are crystalline domains in an organohydrogel, the self-healing also needs to undergo a heat treatment.49
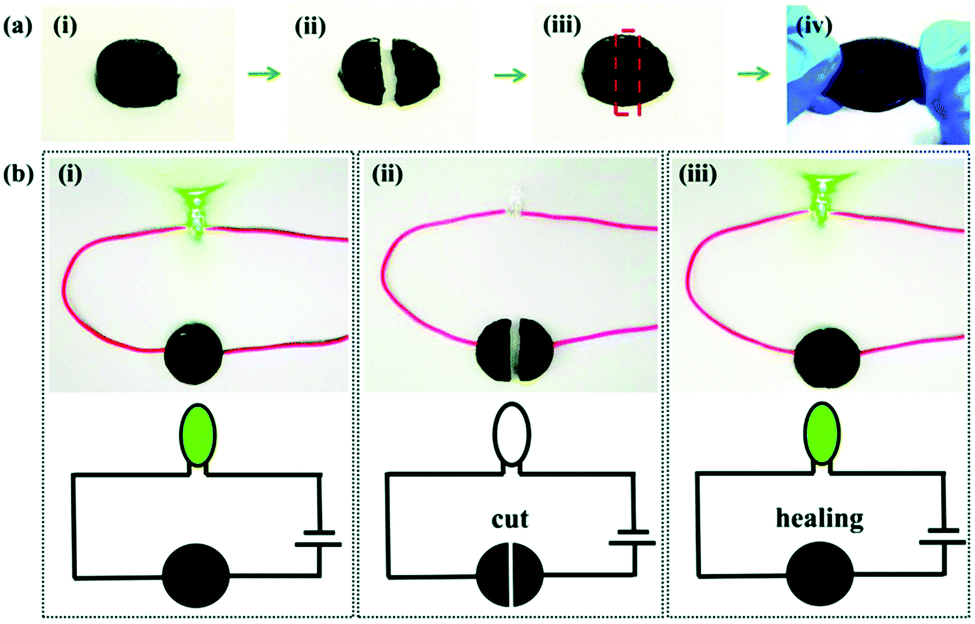 | ||
| Fig. 11 The self-healing ability of organohydrogels: (a) the self-healing process of the mechanical properties for the MXene nanocomposite organohydrogel after cutting. (b) The green LED connected in series with the organohydrogel serves as an indicator of the conductivity in the self-healing process. Reproduced with permission.70 Copyright 2020, Royal Society of Chemistry. | ||
Nowadays, the self-healing ability at low temperatures is very promising, but there are huge challenges to be addressed. The realization of the self-healing ability at sub-zero temperature is limited by the slowing down of molecular chain movement and the retarded reconstruction of interfacial reversible bonds.132,191 Although few antifreeze hydrogels that self-heal through weak hydrogen bonding, host–guest structure, or electrostatic interactions at low temperatures have been reported, their self-healing process is usually time-consuming.132,192 For organohydrogels, their polymer chains still have a good fluidity at sub-zero temperatures, making it possible to self-heal at low temperatures.43,132 Attractively, Borax chemistry is an effective method to achieve remarkable self-healing ability. Due to the incorporation of sufficient, fast-forming, and temperature-insensitive borate bonding sites, it ensures a high interfacial bonding density, and thus achieves a fast self-healing process even at low temperatures.132,193,194 Driven by this, a DN PAA-PVA organohydrogel combined with a high content of borax displayed an outstanding self-healing capability even at an ultralow temperature of −60 °C, and the cut specimens could self-heal within 1 minute of contact due to the cross-linking of borax with PVA and EG solvent.43
3.4. Adhesiveness
The stable contact of an organohydrogel with biological tissues can make it have the potential to be used in portable and wearable electronic devices, which means that good cohesion and adhesion should be displayed in an organohydrogel.195,196 The cohesion of the polymer network is related to its mechanical properties, and the corresponding cohesion strength can be evaluated by measuring the elastic modulus.197,198 In the past, hydrogels needed the help of external adhesives to bond on the substrate, such as bandages, scotch tapes, or 3M adhesives.199,200 They generally possess good stickiness, but they can’t adapt well to uneven surfaces and have poor air permeability, which is not conducive to sticking to the skin for a long time. In particular, when they are applied to the detection of biological signals such as electromyography (EMG) and electrocardiography (ECG), the obvious layered structure will lose the accuracy and durability of detection to a certain extent. Therefore, there is an important requirement for the development of organohydrogels with self-adhesive ability, which requires that the organohydrogels can easily and strongly adhere to various substrates without requiring extra agents, so as to achieve high accuracy of information acquisition.Nowadays, various strategies have been developed to prepare self-adhesive organohydrogels by introducing adhesive components (chitosan, nucleobases, and proteins) or functional groups in the polymer network.84,201,202 These functionalized organohydrogels can form covalent or non-covalent interactions with the surrounding surfaces to achieve durable and repeatable adhesion.15,23,43,55,64,88,132 Generally speaking, many adhesions based on reversible non-covalent interactions can experience multiple adhesion cycles without damaging the material and loss of adhesion strength.203,204 For example, Qin et al. proposed a supramolecular organohydrogel with excellent adhesive properties at various substrate surfaces, which could be attributed to the synergetic interactions of hydrogen bonding, electrostatic interaction, and metal complexation between the organohydrogel and substrates.55 In particular, it can maintain such good adhesiveness even at −70 °C due to its good frost resistance, which is superior to those of current adhesive hydrogels and facilitates its practice applications in actual harsh environments. Also, the adhesiveness at higher temperatures is indispensable in a specific environment, and it is mainly related to the thermal stability of the polymer network. In addition, the adhesion based on covalent interaction is also explored and is generally stronger due to the high energy of chemical bonds, such as Schiff bases205 and disulfide bonds.206 Recently, Niu et al. reported an environmentally compatible gelatin/poly(AA-N-hydrosuccinimide ester) (PAA-NHS ester)-based organohydrogel.207 This organohydrogel demonstrated strong adhesion on the skin even underwater because of the introduction of multiple interactions, including hydrogen bonds, electrostatic interactions, and covalent cross-linking between the NHS ester from the gel and amino groups from human skin (Fig. 12).
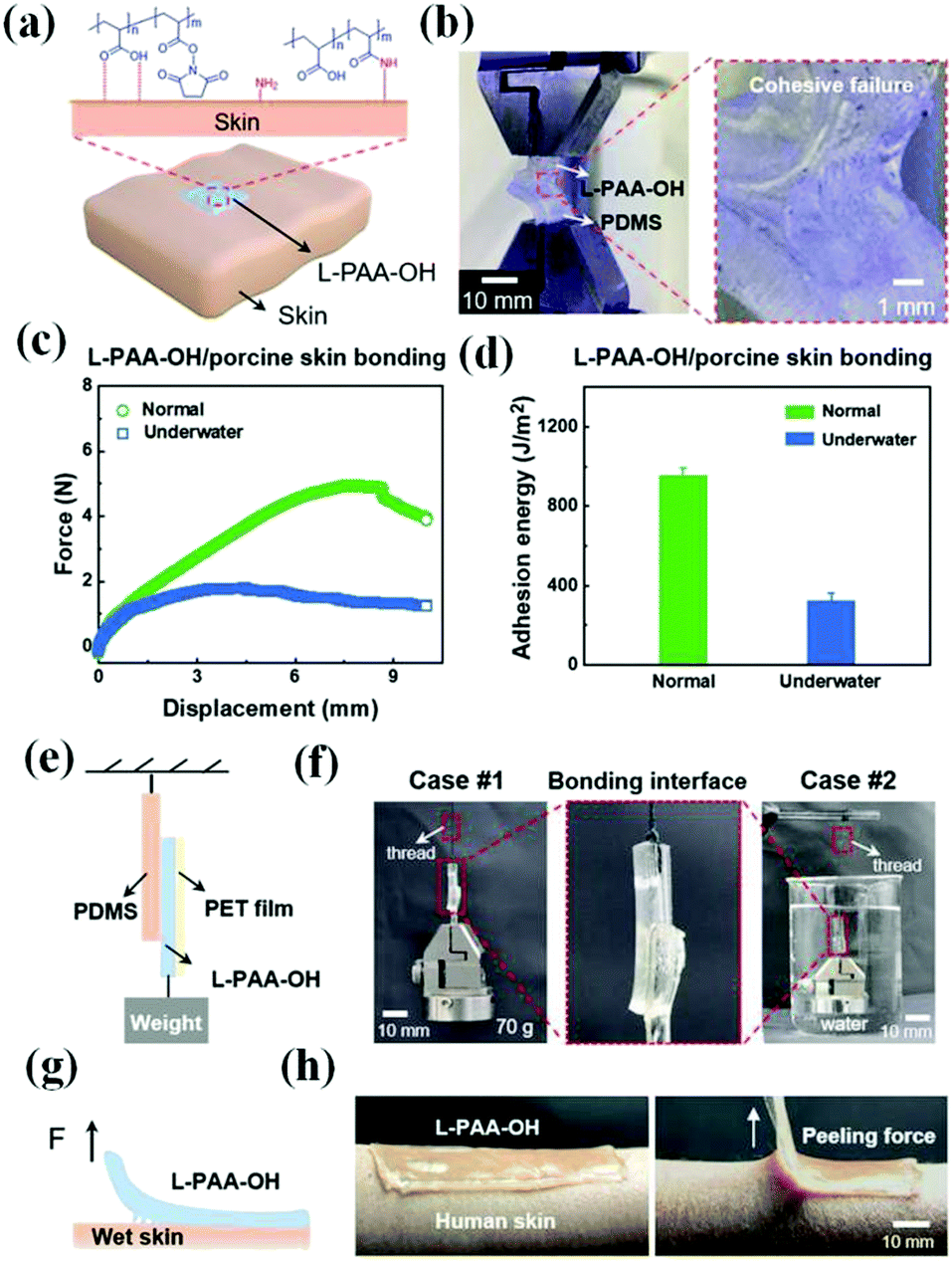 | ||
| Fig. 12 The adhesive performance of organohydrogels: (a) adhesion of an organohydrogel on the skin surface. (b) The T-peeling test shows the presence of strong adhesion between the organohydrogel and PDMS. (c and d) The interfacial adhesion properties between an organohydrogel and porcine skin under normal and underwater conditions. (e) Schematic diagram and (f) photographs of the lap-shear test between an organohydrogel and PDMS. (g) Schematic diagram and (h) photographs of 90° peeling tests between an organohydrogel and a wet human skin. Reproduced with permission.207 Copyright 2021, John Wiley and Sons. | ||
To date, new adhesive materials inspired by nature's biological adhesion properties have been continuously developed. For example, inspired by glutinous rice, Zhou et al. developed a poly(AA-co-AM) organohydrogel by introducing amylopectin (AP) into the polymer network, which showed outstanding adhesion properties in a wide working range (−20 to 80 °C) due to the abundant free hydroxyl groups in AP.208 It is worth noting that underwater adhesion or wet adhesion is the biggest challenge for hydrogel materials. When liquid is present, a hydration layer will be formed on the surface of the adhesive material, which causes the dissolution of the adhesive molecules and prevents them from bonding with other surface groups, resulting in poor adhesion.209,210 Mussels are aquatic organisms with excellent adhesion properties. They secrete byssus composed of viscous proteins so that they can be firmly attached to various surfaces in dry or even in humid environments.84,211,212 The adhesion mechanism of mussels mainly involves the secreted viscous protein rich in dihydroxyphenylalanine (DOPA) residues with catechol groups, which can form chemical bonds and hydrogen bonds with polar surfaces to achieve adhesion. Besides, the catechol group of DOPA tends to be oxidized to reactive quinone moieties, causing cross-linking between proteins and strong cohesion.28,213 Moreover, the oxidized quinone moieties can also form covalent bonds with the diverse nucleophilic groups (e.g., amines, thiols, and imidazoles) on biological tissues to establish a strong adhesion.214,215 Based on this, several mussel-inspired adhesive organohydrogels have been developed by incorporating or modifying additives containing catechol groups in the polymer network. For example, PDA is often incorporated into organohydrogels to enhance self-adhesiveness due to the abundant presence of catechol groups.28,216 Han et al. developed a conductive nanocomposite organohydrogel that exhibited excellent tissue adhesiveness due to the modification of muscle-inspired PDA on CNTs.84 However, DA is difficult to be oxidized to PDA in a binary solvent of organohydrogels, and it requires additional oxidation before gelation, which increases the preparation steps.217
The tunicate mimetic adhesive system can also inspire the preparation of viscous organohydrogels due to the abundant pyrogallol moiety,64,209 which can also form strong interactions with diverse surfaces and provide strong cohesion for polymer networks.218,219 Natural pyrogallol derivatives, such as gallic acid and TA, are cheaper than catecholic compounds and have more abundant reserves.220–222 Compared with the catechol group, the pyrogallol group has a higher binding affinity with the proteins or metal ions,223,224 resulting in better adhesion properties. For example, Wei et al. reported a conductive nanocomposite organohydrogel incorporating TA modified CNTs,23 which exhibited an excellent self-adhesiveness with high adhesive strength applicable to diverse surfaces. Moreover, there are more adhesion mechanisms inspired by nature that can continue to be used in the preparation of organohydrogels with excellent adhesion properties. van der Waals forces and capillary forces are very promising interactions to achieve adhesion, and the gecko is a good example in nature.6,225 Therefore, in addition to the changes in components that need to be considered when designing organohydrogels, microstructure adjustment is also the key to material functionalization.
Taking into account the solvent resistance of heterogeneous organohydrogels, underwater adhesion can also be achieved by introducing hydrophobic polymers without modification of additional surface groups. Liu et al. prepared a solvent-resistant and non-swellable heteronetwork organohydrogel through the copolymerization of MEA and AA in a mixed solvent of H2O/DMSO.45 The organohydrogel exhibited robust adhesion properties in organic and aqueous solutions due to the synergistic hydrophobic and hydrophilic components. Specifically, the hydrophilic segments in the gel can effectively resist the corrosion of organic solvents and achieve underoil adhesion. When the organohydrogel is immersed in water, water molecules can enter the polymer network by displacing the interior DMSO molecules. Meanwhile, the corresponding precipitation and association of the hydrophobic segments can lead to mutual repulsion between water molecules and the organohydrogel and simultaneously increase the cohesion for strong underwater adhesion. Besides, a programmable adaptive adhesion was achieved by introducing thermoelectric components in the heterogeneous organohydrogel to reversibly transform to a more compliant and deformed state to match the rough surface morphology, due to their thermomechanical performance.80 Nevertheless, underwater adhesion and programmable adhesion still need further exploration in the future.
3.5. Others
In addition to the common characteristics mentioned above, organohydrogels need to be developed with more functionalities by modifying some functional molecules to be applied in specific fields, such as antibacterial activity, biocompatibility, degradability, thermoplasticity, transparency, fluorescence, etc.The antibacterial activity appears to be particularly important when the organohydrogel is in contact with biological tissues, because the humid environment of the organohydrogel will inevitably lead to the breeding and growth of a large number of bacteria. Commonly, various antibacterial substances have been introduced into the gel material to obtain antibacterial activity, such as silver nanoparticles,226 antibiotics,227 fullerene,228 and Al3+.119 However, the release of such antibacterial substances will seriously affect the long-term antibacterial properties, and even cause metal-ion toxicities and allergic reactions,229–231 which is adverse to its application in biological tissues. In addition to this, antibacterial activity can also be achieved by changing the surrounding environment of the bacteria, such as temperature232–234 and pH.235 For instance, incorporating photothermal agents in the organohydrogel can cause the denaturation of bacterial proteins by converting light into local heating to impart organohydrogels with an excellent and broad-spectrum photothermal sterilization ability.236,237 Beyond that, positively charged materials are generally considered to possess good antibacterial properties, such as ionic liquids,69 quaternary ammonium salt,238etc., which can be accumulated and adsorbed on the negatively charged bacterial walls through electrostatic force to change the charge of the bacterial cell surface and damage the membrane structure of bacterial cells, thus causing growth inhibition and death of bacteria.239–242 Considering the toxicity of these substances, some non-toxic and inherently antibacterial substances containing numerous positively charged chemically groups, such as chitosan243 and ε-polylysine,53 also show a good long-term antibacterial activity and do not cause additional harm to humans. In particular, biocompatibility is the primary consideration for the application of organohydrogels in vivo, which requires that the materials do not have toxicity or initiate foreign body responses in biological tissues. However, the initiators, crosslinkers, organic solvents, and unreacted monomers frequently used in the preparation of organohydrogels are mostly toxic. Therefore, organohydrogels with excellent biocompatibility need to avoid free radical polymerization and are usually based on natural polymers or FDA-approved polymers.173
There are millions of tons of e-waste accumulated here every year, which is contrary to the concept of sustainable development and causes serious economic and environmental problems.244 In order to alleviate these problems, on the one hand, degradable “green” electronic devices need to be developed. Compared with traditional hydrogels based on chemical cross-linking, many natural material-based hydrogels or physically cross-linked hydrogels (agarose, gelatin, alginate, PVA, etc.) can be entirely degradable without producing any toxic substances.88,245 Similarly, the hydrogel network formed by other weak bonds such as metal-ion coordination or electrostatic interactions is relatively easy to dissociate under certain conditions to achieve degradability.124,246–248 Unfortunately, the introduction of the ability to degradable usually needs to be done at the cost of compromising performance. Degradable hydrogels usually have poor mechanical properties and long-term stability due to weak cross-linking bonds. To address this problem, Baumgartner et al. reported an organohydrogel based on naturally occurring materials that can be completely degraded when treated in wastewater.235 Its mechanical properties can be well maintained for more than one year under ambient conditions by using hydrophobic biodegradable coating strategies based on shellac resins. The degradation of the encapsulation layer is triggered in an alkaline solution, which subsequently leads to the complete degradation of the biogel. This may open the way for the application of degradable gels in water or in vivo. It should be noted that the simple dissolution of polymers cannot reduce environmental problems, and may cause harm through accumulation in filtration systems such as the kidneys.249 On the contrary, a more complete chemical degradation of a single polymer chain is harmless to the human body and the environment.250 On the other hand, reusable electronics can also be developed to reduce e-waste. Generally, organohydrogels with good recyclability or thermoplasticity are composed of reversible non-covalent interactions, which allows them to reform easily into different shapes as required.55,120,121 Unlike degradation, it does not require to destroy the long molecular chains of the polymer. Inevitably, the mechanical properties of the remolded gel network will slightly decrease.
When it comes to optical performance, good transparency can not only realize the visualization of the internal conditions of the wearable electronic devices and the skin during the operating process, but also shows great application potential in military camouflage, optometry products, and display applications.52,251 In the organohydrogel, the incorporation of organic agents can well hinder the phase separation of the polymer or lead to the formation of smaller polymer crystallites,95,121 thereby reducing the interference to light and obtaining higher transparency. Additionally, similar to hydrogels, a fluorescent organohydrogel can also be developed by modifying fluorescent monomers on the polymer network, and it can show a dynamic color or brightness change in response to external stimuli through appropriate molecular design.252,253 For example, Chen et al. developed a heterogeneous organohydrogel with simultaneous fast morphing and fluorescent changes in response to temperature by modifying the fluorescent monomer N-(4-(1,2,2-triphenylvinyl)phenyl)acrylamide (ATPE) and the fluorescent ligand 6-acrylamidopicolinic acid (6APA).252 Specifically, the fluorescence intensity of ATPE on the hydrophobic chains can increase sharply when the temperature increases, showing an aggregation induced emission (AIE) behavior. And 6APA can combine with Eu3+ to form the 6APA–Eu3+ complex that can emit red fluorescence, and its coordination degree also changes with temperature, resulting in a change in fluorescence color.
As demonstrated above, stretchable organohydrogels can be endowed with abundant functionality through unique functional monomers and exquisite structural design, leading to the possibility of their application in various fields. Here we summarize the various properties of some representative organohydrogels reported recently and they are displayed in Table 3. Notably, it is difficult for the existing organohydrogels to achieve a satisfactory balance among the above-mentioned performance. Thus, there are still great challenges in realizing multifunctional organohydrogels. More importantly, in addition to optimizing these properties, some novel functionalities are also expected to be continuously developed in organohydrogels by compounding with other functional materials, such as electromagnetic shielding capabilities, X-ray absorption, photoelectric conversion capabilities, etc. Impressively, considering the coexisting hydrophilic and hydrophobic networks of heterogeneous organohydrogels, they are easier to modify these additives with different hydrophilicities and thus can serve as excellent functional carriers with better manipulation and less restriction.
| Materials | Electro-mechanical properties (maximum strain, tensile strength, conductivity) | Adhesiveness (substrate) | Self-healing (healing efficiency) | Transparency | Antibacterial ability | Biocompatibility/degradability | Thermoplasticity (remoldability) |
|---|---|---|---|---|---|---|---|
| PVA/carbon black (CB)/CNT organohydrogel91 | 643.2%, 4.8 MPa, 105 Ω | — | 64.9% (stress) | — | — | — | Yes |
| 60.1% (strain) | |||||||
| PAM/montmorillonite/CNT organohydrogel66 | 4400%, 170 KPa, 10−5 S cm−1 | — | — | — | — | — | — |
| Gelatin supramolecular organohydrogel55 | 690%, 3 MPa, — | 150 KPa (copper) | 50% (tensile strength) | Yes | — | — | Yes |
| TA@CNF/MXene/PAM organohydrogel23 | 1500%, 156.5 KPa, 2 mS cm−1 | 117 N m−1 (wood) | Yes | — | — | — | — |
| PAM/carrageenan DN organohydrogel87 | 950%, 417 KPa, 15 kΩ | — | Yes | Yes | — | — | — |
| Nanofibrillar PVA organohydrogel52 | 390%, 1.4 MPa, — | — | — | 90% | — | Biocompatibility | — |
| P(MAANa-DMC) organohydrogel15 | 2000%, 56 KPa, 10−2 S cm−1 | 2.5 kg (PTFE) | 96% (conductivity) | — | — | — | — |
| PAAM-Clay organohydrogel256 | 3310%, 0.25 MPa, ∼105 Ω | — | 95% (conductivity) | — | — | — | — |
| PDA-CNTs/PAM/PAA organohydrogel84 | 700%, 70 KPa, 8 S m−1 | 57 KPa (porcine skin) | — | — | — | — | — |
| PVA/SNF/CN organohydrogel119 | 585.8%, 1.39 MPa, — | — | — | — | Yes | — | Yes |
| PVA/CNF organohydrogel95 | 660%, 2.1 MPa, 3.2 S m−1 | — | — | 90% | — | — | — |
| Alginate/PEGDA organohydrogel fibers74 | 400%, 200 KPa, 0.765 S m−1 | — | — | Yes | — | — | — |
| PVA-TA@talc organohydrogel71 | 750%, 550 KPa, — | — | — | UV-filtering | — | — | — |
| PVA-borate-TA-CNTs organohydrogel120 | 180%, 450 kPa, — | — | 98% (strain) | — | — | — | Yes |
| Gelatin-based biogel237 | 254%, 1.86 MPa, — | Yes | Yes | — | Yes | Degradability | — |
| ε-PL-gelatin-PAM organohydrogel53 | 1400%, 300 KPa, — | 181.6 KPa (hogskin) | — | — | Yes | Biocompatibility | — |
| Poly(AA-co-AM)/AP organohydrogel208 | 1089%, 200 KPa, 0.2 S m−1 | 22.18 KPa (copper) | — | 90% | — | — | — |
4. Application for organohydrogels
4.1. Sensors
In the past, sensor research was mainly based on inorganic nanomaterials, such as metal oxide semiconductors,254–256 transition metal dichalcogenides,257–259 and carbon nanomaterials.260–262 Despite achieving excellent sensitivity, their stretchability is limited. In contrast, a conductive organohydrogel also has the ability to convert specific external stimuli (such as strain, pressure, temperature, humidity, gas, etc.) into electrical signals that can be detected, resulting in a significant change in resistance or capacitance in the macroscopic view. This means that organohydrogel can be widely exploited as a multi-functional wearable sensory system in human–machine interface, human health detection, motion detection, and other fields, showing a wide operating temperature range.Strain sensor is a very important type of sensor, which can respond to mechanical deformation and be used for real-time monitoring of various human motions (Fig. 13).95,121 So far, a large number of antifreeze and durable strain sensors based on organohydrogels have been developed. For ion-conductive organohydrogels, the change in electrical signal during stretching is mainly due to the elongated ion transmission pathway.4 In addition, the density of the polymer chain also has an impact on the transport of ions. When a moderate deformation is applied, the polymer chain will unfold accordingly to weaken the hindering effect of the polymer network on ion transportation. However, when excessive stress is applied, the partial rupture of the polymer network will hinder the transport of ions, resulting in a decrease in electrical resistance. For electron-conductive nanofiller incorporated organohydrogels, electrons are transported by forming an interconnected three-dimensional conductive pathway in the polymer network. During the stretching process, the variation of the electrical signal is closely related to the rearrangement of the conductive network.66 Under small tensile strains, the distance between nanofillers will gradually increase, increasing the electrical resistance. As the strain gradually increases, part of the conductive components will gradually separate and obviously damage the conductive channels, leading to a sharp increase in resistance. For organohydrogels, in which both ions and electrons participate in conduction, the change in resistance is related to the changes of both the ion transporting pathway and the electronic conduction channel during stretching. It is worth noting that the degree of resistance change caused by stretching under different strain ranges is different due to the domination of different response mechanisms. Thus, the response of organohydrogels versus strain relationship generally shows two or three linear regions.
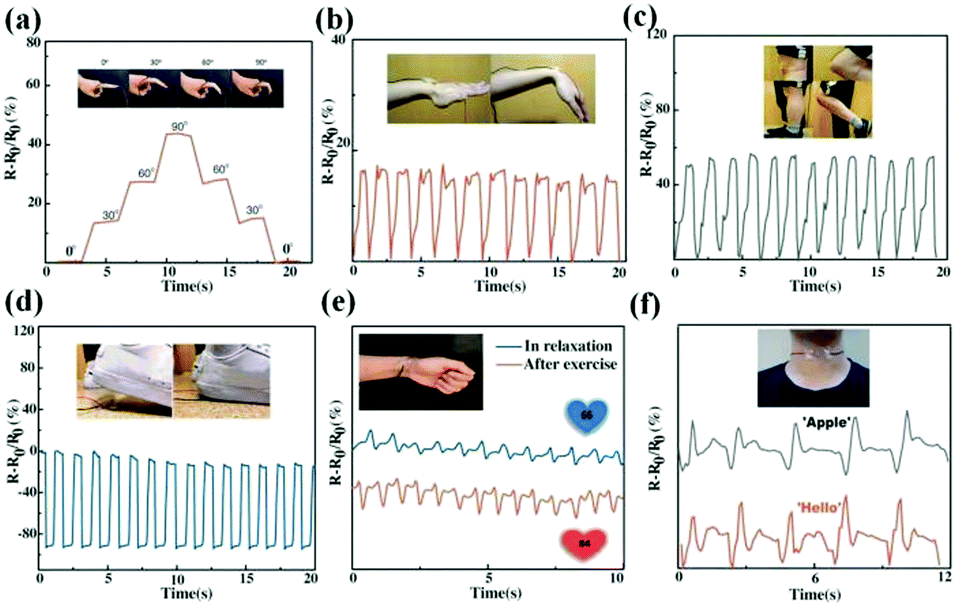 | ||
| Fig. 13 Strain sensors for real-time monitoring of various human motions: recognition of different bending states of (a) fingers, (b) elbows and (c) knees; (d) walking monitoring; (e) pulse detection; and (f) voice recognition. Reproduced with permission.95 Copyright 2020, John Wiley and Sons. | ||
Nowadays, strain sensors based on conductive organohydrogels have two main challenges, including the realization of a wide linear strain range and high sensitivity. The detection of a small strain endows the sensor the ability to detect subtle human motions, such as pulse, micro-expression, vocalization, etc.263 Generally, the polymer network incorporating nanofillers has better sensitivity under a small strain, because the contact of rigid nanofillers will be quickly destroyed under tension.264 Equally important, the detection of moderate and high strains is also of great significance in practical applications, and a wide linear strain range is usually achieved by improving the mechanical properties of the gel. Recently, Sun et al. reported a conductive nanocomposite organohydrogel as a wearable strain sensor with good environmental tolerance,66 which exhibits satisfactory sensitivity and an ultrawide strain sensing range (0–4196%) due to the remarkable mechanical and electrical properties (Fig. 14a and b). The sensitivity of an organohydrogel to strain is evaluated by the gauge factor (GF), which is calculated from the slope of the relative resistance change ((R − R0)/R0) versus the applied strain curve.23,95
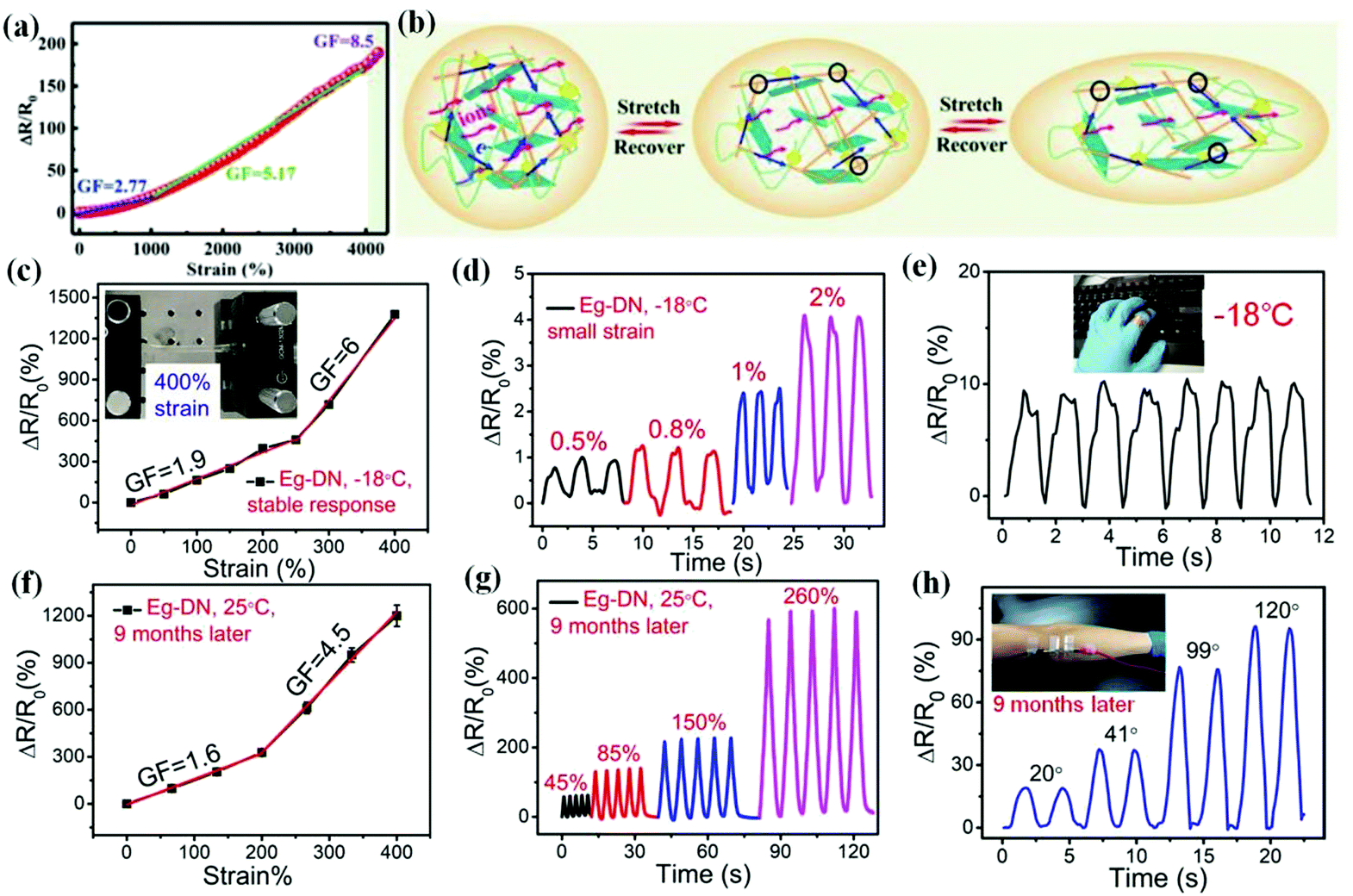 | ||
| Fig. 14 The strain sensing performance of an organohydrogel-based sensor: (a) the relationship between the ΔR/R0 value and the applied strain and (b) the schematic diagram of the sensing mechanism for a conductive nanocomposite organohydrogel-based strain sensor. Reproduced with permission.66 Copyright 2021, John Wiley and Sons. (c) Response–strain curve of the PAM/carrageenan/KCl organohydrogel-based strain sensor at –18 °C. (d and e) The dynamic response curve of the sensor to different small strains (0.5–2%) or finger tapping on the keyboard at −18 °C. (f) Response–strain curve of the PAM/carrageenan/KCl organohydrogel-based strain sensor after placing in ambient air for 9 months. (g and h) The dynamic response curve of the sensor to different strains (45–260%) or elbow joint bending after exposure to ambient air for 9 months, showing an excellent environmental adaptability and durability. Reproduced with permission.87 Copyright 2019, American Chemical Society. | ||
Generally speaking, higher GF values are obtained by increasing the original conductivity of organohydrogels, which results in more excellent signal acquisition capabilities. Based on this, various salts and conductive nanofillers should be introduced into the gel to construct conductive channels that are more easily altered. Representatively, Wu et al. used PAM/carrageenan/KCl organohydrogels to construct an ultrastretchable strain sensor with a GF of 6; various human motions can be detected even at −18 °C or after exposure to ambient air for as long as nine months (Fig. 14c–h).87 Wei et al. developed a nanocomposite organohydrogel based strain sensor with a GF of 8.5 by introducing a conductive network framework formed by interconnecting MXene nanosheets,23 while most of the GF values of hydrogel-based strain sensors reported in the past are only around 2.265,266 Attractively, Ge et al. reported a muscle-inspired organohydrogel-based strain sensor with a high GF of 18.28 by mimicking the fiber-reinforced laminated nanocomposites and “sense-transfer-process” pattern,132 and this superior electromechanical response is attributed to a disconnection mechanism and tunneling effect in sensors. Moreover, Han et al. prepared an ultra-sensitive strain sensor (GF = 343) with anti-freezing and anti-drying ability by introducing tightly cross-linked CNT films on the surface of organohydrogels; this strategy had great advantages of combining high sensitivity and a relatively wide detection range.267 Inspired by this, the replacement of CNTs with other conductive one-dimensional nanomaterials such as Ag nanowires can also lead to an ultra-high response.
Similarly, the compression of organohydrogel will also cause changes in electrical signals, which means that it can also be used in practice as a flexible pressure sensor. Distinct from the traditional electron-conductive metals and inorganic semiconductors, a pure ion-conductive organohydrogel exhibits a smaller change in resistance under compression due to the limited changes in the ion transport pathway.157,268,269 Hence, flexible pressure sensors are usually constructed using electron-conductive nanofiller incorporated organohydrogels to realize higher pressure sensitivity. For instance, Zheng et al. developed a conductive organohydrogel composed of thioctic acid, borate, CNTs, and PVA, which presented a high conductive sensitivity (0.625 KPa−1 for 0–6 KPa) even at −40 °C.120 To further improve the pressure sensitivity, piezoelectric materials such as polyvinylidene fluoride (PVDF) and BaTiO3 nanoparticles,270 can be incorporated into organohydrogels to obtain additional electrical responses derived from the piezoelectric effect.271,272 Structurally, the pressure response of the organohydrogel with a nanofiber stacking structure is significantly improved compared to that of the conventional thin-film structure due to stress concentration.273,274 The broad pressure detection range is also another important performance indicator that the flexible pressure sensor needs to continuously strive to achieve. The thought is to build an elastic gel network to improve the deformation-tolerant performance of the material so as to achieve a wide pressure range.35 Moderate compressive strength and modulus are required to make the material neither too brittle nor too soft.
Furthermore, there are some other performance indicators in strain or pressure sensors that need to be optimized, including response/recovery speeds, reversibility, stability, and durability. On the one hand, the fast response speed is conducive to real-time and accurate sensing. However, most of the existing strain sensors cannot meet this detection requirement due to the hysteresis, and are unsuitable for continuous monitoring of high-speed and high-frequency motion. To address this problem, Song et al. prepared organohydrogel fibers with excellent elasticity resulting from the predominantly covalently crosslinked network, which exhibits negligible hysteresis during the tensile test and can accurately capture high-frequency (4 Hz) and high-speed (24 cm s−1) motion of a simulated engine.74 On the other hand, the reversible, stable, and long-lasting response of the sensor to strain/pressure is also the key to considering whether it can be used in actual production. In general, thanks to the organic solvent contained in the organohydrogel, the organohydrogel-based strain/pressure sensors can exhibit good durability and operate under various harsh conditions.64,73,91,119
Temperature sensor is another essential wearable device that can be used for real-time monitoring of human physiological signals.275,276 In general, for ion-conductive gels, the variation of temperature will change ion mobility and correspondingly cause changes in resistance. Additionally, ion concentration in the migration pathway will increase with temperature due to the dissociation of the ion pair bonded on the polymer chain, resulting in an increase in conductivity.277 Moreover, the thermal movement of the polymer chain accelerates as the temperature increases, resulting in a weakened polymer's hindering effect on ion transport and can also significantly reduce electrical resistance. Based on these mechanisms, Wu et al. first reported a stretchable temperature sensor based on PAM/carrageenan DN hydrogel in 2018, which displayed a sensitivity as high as 2.6%/°C at an extreme tensile strain of 200%.124 Then, they found that the dehydration treatment of the hydrogel can further improve its thermal sensing ability, that is, the increased temperature can lead to a greater resistance change due to the better blocking and trapping of ions caused by the dehydrated high-density polymer network at room temperature.277 Meanwhile, the organohydrogel thus evolved can also achieve ultra-sensitive temperature detection (19.6% °C−1) and a wide detection range (0–102 °C), which is attributed to the higher initial resistance, smaller heat capacity, and stronger hydrogen bonds formed between organic molecules and water molecules in the organohydrogel. Among various flexible materials, organohydrogels neither freeze at sub-zero temperatures like hydrogels nor dissolve at higher temperatures like organogels, which makes them a good material for manufacturing temperature sensors. In consideration of these, various flexible temperature sensors based on organohydrogels have been developed,91,132 but their thermal sensitivity still needs further breakthroughs.
Humidity and gas sensors also have important practical significance, but they have rarely been reported in flexible devices made of intrinsically stretchable materials. Humidity and gas have a standard concentration in the production and living environment; beyond this range, human health will be threatened. In addition, they are also important biomarkers in human exhaled breath and are closely related to human health.261 Therefore, the development of stretchable organohydrogel-based wearable humidity or gas sensors is of great significance to medicine. And the real-time monitoring of humidity and gas can not only prevent but also diagnose diseases. The response of organohydrogels to humidity is mainly related to changes in the concentration and mobility of charge carriers caused by the physical or chemical adsorption of water molecules in the gel network. Wu et al. fabricated highly sensitive humidity sensors by using intrinsically ultrastretchable k-carrageenan/PAM organohydrogels.63 The high response of this organohydrogel to humidity is attributed to the formation of hydrogen bonds between water molecules and a large number of hydrophilic groups in the gel network, including –OH and SO3− on k-carrageenan, –NH2 on PAM, and abundant –OH on organic molecules (Fig. 15). Impressively, the incorporation of organic agents not only boosts the sensitivity by increasing the adsorption sites, but also improves the long-term stability and detection range due to the enhanced moisture-holding ability of the organohydrogel, demonstrating the advantages of organohydrogels in fabricating stretchable humidity sensors. Subsequently, Wu et al. showed that this organohydrogel can also detect NH3 and NO2 due to the effective adsorption of gas molecules on the abundant functional groups of the gel network.278 This organohydrogel gas sensor exhibited high sensitivity (8.4 and 1.4 ppm−1 for NO2 and NH3, respectively) and ultralow theoretical limit of detection (91.6 and 3.5 ppb for NH3 and NO2, respectively) at room temperature without thermal or light activation, which has obvious advantages compared with other types of traditional gas sensors (such as graphene, reduced graphene oxide (rGO), MXene, metal oxide, etc.), resulting in low energy consumption and better safety. The in-depth gas sensing mechanism of ion-conducting hydrogel/organohydrogel may also relate to the redox reactions that occurred at the gel–electrode interface,279,280, which requires further investigation. Considering that humidity and gas sensing are mainly related to surface reactions, the development of some micro/nanostructures with a higher specific surface area (such as a three-dimensional structure) is an important strategy for improving the sensitivity of organohydrogels to gas or humidity, which can be investigated in the near future.
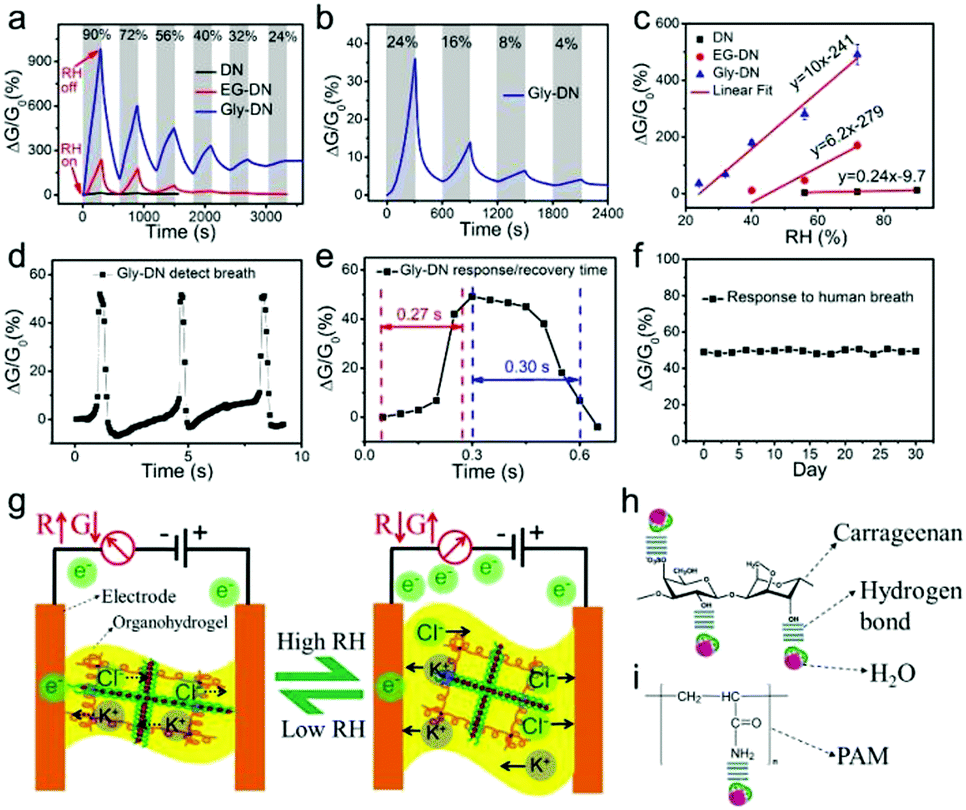 | ||
| Fig. 15 The humidity sensing performance and mechanism of an organohydrogel. (a) Dynamic response curves of the DN hydrogel and EG/Gly-DN organohydrogels to different humidity ranging from 90% to 24% RH. (b) Dynamic response curves of the Gly-DN organohydrogel to lower humidity ranging from 24% to 4% RH. (c) Relationship between the humidity and responses of these sensors. (d) Repeatability test, (e) response–recovery curve and (f) long-term stability of the Gly-DN organohydrogel-based sensor to human respiration. (g–i) Schematic diagram of the sensing mechanism of an organohydrogel to humidity, the response is mainly related to the formation of hydrogen bonds between water molecules and the hydrophilic groups on carrageenan and PAM, promoting moisture adsorption. Reproduced with permission.63 Copyright 2019, Royal Society of Chemistry. | ||
4.2. Energy storage devices
Electrochemical energy storage devices have attracted much attention due to the energy crisis and the increasing demand for sustainable energy, mainly including supercapacitors and metal-ion batteries.5,281 The traditional structure of energy storage devices mainly includes electrodes, electrolytes, and current collectors. Metal-ion batteries realize energy storage and release through the intercalation and de-intercalation of metal ions between the positive and negative electrodes. Reversible electrochemical reactions occur on the surface and inside of the electrodes during the charge and discharge process,282,283 while supercapacitors mainly rely on the electrostatic adsorption and desorption of electrolyte ions on the electrode surface or the occurrence of reversible redox reactions to achieve charge storage and release.284,285 Nowadays, the development of flexible energy storage devices is imperative, which is conducive to reducing their mechanical damage, increasing their service life and serving as a power supply unit for wearable devices. Its realization requires the cooperation of various components. However, traditional electrode materials such as activated carbon and transition metal composites286 are rigid powders and lack flexibility. New electrode materials, including CNTs, carbon fibers, graphene, and conductive polymers,287–289 have both considerable electrochemical properties and varying degrees of flexibility, making it possible to realize flexible electrodes.The traditional liquid electrolyte is volatile, flammable, and easy to leak under mechanical external force, resulting in poor safety and unsatisfactory long-term stability of the device. Therefore, more attention has recently been paid to the development of solid-state polymer electrolytes. Generally, the solid-state polymer electrolyte needs to have sufficient ionic conductivity and good mechanical properties to achieve flexible energy storage devices with better electrochemical performance. In some instances, good frost resistance, thermal stability, and flame retardancy should be achieved in the solid-state polymer electrolyte to enable it to work in harsh environments. It is obvious that simple hydrogels cannot meet these requirements due to their own limitations, and the addition of ionic liquids to the hydrogels will correspondingly cause higher costs and toxicity.44,84,85,290–295 Satisfactorily, organohydrogels show great advantages to prepare all-temperature flexible electrolytes due to their excellent anti-freezing and moisturizing ability. Nevertheless, the lower ionic conductivity of the organohydrogel electrolytes compared to that of the liquid electrolytes will lead to high equivalent series resistance (ESR) and subsequent low power output, which is not conducive to their practical applications. Considering this, the ionic conductivity of the organohydrogel electrolyte is generally improved by adding some metal salt.
Among various polymers, PVA has good mechanical properties and hydrophilicity; it is currently the most common polymer for preparing organohydrogel electrolytes due to its high adsorption capacity of electrolytes and low commercial price.296–299 For example, Rong et al. reported a low temperature tolerant PVA-based organohydrogel electrolyte by using the water/EG binary solvent.42 The high ionic conductivity (2.38 mS cm−1 at −40 °C) of the organohydrogel electrolyte was achieved by adding LiCl as the electrolyte salt. Additionally, Lu and Chen constructed a flexible supercapacitor with high energy density and long cycling life based on an anti-freezing and thermally stable montmorillonite (MMT)/PVA organohydrogel electrolyte.300 This device can not only deliver a stable energy supply under bending, twisting, and stretching states, but also exhibit good mechanical and electrochemical properties under a wide temperature range from −50 to 90 °C (Fig. 16). Specifically, lamellar structural MMT materials are not only employed as flame retardants due to the strong hydrogen bond formation with PVA chains, but also increase the ionic conductivity by providing oriented channels in the PVA matrix for more efficient ion migration. Considering the relatively weak hydrogen bond interaction in PVA gel, Liu et al. demonstrated a thermally stable and anti-freezing organohydrogel synthesized by the copolymerization of AM and 2-acrylamido-2-methylpropane sulfonic acid (AMPS) in a water/DMSO binary solvent system containing LiCl conductive additives,301 which is a promising candidate to be used as an electrolyte for flexible supercapacitors with high electrochemical performance and wide temperature adaptability. Meanwhile, the resulting supercapacitors also exhibited excellent flexibility and mechanically bending stability. Likewise, benefiting from the excellent mechanical properties and exceptional freeze resistance of the organohydrogel electrolyte, a freeze-resistant flexible zinc manganese-dioxide battery constructed by sandwiching an EG based waterborne anionic polyurethane acrylate (EG-waPUA)/PAM organohydrogel electrolyte containing 2 mol L−1 ZnSO4 and 0.1 mol L−1 MnSO4 between the fabricated flexible zinc anode and α-MnO2/CNT cathode was also developed.118 As a result, this solid-state battery still exhibits good electrochemical performance at sub-zero temperatures or under various deformations, owing to the outstanding flexibility and conductive stability.
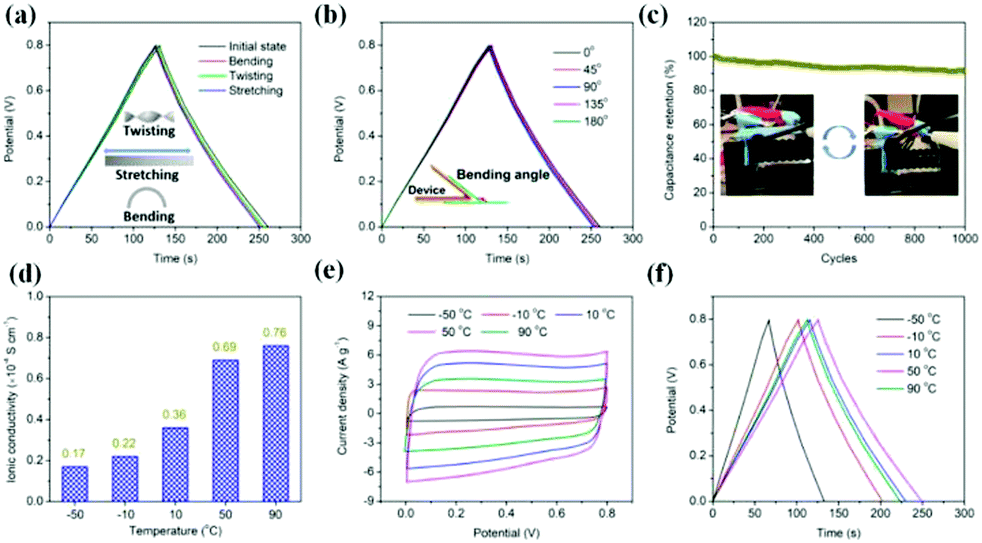 | ||
| Fig. 16 Stability of flexible supercapacitors based on an organohydrogel electrolyte. Galvanostatic charge–discharge (GCD) curves of the device (a) under different flexible conditions and (b) under different bending angles. (c) Bending tests of flexible supercapacitors. (d) Ionic conductivity, (e) cyclic voltammogram (CV) curves and (f) GCD curves of the device under different operation temperatures. Reproduced with permission.300 Copyright 2020, American Chemical Society. | ||
Furthermore, the undesirable electrochemical performance of the device can also be attributed to insufficient interface contact between the electrode material and the gel electrolyte, and these interfaces may partially detach after long-term cycling or at low temperatures due to their different thermal expansion coefficients. To address this problem, Wang et al. designed an all-in-one supercapacitor by infusing the PVA-KOH organohydrogel electrolyte into the 3D multiwall carbon nanotube (MWCNT) network as electrodes,302 which exhibited superior capacitive behavior at sub-zero temperatures and excellent cycling performance due to the stable electrode–electrolyte interfaces. Also, it showed a good mechanical performance due to the synergistic effect of flexible electrolytes and MWCNTs. Hou et al. reported a flexible solid supercapacitor by combining the rGO electrode with the PVA-based organohydrogel electrolyte without the usage of binders;148 the excellent flexibility and good electrochemical performance of the organohydrogel electrolyte even at low temperature enable this device to be used in harsh environments. The good contact between the electrode and the organohydrogel electrolyte stems from the excellent self-adhesiveness of the organohydrogel, ensuring lower interfacial resistance and better electrochemical stability of this supercapacitor. Additionally, the flexibility of the rGO electrode is greatly increased by heating the GO membranes to form a honeycomb-like porous microstructure, thereby achieving the overall flexibility of the device. Although these flexible energy storage devices have been successfully implemented, their electrochemical performance is still unsatisfactory when compared with those of the traditional ones, and needs to be improved in the future.
4.3. Nanogenerator
The self-powered ability is a promising strategy to solve the current problem of continuous energy supply of wearable and implantable electronic devices, which shows great practical significance. Considering the sensing and energy storage capabilities of organohydrogels, a self-powered sensing system integrating strain sensors and supercapacitors was proposed by Huang et al.303,304 In these cases, the strain sensor was powered by the supercapacitor, and the sensing materials and electrolytes in the system were composed of the same ionic organohydrogel. Although the output voltage of the supercapacitor and the sensor was gradually decreasing due to the self-discharge behavior, the resistance change caused by the applied strain remained unchanged, showing a stable sensor output. More importantly, thanks to the excellent mechanical flexibility and wide environmental tolerance of the organohydrogel, this integrated system can be directly attached to the human body to detect human movement without external power supply under various environments. However, the supercapacitor still requires an additional charging process, which is not conducive to the sustainable use of the device. In comparison, nanogenerators can realize continuous energy supply by converting mechanical, light, heat, and chemical energy into electrical energy, which is currently the main research focus for constructing self-powered devices.305Nowadays, flexible triboelectric nanogenerator (TENG) based on the occurrence of triboelectricity between various materials with different electron affinity and electrostatic induction (Fig. 17a) is considered to be a promising power supply for wearable electronics through the harvest of biomechanical energy.306 Hydrogels have been widely used in wearable TENG due to their ultrahigh stretchability, excellent conductivity, good self-healing capability, and adhesion abilities that can be imparted, which are conducive to their direct attachment to organisms and prevent various mechanical impacts during operation, but they cannot work stably as the environment changes.295,307 In this context, TENG constructed using organohydrogel as the electrode layer shows an excellent environmental tolerance and stability and has significant advantages in practical applications. For example, Sun et al. prepared a sustainable TENG using a conductive nanocomposite organohydrogel as the electrode material, which could effectively collect mechanical energy from human activities within a wide temperature range (−60 to 60 °C), with a maximum output power density of 41.2 mW m−2.66 The resulting TENG could be connected with various capacitors to build a self-charging power system and directly power different portable electronics. Huang et al. reported a sandwich structured TENG constructed by two IU-PDMS membranes and a single PAM-clay organohydrogel layer, which exhibited a maximum output power density of 710 mW m−2.308 Compared with the hydrogel-based TENG, the output performance of this obtained TENG was very stable in a wide temperature range (−30 to 80 °C), thanks to the ultra-fast self-healing ability and excellent environment tolerance of the organohydrogel. Since the open-circuit voltage (Voc) of TENG is related to the contact area between tribomaterials, this TENG can be exploited as a self-powered sensor to detect different human motion modes. Notably, the output performance of TENG is positively related to the ionic conductivity in organohydrogel. To this end, Wu et al. developed a single-electrode TENG based on DN ionic conductive organohydrogels, which can maintain a stable output in a broad temperature range from −50 to 100 °C and after four months of storage, showing excellent environmental stability.309 These organohydrogels can maintain a stable charge channel after solvent replacement because more ions can be bonded to the polymer chains of the DN through dipole interactions and stronger sodium bonds can be formed through electrostatic interaction, which subsequently results in a higher conductivity and output power density (1.02–1.81 W m−2).
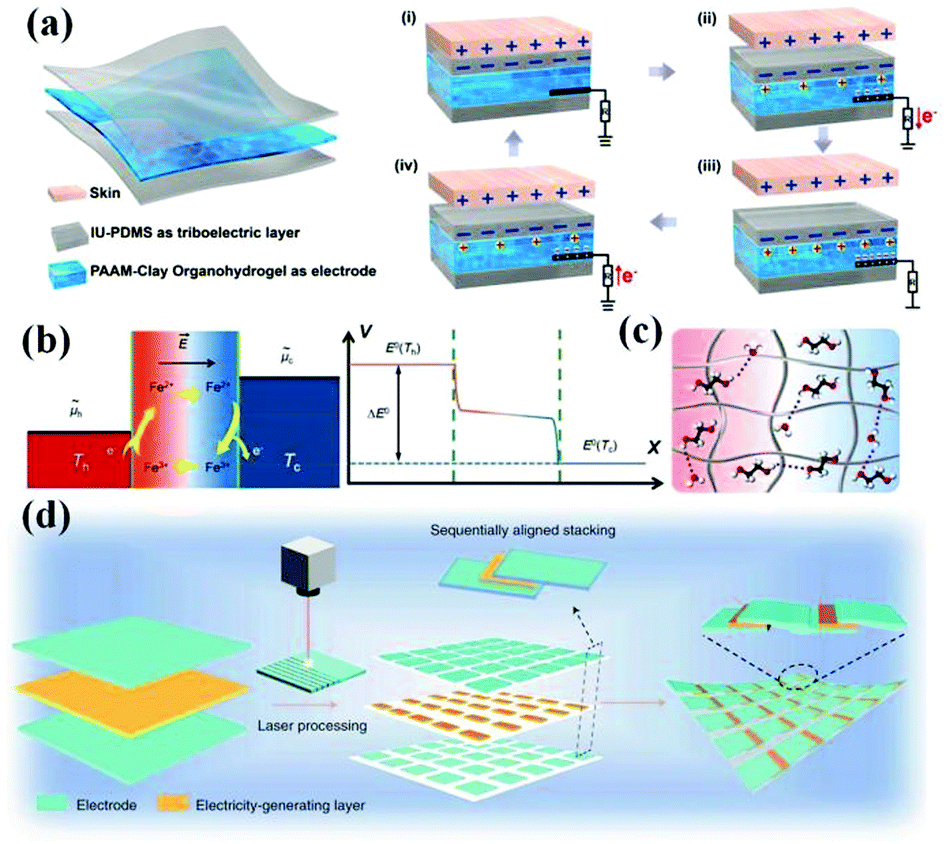 | ||
| Fig. 17 The mechanism of an organohydrogel-based nanogenerator and the corresponding scalable integration method. (a) Schematic diagram of the structure and electricity generation mechanism of the organohydrogel-based TENG. Reproduced with permission.308 Copyright 2021, Elsevier. (b) Schematic diagram of the thermoelectric effect of the organohydrogel with the addition of Fe3+/2+ redox ions. (c) Structure diagram of organic hydrogel under temperature gradient. Reproduced with permission.310 Copyright 2021, John Wiley and Sons. (d) Schematic diagram of a sequentially aligned stacking method that integrates nanogenerator units in series. Reproduced with permission.311 Copyright 2021, Springer Nature. | ||
Beyond that, energy harvesting technology that converts the abundant green energy in nature into electricity is equally important in the sustainable energy supply. For example, thermoelectric materials based on the Seebeck effect can realize the conversion of heat to electricity so as to maintain the continuous operation of electronic devices.312 Taking into account the poor mechanical flexibility of traditional inorganic thermoelectric materials, organohydrogels added with thermogalvanic ions have greater advantages in the preparation of environmentally tolerant flexible thermoelectric generators. Gao et al. designed a stretchable organohydrogel thermocell, which can still maintain relatively good thermoelectric performance even at sub-zero temperatures and has the potential to be used in polar and space exploration.310 In this case, a relatively high power density (0.1 mW m−2 K−2) was achieved by adding the redox couple of Fe3+/2+ as thermogalvanic ions and methyl chloride quaternized N,N-dimethylamino ethylacrylate (DMAEA-Q) as a chaotropic comonomer. As shown in Fig. 17b and c, the reduction of Fe3+ at the high-temperature electrode and the oxidation of Fe2+ at the low-temperature electrode could cause a difference in the electrode potential on both sides of the organohydrogel, resulting in a voltage output. Subsequently, the consumption of iron ions at one electrode can lead to the diffusion and replenishment of the same ions on the other side, realizing the continuous power supply of this system. DMAEA-Q can promote the increase of the entropy difference of redox ions by combining with Fe3+ and thus enhance the thermoelectric effect. Furthermore, the moist-electric generator also has huge application potential as an energy supply. He et al. utilized organohydrogels to realize the conversion of ambient moisture to electricity, with an output voltage of 80 mV under constant moisture flow.16 In detail, the electrical output is due to the release of protons produced by the dissociation of the hydroxyl groups on the side close to the humid air and the movement of these protons to the other side driven by the concentration gradient. Equally important, given the small output voltage of these generators, their scalable integration is essential for the realization of power supply for electronic devices in practice. For instance, a sequentially aligned stacking method was developed to integrate the moist-electric generator unit on various flexible substrates by laser processing (Fig. 17d).311 The integrated device formed by 1600 units in series could achieve a high output voltage of 1000 V, demonstrating the feasibility of nanogenerators in practice.
4.4. Biomedicine
An organohydrogel is an ideal material for wearable dressing or wound dressing due to its flexibility and adaptability. Considering the advantages of good adhesiveness and anti-freezing ability, an organohydrogel can serve as an excellent wearable dressing to protect skin from frostbite.84 Meanwhile, imparting ultraviolet (UV)-blocking capabilities to organohydrogel dressings can protect human skin from exposure to UV radiation that may induce skin cancer,313 which is also important in other wearable electronic devices. Therefore, a PVA-TA@talc organohydrogel with remarkable UV-filtering capabilities was prepared, owing to the UV reflection of talc and the UV absorption of TA in the organohydrogel.71 Sun et al. also prepared a PVA-based organohydrogel with UV blocking capabilities by adding TA, demonstrating the effectiveness of TA in absorbing and shielding UV light.73Wound dressings are widely used in clinics. At present, the traditional wound dressings commonly used in the market are usually gauze, bandages, or sponges, and they usually only have the function of forming a physical barrier and absorbing tissue exudate. However, in addition to these basic functions, an ideal wound dressing also needs to have excellent antibacterial ability to prevent infection or inflammation and maintain high humidity at the wound surface to facilitate the regeneration of wound granulation tissue and epithelial cells and subsequently promote wound healing.314–316 In addition, wound dressing should exhibit good adaptability to biological tissues and self-adhesiveness so that it can meet the healing requirements of different wounds and cannot cause secondary damage to the wound during the removal process. To date, various wound dressings based on hydrogels cross-linked by different biocompatible polymers (such as PVA,317,318 PAM,85,319 and chitosan320,321) have been studied. However, the hydrogel is easy to dry in the air, leading to the loss of various properties and even damaging the wound. Significantly, organohydrogels can mimic biological tissues well due to their high softness, elasticity, and water content similar to that of tissues, and the specific functional design can impart them with high self-adhesiveness, antibacterial properties, and admirable biocompatibility. More importantly, the unique moisturizing ability and frost resistance of organohydrogel can extend the service life of the corresponding wound dressing and broaden its application environment, making it more suitable as a wound dressing material. Note that most organic cryoprotectants are harmful to organisms, Gly has good biocompatibility and cannot cause secondary damage to wounds. Driven by this, Liu et al. utilized a water/Gly binary solvent to fabricate organohydrogels containing PAM, gelatin, and non-biotoxic antibacterial ε-PL chains.53 This organohydrogel exhibited excellent mechanical properties, high adhesiveness, and good antibacterial properties in a wide temperature range (−20 to 60 °C), which could efficiently promote the healing of diabetic foot ulcers (DFUs) by inducing cell proliferation, accelerating collagen deposition, promoting angiogenesis, and inhibiting bacterial breed as a wound dressing (Fig. 18).
 | ||
| Fig. 18 An organohydrogel for wound dressing. (a) Cell proliferation of L929 cells in W-PAGL (hydrogel) and G-PAGL (organohydrogel) for different days. The therapeutic effects of the phosphate-buffered saline (PBS, control), PAM-GT hydrogel, and G-PAGL (organohydrogel) on infectious DFUs: (b) representative photographs and (c) quantification of the wound residual area at different time points. (d) Characterization of granulation tissue formation at the wound site by hematoxylin and eosin (H&E) staining. (e) Characterization of the newly formed collagen deposition in the regenerated skin tissue by Masson's trichrome staining and (f) the corresponding quantification of collagen deposition percentage via measuring the intensity of the blue-stained areas. (g) Characterization of neovascularization in granulation tissue via CD31 immunohistochemical staining and (f) the corresponding quantification of blood vessels. The wound healing process treated with an organohydrogel produces more granulation tissue, collagen deposition and new blood vessels, showing better wound healing effectiveness. Reproduced with permission.53 Copyright 2021, American Chemical Society. | ||
Based on the above discussion, organohydrogels can be well used to construct various flexible devices by imparting corresponding functionality. However, when considering the application of these devices in practice, the performance and stability of the devices are the most important, which need to be continuously optimized in subsequent research studies. Simultaneously, the application of organohydrogels in some new fields should be explored.
5. Conclusions and outlook
In this review, we summarized recent developments of organohydrogels from various aspects, including preparation strategies, performance optimization, and applications. Owing to their unique anti-freezing and water retention ability, solvent resistance, adjustable surface wettability, and shape memory effect, organohydrogels are ideal candidates for the preparation of flexible devices that can operate under various harsh conditions or be used in intelligent systems. In addition, organohydrogels can be easily endowed with various functionalities due to the reconfigurable polymer network structure, which is conducive to more flexible and stable applications. Considering these excellent electro-mechanical properties and various functionalities in organohydrogels, they have been widely applied in sensors, energy storage devices, nanogenerators, and biomedical fields.Although great progress has been made in the development of organohydrogels and related devices, a huge gap still exists compared with the research history of inorganic materials such as metals or semiconductors. Many challenges still need to be addressed in the future research of organohydrogels:
First of all, the current diversity and synthesis techniques of organohydrogels are still insufficient, mainly limited by the solubility of organic substances and the elusive polymerization conditions; more simple and unrestricted preparation methods for different organohydrogels need to be developed. In particular, organohydrogels with different polymer networks urgently need to be developed so as to achieve some fascinating intrinsic properties. For example, fluorine-rich polymers can exist stably in strong acids and alkalis due to the high electronegativity of fluorine, so they can be used for developing organohydrogels with wide pH adaptability. Also, these polymers can impart good self-healing ability to the resulting organohydrogels.
Secondly, current flexible devices are mainly constructed based on bulk materials, which limits their large-scale integration. In particular, the preparation of micro-/nano-sized or microstructured organohydrogels also plays an important role in the optimization of their properties. Furthermore, the miniaturization and integration of organohydrogels are quite necessary for their development in flexible integrated electronics, but they are currently rarely involved due to their poor controllability. Recently, 3D printing technology has made good progress in patterning and miniaturization of hydrogels, and it is conceivable that it will also be dedicated to the development of miniaturized organohydrogels.
Thirdly, although good freezing resistance and moisturizing ability are realized in organohydrogels, their environmental tolerance still needs to be further optimized. At sub-zero temperatures, the crystallization of water molecules in the organohydrogel is well prevented, but the freezing of water molecules adsorbed on the surface still occurs. Therefore, surface hydrophobic treatment becomes indispensable in the preparation process of an organohydrogel. In addition, dehydration still inevitably occurs especially after size reduction due to the exposure of the more specific surface area.
Fourthly, in addition to maintaining structural integrity, it is equally important and more challenging to achieve functional stability of organohydrogels in various environments, such as underwater adhesion and self-healing at sub-zero temperatures. Thus, more stable adhesion or self-healing mechanisms need to be developed and used in organohydrogels. Also, the various properties of the organoydrogels require further optimization. For example, the decrease in conductivity of organohydrogels synthesized by solvent replacement needs to be addressed in the future. Moreover, considering the limited functionality of current organohydrogels due to the dilemma between some properties, a prominent organohydrogel with multiple functions is expected to be developed in the future based on functionality balance.
Fifthly, although organohydrogels have been proven to be applied in various fields, device performance and stability still need to be further improved. Interestingly, some organohydrogels also show great potential to be developed and applied to smart materials, oil–water separation, or underwater electronics, and their feasibility needs further verification. Moreover, lots of efforts can be put into the full use of organohydrogels and the development of multifunctional integrated systems with lower costs.
Finally, the current in situ characterization methods have made great progress, such as Raman spectroscopy, infrared spectroscopy, electrochemical impedance spectroscopy, etc. Correspondingly, these will enable more in-depth mechanism exploration of organohydrogels under different behaviors that are not completely clear.
Author contributions
Q. L. Ding: conceptualization, methodology, investigation, writing – original draft, and review & editing. Z. X. Wu: conceptualization, investigation, and writing – review & editing. K. Tao, W. Y. Wang, Y. M. Wei, B. R. Yang, and X. Xie: writing – review & editing. J. Wu: conceived this research, supervision, investigation, writing –original draft, and review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. W. acknowledges financial support from the National Natural Science Foundation of China (61801525), the Guangdong Basic and Applied Basic Research Foundation (2020A1515010693), and the Science and Technology Program of Guangzhou (201904010456).Notes and references
- C. Yang and Z. Suo, Nat. Rev. Mater., 2018, 3, 125–142 CrossRef CAS
.
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 CrossRef CAS PubMed
- J. Y. Sun, C. Keplinger, G. M. Whitesides and Z. Suo, Adv. Mater., 2014, 26, 7608–7614 CrossRef CAS PubMed
- Z. Wu, X. Yang and J. Wu, ACS Appl. Mater. Interfaces, 2021, 13, 2128–2144 CrossRef CAS PubMed
- X. Tong, Z. Tian, J. Sun, V. Tung, R. B. Kaner and Y. Shao, Mater. Today, 2021, 44, 78 CrossRef CAS
- S. Xia, Y. Chen, J. Tian, J. Shi, C. Geng, H. Zou, M. Liang and Z. Li, Adv. Funct. Mater., 2021, 2101143 CrossRef CAS
- H. E. Jeong, S. H. Lee, P. Kim and K. Y. Suh, Nano Lett., 2006, 6, 1508–1513 CrossRef CAS PubMed
- H. Liu, J. Gao, W. Huang, K. Dai, G. Zheng, C. Liu, C. Shen, X. Yan, J. Guo and Z. Guo, Nanoscale, 2016, 8, 12977–12989 RSC
- Y. Yang, G. Zhao, X. Cheng, H. Deng and Q. Fu, ACS Appl. Mater. Interfaces, 2021, 13, 14599–14611 CrossRef CAS PubMed
- Y. Ma, Y. Yu, P. She, J. Lu, S. Liu, W. Huang and Q. Zhao, Sci. Adv., 2020, 6, eaaz2386 CrossRef CAS PubMed
- Y. Jian, S. Handschuh-Wang, J. Zhang, W. Lu, X. Zhou and T. Chen, Mater. Horiz., 2021, 8, 351–369 RSC
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS
- H. Yuk, S. Lin, C. Ma, M. Takaffoli, N. X. Fang and X. Zhao, Nat. Commun., 2017, 8, 1–12 CrossRef PubMed
- Y. Yang, L. Guan, X. Li, Z. Gao, X. Ren and G. Gao, ACS Appl. Mater. Interfaces, 2018, 11, 3428–3437 CrossRef PubMed
- P. He, J. Wu, X. Pan, L. Chen, K. Liu, H. Gao, H. Wu, S. Cao, L. Huang and Y. Ni, J. Mater. Chem. A, 2020, 8, 3109–3118 RSC
- M. K. Jaiswal, J. R. Xavier, J. K. Carrow, P. Desai, D. Alge and A. K. Gaharwar, ACS Nano, 2015, 10, 246–256 CrossRef PubMed
- M. Hua, S. Wu, Y. Ma, Y. Zhao, Z. Chen, I. Frenkel, J. Strzalka, H. Zhou, X. Zhu and X. He, Nature, 2021, 590, 594–599 CrossRef CAS PubMed
- X.-J. Zha, S.-T. Zhang, J.-H. Pu, X. Zhao, K. Ke, R.-Y. Bao, L. Bai, Z.-Y. Liu, M.-B. Yang and W. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 23514–23522 CrossRef CAS PubMed
- G. Chen, J. Huang, J. Gu, S. Peng, X. Xiang, K. Chen, X. Yang, L. Guan, X. Jiang and L. Hou, J. Mater. Chem. A, 2020, 8, 6776–6784 RSC
- W. Xue, M. B. Huglin and E. Khoshdel, Polym. Int., 1999, 48, 8–14 CrossRef CAS
- K.-T. Huang, K. Ishihara and C.-J. Huang, Biomacromolecules, 2019, 20, 3524–3534 CrossRef CAS PubMed
- Y. Wei, L. Xiang, H. Ou, F. Li, Y. Zhang, Y. Qian, L. Hao, J. Diao, M. Zhang and P. Zhu, Adv. Funct. Mater., 2020, 30, 2005135 CrossRef CAS
- X. P. Morelle, W. R. Illeperuma, K. Tian, R. Bai, Z. Suo and J. J. Vlassak, Adv. Mater., 2018, 30, 1801541 CrossRef PubMed
- S. Li, L. Wang, W. Zheng, G. Yang and X. Jiang, Adv. Funct. Mater., 2020, 30, 2002370 CrossRef CAS
- X. Liu, Q. Zhang, L. Duan and G. Gao, ACS Appl. Mater. Interfaces, 2019, 11, 6644–6651 CrossRef CAS PubMed
- Y. Ma, J. Yao, Q. Liu, T. Han, J. Zhao, X. Ma, Y. Tong, G. Jin, K. Qu and B. Li, Adv. Funct. Mater., 2020, 30, 2001820 CrossRef CAS
- M. Lo Presti, G. Rizzo, G. M. Farinola and F. G. Omenetto, Adv. Sci., 2021, 2004786 CrossRef CAS PubMed
- N. D. Catron, H. Lee and P. B. Messersmith, Biointerphases, 2006, 1, 134–141 CrossRef CAS PubMed
- L.-D. Koh, Y. Cheng, C.-P. Teng, Y.-W. Khin, X.-J. Loh, S.-Y. Tee, M. Low, E. Ye, H.-D. Yu and Y.-W. Zhang, Prog. Polym. Sci., 2015, 46, 86–110 CrossRef CAS
- C. Zhong, T. Gurry, A. A. Cheng, J. Downey, Z. Deng, C. M. Stultz and T. K. Lu, Nat. Nanotechnol., 2014, 9, 858–866 CrossRef CAS PubMed
-
S. M. Roopan and D. Devipriya, Polymer Gels, Springer, 2018, pp. 285–310 Search PubMed
- Y. Gao, L. Shi, S. Lu, T. Zhu, X. Da, Y. Li, H. Bu, G. Gao and S. Ding, Chem. Mater., 2019, 31, 3257–3264 CrossRef CAS
- J. Ryu, S. H. Ku, H. Lee and C. B. Park, Adv. Funct. Mater., 2010, 20, 2132–2139 CrossRef CAS
- A. Wang, Y. Wang, B. Zhang, K. Wan, J. Zhu, J. Xu, C. Zhang and T. Liu, Chem. Eng. J., 2021, 411, 128506 CrossRef CAS
- Z. Wang, Y. Cong and J. Fu, J. Mater. Chem. B, 2020, 8, 3437–3459 RSC
- T. Zhao, G. Wang, D. Hao, L. Chen, K. Liu and M. Liu, Adv. Funct. Mater., 2018, 28, 1800793 CrossRef
- C. Wang, K. Hu, C. Zhao, Y. Zou, Y. Liu, X. Qu, D. Jiang, Z. Li, M. R. Zhang and Z. Li, Small, 2020, 16, 1904758 CrossRef CAS PubMed
- D. Zhou, F. Chen, S. Handschuh-Wang, T. Gan, X. Zhou and X. Zhou, ChemPhysChem, 2019, 20, 2139–2154 CrossRef CAS PubMed
- T. Wang, Y. Zhang, Q. Liu, W. Cheng, X. Wang, L. Pan, B. Xu and H. Xu, Adv. Funct. Mater., 2018, 28, 1705551 CrossRef
- Y. Guo and S. Dong, Anal. Chem., 1997, 69, 1904–1908 CrossRef CAS
- Q. Rong, W. Lei, J. Huang and M. Liu, Adv. Energy Mater., 2018, 8, 1801967 CrossRef
- X. Su, H. Wang, Z. Tian, X. Duan, Z. Chai, Y. Feng, Y. Wang, Y. Fan and J. Huang, ACS Appl. Mater. Interfaces, 2020, 12, 29757–29766 CAS
- H. Gao, Z. Zhao, Y. Cai, J. Zhou, W. Hua, L. Chen, L. Wang, J. Zhang, D. Han and M. Liu, Nat. Commun., 2017, 8, 1–8 CrossRef CAS PubMed
- X. Liu, Q. Zhang and G. Gao, ACS Nano, 2020, 14, 13709–13717 CrossRef CAS PubMed
- Z. Zhao, C. Li, Z. Dong, Y. Yang, L. Zhang, S. Zhuo, X. Zhou, Y. Xu, L. Jiang and M. Liu, Adv. Funct. Mater., 2019, 29, 1807858 CrossRef
- Z. Zhao, K. Zhang, Y. Liu, J. Zhou and M. Liu, Adv. Mater., 2017, 29, 1701695 CrossRef PubMed
- J. Chen, Q. Dong, X. Ma, T.-H. Fan and Y. Lei, Sci. Rep., 2016, 6, 1–9 CrossRef CAS PubMed
- Q. Rong, W. Lei, L. Chen, Y. Yin, J. Zhou and M. Liu, Angew. Chem., Int. Ed., 2017, 56, 14159–14163 CrossRef CAS PubMed
- R. Ricciardi, F. Auriemma, C. Gaillet, C. De Rosa and F. Lauprêtre, Macromolecules, 2004, 37, 9510–9516 CrossRef CAS
-
J. Y. Chang, D. Godovsky, M. Han, C. Hassan, J. Kim, B. Lee, Y. Lee, N. Peppas, R. Quirk and T. Yoo, Biopolymers· PVA Hydrogels Anionic Polymerisation Nanocomposites, Springer Science & Business Media, 2000 Search PubMed
- X.-J. Zha, S.-T. Zhang, J.-H. Pu, X. Zhao, K. Ke, R.-Y. Bao, L. Bai, Z.-Y. Liu, M.-B. Yang and W. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 23514–23522 CrossRef CAS PubMed
- H. Liu, Z. Li, Y. Zhao, Y. Feng, A. V. Zvyagin, J. Wang, X. Yang, B. Yang and Q. Lin, ACS Appl. Mater. Interfaces, 2021, 13, 26770–26781 CrossRef CAS PubMed
- L. Guo, R. H. Colby, C. P. Lusignan and T. H. Whitesides, Macromolecules, 2003, 36, 9999–10008 CrossRef CAS
- Z. Qin, D. Dong, M. Yao, Q. Yu, X. Sun, Q. Guo, H. Zhang, F. Yao and J. Li, ACS Appl. Mater. Interfaces, 2019, 11, 21184–21193 CrossRef CAS PubMed
- B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2012, 64, 154–162 CrossRef
- R. Yoshida, Adv. Mater., 2010, 22, 3463–3483 CrossRef CAS PubMed
- K. Murata, M. Aoki, T. Suzuki, T. Harada, H. Kawabata, T. Komori, F. Ohseto, K. Ueda and S. Shinkai, J. Am. Chem. Soc., 1994, 116, 6664–6676 CrossRef CAS
- C. T. Lee, K. A. Smith and T. A. Hatton, Macromolecules, 2004, 37, 5397–5405 CrossRef CAS
- I. Tomatsu, A. Hashidzume and A. Harada, J. Am. Chem. Soc., 2006, 128, 2226–2227 CrossRef CAS PubMed
- S. Ahmed, X. Sallenave, F. Fages and G. Mieden-Gundert, Langmuir, 2002, 18, 7096–7101 CrossRef CAS
- S.-i. Kawano, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2004, 126, 8592–8593 CrossRef CAS PubMed
- J. Wu, Z. Wu, H. Xu, Q. Wu, C. Liu, B.-R. Yang, X. Gui, X. Xie, K. Tao and Y. Shen, Mater. Horiz., 2019, 6, 595–603 RSC
- Z. He and W. Yuan, ACS Appl. Mater. Interfaces, 2021, 13, 1474–1485 CrossRef CAS PubMed
- X. Wan, X. Xu, X. Liu, L. Jia, X. He and S. Wang, Adv. Mater., 2021, 33, 2008557 CrossRef CAS PubMed
- H. Sun, Y. Zhao, S. Jiao, C. Wang, Y. Jia, K. Dai, G. Zheng, C. Liu, P. Wan and C. Shen, Adv. Funct. Mater., 2021, 31, 2101696 CrossRef CAS
- J. Liu, Z. Chen, Y. Chen, H. U. Rehman, Y. Guo, H. Li and H. Liu, Adv. Funct. Mater., 2021, 31, 2101464 CrossRef CAS
- X. Sui, H. Guo, C. Cai, Q. Li, C. Wen, X. Zhang, X. Wang, J. Yang and L. Zhang, Chem. Eng. J., 2021, 419, 129478 CrossRef CAS
- K. Wang, J. Wang, L. Li, L. Xu, N. Feng, Y. Wang, X. Fei, J. Tian and Y. Li, Chem. Eng. J., 2019, 372, 216–225 CrossRef CAS
- X. Wu, H. Liao, D. Ma, M. Chao, Y. Wang, X. Jia, P. Wan and L. Zhang, J. Mater. Chem. C, 2020, 8, 1788–1795 RSC
- X. Pan, Q. Wang, R. Guo, Y. Ni, K. Liu, X. Ouyang, L. Chen, L. Huang, S. Cao and M. Xie, J. Mater. Chem. A, 2019, 7, 4525–4535 RSC
- G. Ge, Y.-Z. Zhang, W. Zhang, W. Yuan, J. K. El-Demellawi, P. Zhang, E. Di Fabrizio, X. Dong and H. N. Alshareef, ACS Nano, 2021, 15, 2698–2706 CrossRef CAS PubMed
- C. Sun, C. Hou, H. Zhang, Y. Li, Q. Zhang and H. Wang, APL Mater., 2021, 9, 011101 CrossRef CAS
- J. Song, S. Chen, L. Sun, Y. Guo, L. Zhang, S. Wang, H. Xuan, Q. Guan and Z. You, Adv. Mater., 2020, 32, 1906994 CrossRef CAS PubMed
- K. Tian, J. Bae, S. E. Bakarich, C. Yang, R. D. Gately, G. M. Spinks, M. in het Panhuis, Z. Suo and J. J. Vlassak, Adv. Mater., 2017, 29, 1604827 CrossRef PubMed
- H. Liu, W. Zhao, G. Gao and X. Ren, Mater. Today Commun., 2019, 21, 100609 CrossRef CAS
- M. Amjadi, A. Pichitpajongkit, S. Lee, S. Ryu and I. Park, ACS Nano, 2014, 8, 5154–5163 CrossRef CAS PubMed
- J. Chen and E. Dickinson, Colloids Surf., B, 1999, 12, 373–381 CrossRef CAS
- T. B. Blijdenstein, W. P. Hendriks, E. van der Linden, T. van Vliet and G. A. van Aken, Langmuir, 2003, 19, 6657–6663 CrossRef CAS
- S. Zhuo, Z. Zhao, Z. Xie, Y. Hao, Y. Xu, T. Zhao, H. Li, E. M. Knubben, L. Wen and L. Jiang, Sci. Adv., 2020, 6, eaax1464 CrossRef CAS PubMed
- C. Li, S. Feng, C. Li, Y. Sui, J. Shen, C. Huang, Y. Wu and W. Huang, Adv. Funct. Mater., 2020, 30, 2002163 CrossRef CAS
- Y. Liu, L. Wang, H. Lu and Z. Huang, Langmuir, 2021, 37, 6711 CrossRef CAS PubMed
- S. Shi, X. Peng, T. Liu, Y.-N. Chen, C. He and H. Wang, Polymer, 2017, 111, 168–176 CrossRef CAS
- L. Han, K. Liu, M. Wang, K. Wang, L. Fang, H. Chen, J. Zhou and X. Lu, Adv. Funct. Mater., 2018, 28, 1704195 CrossRef
- F. Chen, D. Zhou, J. Wang, T. Li, X. Zhou, T. Gan, S. Handschuh-Wang and X. Zhou, Angew. Chem., 2018, 130, 6678–6681 CrossRef
- J. W. Zhang, D. D. Dong, X. Y. Guan, E. M. Zhang, Y. M. Chen, K. Yang, Y. X. Zhang, M. M. B. Khan, Y. Arfat and Y. Aziz, Front. Chem., 2020, 8, 102 CrossRef CAS PubMed
- J. Wu, Z. Wu, X. Lu, S. Han, B.-R. Yang, X. Gui, K. Tao, J. Miao and C. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 9405–9414 CrossRef CAS PubMed
- L. Fang, J. Zhang, W. Wang, Y. Zhang, F. Chen, J. Zhou, F. Chen, R. Li, X. Zhou and Z. Xie, ACS Appl. Mater. Interfaces, 2020, 12, 56393–56402 CrossRef CAS PubMed
- M. Matsugami, T. Takamuku, T. Otomo and T. Yamaguchi, J. Phys. Chem. B, 2006, 110, 12372–12379 CrossRef CAS PubMed
- W. P. Williams, P. J. Quinn, L. I. Tsonev and R. D. Koynova, Biochim. Biophys. Acta, Biomembr., 1991, 1062, 123–132 CrossRef CAS
- J. Gu, J. Huang, G. Chen, L. Hou, J. Zhang, X. Zhang, X. Yang, L. Guan, X. Jiang and H. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 40815–40827 CrossRef CAS PubMed
- D. Rasmussen and A. MacKenzie, Nature, 1968, 220, 1315–1317 CrossRef CAS PubMed
- J. F. Abrahamsen, A. M. Bakken and Ø. Bruserud, Transfusion, 2002, 42, 1573–1580 CrossRef CAS PubMed
- X. Zhao, F. Chen, Y. Li, H. Lu, N. Zhang and M. Ma, Nat. Commun., 2018, 9, 1–8 CrossRef PubMed
- Y. Ye, Y. Zhang, Y. Chen, X. Han and F. Jiang, Adv. Funct. Mater., 2020, 30, 2003430 CrossRef CAS
- S. Cerveny, J. Colmenero and A. Alegria, Macromolecules, 2005, 38, 7056–7063 CrossRef CAS
- H. Kiani and D.-W. Sun, Trends Food Sci. Technol., 2011, 22, 407–426 CrossRef CAS
- H. Yuk, T. Zhang, G. A. Parada, X. Liu and X. Zhao, Nat. Commun., 2016, 7, 1–11 Search PubMed
- X. Liu, Q. Zhang, L. Duan and G. Gao, Adv. Funct. Mater., 2019, 29, 1900450 CrossRef
- Y.-Q. Wang, Y. Zhu, J.-H. Wang, X.-N. Li, X.-G. Wu, Y.-X. Qin and W.-Y. Chen, Compos. Sci. Technol., 2021, 206, 108653 CrossRef CAS
- B. Q. Y. Chan, Z. W. K. Low, S. J. W. Heng, S. Y. Chan, C. Owh and X. J. Loh, ACS Appl. Mater. Interfaces, 2016, 8, 10070–10087 CrossRef CAS PubMed
- Y. Wong, J. Kong, L. K. Widjaja and S. S. Venkatraman, Sci. China: Chem., 2014, 57, 476–489 CrossRef CAS
- G. Li, G. Fei, H. Xia, J. Han and Y. Zhao, J. Mater. Chem., 2012, 22, 7692–7696 RSC
- R. Liang, L. Wang, H. Yu, A. Khan, B. U. Amin and R. U. Khan, Eur. Polym. J., 2019, 114, 380–396 CrossRef CAS
- C.-H. Lu, W. Guo, Y. Hu, X.-J. Qi and I. Willner, J. Am. Chem. Soc., 2015, 137, 15723–15731 CrossRef CAS PubMed
- Y. Liu, H. Du, L. Liu and J. Leng, Smart Mater. Struct., 2014, 23, 023001 CrossRef CAS
- H. Meng and G. Li, Polymer, 2013, 54, 2199–2221 CrossRef CAS
- Y. Zhang, J. Liao, T. Wang, W. Sun and Z. Tong, Adv. Funct. Mater., 2018, 28, 1707245 CrossRef
- J. G. Hardy, M. Palma, S. J. Wind and M. J. Biggs, Adv. Mater., 2016, 28, 5717–5724 CrossRef CAS PubMed
- A. T. Neffe, B. D. Hanh, S. Steuer and A. Lendlein, Adv. Mater., 2009, 21, 3394–3398 CrossRef CAS PubMed
- T. Xie, Polymer, 2011, 52, 4985–5000 CrossRef CAS
- W. Voit, T. Ware, R. R. Dasari, P. Smith, L. Danz, D. Simon, S. Barlow, S. R. Marder and K. Gall, Adv. Funct. Mater., 2010, 20, 162–171 CrossRef CAS
- N. Zheng, G. Fang, Z. Cao, Q. Zhao and T. Xie, Polym. Chem., 2015, 6, 3046–3053 RSC
- Y. Osada and A. Matsuda, Nature, 1995, 376, 219–219 CrossRef CAS PubMed
- J. Hao and R. Weiss, ACS Macro Lett., 2013, 2, 86–89 CrossRef CAS
- Y. Zhang, Y. Li and W. Liu, Adv. Funct. Mater., 2015, 25, 471–480 CrossRef CAS
- Z. Zhao, S. Zhuo, R. Fang, L. Zhang, X. Zhou, Y. Xu, J. Zhang, Z. Dong, L. Jiang and M. Liu, Adv. Mater., 2018, 30, 1804435 CrossRef PubMed
- F. Mo, G. Liang, Q. Meng, Z. Liu, H. Li, J. Fan and C. Zhi, Energy Environ. Sci., 2019, 12, 706–715 RSC
- S. Bao, J. Gao, T. Xu, N. Li, W. Chen and W. Lu, Chem. Eng. J., 2021, 411, 128470 CrossRef CAS
- W. Zheng, L. Xu, Y. Li, Y. Huang, B. Li, Z. Jiang and G. Gao, J. Colloid Interface Sci., 2021, 594, 584–592 CrossRef CAS PubMed
- Z. Qin, X. Sun, H. Zhang, Q. Yu, X. Wang, S. He, F. Yao and J. Li, J. Mater. Chem. A, 2020, 8, 4447–4456 RSC
- J.-Y. Sun, X. Zhao, W. R. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed
- L. Fang, Z. Cai, Z. Ding, T. Chen, J. Zhang, F. Chen, J. Shen, F. Chen, R. Li and X. Zhou, ACS Appl. Mater. Interfaces, 2019, 11, 21895–21903 CrossRef CAS PubMed
- J. Wu, S. Han, T. Yang, Z. Li, Z. Wu, X. Gui, K. Tao, J. Miao, L. K. Norford and C. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 19097–19105 CrossRef CAS PubMed
- G. Ramorino, F. Bignotti, S. Pandini and T. Riccò, Compos. Sci. Technol., 2009, 69, 1206–1211 CrossRef CAS
- T. Bai, B. Zhu, H. Liu, Y. Wang, G. Song, C. Liu and C. Shen, Int. J. Biol. Macromol., 2020, 151, 628–634 CrossRef CAS PubMed
- O. Lourie, D. Cox and H. Wagner, Phys. Rev. Lett., 1998, 81, 1638 CrossRef CAS
- J. Y. Kim, Materials, 2009, 2, 1955–1974 CrossRef CAS
- J. H. Xu, S. Ye, C. Di Ding, L. H. Tan and J. J. Fu, J. Mater. Chem. A, 2018, 6, 5887–5898 RSC
- X. Gong, J. Liu, S. Baskaran, R. D. Voise and J. S. Young, Chem. Mater., 2000, 12, 1049–1052 CrossRef CAS
- M. K. Jaiswal, J. R. Xavier, J. K. Carrow, P. Desai, D. Alge and A. K. Gaharwar, ACS Nano, 2016, 10, 246–256 CrossRef CAS PubMed
- G. Ge, Y. Lu, X. Qu, W. Zhao, Y. Ren, W. Wang, Q. Wang, W. Huang and X. Dong, ACS Nano, 2019, 14, 218–228 CrossRef PubMed
- S. Wu, M. Hua, Y. Alsaid, Y. Du, Y. Ma, Y. Zhao, C. Y. Lo, C. Wang, D. Wu and B. Yao, Adv. Mater., 2021, 33, 2007829 CrossRef CAS PubMed
- P. Lin, S. Ma, X. Wang and F. Zhou, Adv. Mater., 2015, 27, 2054–2059 CrossRef CAS PubMed
- P. Lin, T. Zhang, X. Wang, B. Yu and F. Zhou, Small, 2016, 12, 4386–4392 CrossRef CAS PubMed
- C. Xiang, Z. Wang, C. Yang, X. Yao, Y. Wang and Z. Suo, Mater. Today, 2020, 34, 7–16 CrossRef CAS
- H. Zhang, I. Hussain, M. Brust, M. F. Butler, S. P. Rannard and A. I. Cooper, Nat. Mater., 2005, 4, 787–793 CrossRef CAS PubMed
- M. T. I. Mredha, H. H. Le, P. Trtik, J. Cui and I. Jeon, Mater. Horiz., 2019, 6, 1504–1511 RSC
- M. T. I. Mredha, Y. Z. Guo, T. Nonoyama, T. Nakajima, T. Kurokawa and J. P. Gong, Adv. Mater., 2018, 30, 1704937 CrossRef PubMed
- Y. Huang, D. R. King, T. L. Sun, T. Nonoyama, T. Kurokawa, T. Nakajima and J. P. Gong, Adv. Funct. Mater., 2017, 27, 1605350 CrossRef
- X. Hu, M. Vatankhah-Varnoosfaderani, J. Zhou, Q. Li and S. S. Sheiko, Adv. Mater., 2015, 27, 6899–6905 CrossRef CAS PubMed
- Q. He, Y. Huang and S. Wang, Adv. Funct. Mater., 2018, 28, 1705069 CrossRef
- R. Bai, J. Yang, X. P. Morelle and Z. Suo, Macromol. Rapid Commun., 2019, 40, 1800883 CrossRef PubMed
- C. N. Maganaris and J. P. Paul, J. Physiol., 1999, 521, 307 CrossRef CAS PubMed
- C. Keplinger, J.-Y. Sun, C. C. Foo, P. Rothemund, G. M. Whitesides and Z. Suo, Science, 2013, 341, 984–987 CrossRef CAS PubMed
- H. R. Lee, C. C. Kim and J. Y. Sun, Adv. Mater., 2018, 30, 1704403 CrossRef PubMed
- C. Liu, S. Han, H. Xu, J. Wu and C. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 31716–31724 CrossRef CAS PubMed
-
X. Hou, Q. Zhang, L. Wang, G. Gao and W. Lü, ACS Appl. Mater. Interfaces, 2021, 13, 12432–12441 Search PubMed
- H. Chen, X. Ren and G. Gao, ACS Appl. Mater. Interfaces, 2019, 11, 28336–28344 CrossRef CAS PubMed
- Y. Shi, L. Pan, B. Liu, Y. Wang, Y. Cui, Z. Bao and G. Yu, J. Mater. Chem. A, 2014, 2, 6086–6091 RSC
- D. Mawad, E. Stewart, D. L. Officer, T. Romeo, P. Wagner, K. Wagner and G. G. Wallace, Adv. Funct. Mater., 2012, 22, 2692–2699 CrossRef CAS
- Y. Zhao, B. Liu, L. Pan and G. Yu, Energy Environ. Sci., 2013, 6, 2856–2870 RSC
- S. Naficy, J. M. Razal, G. M. Spinks, G. G. Wallace and P. G. Whitten, Chem. Mater., 2012, 24, 3425–3433 CrossRef CAS
- C. Wang, S. Wang, G. Chen, W. Kong, W. Ping, J. Dai, G. Pastel, H. Xie, S. He and S. Das, Chem. Mater., 2018, 30, 7707–7713 CrossRef CAS
- T. Li, S. X. Li, W. Kong, C. Chen, E. Hitz, C. Jia, J. Dai, X. Zhang, R. Briber and Z. Siwy, Sci. Adv., 2019, 5, eaau4238 CrossRef CAS PubMed
- X. Tian, Y. Yi, P. Yang, P. Liu, L. Qu, M. Li, Y.-s. Hu and B. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 4001–4010 CrossRef CAS PubMed
- H. Liu, X. Chen, Y. Zheng, D. Zhang, Y. Zhao, C. Wang, C. Pan, C. Liu and C. Shen, Adv. Funct. Mater., 2021, 31, 2008006 CrossRef CAS
- S. Sharma, A. Chhetry, S. Zhang, H. Yoon, C. Park, H. Kim, M. Sharifuzzaman, X. Hui and J. Y. Park, ACS Nano, 2021, 15, 4380–4393 CrossRef CAS PubMed
- K. Maleski, V. N. Mochalin and Y. Gogotsi, Chem. Mater., 2017, 29, 1632–1640 CrossRef CAS
- B. J. Blaiszik, S. L. Kramer, S. C. Olugebefola, J. S. Moore, N. R. Sottos and S. R. White, Annu. Rev. Mater. Res., 2010, 40, 179–211 CrossRef CAS
- Y. Huang, M. Zhu, Y. Huang, Z. Pei, H. Li, Z. Wang, Q. Xue and C. Zhi, Adv. Mater., 2016, 28, 8344–8364 CrossRef CAS PubMed
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC
- M. Q. Zhang and M. Z. Rong, J. Polym. Sci., Part B: Polym. Phys, 2012, 50, 229–241 CrossRef CAS
- X. He, C. Zhang, M. Wang, Y. Zhang, L. Liu and W. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 11134–11143 CrossRef CAS PubMed
- Y. Cao, T. G. Morrissey, E. Acome, S. I. Allec, B. M. Wong, C. Keplinger and C. Wang, Adv. Mater., 2017, 29, 1605099 CrossRef PubMed
- H. Qin, T. Zhang, H.-N. Li, H.-P. Cong, M. Antonietti and S.-H. Yu, Chem, 2017, 3, 691–705 CAS
- L. Shi, H. Carstensen, K. Hölzl, M. Lunzer, H. Li, J. Hilborn, A. Ovsianikov and D. A. Ossipov, Chem. Mater., 2017, 29, 5816–5823 CrossRef CAS
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed
- Y. Guo, K. Zheng and P. Wan, Small, 2018, 14, 1704497 CrossRef PubMed
- G. Li, H. Zhang, D. Fortin, H. Xia and Y. Zhao, Langmuir, 2015, 31, 11709–11716 CrossRef CAS PubMed
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 1–6 Search PubMed
- Q. Yang, P. Wang, C. Zhao, W. Wang, J. Yang and Q. Liu, Macromol. Rapid Commun., 2017, 38, 1600741 CrossRef PubMed
- Z. Wang, Y. Ren, Y. Zhu, L. Hao, Y. Chen, G. An, H. Wu, X. Shi and C. Mao, Angew. Chem., 2018, 130, 9146–9150 CrossRef
- Y. Shi, M. Wang, C. Ma, Y. Wang, X. Li and G. Yu, Nano Lett., 2015, 15, 6276–6281 CrossRef CAS PubMed
- M. A. Darabi, A. Khosrozadeh, R. Mbeleck, Y. Liu, Q. Chang, J. Jiang, J. Cai, Q. Wang, G. Luo and M. Xing, Adv. Mater., 2017, 29, 1700533 CrossRef PubMed
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed
- D. C. Tuncaboylu, A. Argun, M. Sahin, M. Sari and O. Okay, Polymer, 2012, 53, 5513–5522 CrossRef CAS
- V. Can, Z. Kochovski, V. Reiter, N. Severin, M. Siebenbürger, B. Kent, J. Just, J. r. P. Rabe, M. Ballauff and O. Okay, Macromolecules, 2016, 49, 2281–2287 CrossRef CAS
- Y. Ren, R. Lou, X. Liu, M. Gao, H. Zheng, T. Yang, H. Xie, W. Yu and X. Ma, Chem. Commun., 2016, 52, 6273–6276 RSC
- M. W. England, C. Urata, G. J. Dunderdale and A. Hozumi, ACS Appl. Mater. Interfaces, 2016, 8, 4318–4322 CrossRef CAS PubMed
- Y. Xu, Q. Wu, Y. Sun, H. Bai and G. Shi, ACS Nano, 2010, 4, 7358–7362 CrossRef CAS PubMed
- A. J. Patil, J. L. Vickery, T. B. Scott and S. Mann, Adv. Mater., 2009, 21, 3159–3164 CrossRef CAS
- S. Burattini, B. W. Greenland, W. Hayes, M. E. Mackay, S. J. Rowan and H. M. Colquhoun, Chem. Mater., 2011, 23, 6–8 CrossRef CAS
- H. Guo, Y. J. Tan, G. Chen, Z. Wang, G. J. Susanto, H. H. See, Z. Yang, Z. W. Lim, L. Yang and B. C. Tee, Nat. Commun., 2020, 11, 1–10 Search PubMed
- G. E. Fantner, E. Oroudjev, G. Schitter, L. S. Golde, P. Thurner, M. M. Finch, P. Turner, T. Gutsmann, D. E. Morse and H. Hansma, Biophys. J., 2006, 90, 1411–1418 CrossRef CAS PubMed
- F.-Y. Hsieh, H.-W. Han, X.-R. Chen, C.-S. Yang, Y. Wei and S.-H. Hsu, Biomaterials, 2018, 174, 31–40 CrossRef CAS PubMed
- F.-Y. Hsieh, L. Tao, Y. Wei and S.-H. Hsu, NPG Asia Mater., 2017, 9, e363–e363 CrossRef CAS
- C. e. Yuan, M. Z. Rong, M. Q. Zhang, Z. P. Zhang and Y. C. Yuan, Chem. Mater., 2011, 23, 5076–5081 CrossRef CAS
- G. Deng, F. Li, H. Yu, F. Liu, C. Liu, W. Sun, H. Jiang and Y. Chen, ACS Macro Lett., 2012, 1, 275–279 CrossRef CAS
- P. Casuso, I. Odriozola, A. N. Pérez-San Vicente, I. Loinaz, G. N. Cabañero, H.-J. R. Grande and D. Dupin, Biomacromolecules, 2015, 16, 3552–3561 CrossRef CAS PubMed
- Z. Liu, Y. Wang, Y. Ren, G. Jin, C. Zhang, W. Chen and F. Yan, Mater. Horiz., 2020, 7, 919–927 RSC
- D. L. Taylor and M. in het Panhuis, Adv. Mater., 2016, 28, 9060–9093 CrossRef CAS PubMed
- W.-P. Chen, D.-Z. Hao, W.-J. Hao, X.-L. Guo and L. Jiang, ACS Appl. Mater. Interfaces, 2018, 10, 1258–1265 CrossRef CAS PubMed
- V. K. Thakur and M. R. Kessler, Polymer, 2015, 69, 369–383 CrossRef CAS
- B. J. Kim, D. X. Oh, S. Kim, J. H. Seo, D. S. Hwang, A. Masic, D. K. Han and H. J. Cha, Biomacromolecules, 2014, 15, 1579–1585 CrossRef CAS PubMed
- Q. Lu, E. Danner, J. H. Waite, J. N. Israelachvili, H. Zeng and D. S. Hwang, J. R. Soc., Interface, 2013, 10, 20120759 CrossRef PubMed
- S. A. Burke, M. Ritter-Jones, B. P. Lee and P. B. Messersmith, Biomed. Mater., 2007, 2, 203 CrossRef CAS PubMed
- N. Holten-Andersen, A. Jaishankar, M. J. Harrington, D. E. Fullenkamp, G. DiMarco, L. He, G. H. McKinley, P. B. Messersmith and K. Y. C. Lee, J. Mater. Chem. B, 2014, 2, 2467–2472 RSC
- T. Yamada, Y. Hayamizu, Y. Yamamoto, Y. Yomogida, A. Izadi-Najafabadi, D. N. Futaba and K. Hata, Nat. Nanotechnol., 2011, 6, 296–301 CrossRef CAS PubMed
- H. Yuk, T. Zhang, S. Lin, G. A. Parada and X. Zhao, Nat. Mater., 2016, 15, 190–196 CrossRef CAS PubMed
- H. Zhou, J. Lai, X. Jin, H. Liu, X. Li, W. Chen, A. Ma and X. Zhou, Chem. Eng. J., 2021, 413, 127544 CrossRef CAS
- J. Xu, R. Jin, X. Ren and G. Gao, Chem. Eng. J., 2021, 413, 127446 CrossRef CAS
- J. Tang, J. Li, J. J. Vlassak and Z. Suo, Soft Matter, 2016, 12, 1093–1099 RSC
- Q. Liu, G. Nian, C. Yang, S. Qu and Z. Suo, Nat. Commun., 2018, 9, 1–11 CrossRef PubMed
- L. Zhou, C. Dai, L. Fan, Y. Jiang, C. Liu, Z. Zhou, P. Guan, Y. Tian, J. Xing and X. Li, Adv. Funct. Mater., 2021, 31, 2007457 CrossRef CAS
- G. Ye, J. Qiu, X. Fang, T. Yu, Y. Xie, Y. Zhao, D. Yan, C. He and N. Liu, Mater. Horiz., 2021, 8, 1047–1057 RSC
- Y. Niu, H. Liu, R. He, M. Luo, M. Shu and F. Xu, Small, 2021, 17, 2101151 CrossRef CAS PubMed
- H. Zhou, J. Lai, B. Zheng, X. Jin, G. Zhao, H. Liu, W. Chen, A. Ma, X. Li and Y. Wu, Adv. Funct. Mater., 2021, 2108423 Search PubMed
- S. Lim, Y. S. Choi, D. G. Kang, Y. H. Song and H. J. Cha, Biomaterials, 2010, 31, 3715–3722 CrossRef CAS PubMed
- Y. Chen, J. Meng, Z. Gu, X. Wan, L. Jiang and S. Wang, Adv. Funct. Mater., 2020, 30, 1905287 CrossRef CAS
- J. H. Waite, N. H. Andersen, S. Jewhurst and C. Sun, J. Adhes., 2005, 81, 297–317 CrossRef CAS
- T. Priemel, E. Degtyar, M. N. Dean and M. J. Harrington, Nat. Commun., 2017, 8, 1–12 CrossRef PubMed
- X. Pei, J. Wang, Y. Cong and J. Fu, J. Polym. Sci., 2021, 59, 1312 CrossRef CAS
- S. Wu, Z. Zhao, J. R. Rzasa, E. Kim, J. Li, E. VanArsdale, W. E. Bentley, X. Shi and G. F. Payne, Adv. Funct. Mater., 2021, 31, 2007709 CrossRef CAS
- C. Zhang, B. Wu, Y. Zhou, F. Zhou, W. Liu and Z. Wang, Chem. Soc. Rev., 2020, 49, 3605–3637 RSC
- L. Han, M. Wang, P. Li, D. Gan, L. Yan, J. Xu, K. Wang, L. Fang, C. W. Chan and H. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 28015–28026 CrossRef CAS PubMed
- L. Han, L. Yan, K. Wang, L. Fang, H. Zhang, Y. Tang, Y. Ding, L.-T. Weng, J. Xu and J. Weng, NPG Asia Mater., 2017, 9, e372–e372 CrossRef CAS
- H. Ejima, J. J. Richardson, K. Liang, J. P. Best, M. P. van Koeverden, G. K. Such, J. Cui and F. Caruso, Science, 2013, 341, 154–157 CrossRef CAS PubMed
- T. S. Sileika, D. G. Barrett, R. Zhang, K. H. A. Lau and P. B. Messersmith, Angew. Chem., 2013, 125, 10966–10970 CrossRef
- D. X. Oh, S. Kim, D. Lee and D. S. Hwang, Acta Biomater., 2015, 20, 104–112 CrossRef CAS PubMed
- A. S. Lee, P. J. Mahon and D. C. Creagh, Vib. Spectrosc., 2006, 41, 170–175 CrossRef CAS
- H. Kumari, S. R. Kline, C. L. Dennis, A. V. Mossine, R. L. Paul, C. A. Deakyne and J. L. Atwood, Angew. Chem., 2012, 124, 9397–9400 CrossRef
- J. P. McManus, K. G. Davis, J. E. Beart, S. H. Gaffney, T. H. Lilley and E. Haslam, J. Chem. Soc., Perkin Trans. 2, 1985, 1429–1438 RSC
- K. Kustin, S. Liu, C. Nicolini and D. Toppen, J. Am. Chem. Soc., 1974, 96, 7410–7415 CrossRef CAS
- G. Huber, H. Mantz, R. Spolenak, K. Mecke, K. Jacobs, S. N. Gorb and E. Arzt, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16293–16296 CrossRef CAS PubMed
- A. L. Urzedo, M. C. Gonçalves, M. H. Nascimento, C. B. Lombello, G. Nakazato and A. B. Seabra, ACS Biomater. Sci. Eng., 2020, 6, 2117–2134 CrossRef CAS PubMed
- A. Y. Gahane, V. Singh, A. Kumar and A. K. Thakur, Biomater. Sci., 2020, 8, 1996–2006 RSC
- H. Ji, H. Sun and X. Qu, Adv. Drug Delivery Rev., 2016, 105, 176–189 CrossRef CAS PubMed
- G. He, X. Chen, Y. Yin, W. Cai, W. Ke, Y. Kong and H. Zheng, J. Biomater. Sci., Polym. Ed., 2016, 27, 370–384 CrossRef CAS PubMed
- S. Li, S. Dong, W. Xu, S. Tu, L. Yan, C. Zhao, J. Ding and X. Chen, Adv. Sci., 2018, 5, 1700527 CrossRef PubMed
- P. AshaRani, G. Low Kah Mun, M. P. Hande and S. Valiyaveettil, ACS Nano, 2009, 3, 279–290 CrossRef CAS PubMed
- J. He, M. Shi, Y. Liang and B. Guo, Chem. Eng. J., 2020, 394, 124888 CrossRef CAS
- M. Li, X. Liu, L. Tan, Z. Cui, X. Yang, Z. Li, Y. Zheng, K. W. K. Yeung, P. K. Chu and S. Wu, Biomater. Sci., 2018, 6, 2110–2121 RSC
- Z. Zhao, R. Yan, X. Yi, J. Li, J. Rao, Z. Guo, Y. Yang, W. Li, Y.-Q. Li and C. Chen, ACS Nano, 2017, 11, 4428–4438 CrossRef CAS PubMed
- M. Baumgartner, F. Hartmann, M. Drack, D. Preninger, D. Wirthl, R. Gerstmayr, L. Lehner, G. Mao, R. Pruckner and S. Demchyshyn, Nat. Mater., 2020, 19, 1102–1109 CrossRef CAS PubMed
- L. Feng, W. Shi, Q. Chen, H. Cheng, J. Bao, C. Jiang, W. Zhao and C. Zhao, Adv. Healthcare Mater., 2021, 2100784 CrossRef CAS PubMed
- X. Zhang, L.-Y. Xia, X. Chen, Z. Chen and F.-G. Wu, Sci. China Mater., 2017, 60, 487–503 CrossRef CAS
- Z. Yang, R. Huang, B. Zheng, W. Guo, C. Li, W. He, Y. Wei, Y. Du, H. Wang and D. Wu, Adv. Sci., 2021, 8, 2003627 CrossRef CAS PubMed
- X. Zhao, B. Guo, H. Wu, Y. Liang and P. X. Ma, Nat. Commun., 2018, 9, 1–17 CrossRef PubMed
- J. Qu, X. Zhao, Y. Liang, T. Zhang, P. X. Ma and B. Guo, Biomaterials, 2018, 183, 185–199 CrossRef CAS PubMed
- C. Wang, H. Niu, X. Ma, H. Hong, Y. Yuan and C. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 34595–34608 CrossRef CAS PubMed
- Y. Su, L. Tian, M. Yu, Q. Gao, D. Wang, Y. Xi, P. Yang, B. Lei, P. X. Ma and P. Li, Polym. Chem., 2017, 8, 3788–3800 RSC
- Y. Zhang, L. Tao, S. Li and Y. Wei, Biomacromolecules, 2011, 12, 2894–2901 CrossRef CAS PubMed
- K. Zhang, J. L. Schnoor and E. Y. Zeng, Environ. Sci. Technol., 2012, 46, 10861–10867 CrossRef CAS PubMed
- J. Wei, G. Wei, Y. Shang, J. Zhou, C. Wu and Q. Wang, Adv. Mater., 2019, 31, 1900248 CrossRef PubMed
- Y. Huang, M. Zhong, Y. Huang, M. Zhu, Z. Pei, Z. Wang, Q. Xue, X. Xie and C. Zhi, Nat. Commun., 2015, 6, 1–8 Search PubMed
- Y. Huang, J. Liu, J. Wang, M. Hu, F. Mo, G. Liang and C. Zhi, Angew. Chem., Int. Ed., 2018, 57, 9810–9813 CrossRef CAS PubMed
- Y. Guo, X. Zhou, Q. Tang, H. Bao, G. Wang and P. Saha, J. Mater. Chem. A, 2016, 4, 8769–8776 RSC
- Y. Deng, Y. Zhang, B. Lemos and H. Ren, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed
- V. R. Feig, H. Tran and Z. Bao, ACS Cent. Sci., 2018, 4, 337–348 CrossRef CAS PubMed
- Z. Yu and P. Wu, Adv. Mater., 2021, 33, 2008479 CrossRef CAS PubMed
- H. Shang, X. Le, M. Si, S. Wu, Y. Peng, F. Shan, S. Wu and T. Chen, Chem. Eng. J., 2022, 429, 132290 CrossRef CAS
- X. Le, H. Shang, S. Wu, J. Zhang, M. Liu, Y. Zheng and T. Chen, Adv. Funct. Mater., 2021, 2108365 CrossRef CAS
- V. Solanki, S. Krupanidhi and K. Nanda, ACS Appl. Mater. Interfaces, 2017, 9, 41428–41434 CrossRef CAS PubMed
- P. M. Faia and C. S. Furtado, Sens. Actuators, B, 2013, 181, 720–729 CrossRef CAS
- J. Qi, S. Gao, K. Chen, J. Yang, H. Zhao, L. Guo and S. Yang, J. Mater. Chem. A, 2015, 3, 18019–18026 RSC
- L. Liu, M. Ikram, L. Ma, X. Zhang, H. Lv, M. Ullah, M. Khan, H. Yu and K. Shi, J. Hazard. Mater., 2020, 393, 122325 CrossRef CAS PubMed
- Z. Qin, K. Xu, H. Yue, H. Wang, J. Zhang, C. Ouyang, C. Xie and D. Zeng, Sens. Actuators, B, 2018, 262, 771–779 CrossRef CAS
- R. Kumar, W. Zheng, X. Liu, J. Zhang and M. Kumar, Adv. Mater. Technol., 2020, 5, 1901062 CrossRef CAS
- N. Wang, M. Lin, H. Dai and H. Ma, Biosens. Bioelectron., 2016, 79, 320–326 CrossRef CAS PubMed
- Y. Pang, J. Jian, T. Tu, Z. Yang, J. Ling, Y. Li, X. Wang, Y. Qiao, H. Tian and Y. Yang, Biosens. Bioelectron., 2018, 116, 123–129 CrossRef CAS PubMed
- D. D. Han, Y. L. Zhang, J. N. Ma, Y. Liu, J. W. Mao, C. H. Han, K. Jiang, H. R. Zhao, T. Zhang and H. L. Xu, Adv. Mater. Technol., 2017, 2, 1700045 CrossRef
- H. Li, J. Chen, X. Chang, Y. Xu, G. Zhao, Y. Zhu and Y. Li, J. Mater. Chem. A, 2021, 9, 1795–1802 RSC
- Y. Yang, G. Zhao, X. Cheng, H. Deng and Q. Fu, ACS Appl. Mater. Interfaces, 2021, 13, 14599–14611 CrossRef CAS PubMed
- G. Cai, J. Wang, K. Qian, J. Chen, S. Li and P. S. Lee, Adv. Sci., 2017, 4, 1600190 CrossRef PubMed
- W. Xu, J. Ma and E. Jabbari, Acta Biomater., 2010, 6, 1992–2002 CrossRef CAS PubMed
- S. Han, C. Liu, X. Lin, J. Zheng, J. Wu and C. Liu, ACS Appl. Polym. Mater., 2020, 2, 996–1005 CrossRef CAS
- R. Yu, C. Pan, J. Chen, G. Zhu and Z. L. Wang, Adv. Funct. Mater., 2013, 23, 5868–5874 CrossRef CAS
- S. Gong, W. Schwalb, Y. Wang, Y. Chen, Y. Tang, J. Si, B. Shirinzadeh and W. Cheng, Nat. Commun., 2014, 5, 1–8 Search PubMed
- M. Zhu, M. Lou, I. Abdalla, J. Yu, Z. Li and B. Ding, Nano Energy, 2020, 69, 104429 CrossRef CAS
- X. Wang, W.-Z. Song, M.-H. You, J. Zhang, M. Yu, Z. Fan, S. Ramakrishna and Y.-Z. Long, ACS Nano, 2018, 12, 8588–8596 CrossRef CAS PubMed
- M.-H. You, X.-X. Wang, X. Yan, J. Zhang, W.-Z. Song, M. Yu, Z.-Y. Fan, S. Ramakrishna and Y.-Z. Long, J. Mater. Chem. A, 2018, 6, 3500–3509 RSC
- S. Lee, A. Reuveny, J. Reeder, S. Lee, H. Jin, Q. Liu, T. Yokota, T. Sekitani, T. Isoyama and Y. Abe, Nat. Nanotechnol., 2016, 11, 472–478 CrossRef CAS PubMed
- C. Pang, G.-Y. Lee, T.-I. Kim, S. M. Kim, H. N. Kim, S.-H. Ahn and K.-Y. Suh, Nat. Mater., 2012, 11, 795–801 CrossRef CAS PubMed
- Z. Wu, H. Ding, K. Tao, Y. Wei, X. Gui, W. Shi, X. Xie and J. Wu, ACS Appl. Mater. Interfaces, 2021, 13, 21854–21864 CrossRef CAS PubMed
- Z. Wu, W. Shi, H. Ding, B. Zhong, W. Huang, Y. Zhou, X. Gui, X. Xie and J. Wu, J. Mater. Chem. C, 2021, 9, 13668–13679 RSC
- J. Wu, Z. Wu, Y. Wei, H. Ding, W. Huang, X. Gui, W. Shi, Y. Shen, K. Tao and X. Xie, ACS Appl. Mater. Interfaces, 2020, 12, 19069–19079 CrossRef CAS PubMed
- J. Wu, Z. Wu, W. Huang, X. Yang, Y. Liang, K. Tao, B.-R. Yang, W. Shi and X. Xie, ACS Appl. Mater. Interfaces, 2020, 12, 52070–52081 CrossRef CAS PubMed
- Z. Wu, L. Rong, J. Yang, Y. Wei, K. Tao, Y. Zhou, B. R. Yang, X. Xie and J. Wu, Small, 2021, 2104997 CrossRef CAS PubMed
- Y. Liang, Z. Wu, Y. Wei, Q. Ding, K. Tao, X. Xie and j. Wu, Nano-Micro Lett., 2022, 14, 52 CrossRef PubMed
- H. Lin, S. Zhang, T. Zhang, H. Ye, Q. Yao, G. W. Zheng and J. Y. Lee, Adv. Energy Mater., 2018, 8, 1801868 CrossRef
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed
- P. Guo, D. Liu, Z. Liu, X. Shang, Q. Liu and D. He, Electrochim. Acta, 2017, 256, 28–36 CrossRef CAS
- J. Chmiola, C. Largeot, P.-L. Taberna, P. Simon and Y. Gogotsi, Science, 2010, 328, 480–483 CrossRef CAS PubMed
- C. Lu and X. Chen, J. Mater. Chem. A, 2019, 7, 20158–20161 RSC
- Z. Zhang, M. Liao, H. Lou, Y. Hu, X. Sun and H. Peng, Adv. Mater., 2018, 30, 1704261 CrossRef PubMed
- Y. Xu, Y. Zhang, Z. Guo, J. Ren, Y. Wang and H. Peng, Angew. Chem., 2015, 127, 15610–15614 CrossRef
- H. Jin, L. Zhou, C. L. Mak, H. Huang, W. M. Tang and H. L. W. Chan, Nano Energy, 2015, 11, 662–670 CrossRef CAS
- M. Kunitski, N. Eicke, P. Huber, J. Köhler, S. Zeller, J. Voigtsberger, N. Schlott, K. Henrichs, H. Sann and F. Trinter, Nat. Commun., 2019, 10, 1–7 CrossRef CAS PubMed
- S. Wang, J. Xu, W. Wang, G.-J. N. Wang, R. Rastak, F. Molina-Lopez, J. W. Chung, S. Niu, V. R. Feig and J. Lopez, Nature, 2018, 555, 83–88 CrossRef CAS PubMed
- N. Chen, Y. Li, Y. Dai, W. Qu, Y. Xing, Y. Ye, Z. Wen, C. Guo, F. Wu and R. Chen, J. Mater. Chem. A, 2019, 7, 9530–9536 RSC
- W. Wang, Y. Liu, S. Wang, X. Fu, T. Zhao, X. Chen and Z. Shao, ACS Appl. Mater. Interfaces, 2020, 12, 25353–25362 CrossRef CAS PubMed
- X. F. Zhang, X. Ma, T. Hou, K. Guo, J. Yin, Z. Wang, L. Shu, M. He and J. Yao, Angew. Chem., Int. Ed., 2019, 58, 7366–7370 CrossRef CAS PubMed
- J. Le Bideau, L. Viau and A. Vioux, Chem. Soc. Rev., 2011, 40, 907–925 RSC
- L. Sun, S. Chen, Y. Guo, J. Song, L. Zhang, L. Xiao, Q. Guan and Z. You, Nano Energy, 2019, 63, 103847 CrossRef CAS
- Y. Yu, J. Zhong, W. Sun, R. Kumar and N. Koratkar, Adv. Funct. Mater., 2017, 27, 1606461 CrossRef
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC
- C. Meng, C. Liu, L. Chen, C. Hu and S. Fan, Nano Lett., 2010, 10, 4025–4031 CrossRef CAS PubMed
- W. Li, F. Gao, X. Wang, N. Zhang and M. Ma, Angew. Chem., 2016, 128, 9342–9347 CrossRef
- C. Lu and X. Chen, Nano Lett., 2020, 20, 1907–1914 CrossRef CAS PubMed
- Y. Liu, H. Li, X. Wang, T. Lv, K. Dong, Z. Chen, Y. Yang, S. Cao and T. Chen, J. Mater. Chem. A, 2021, 9, 12051–12059 RSC
- Y. Wang, H. Yuan, Y. Zhu, Z. Wang, Z. Hu, J. Xie, C. Liao, H. Cheng, F. Zhang and Z. Lu, Sci. China Mater., 2020, 63, 660–666 CrossRef
- J. Huang, S. Peng, J. Gu, G. Chen, J. Gao, J. Zhang, L. Hou, X. Yang, X. Jiang and L. Guan, Mater. Horiz., 2020, 7, 2085–2096 RSC
- J. Huang, J. Gu, J. Liu, J. Guo, H. Liu, K. Hou, X. Jiang, X. Yang and L. Guan, J. Mater. Chem. A, 2021, 9, 16345–16358 RSC
- B. Sun, G. Zhou, K. Xu, Q. Yu and S. Duan, ACS Mater. Lett., 2020, 2, 1669–1690 CrossRef CAS
- Y. Zou, V. Raveendran and J. Chen, Nano Energy, 2020, 77, 105303 CrossRef CAS
- K. Tao, Z. Chen, J. Yu, H. Zeng, J. Wu, Z. Wu, Q. Jia, P. Li, Y. Fu, H. Chang and W. Yuan, Advanced Science, 2022, 2104168 CrossRef PubMed
- L.-B. Huang, X. Dai, Z. Sun, M.-C. Wong, S.-Y. Pang, J. Han, Q. Zheng, C.-H. Zhao, J. Kong and J. Hao, Nano Energy, 2021, 82, 105724 CrossRef CAS
- Y. Wu, J. Qu, X. Zhang, K. Ao, Z. Zhou, Z. Zheng, Y. Mu, X. Wu, Y. Luo and S.-P. Feng, ACS Nano, 2021, 15, 13427 CrossRef CAS PubMed
- W. Gao, Z. Lei, C. Zhang, X. Liu and Y. Chen, Adv. Funct. Mater., 2021, 2104071 CrossRef CAS
- H. Wang, Y. Sun, T. He, Y. Huang, H. Cheng, C. Li, D. Xie, P. Yang, Y. Zhang and L. Qu, Nat. Nanotechnol., 2021, 16, 1–9 Search PubMed
- J. Duan, G. Feng, B. Yu, J. Li, M. Chen, P. Yang, J. Feng, K. Liu and J. Zhou, Nat. Commun., 2018, 9, 1–8 CrossRef PubMed
- D. J. Moran and F. C. Hollows, Br. J. Ophthalmol., 1984, 68, 343–346 CrossRef CAS PubMed
- D. Liang, Z. Lu, H. Yang, J. Gao and R. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 3958–3968 CrossRef CAS PubMed
- Y. Hu, Z. Zhang, Y. Li, X. Ding, D. Li, C. Shen and F. J. Xu, Macromol. Rapid Commun., 2018, 39, 1800069 CrossRef PubMed
- M. Yin, X. Wang, Z. Yu, Y. Wang, X. Wang, M. Deng, D. Zhao, S. Ji, N. Jia and W. Zhang, J. Mater. Chem. B, 2020, 8, 8395–8404 RSC
- H. Zhi, X. Fei, J. Tian, M. Jing, L. Xu, X. Wang, D. Liu, Y. Wang and J. Liu, J. Mater. Chem. B, 2017, 5, 5738–5744 RSC
- H. Zhi, X. Fei, J. Tian, L. Zhao, H. Zhang, M. Jing, L. Xu, Y. Wang and Y. Li, Biomater. Sci., 2018, 6, 2320–2326 RSC
- W. J. Yang, X. Tao, T. Zhao, L. Weng, E.-T. Kang and L. Wang, Polym. Chem., 2015, 6, 7027–7035 RSC
- Z. Cao, X. Luo, H. Zhang, Z. Fu, Z. Shen, N. Cai, Y. Xue and F. Yu, Cellulose, 2016, 23, 1349–1361 CrossRef CAS
- K. Bankoti, A. P. Rameshbabu, S. Datta, P. P. Maity, P. Goswami, P. Datta, S. K. Ghosh, A. Mitra and S. Dhara, Mater. Sci. Eng., C, 2017, 81, 133–143 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2022 |