
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo-dimensional benzo[1,2-b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) :4,5-b′]difurans as donor building blocks for the formation of novel donor–acceptor copolymers†
:4,5-b′]difurans as donor building blocks for the formation of novel donor–acceptor copolymers†
Carmen L.
Gott-Betts
,
Alfred A.
Burney-Allen
,
David L.
Wheeler
and
Malika
Jeffries-EL
 *
*
Department of Chemistry, Division of Materials Science and Engineering, Boston University, Boston, MA 02115, USA. E-mail: malikaj@bu.edu
First published on 12th May 2022
Abstract
Four donor–acceptor copolymers combining the novel electron-donating moiety, 2,3,6,7-tetra(thiophen-2-yl)benzo[1,2-b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4,5-b′]difuran (BDF) with either 2,3,1-benzothiadiazole (BT) or 2-octyl-2H-benzo[d][1,2,3]triazole (BTz) as the electron-accepting unit were prepared via the Stille cross-coupling reaction. Two different alkyl chains were attached on the BDF monomers and the impact on the polymers’ solubility and thin film morphology was evaluated. The resulting polymers had optical bandgaps between 1.7–1.9 eV and HOMO levels that ranged from −5.60 to −5.85 eV. In thin films, the BTz copolymer showed absorption bands in the range of 400–625 nm. The BT copolymer exhibited two absorption bands: one from 350–450 nm and a broad absorption in the range of 450–750 nm. When the polymers were used as donor materials with the electron-acceptor PC71BM in bulk-heterojunction photovoltaic cells, power conversion efficiencies of up to 2.93% were obtained. These results indicate that BDF is a promising building block for the synthesis of donor materials for use in organic solar cells, due to the favorable energy levels. Although, additional modifications are needed to improve device performance.
:4,5-b′]difuran (BDF) with either 2,3,1-benzothiadiazole (BT) or 2-octyl-2H-benzo[d][1,2,3]triazole (BTz) as the electron-accepting unit were prepared via the Stille cross-coupling reaction. Two different alkyl chains were attached on the BDF monomers and the impact on the polymers’ solubility and thin film morphology was evaluated. The resulting polymers had optical bandgaps between 1.7–1.9 eV and HOMO levels that ranged from −5.60 to −5.85 eV. In thin films, the BTz copolymer showed absorption bands in the range of 400–625 nm. The BT copolymer exhibited two absorption bands: one from 350–450 nm and a broad absorption in the range of 450–750 nm. When the polymers were used as donor materials with the electron-acceptor PC71BM in bulk-heterojunction photovoltaic cells, power conversion efficiencies of up to 2.93% were obtained. These results indicate that BDF is a promising building block for the synthesis of donor materials for use in organic solar cells, due to the favorable energy levels. Although, additional modifications are needed to improve device performance.
Introduction
The development of new conjugated polymers remains at the forefront of the field of materials chemistry due to their favourable optical and electronic properties which can be modulated through chemical synthesis.1–3 Additionally, these organic semiconductors can be fabricated into large area films using solution processing techniques, which would lower the cost of device fabrication.4,5 With a virtually infinite number of possible structural variants, synthetic chemists strive to achieve molecular perfection to optimize key figures of merit such as optical absorption, luminescence, frontier molecular orbital energy levels, charge transport and exciton dissociation.6,7 A common approach for tuning the optical and electronic properties of an organic semi-conductor is to alternate electron-donating and electron-accepting arenes within the pi-conjugated backbone, thus creating a “push–pull” system. The hybridized frontier molecular orbitals that result from this arrangement allow for facile tuning of the position of the lowest unoccupied molecular orbital (LUMO), highest occupied molecular orbital (HOMO), and the optical gap.6,8,9 Using this approach, it is possible to develop materials with broad absorption profiles, low-lying HOMOs, and high charge carrier mobilities all of which are ideal for use in bulk-heterojunction (BHJ) organic photovoltaics (OPV)s.10–12The benzo[1,2-b:4,5-b′]dithiophene (BDT) moiety has been comprehensively investigated as the electron-donating species in donor–acceptor (D/A) conjugated polymers.13–16 BDT has a planar backbone that facilitates pi-stacking and enhances charge carrier mobilities. As a result, BDT-based organic semiconductors are among the highest performing materials in the field of OPVs with photocurrent efficiencies (PCE) of ∼16.0%,17,18 and hole mobilities exceeding 0.1cm2V−1 s−1.19 Due to the success of BDT-based materials in these applications, our group has turned its focus towards the development its oxygen containing analog, benzo[1,2-b:4,5-b′]difuran (BDF). Furan is isoelectronic to thiophene, but oxygen has a smaller atomic radius than sulfur. Thus, the steric hindrance between adjacent heterocycles is reduced and the planarity between adjacent rings is increased.20,21 At the same time, BDF-based materials have been shown to exhibit improved solubility compared to their BDT analogs.20,22 Furthermore, the lower Dewar resonance energy of furan relative to thiophene makes the formation of quinoidal structures more favorable.23 Consequently, BDF containing materials have a narrower bandgap and more stabilized HOMO level in comparison to their BDT analogs.24–26
Previously we published the synthesis of a new BDF-based building block, 5,5′-(3,7-didecylbenzo[1,2-b:4,5-b′]difuran-2,6-diyl)bis(4-alkylthiophene-5,2-diyl)bis(trimethyl-stannane) and utilized it in D–A copolymers with 6,6′-dibromo-N,N′-(2-octyldodecanyl)-isoindigo.27 The resulting polymers exhibited two main absorption bands: a high energy band due to the π–π* transition, and a low energy band due to intramolecular charge transfer between the donor and acceptor units. However, the poor solubility of the copolymer adversely impacted the film morphology. As a result, the OPVs fabricated from these polymers exhibited low PCEs of ∼1.0%. In another report, we detailed the copolymerization of BDF with the electron deficient 1,4-diketopyrrolo[3,4-c]pyrrole (DPP) moiety.28 Although the absorption profile was strong and broad within the 600–800 nm range, like many DPP containing polymers, these materials also had poor solubility. As a result of the film morphology, modest device performances of ∼3.0% were attained.
In this report, to further improve upon our BDF building block we introduced thienyl substituents onto the BDF moiety. Using aromatic substituents instead of alkyl or alkoxy-chains broadens absorption profiles and improves the intramolecular charge transfer (ICT) within the material.24 Our functional BDF has reactive handles at the 3- and 7-positions. Thus, this molecule can be used to produce novel two-dimensional BDF monomers as most of the previous examples of two-dimensional BDT/BDF polymers have the aryl substituents at the 4- and 8-positions.19,22,29 Herein, We synthesized four donor–acceptor copolymers using a novel two-dimensional monomer 2,3,6,7-tetra(thiophen-2-yl)benzo[1,2-b:4,5-b′]difuran with either 2,3,1benzothiadiazole (BT) or 2-octyl-2H-benzo[d][1,2,3]triazole (BTz) as the comonomer. Here, BT and BTz were chosen for their facile synthesis and their demonstrated utility in high performing OPV materials.23,24 For the two-dimensional axillary thiophenes, alkyl side chains were attached to enhance solubility. The alkyl chain length was also varied to evaluate its impact on device performance.
Results and discussion
The synthesis of the benzodifuran monomers is shown in Scheme 1. Compound 2 was obtained from the Sonogashira cross-coupling reaction of 2-iodothiophene and 1,4-diethynyl-2,5-dimethoxybenzene.30 The 5 and 5′ positions of the thiophenes were protected with triisopropylsilyl (TIPS) groups to produce 3, which upon iodine mediated cyclization afforded the benzodifuran core 4. The subsequent Suzuki cross-coupling reaction of 4 with either 2-ethylhexyl (EH) 5a31 or 2-octyldodecyl (OD) 5b31 thiophenepinacolborane yielded 6a and 6b, respectively. Removal of the TIPS groups from 6a and 6b afforded 7a and 7b, respectively. The lithiation of 7a and 7b and subsequent quenching with trimethyltinchloride produced the target monomers 8a and 8b. A highlight of the monomer synthesis detailed here is that all intermediates and products were obtained in relatively high isolated yields ranging from 70%–93%. | ||
| Scheme 1 Synthesis of BDF monomers. | ||
The synthesis for the polymers is shown in Scheme 2. The polymers were all synthesized via Stille cross-coupling reaction of either 8a or 8b with the corresponding comonomers to yield polymers P1–P4. The yields ranged from 51–84% after purification via Sohxlet extraction. All polymers were soluble in chloroform at room temperature enabling characterization by proton nuclear magnetic resonance spectroscopy (1H NMR).
 | ||
| Scheme 2 Synthesis of D–A Copolymers P1–P4. | ||
The spectra obtained are consistent with the expected polymer structures. The molecular weights were assessed using gel permeation chromatography (GPC) at 25 °C using THF as the eluent. The resulting characterization data is summarized in Table 1. Comparing the molecular weights of polymers with the same alkyl chain (P1vs.P2, P3vs.P4), P2 has a slightly lower molecular weight (Mn and Mw) when compared to its BTz analog P1. Further, both polymers have low degrees of polymerization, indicating that the ethyl hexyl side chain is not adequate to impart solubility. Conversely, P3 has a lower molecular weight compared to P4. Assuming equal reactivity, this suggests that the additional alkyl chain on the BTz unit did not increase the solubility of the polymer. When comparing the polymers with the same acceptor units (P1vs.P3, P2vs.P4), the 2-ethylhexyl substituted polymers have lower molecular weights and degrees of polymerization than the 2-octyldodecyl variants. Assuming equal reactivity, this suggests that the longer branched alkyl substituents on the BDF core are better for improving solubility and increasing molecular weight.
| Polymer | Yielda (%) | M n (Da) | M w (Da) | Đ | DPn | T d (°C) |
|---|---|---|---|---|---|---|
| a Isolated yield after Soxhlet extraction. b Molecular weight data was obtained by GPC (see ESI). c 5% weight loss temperature determined by TGA. | ||||||
| P1 | 65 | 6155 | 9045 | 1.47 | 10 | 393 |
| P2 | 84 | 4898 | 6246 | 1.28 | 7 | 363 |
| P3 | 76 | 11453 |
17305 |
1.51 | 14 | 379 |
| P4 | 51 | 20054 |
29558 |
1.47 | 26 | 389 |
The thermal properties of the polymers were evaluated using differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA). DSC did not reveal any observable phase transitions for temperatures up to 250 °C. The TGA results are summarized in Table 1 and indicate that 5% weight loss onsets occurred between 363–393 °C. This thermal data supports that these polymer materials are thermally stable well above the operating temperature threshold for organic photovoltaic devices.32
Optical and electrochemical properties
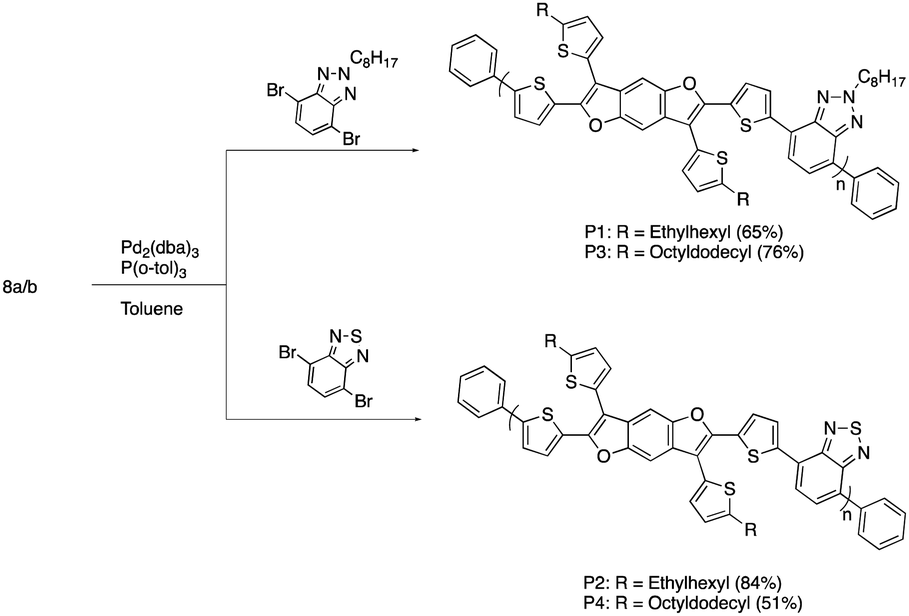
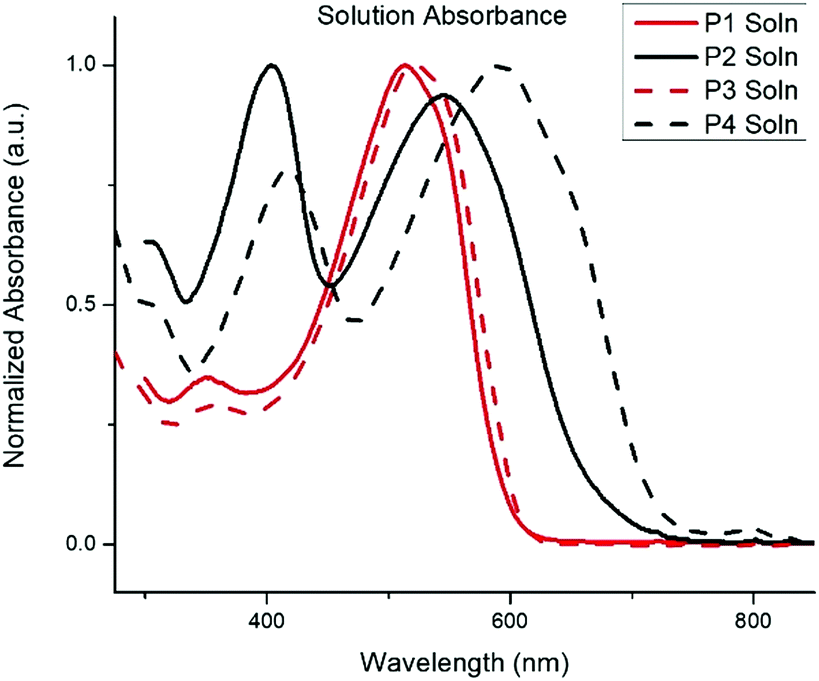
The normalized absorption spectra for P1–P4 in dilute CHCl3 solutions and thin films are shown in Fig. 1 and 2, respectively. The corresponding data is summarized in Table 2. The spectra for both BTz polymers P1 and P3 exhibit a weak high energy peak and a strong peak with a λmax at 539 and 546nm respectively. Whereas the spectra for the BT polymers P2 and P4 have two distinct absorption bands as is typically seen in D/A copolymers: a low energy peak resulting from intramolecular charge transfer (ICT) and a higher energy peak associated with the π–π* transition.33 In the solid-state spectra, the small high energy bands for P1 and P3 have almost disappeared, and the lower energy peaks now display small shoulders, slightly broadened peak widths, and concurrent red-shifting of the λmax. The side chains did not have any impact on the solid-state packing as the λmax for both P1 and P3 shifted by 26 nm from solution to film. Conversely, the spectra for the BT polymers showed a significant peak broadening between the solution and solid state along with a hypsochromic shift of all peaks. The extent of spectral broadening and shifting was dependent on the length of the side chains, with P4 being the broadest.
 | ||
| Fig. 1 Solution absorption profile of P1–P4 with the concentration of 10−5 M in chloroform. | ||
 | ||
| Fig. 2 Thin film absorption spectra of P1–P4. Films spun from CHCl3 solutions. | ||
| Polymer | λ max (nm) Solution | λ max (nm) Film |
E
OPTga (eV) |
HOMOb (eV) | LUMOc (eV) |
|---|---|---|---|---|---|
| a Optical bandgap as measured by the optical onset (Eg = 1240/λoffset). b HOMO levels measured by CV (see ESI). c Estimated LUMO = HOMO+Eg. | |||||
| P1 | 513 | 539 | 1.9 | −5.85 | −4.0 |
| P2 | 403, 545 | 421, 585 | 1.7 | −5.60 | −3.9 |
| P3 | 520 | 546 | 1.9 | −5.71 | −3.8 |
| P4 | 417, 592 | 437, 622 | 1.7 | −5.82 | −4.1 |
To evaluate the electrochemical properties of the polymers, the redox profile was evaluated using cyclic voltammetry, see ESI.† All four polymers exhibit measurable and reproducible oxidation processes, but reduction curves were not observed. The HOMO levels were estimated from the onset of oxidation using the absolute energy level of ferrocene/ferrocenium (Fc/Fc+) as 5.1 eV under vacuum and are summarized in Table 2. The polymers had calculated HOMO values between −5.60 eV and −5.85 eV affording good stability in air, and open circuit voltage (VOC), making them suitable for use in OPVs.34 The LUMO values were estimated from the HOMO and the optical bandgap and ranged from −3.8 eV to −4.1 eV. As a result, the offset between the polymer LUMO and that of the fullerene (−4.1 eV) acceptor is small, which can negatively impact device performance.35
Photovoltaic devices
The performance of all four polymers in OPV devices were evaluated using [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM) as the n-type electron acceptor with a device configuration of ITO/PEDOT:PSS/polymer:PC71BM/Ca/Al. The active layers were deposited from 30 mg mL−1o-DCB with 0–2% of 1-chloronapthalene (CN) as a high-boiling solvent additive to improve polymer/PCBM blend morphology. The current density–voltage curves are shown in the ESI.† The resulting short circuit current density (JSC), open circuit voltage (VOC), fill factor (FF), and power conversion efficiency (PCE) for the devices are shown in Table 3.| Polymer | Additive (%) | J SC (mA cm−2) | V OC (V) | FF | PCE (%) | RMS |
|---|---|---|---|---|---|---|
| Average of 8 devices. | ||||||
| P1 | None | 8.47 ± 0.40 | 0.50 ± 0.01 | 34.77 ± 1.31 | 1.67 ± 0.10 | — |
| 2% CN | 8.80 ± 0.79 | 0.60 ± 0.01 | 37.19 ± 1.03 | 1.98 ± 0.21 | 0.512 | |
| P2 | None | 6.69 ± 0.33 | 0.64 ± 0.02 | 35.10 ± 0.79 | 1.51 ± 0.09 | — |
| 1% CN | 6.99 ± 0.52 | 0.64 ± 0.4 | 33.62 ± 1.15 | 1.53 ± 0.15 | 0.558 | |
| P3 | None | 4.60 ± 0.19 | 0.81 ± 0.00 | 58.89 ± 1.10 | 2.22 ± 0.08 | — |
| 1% CN | 5.21 ± 0.27 | 0.83 ± 0.00 | 56.15 ± 1.05 | 2.45 ± 0.15 | 9.916 | |
| P4 | None | 5.79 ± 0.34 | 0.72 ± 0.01 | 65.36 ± 0.90 | 2.75 ± 0.15 | 0.739 |
| 1% CN | 5.47 ± 0.40 | 0.73 ± 0.00 | 57.90 ± 2.35 | 2.33 ± 0.18 | — | |
Among the devices fabricated without solvent additives, P4 gave the highest PCE at 2.93% with the highest FF of 64.86. P4 also demonstrated a moderate VOC of 0.73 V and moderate JSC of 6.18 mA cm−2. P1 and P2 gave lower efficiencies at 1.81% and 1.63% respectively. The lowest performing polymer, P2, had a modest VOC at 0.67 V but a lower FF of 35.07 and JSC of 6.93 mA cm−2. When CN was added, there was an increase in performance for all the polymers except for P4, due to increases in JSC and/or VOC. P3 had the highest performing device at 2.67% under these conditions. The highest performing device for P1 required a 2% CN additive compared to 1% for the other three polymers. The decrease in the P4 device performance with the addition of CN additive is likely a result of its higher solubility than the other polymers.
A comparison of the alkyl chains shows that the ethylhexyl polymers P1 and P2 had significantly lower FF than their octyldodecyl variants, P3 and P4. Conversely, P1 and P2 showed higher JSC compared to the octyldodecyl polymers, P3 and P4. Overall polymers with ethylhexyl side chains had poorer performance when compared to their octyldodecyl counterparts. Next, when comparing polymers with different electron withdrawing units, the nature of the electron deficient comonomer was not significant as P1 performed better than P2, but P4 performed better than P3.
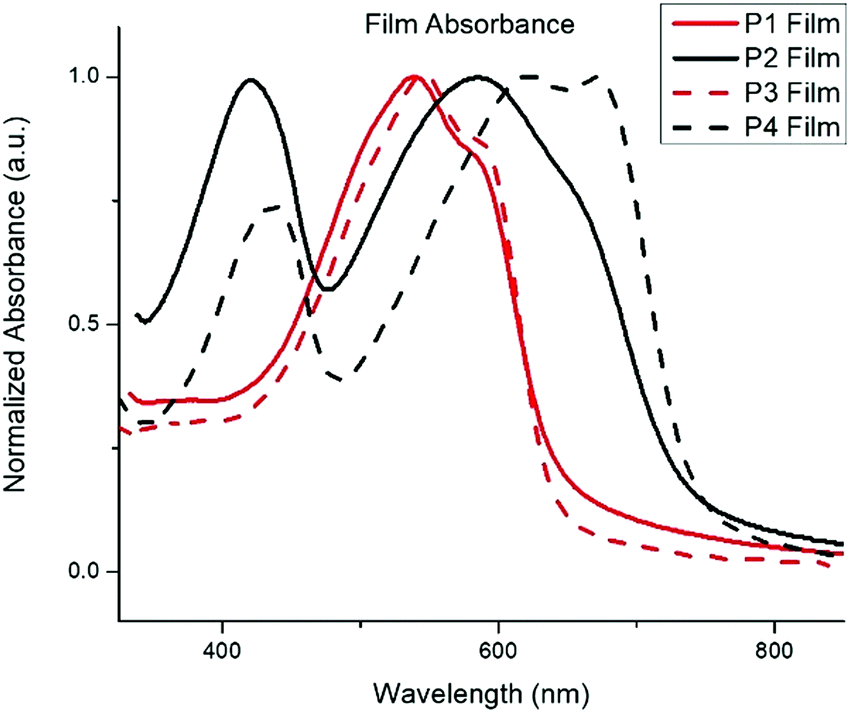
To further investigate the performance trends, the morphology of the highest performing polymer/PC71BM devices was evaluated using atomic force microscopy (AFM) as shown in Fig. 3. The root mean-square (RMS) values are shown in Table 3. When first evaluating the effect of alkyl chain length on the BDF donor, it was observed that P2 and P4 formed relatively smooth films with RMS roughness values of 0.56 and 0.74. The OPV performance increased from 1.75% for P2 to 2.93% with P4 even though P4 had a higher roughness value. Analogously, P1 has a very smooth film compared to P3 with RMS values of 0.51 and 9.9, respectively. Although P3 has a very large RMS value, the overall device performance was like P1 with PCE values of 2.67% and 2.33% respectively. While it has been reported that a higher RMS value may lead to better photovoltaic performance,36 high RMS values indicate that the device films made with P3 have pinholes and/or completely void areas. In comparing the electron accepting unit within the D–A polymer it was observed that P1 and P2 have similar RMS values of 0.51 and 0.56 respectively. Thus, the heterocycles have less of an impact on film morphology than the side chains do. Whereas, for P3 and P4, the RMS values were too different to determine if the difference in performance was due to the smoother film or to the intrinsic optical properties of the materials. However, it is impressive that P3 has such a high PCE considering the severe film defects. These results suggest that a mid-range side chain may be beneficial to improve device properties. Further optimization of the P3 device film would be necessary to produce truly optimized devices.
 | ||
| Fig. 3 AFM images of highest performing device active layers for P1–P4. | ||
Conclusions
A series of donor–acceptor copolymers based on electron withdrawing 4,7-dibromo-2-octyl-2H-benzo[d][1,2,3]triazole or 4,7-dibromobenzo[c][1,2,5]thiadiazole units and electron donating 2,3,6,7-tetra(thiophen-2-yl)benzo[1,2-b:4,5-b′]difurans bearing different alkyl chains were synthesized. Polymer pairings with the same comonomer had similar absorption profiles and optical bandgaps, regardless of the side-chain length. Whereas the polymer sets with the same alkyl chain had different optical profiles due to the relative difference in the strength of the electron acceptor unit. It was hypothesized that the two-dimensional BDF monomer would lead to improvements in OPV performance. The resulting polymers had low-lying LUMO levels, and deep HOMO-levels both of which were favorable for a novel OPV donor materials. However, the band gaps ranging from 1.7–1.9 eV, which combined with the poor film forming properties lead to modest OPV performances. Despite these outcomes, the aforementioned synthetic ease and oxidative stability make the BDT moiety a promising building block for designing new materials.37 In the future, we will investigate the BDF moiety with stronger electron-accepting comonomers. Doing this would both reduce the polymer bandgap and allow for the use of additional alkyl character to improve solubility and improve film formation.
Experimental methods
Materials
Air and moisture sensitive reactions were completed using standard Schlenk techniques. Solvents used for metal catalyzed reactions were deoxygenated prior to use by sparging with nitrogen while vigorously stirring for 30 minutes. SiliaFlash Irregular Silica Gels F60 was purchased from Silicyle for purification of materials using silica columns. All other chemical reagents were purchased from commercial sources. The molecules 4,7-dibromo-2-octyl-2H-benzo[d][1,2,3]triazole,38 4,7-dibromobenzo[c][1,2,5]thiadiazole,39 1,4-diethynyl-2,5-dimethoxybenzene (1),30 and 4,4,5,5-tetramethyl-2-(5-(2-octyldodecyl)thiophen-2-yl)-1,3,2-dioxaborolane31 were prepared according to literature procedures. The synthesis of 2-[5-(2-ethylhexyl)thien-2-yl]-4,4,5,5-tetramethyl-1,3,2-dioxaborolane is provided in the ESI.†Characterization
Nuclear magnetic resonance (NMR) spectra were obtained using chloroform-d unless otherwise noted and recorded on an Agilent 500 MHz VNMRS spectrometer with a Varian ultra-shielded magnet or an Agilent 400 MHz VNMRS unity plus spectrometer with an Oxford Instruments superconducting magnet. The 1H NMR spectra were internally referenced to the residual protonated solvent peak and all chemical shifts are given in ppm (δ) relative to the solvent, 7.26 for chloroform. The gel permeation chromatography (GPC) measurements were conducted at 25 °C using THF and a flow rate of 1.0 mL min−1. Calibration was based on polystyrene standards. Thermogravimetric analysis (TGA) measurements were performed over an interval of 100–500 °C at a heating rate of 10 °C min−1 under nitrogen atmosphere. Differential scanning calorimetry (DSC) was obtained using a scan heating rate of 10 °C min−1 between −30 °C and 250 °C. Cyclic voltammetry (CV) was performed using an e-DAQ e-corder 410 potentiostat with a scanning rate of 100 mV s−1. The polymer solutions (5 mg mL−1 in chloroform) were drop-cast on a platinum electrode and Ag/Ag+ was used as the reference electrode and a platinum wire as the auxiliary electrode. The reported values are referenced to Fc/Fc+ (−5.1 eV versus vacuum).40 All cyclic voltammetry experiments were performed in deoxygenated acetonitrile under an argon atmosphere using 0.1 M tetrabutylammonium hexafluorophosphate as the electrolyte. Absorption spectra were obtained on a photodiode-array Shimadzu UV-1800 ultraviolet spectrophotometer using polymer solutions in chloroform and thin films. The thin films were made by spin-coating 25 × 25 × 1 mm glass slides using solutions of polymer (5 mg mL−1) in chloroform at a spin rate of 1200 rpm.Fabrication of photovoltaic devices
All organic photovoltaic devices were performed under inert atmosphere using solution-based spin casting. The indium tin oxide (ITO) substrates had a resistance of 40 Ohm. The device size was 25 mm. Conventional architecture was used to fabricate organic photovoltaic devices via (ITO)/Poly(3,4-ethylenedioxythiophene): poly(styrene sulfonate) (PEDOT:PSS)/polymer:[6,6]-phenyl-C71-butyric acid methyl ester (PC71BM)/Ca/Al). ITO glass substrates were cleaned via sonication in (1) Mucasol, (2) deionized water, (3) acetone, and (4) isopropanol for 10 minutes each respectively. Slides were dried in an oven followed by UV-ozone treatment for 15 minutes. PEDOT:PSS layers (Clevios P VP Al 4083) were filtered (0.45 μm) and spin coated onto ITO substrates at 3500 rpm for 2 minutes and annealed for 100 °C for 30 min in air. After cooling, substrates were transferred into a nitrogen filled glove box. All polymer:PC71BM solutions were prepared in 1:2–3 and 1:1 blend ratio (20 mg mL−1), respectively, with chlorobenzene as the processing solvent. The prepared solutions were allowed to stir for 24 h at 80 °C prior to spin coating onto the PEDOT:PSS layer at 1000 rpm for 2 minutes. Following spin coating, Calcium (15 nm) and Aluminum (100 nm) electrodes underwent successful deposition via thermal evaporation. Films were also evaluated under post thermal annealing conditions at 100 °C for 10 minutes. Current–density (J–V) data were obtained via Keithley 2400 source meter and simulated AM 1.5 G illumination (100 mW cm−2, Newport 91160) calibrated using a KG-5 filter Silicon reference cell.
Synthesis
:1) and 2-iodothiophene (7.10 mL, 64.27 mmol) then stirred overnight at room temperature. The reaction mixture was poured into ice and extracted three times with dichloromethane. The combined organic layers were washed with saturated aqueous NH4Cl, water, and brine. The organic layer was dried over MgSO4, evaporated, and purified using a silica plug (hex/DCM 4:1). The resulting solid was recrystallized from ethyl acetate/hexanes (2:1) to yield 2 as a yellow solid (8.09 g, 79% yield). 1H NMR (500 MHz, CDCl3) δ 7.31 (m, 4H), 7.02 (m, 4H), 3.90 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 153.87, 132.37, 127.75, 127.27, 123.28, 115.44, 113.28, 89.47, 88.40, 77.41, 77.16, 76.91, 56.58. LRMS (ESI) m/z: [M+H]+ calculated for C20H14O2S2:350.04; found: 351.0513 m/z, error 1.0113 ppm.
:1) (5.68 g, 70% yield). 1H NMR (500 MHz, CDCl3) δ 7.39 (d, J = 3.53 Hz, 2H), δ 7.15 (d, J = 3.5 Hz, 2H), δ 6.99 (s, 2H), δ 3.88 (s, 6H), δ 1.34 (m, 6H), 1.11 (d, J= 7.44 Hz, 36H). 13C NMR (125 MHz, CDCl3) δ 153.57, 138.10, 135.28, 132.75, 127.74, 115.13, 113.13, 90.41, 88.22, 77.16, 76.91, 76.65, 56.33, 18.41, 11.65. HRMS (ESI) m/z: [M+H]+ calculated for C38H54O2S2Si2:663.3182; found: 663.3209 m/z, error 4.0704 ppm.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :4,5-b′]difuran-2,6-diyl)bis(thiophene-5,2-diyl))bis(triisopropylsilane) (4).
A 500 mL Erlenmeyer flask was charged with 3 (5.30 g, 8 mmol) which was dissolved in CH2Cl2 and cooled to 0 °C. The solution was rapidly stirred as iodine (6.09 g, 23.98 mmol) in 200 mL CH2Cl2/hexanes (3:1) was added dropwise over 10 minutes. Upon completion of the addition, the reaction mixture was stirred at 0 °C for 4 hours. The cold reaction mixture was then filtered to collect the crude product. The solid was stirred in hot hexanes, cooled, and filtered. The yellow solid was then recrystallized from toluene to afford yellow crystals (5.18 g, 73% yield). 1H NMR (500 MHz, CDCl3) δ 8.08 (d, J = 3.6 Hz, 2H), δ 7.45 (s, 2H), δ 7.33 (d, J = 3.6 Hz, 2H), δ 1.40 (m, 6H), 1.15 (d, J = 7.4 Hz, 36H). HRMS (ESI) m/z: [M+H]+ calculated for C36H48I2O2S2Si2:886.07; found: 887.0829, error 3.0437 ppm.
:1) under nitrogen. The reaction mixture was heated at 80 °C overnight. Next, the mixture was poured into 100 mL of water and extracted three times with dichloromethane. The combined organic layers were washed with deionized water, NH4Cl, brine, and dried over MgSO4. The solvent was removed in vacuo and the residue was sent through a silica plug using hexanes then used without further purification as a viscous, orange semi-solid. (6a, 86%): 1H NMR (500 MHz, CDCl3) δ 7.60 (s, 2H), δ 7.57 (d, J = 3.60 Hz, 2H), δ 7.17 (d, J = 3.61 Hz, 2H), δ 7.12 (d, J = 3.40 Hz, 2H), δ 6.86 (d, J = 3.39 Hz, 2H), δ 2.85 (d, J = 6.69 Hz, 4H), δ 1.67 (m, 2H), δ 1.30–1.45 (m, 22H), δ 1.11 (d, J = 7.4 Hz, 36H), δ 0.95 (m, 12H). HRMS (ESI) m/z: [M+H]+ calculated for C60H86O2S4Si2:1022.50; found: 1023.5139, error 1.0747 ppm. (6b, 81%): 1H NMR (400 MHz, CDCl3) δ 7.60 (s, 2H), 7.56 (d, J = 3.6 Hz, 2H), 7.17 (d, J = 3.7 Hz, 2H), 7.12 (d, J = 3.3 Hz, 2H), 6.85 (d, J = 3.3 Hz, 2H), 2.85 (d, J = 6.6 Hz, 4H), 1.71 (m, 2H), 1.36 (m, 70H), 1.11 (d, J = 7.3 Hz, 36H), 0.89 (m, 12H).
:710.24; found:: 711.2477, error 2.5308 ppm. (7b, 93%): 1H NMR (500 MHz, CDCl3) δ 7.58 (d, J = 0.6 Hz, 2H), 7.51 (d, J = 3.7 Hz, 2H), 7.30 (d, J = 6.1 Hz, 2H), 7.11 (d, J = 3.4 Hz, 2H), 7.07–7.00 (m, 2H), 6.86 (d, J = 3.4 Hz, 2H), 2.85 (d, J = 6.7 Hz, 4H), 1.74–1.69 (m, 2H), 1.36–1.21 (m, 64H), 0.88 (d, J = 5.1 Hz, 12H). 13C NMR (125 MHz, CDCl3) δ 151.20, 148.68, 146.51, 132.61, 129.59, 129.36, 128.24, 127.50, 126.64, 110.83, 40.25, 34.93, 33.51, 32.08, 31.75, 30.19, 29.87, 29.53, 26.84, 25.44, 22.85, 14.27. HRMS (ESI) m/z: [M+H]+ calculated for C66H94O2S4:1046.61; found: 1047.6223, error 0.7636 ppm.
:1H NMR (500 MHz, CDCl3) δ 8.04 (br, 2H), δ 7.82 (br, 1H), δ 7.45 (br, 4H), δ 7.12 (br, 2H), δ 6.88 (br, 2H), δ 6.78 (br, 1H), δ 4.79 (br, 2H), δ 2.74–2.90 (br, 4H), δ 2.20 (br, 2H), δ 1.28–1.71 (br, 28H), δ 0.87–0.98 (br, 15H). P3:1H NMR (500 MHz, CDCl3, δ) 8.08 (br, 2H), 7.84 (br, 1H), 7.58 (br, 2H), 7.43 (br, 2H), 7.12 (br, 2H), 6.92 (br, 2H), 6.77 (br, 1H), 4.80 (br, 2H), 2.89 (br, 4H), 2.21 (br, 2H), 1.24–1.39 (br, 76H), 0.86 (br, 15H).
:1H NMR (500 MHz, CDCl3) δ 8.03 (br, 1H), 7.76 (br, 2H), 7.49 (br, 3H), 7.10 (br, 3H), 6.92 (br, 2H), 6.77 (br, 1H), 2.75–2.90 (br, 6H), 1.26–1.73 (br, 16H), 0.89–1.06 (br, 12H). P4:1H NMR (500 MHz, CDCl3, δ) 8.08 (br, 1H), 7.81 (br, 2H), 7.55 (br, 3H), 7.15 (br, 3H), 6.93 (br, 1H), 2.92 (br, 4H), 2.74 (br, 2H), 1.26–1.57 (br, 63H), 0.86 (br, 12H).
:4,5-b′]difuran-2,6-diyl)bis(thiophene-5,2-diyl))bis(triisopropylsilane) (4).
A 500 mL Erlenmeyer flask was charged with 3 (5.30 g, 8 mmol) which was dissolved in CH2Cl2 and cooled to 0 °C. The solution was rapidly stirred as iodine (6.09 g, 23.98 mmol) in 200 mL CH2Cl2/hexanes (3:1) was added dropwise over 10 minutes. Upon completion of the addition, the reaction mixture was stirred at 0 °C for 4 hours. The cold reaction mixture was then filtered to collect the crude product. The solid was stirred in hot hexanes, cooled, and filtered. The yellow solid was then recrystallized from toluene to afford yellow crystals (5.18 g, 73% yield). 1H NMR (500 MHz, CDCl3) δ 8.08 (d, J = 3.6 Hz, 2H), δ 7.45 (s, 2H), δ 7.33 (d, J = 3.6 Hz, 2H), δ 1.40 (m, 6H), 1.15 (d, J = 7.4 Hz, 36H). HRMS (ESI) m/z: [M+H]+ calculated for C36H48I2O2S2Si2:886.07; found: 887.0829, error 3.0437 ppm.
:1) under nitrogen. The reaction mixture was heated at 80 °C overnight. Next, the mixture was poured into 100 mL of water and extracted three times with dichloromethane. The combined organic layers were washed with deionized water, NH4Cl, brine, and dried over MgSO4. The solvent was removed in vacuo and the residue was sent through a silica plug using hexanes then used without further purification as a viscous, orange semi-solid. (6a, 86%): 1H NMR (500 MHz, CDCl3) δ 7.60 (s, 2H), δ 7.57 (d, J = 3.60 Hz, 2H), δ 7.17 (d, J = 3.61 Hz, 2H), δ 7.12 (d, J = 3.40 Hz, 2H), δ 6.86 (d, J = 3.39 Hz, 2H), δ 2.85 (d, J = 6.69 Hz, 4H), δ 1.67 (m, 2H), δ 1.30–1.45 (m, 22H), δ 1.11 (d, J = 7.4 Hz, 36H), δ 0.95 (m, 12H). HRMS (ESI) m/z: [M+H]+ calculated for C60H86O2S4Si2:1022.50; found: 1023.5139, error 1.0747 ppm. (6b, 81%): 1H NMR (400 MHz, CDCl3) δ 7.60 (s, 2H), 7.56 (d, J = 3.6 Hz, 2H), 7.17 (d, J = 3.7 Hz, 2H), 7.12 (d, J = 3.3 Hz, 2H), 6.85 (d, J = 3.3 Hz, 2H), 2.85 (d, J = 6.6 Hz, 4H), 1.71 (m, 2H), 1.36 (m, 70H), 1.11 (d, J = 7.3 Hz, 36H), 0.89 (m, 12H).
:710.24; found:: 711.2477, error 2.5308 ppm. (7b, 93%): 1H NMR (500 MHz, CDCl3) δ 7.58 (d, J = 0.6 Hz, 2H), 7.51 (d, J = 3.7 Hz, 2H), 7.30 (d, J = 6.1 Hz, 2H), 7.11 (d, J = 3.4 Hz, 2H), 7.07–7.00 (m, 2H), 6.86 (d, J = 3.4 Hz, 2H), 2.85 (d, J = 6.7 Hz, 4H), 1.74–1.69 (m, 2H), 1.36–1.21 (m, 64H), 0.88 (d, J = 5.1 Hz, 12H). 13C NMR (125 MHz, CDCl3) δ 151.20, 148.68, 146.51, 132.61, 129.59, 129.36, 128.24, 127.50, 126.64, 110.83, 40.25, 34.93, 33.51, 32.08, 31.75, 30.19, 29.87, 29.53, 26.84, 25.44, 22.85, 14.27. HRMS (ESI) m/z: [M+H]+ calculated for C66H94O2S4:1046.61; found: 1047.6223, error 0.7636 ppm.
:1H NMR (500 MHz, CDCl3) δ 8.04 (br, 2H), δ 7.82 (br, 1H), δ 7.45 (br, 4H), δ 7.12 (br, 2H), δ 6.88 (br, 2H), δ 6.78 (br, 1H), δ 4.79 (br, 2H), δ 2.74–2.90 (br, 4H), δ 2.20 (br, 2H), δ 1.28–1.71 (br, 28H), δ 0.87–0.98 (br, 15H). P3:1H NMR (500 MHz, CDCl3, δ) 8.08 (br, 2H), 7.84 (br, 1H), 7.58 (br, 2H), 7.43 (br, 2H), 7.12 (br, 2H), 6.92 (br, 2H), 6.77 (br, 1H), 4.80 (br, 2H), 2.89 (br, 4H), 2.21 (br, 2H), 1.24–1.39 (br, 76H), 0.86 (br, 15H).
:1H NMR (500 MHz, CDCl3) δ 8.03 (br, 1H), 7.76 (br, 2H), 7.49 (br, 3H), 7.10 (br, 3H), 6.92 (br, 2H), 6.77 (br, 1H), 2.75–2.90 (br, 6H), 1.26–1.73 (br, 16H), 0.89–1.06 (br, 12H). P4:1H NMR (500 MHz, CDCl3, δ) 8.08 (br, 1H), 7.81 (br, 2H), 7.55 (br, 3H), 7.15 (br, 3H), 6.93 (br, 1H), 2.92 (br, 4H), 2.74 (br, 2H), 1.26–1.57 (br, 63H), 0.86 (br, 12H).
Author contributions
Carmen Gott-Betts: conceptualization, methodology, investigation, writing – original draft, writing – review & editing, and visualization; Alfred Burney Allen: methodology, investigation, writing – original draft, and writing – review & editing; David Wheeler: methodology, investigation, writing – original draft, and writing – review & editing; Malika Jeffries-EL: conceptualization, methodology, investigation, writing – original draft, writing – review & editing, visualization supervision, project administration and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Norman Lee and Dr Paul Ralifo from the Chemical Instrumentation Facility at Boston University for analysis of our molecules and polymers. We also thank Dr Anlee Krupp for help with AFM analysis of polymer films. Next, the authors would like to thank the Center for Electronic Materials and Devices at the University of Massachusetts-Amherst for device fabrication. We also thank Professor Mark Grinstaff for usage of thermal gravimetric analysis equipment and Aidan Murphy and Sara El-Arid for help with gel permeation chromatography analysis. Lastly, we thank the National Science Foundation (DMR-1410088/1640297) for and Boston University for financial support of this work.References
- S. C. Rasmussen, ChemPlusChem, 2020, 85, 1412–1429 CrossRef CAS PubMed.
- R. M. Pankow and B. C. Thompson, Polymer, 2020, 207, 122874 CrossRef CAS.
- Z. Qiu, B. A.-G. Hammer and K. Müllen, Prog. Polym. Sci., 2020, 100, 101179 CrossRef CAS.
- I. Burgues-Ceballos, M. Stella, P. Lacharmoise and E. Martinez-Ferrero, J. Mater. Chem. A, 2014, 2, 17711–17722 RSC.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- T. Mikie and I. Osaka, J. Mater. Chem. C, 2020, 8, 14262–14288 RSC.
- I. Osaka, in Organic Solar Cells: Energetic and Nanostructural Design, ed. M. Hiramoto and S. Izawa, Springer, Singapore, 2021, pp. 89–121, DOI:10.1007/978-981-15-9113-6_5.
- E. E. Havinga, W. ten Hoeve and H. Wynberg, Polym. Bull., 1992, 29, 119–126 CrossRef CAS.
- E. E. Havinga, W. ten Hoeve and H. Wynberg, Synth. Met., 1993, 55, 299–306 CrossRef CAS.
- W. Chen, G. Huang, X. Li, Y. Li, H. Wang, H. Jiang, Z. Zhao, D. Yu, E. Wang and R. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 33173–33178 CrossRef CAS PubMed.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- C. Lee, S. Lee, G.-U. Kim, W. Lee and B. J. Kim, Chem. Rev., 2019, 119, 8028–8086 CrossRef CAS PubMed.
- J. G. Laquindanum, H. E. Katz, A. J. Lovinger and A. Dodabalapur, Adv. Mater., 1997, 9, 36–39 CrossRef CAS.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS PubMed.
- Y. Liang and L. Yu, Acc. Chem. Res., 2010, 43, 1227–1236 CrossRef CAS PubMed.
- Y. Che, Y. Zhang, Y. Yang, C.-H. Liu, R. Izquierdo, S. S. Xiao and D. F. Perepichka, J. Org. Chem., 2020, 85, 52–61 CrossRef CAS PubMed.
- S. Liu, J. Yuan, W. Deng, M. Luo, Y. Xie, Q. Liang, Y. Zou, Z. He, H. Wu and Y. Cao, Nat. Photonics, 2020, 14, 300–305 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, T. Zhang, Y. Wang, L. Hong, K. Xian, B. Xu, S. Zhang, J. Peng, Z. Wei, F. Gao and J. Hou, Nat. Commun., 2019, 10, 2515 CrossRef PubMed.
- Y.-W. Huang, Y.-C. Lin, Y.-S. Wu, Y.-T. Wong, M.-Y. Kuo, W.-C. Chen and C.-C. Chueh, Ind. Eng. Chem. Res., 2020, 59, 9105–9115 CrossRef CAS.
- O. Gidron and M. Bendikov, Angew. Chem., Int. Ed., 2014, 53, 2546–2555 CrossRef CAS PubMed.
- M. Jeffries-El, B. M. Kobilka and B. J. Hale, Macromolecules, 2014, 47, 7253–7271 CrossRef CAS.
- L. Liang, J.-T. Wang, X. Xiang, J. Ling, F.-G. Zhao and W.-S. Li, J. Mater. Chem. A, 2014, 2, 15396–15405 RSC.
- R. S. Hosmane and J. F. Liebman, Tetrahedron Lett., 1992, 33, 2303–2306 CrossRef CAS.
- L. Huo, T. Liu, B. Fan, Z. Zhao, X. Sun, D. Wei, M. Yu, Y. Liu and Y. Sun, Adv. Mater., 2015, 27, 6969–6975 CrossRef CAS PubMed.
- J. Warnan, C. Cabanetos, A. E. Labban, M. R. Hansen, C. Tassone, M. F. Toney and P. M. Beaujuge, Adv. Mater., 2014, 4357–4362, DOI:10.1002/adma.201305344.
- E. He, Y. Lu, Z. Zheng, F. Guo, S. Gao, L. Zhao and Y. Zhang, J. Mater. Chem. C, 2020, 8, 139–146 RSC.
- B. M. Kobilka, A. V. Dubrovskiy, M. D. Ewan, A. L. Tomlinson, R. C. Larock, S. Chaudhary and M. Jeffries-EL, Chem. Commun., 2012, 48, 8919–8921 RSC.
- B. M. Kobilka, B. J. Hale, M. D. Ewan, A. V. Dubrovskiy and M. Jeffries-El, Polym. Chem., 2013, 3, 5329–5336 RSC.
- C. Gu, D. Liu, J. Wang, Q. Niu, C. Gu, B. Shahid, B. Yu, H. Cong and R. Yang, J. Mater. Chem. A, 2018, 6, 2371–2378 RSC.
- J. L. Novotney and W. R. Dichtel, ACS Macro Lett., 2013, 2, 423–426 CrossRef CAS.
- M. Scheuble, Y. M. Gross, D. Trefz, M. Brinkmann, J. T. López Navarrete, M. C. Ruiz Delgado and S. Ludwigs, Macromolecules, 2015, 48, 7049–7059 CrossRef CAS.
- G. Bardizza, E. Salis, C. Toledo and E. D. Dunlop, Prog. Photovoltaics, 2020, 28, 593–600 Search PubMed.
- B. C. Thompson, Y.-G. Kim and J. R. Reynolds, Macromolecules, 2005, 38, 5359–5362 CrossRef CAS.
- C. G. Tang, M. C.-Y. Ang, K.-K. Choo, V. Keerthi, J.-K. Tan, M. N. Syafiqah, T. Kugler, J. H. Burroughes, R.-Q. Png, L.-L. Chua and P. K.-H. Ho, Nature, 2016, 539, 536–540 CrossRef CAS PubMed.
- N. A. Ran, J. A. Love, M. C. Heiber, X. Jiao, M. P. Hughes, A. Karki, M. Wang, V. V. Brus, H. Wang, D. Neher, H. Ade, G. C. Bazan and T.-Q. Nguyen, Adv. Energy Mater., 2018, 8, 1701073 CrossRef.
- M. He, W. Han, J. Ge, Y. Yang, F. Qiu and Z. Lin, Energy Environ. Sci., 2011, 4, 2894–2902 RSC.
- X. Li, Y. Li, Y. Zhang and Y. Sun, Small Sci., 2022, 2200006 CrossRef.
- A. Balan, G. Gunbas, A. Durmus and L. Toppare, Chem. Mater., 2008, 20, 7510–7513 CrossRef CAS.
- B. Burkhart, P. P. Khlyabich, T. Cakir Canak, T. W. LaJoie and B. C. Thompson, Macromolecules, 2011, 44, 1242–1246 CrossRef CAS.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of intermediates, 1H NMR spectra, and CV traces. See DOI: https://doi.org/10.1039/d2ma00116k |
| This journal is © The Royal Society of Chemistry 2022 |