
A chemical approach for the future of PLA upcycling: from plastic wastes to new 3D printing materials†
Lin
Shao
,
Yu-Chung
Chang
 *,
Cheng
Hao
,
Ming-en
Fei
,
Baoming
Zhao
,
Brian J.
Bliss
and
Jinwen
Zhang
*
*,
Cheng
Hao
,
Ming-en
Fei
,
Baoming
Zhao
,
Brian J.
Bliss
and
Jinwen
Zhang
*
School of Mechanical and Materials Engineering, Composite Materials and Engineering Center, Washington State University, 2001 Grimes Way, Pullman, Washington 99164, USA. E-mail: yu-chung.chang@wsu.edu; jwzhang@wsu.edu
First published on 12th July 2022
Abstract
As the demand for PLA increases, post-consumer disposal strategies must be carefully considered. While we would love to embrace a bioplastic future, we also need to tread carefully. Though PLA is widely claimed to be biodegradable, full degradation often requires conditions not typically found in landfills or industrial composting. Therefore, it will negatively impact the environment if treated carelessly. In this work, we report a simple PLA upcycling path to turn existing PLA wastes into new 3D printable materials within 48 hours. The ester bonds of PLA can be cleaved efficiently via aminolysis. The obtained monomeric compound was derivatized with methacrylic anhydride, which introduces double bonds and thus a cross-linkable monomer is obtained. In combination with a comonomer and initiator, a photocurable resin is produced. The resin can be fed into any commercially available photocuring 3D printer. The 3D printed parts derived from PLA wastes exhibit impressive performances with a tensile strength of 58.6 MPa, Young's modulus of 2.8 GPa, and glass transition at ∼180 °C. Our work demonstrates a new route to active upcycling of PLA while minimizing the need for disposal.
1. Introduction
The exponential growth of polylactic acid (PLA) can be attributed to the following causes: the rapid expansion of consumer desktop 3D printing, namely the fused deposition modeling (FDM) or the fused filament fabrication (FFF), and the rise in environmental awareness which drives the pursuit of biobased and biodegradable plastics. Of all the bioplastics in production, sugar or corn starch-based PLA is one of the most promising alternatives to petroleum-based plastics such as polyethylene (PE) and polypropylene (PP). PLA was first synthesized by Theophile-Jules Pelouze in 1845 but was not fully utilized until 1954 by DuPont.1 The mechanical properties of virgin PLA are fairly impressive with a high modulus of 3 GPa and a high strength of 50–70 MPa.2 Today PLA has a wide variety of applications ranging from consumer 3D printing, food packaging/cutlery, and agricultural films.3–5In the eyes of the public, PLA is often regarded as a “biodegradable” plastic and is believed to have less environmental impact than conventional petroleum polymer materials. However, this is not entirely accurate. According to the International Universal Recycling Codes (IURC), PLA is categorized as #7. The #7 category is considered the “everything else” sector and minimal to no recycling effort at all is given to these plastics. Therefore, PLA itself lacks the proper infrastructure and recycling loop for a sustainable cycle. Also, improper dumping could also create a sorting problem with plastics from other categories due to contamination issues. Although PLA is known for its biodegradability, this is only true under specific conditions. For PLA to naturally degrade under typical landfill conditions, it could take up to 100 years.6 In the industrial composting plant, PLA can be fully decomposed if maintained at a temperature of 60 °C and with a constant feeding of micro-organism for degradation assistance.7 Both landfill and industrial composting require considerable amounts of time and energy. Researchers also reported that PLA in fresh and sea water immersion results in little to no mass loss even after 1 year.8 Therefore, direct disposal of PLA wastes will create a serious environmental burden and potentially hazardous conditions to wildlife and ecosystems itself.
Traditionally, thermoplastics like PLA can be melted and reformed into new shapes, hence follow the physical recycling route. However, such methods have an inherent limitation. The repeating melt processes cause the thermal and mechanical degradation of polymer in each cycle, resulting in lower overall performance each time. Tensile strength of PLA decreased from 66 to 25 MPa after 7 cycles of injection moulding.9 Ultimately, after multiple cycles, the materials become oxidized and unusable. On the other hand, upcycling via chemical recycling of PLA has started to attract attention. Chemical recycling is mainly divided into four types: hydrolysis, alcoholysis, ammonolysis, and aminolysis. Hydrolysis has attracted much attention from researchers because the final product of PLA after hydrolysis is lactic acid. A closed loop can be realized from lactic acid to PLA. However, hydrolysis usually requires harsh degradation conditions, like large quantities of concentrated acids or bases, and relatively high temperature and pressure (120–350 °C, ∼100 bar).10–13 Alcoholysis is also an efficient method to decompose PLA, but transesterification catalysts are necessary to promote the degradation. Lewis acid is an effective catalyst, which is commonly used in transesterification reactions. Collinson et al. demonstrated Zn(OAc)2 as a catalyst for PLA alcoholysis in methanol and ethanol.14 PLA alcoholysis in methanol showed a higher yield (∼70%) after 15 h. Liu et al. utilized a series of Lewis acids to degrade PLA pellets in methanol.15 FeCl3 showed the best catalytic efficiency among the investigated Lewis acids. The methyl lactate yield was as high as 87.2% at 130 °C for 4 h under the catalysis of FeCl3. Other catalytic systems, like ionic liquids and organic catalysts, show fair efficiency in alcoholysis.16–19 However, considering their high cost, they are not suitable for a large scale and industry production. Besides, alcohol type solvents, like methanol and ethanol, usually exhibit low boiling points, so high pressure is also required, which could create safety concerns. Moreover, the toxic catalysts retained in the decomposed polymer matrix are difficult to remove. Ammonolysis is a chemical reaction in which ammonia acts as a reactant. Ma et al. proposed a novel transformation process based on ammonolysis upcycling of PLA waste into value-added products (alanine) with high selectivity (94%).20 However, the process requires a noble metal catalyst and the low boiling point NH3·H2O, making the potential industrial scale-up more demanding. Aminolysis is another effective and efficient method to break up ester bonds, which has been widely studied in PET degradation.21,22 To the best of our knowledge, there has been no research on PLA aminolysis in the literature. Amine is a nucleophilic group which can easily attack ester bonds to form amide bonds without the need for catalysts. Therefore, aminolysis is an efficient and effective method to decompose polymers containing plenty of ester bonds.
In this study, we demonstrate a facile, mild, fast, and catalyst free PLA recycling method via aminolysis. The degradation product is a distinct amide diol compound. Moreover, by modifying the diol compound with methacrylic anhydride (MAh), a functional photo-curable reactive monomer/crosslinker was synthesized and used in 3D printing application. To the best of our knowledge, this is the first report on the aminolysis of PLA into N-lactoyl ethanolamine (N-LEA), converting an acrylic decomposed monomer into a photo-curable 3D printing ink by mixing with 4-acryloylmorpholine (ACMO). Finally, a high Tg, high tensile strength, and high tensile modulus crosslinked thermosetting polymer was developed.
2. Experimental section
2.1. Materials and chemicals
Injection molded PLA was made of virgin PLA 4043D (NatureWorks®, Minnetonka, MN), which is commonly used for commercial 3D printing filament production, by our lab using minijet (Thermo Scientific) without adding any additives. PLA 3D printed wastes, including supporting structures from 3D printing and post-consumer 3D printing parts, were collected from our lab (generic brands). 2-Aminoethanol (EA, 99+%, TCI), methacrylic anhydride (MAh, Sigma-Aldrich, 94%), dimethylaminopyridine (DMAP, 98%, Oakwood Chemical), 4-acryloylmorpholine (ACMO, 99+%, CHEM IMPEX), and (phenylphosphoryl)bis(mesitylmethanone) (99+%, AmBeed) were used as received. All the solvents (GR grade) were used without any purification.2.2. Measurements
Fourier transform infrared spectra were obtained using a NICOLET iS50 FTIR (Thermo Fisher, Waltham, MA) spectrometer in transmission mode with an ATR accessory at room temperature. Nuclear magnetic resonance (NMR) spectra were recorded on a Varian 600 MHz-NMR spectrometer (Varian Medical System, Palo Alto, CA). CDCl3 or DMSO-d was used as the deuterated solvent. Liquid chromatography-mass spectrometry (LC-Mass) was performed using Synapt G2-S, a quadrupole time-of-flight mass spectrometer (Waters) equipped with an Acquity ultra-performance liquid chromatography system and an Acquity PDA detector (Waters). The separation of the samples was achieved on a ZIC-pHILIC column (2.1 × 100 mm, 5 μm particle size, SeQuant® EMD Millipore) using water with 0.1% formic acid as the solvent A and acetonitrile with 0.1% formic acid as solvent B at a flow rate of 400 μL min−1 and the following linear gradient: 0 min, 20% A; 4 min, 20% A; 6 min, 90% A; 7.5 min, 90% A; 10 min, 20% A; 14 min, 20% A. The mass spectrometer was operated in the positive electrospray ionization mode with the following settings: capillary at 2.9 kV, sampling cone at 40 V and source temperature of 120 °C, source offset at 80 V, and 850 L h−1 desolvation gas flow and 250 °C desolvation temperature. Differential scanning calorimetry (DSC) was recorded on a DSC1 (Mettler-Toledo, Columbus, OH) instrument at a ramp rate of 10 °C min−1 for Tg's and the melting point under a nitrogen atmosphere. Thermal stability was evaluated using a thermo-gravimetric analyzer (TGA, Mettler-Toledo, Columbus OH). The sample (∼10 mg) was scanned from 25 to 800 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere.Dynamic mechanical properties were measured on a dynamic mechanical analyzer (DMA, Q800, TA instrument, New Castle, DE) in a single cantilever clamp mode. The specimens with dimensions of 35 mm × 12.5 mm × 3 mm were scanned from 25 to 250 °C at a heating rate of 3 °C min−1. The frequency and oscillating amplitude were set at 1 Hz and 15 μm, respectively. The tensile strengths of 3D printed tensile specimens (type V) were characterized using an Instron 5544 tensile tester (Instron, Norwood, MA) according to ASTM D638. The un-notched impact strengths were measured using a BPI Basic Pendulum impact tester according to ASTM D4812. At least three samples were prepared and tested for each formulation.
The gel content and swelling ratio of 3D printed samples were measured by the solvent extraction method according to ASTM D2765-16. A dried sample (∼0.6 g, w1) was weighted, wrapped in filter paper, and placed in a Soxhlet extractor. 300 mL of toluene was loaded into the extractor and refluxed for 12 h. After extraction, the swollen samples were weighed as wg and dried at 90 °C under vacuum until a constant weight (w2) was reached. The density was measured using a pycnometer with water as the solvent. The gel content and swelling ratio were calculated according to the following equations,
 | (1) |
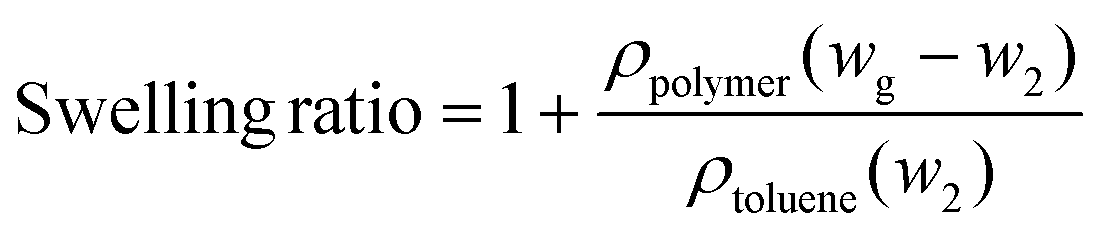 | (2) |
2.3. General procedure for aminolysis of the PLA/PLA waste
Degradation behavior effect on different parameters was characterized using virgin PLA 4043D, because virgin PLA possesses a uniform sample size. Virgin PLA 4043D was first dried in a forced convection oven for 24 h at 50 °C. The dried PLA 4043D and excessive EA were placed in a Schlenk flask. The tube was heated at a predetermined temperature under a nitrogen atmosphere. After aminolysis, the residue was washed, dried, and collected. The degradation degree (Dd) is determined by eqn (3):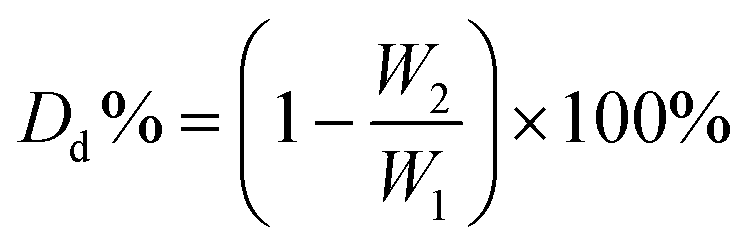 | (3) |
The injection molded PLA and 3D printed PLA waste were cut into small pieces, and added into EA. The general procedure is similar to the virgin PLA degradation as described above. After aminolysis, solid pieces were dissolved in EA. For 3D printed PLA waste, filtration was required to remove pigments and insolubles out.
2.4. Isolation and purification of the degradation product
Aminolysis of injection molded PLA and 3D printed PLA waste in EA gives the same single product, N-lactoyl ethanolamine (N-LEA), as shown in Scheme 1. N-LEA was soluble in EA to form a homogeneous solution. Reduced pressure distillation was employed to remove excess EA, yielding a liquid N-LEA (Scheme 1b).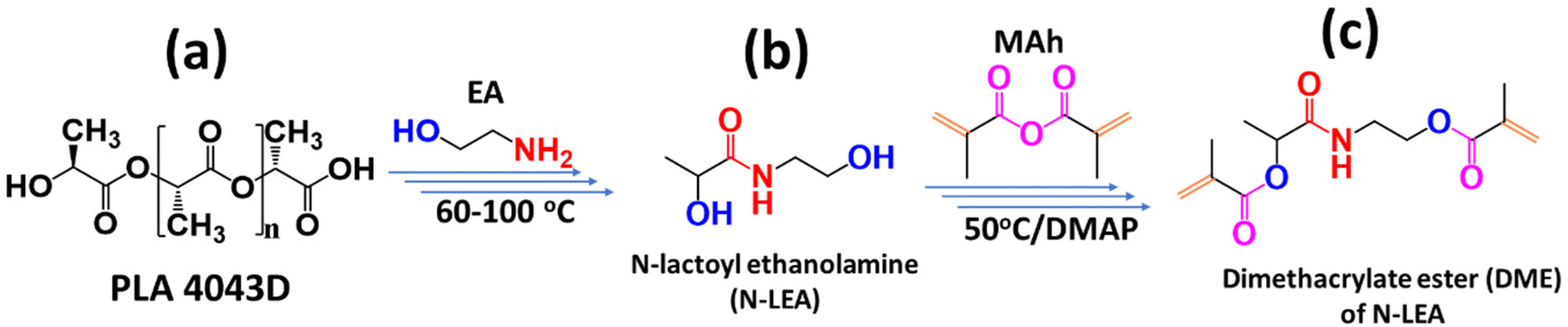 | ||
| Scheme 1 General procedure of synthesis from PLA to diacrylate ester DME, (a) PLA structure, (b) N-LEA, and (c) DME. | ||
2.5. Synthesis of dimethacrylate ester (DME) of N-LEA
N-LEA (1 eq.) and dimethylaminopyridine (0.1 eq., DMAP) were added to a flask and cooled to 0 °C. Then the excess amount of methacrylic anhydride (2.2 eq., MAh) was added dropwise under stirring. Once MAh and N-LEA were mixed into a homogeneous solution, the solution was taken out of the ice bath and allowed to reach room temperature. Afterward, the solution was heated to 50 °C and reacted for 24 h. After the reaction mixture was cooled to room temperature, saturated NaHCO3 solution was added to consume the unreacted MAh. After washing with water three times, a light-yellow powder was obtained, named dimethacrylate ester (DME) of N-LEA (Scheme 1c).2.6. MSLA 3D printing
The photocurable resin was composed of the homogeneous mixture of DME, ACMO, and photo-initiator (1 wt% on the basis of total DME/ACMO mass). The ACMO served as a comonomer and solvent for the solid DME and photo-initiator. The DME/ACMO mass ratio was set at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90, 20:80, and 30:70, respectively. 3D parts and test specimens of the DME–ACMO resin were fabricated using a MSLA 3D printer (Anycubic Photon Mono SE, Shenzhen, China). The dog-bone specimen (ASTM D638 type V), impact specimen (57 × 12 × 3.7 mm), and DMA specimen (35 × 12.5 × 3 mm) were modelled using computer-assisted design (CAD) software SolidWorks™ (Dassault System, Waltham, MA). The printing was performed at a 50 μm layer resolution, with 50 s exposure for the first 6 layers and 4 s for the subsequent layers. The UV power meter (OAI, California) indicated that the printer's radiation intensity is 3.5 mW cm−2 at 405 nm, giving that exposures of 50 s and 4 s would provide a total UV energy dosage of 175 and 14 mJ cm−2 for the corresponding layers, respectively.
:90, 20:80, and 30:70, respectively. 3D parts and test specimens of the DME–ACMO resin were fabricated using a MSLA 3D printer (Anycubic Photon Mono SE, Shenzhen, China). The dog-bone specimen (ASTM D638 type V), impact specimen (57 × 12 × 3.7 mm), and DMA specimen (35 × 12.5 × 3 mm) were modelled using computer-assisted design (CAD) software SolidWorks™ (Dassault System, Waltham, MA). The printing was performed at a 50 μm layer resolution, with 50 s exposure for the first 6 layers and 4 s for the subsequent layers. The UV power meter (OAI, California) indicated that the printer's radiation intensity is 3.5 mW cm−2 at 405 nm, giving that exposures of 50 s and 4 s would provide a total UV energy dosage of 175 and 14 mJ cm−2 for the corresponding layers, respectively.
After printing, the parts were rinsed with 70% ethanol solution for 2 minutes to remove uncured resin on the surface and post-cured using a wash and cure machine (Anycubic, Shenzhen, China) for 6 minutes at the radiation intensity of 14.2 mW cm−2 at 405 nm, giving it a total energy dosage of 2556 mJ cm−2. Subsequently, the specimens were thermally treated in a forced convection oven at 160 °C for 90 min. The specimens were conditioned at standard room temperature and humidity for 1 week prior to physical, thermal and mechanical characterizations. For performance comparison, the commercially available photocuring resins, Anycubic 3D Printing UV Sensitive Resin Basic (ANYCUBIC) and Monoprice Rapid UV Printer Resin (MONOPRICE), were also used to print the same set of specimens using the same processing parameters mentioned above. However, the post-curing conditions were chosen at 70 °C for 3 h, because the selected commercial 3D printing materials cannot sustain higher temperature for post-curing.
A solvent resistance test was employed according to ASTM D543-21. Five solvents were selected: distilled water, methyl alcohol, 5% acetic acid, ethyl acetate, and 10% sodium hydroxide solution, respectively. DME–ACMO 3D printed samples (wi) were placed in 10 mL of solvent for 7 days and moderate manual rotation was performed every 24 hours. After 7 days, the samples were taken out, and the weight was immediately recorded (ws). Then the samples were placed in a vacuum oven at 80 °C for 24 h, the dried samples were re-weighed and recorded as wd. The swelling ratio (SR) and weight remaining (wr) were calculated by the following equations:
 | (4) |
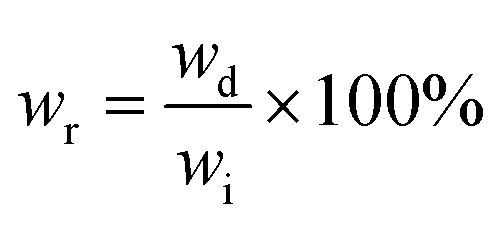 | (5) |
3. Results and discussion
3.1. Aminolysis of PLA
Aminolysis of PLA 4043D (PLA hereafter) was carried out in aminoethanol (EA) without any catalysts. Effects of reaction temperature, time, and feeding ratio on degradation degree were initially investigated by full factorial analysis, indicated by the Pareto chart (Fig. S1†). These three parameters have a statistically significant impact on PLA aminolysis, of which temperature is the most significant factor followed by time and then the feeding ratio. To further analyze the impact of each individual factor on PLA aminolysis, aminolysis by varying experimental factors individually was also investigated (Fig. 1). In Fig. 1a, the reaction temperature and the feeding ratio between PLA and EA were controlled as constants (100 °C, 1:4). The initial degradation rate of aminolysis was expeditious, reaching 53.81% in 10 min. As reaction time increased further, the degradation slowed down, and PLA was completely decomposed in 60 min. In Fig. 1b, when the reaction time and the feeding ratio were fixed as constants (60 min, 1:4), the degradation degree increased almost linearly with the temperature increasing from 60 to 100 °C, and eventually, PLA was totally decomposed at 100 °C. As shown in Fig. 1c, the degradation degree increased with increasing EA content at 100 °C for 60 min. When the molar ratio of PLA repeating units/EA is 1:1 (weight ratio 1:0.8474), the corresponding degradation degree was 64.62%. Since PLA aminolysis is a bulk and heterogenous reaction, excess EA must be added for the reaction to proceed at a pre-determined time and temperature. This is to ensure sufficient contact reaction between the PLA polymer chain and EA. When the feeding ratio increased to 1:2, most of the PLA was decomposed into a monomeric compound, and the degradation degree was 97.78%. Finally, when the feeding ratio was 1:4, PLA was completely depolymerized. The unreacted EA can be recycled by reduced pressure distillation for future use.
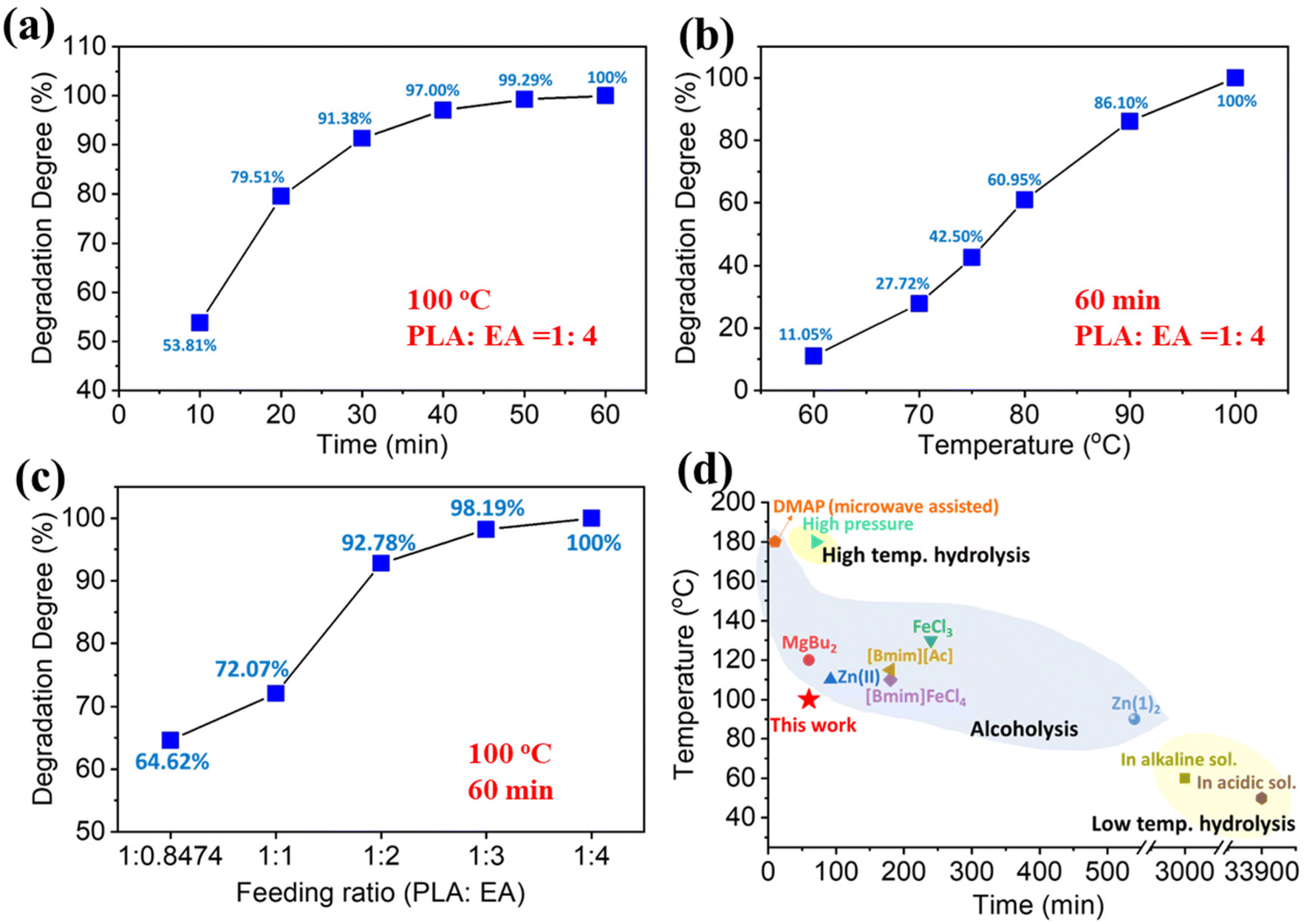 | ||
| Fig. 1 Degradation behavior of three variables: effects of (a) temperature; (b) time; and (c) PLA/EA feeding ratio on the degradation degree (Dd) of PLA, (d) comparison of this work with other reported chemical degradations of PLA through bulk reaction (degradation degree/PLA conversion/yield of degradation products/weight loss ≥90%).12,15,23–30 | ||
Fig. 1d gives a graphical summary of the current PLA degradation methods. In general, PLA aminolysis in EA is conducted under relatively mild conditions (100 °C, 60 min, feeding ratio = 1:4) in a bulk reaction. Currently, PLA degradation routes are divided into three categories, high temperature hydrolysis, low temperature hydrolysis and alcoholysis. Alcoholysis usually requires transesterification catalysts to promote the decomposition. Additionally, the solvents for alcoholysis are mainly methanol and ethanol, which have low boiling points. Therefore, high pressure is required to achieve the necessary temperature. Like alcoholysis, high temperature hydrolysis is usually performed in a high pressure environment. Although low temperature hydrolysis can be carried out at 50 °C or 60 °C, the degradation time is extremely long. For example, in acidic and alkaline environments, the degradation time can be as long as 33900 min and 3000 min, respectively. Unlike alcoholysis and hydrolysis, catalyst-free aminolysis has the advantages of low degradation temperature, short reaction time, and can be performed under ambient pressure.
3.2. Analysis of the degradation product
The degradation product of PLA aminolysis was soluble in EA and was collected after EA was removed by reduced pressure distillation. The appearance of the degradation product was a dark brown liquid. Fig. 2a shows the FTIR spectra of the injection molded PLA and its degradation product. In the FTIR spectra of the degradation product, the peak at ∼1736 cm−1 corresponding to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the ester bonds in PLA disappeared, and instead new peaks at 1652 cm−1 (vCO, amide I), 1557 cm−1 (N–H bending, amide II) and 1179 cm−1 (vC–N, amide III) appeared. This result suggests that the ester bonds from PLA chains were completely cleaved by EA, and amide bonds were formed during the aminolysis. Moreover, the degradation product exhibited a strong and broad peak at ∼3400 cm−1, which was attributed to the stretching vibration of hydroxyl groups (vO–H). This is because the cleavage of ester bond released the hydroxyl group from the lactic acid, and the amide formed with EA introduced another new hydroxyl group. The 1H-NMR spectrum (Fig. 2b) and 13C-NMR spectrum (Fig. S2†) confirmed the degradation product is N-lactoyl ethanolamine (N-LEA). In the 1H-NMR spectrum, the chemical shift at ∼7.6 ppm is associated with the hydrogen of the amide group, and the peaks at ∼4.7 ppm and ∼5.5 ppm are attributed to the two hydroxyl groups. From the 13C-NMR spectrum, the chemical shift at ∼176 ppm is assigned to the carbon of the amide group. LC mass spectrometry was employed to further confirm the structure of N-LEA (Fig. S3†). There is a single, symmetric peak in the selected ion mode (Fig. S3a†), and the m/z of this peak mainly was 134.08 (Fig. S3b†), which is consistent with the molecular weight of N-LEA (Mw = 133.15 g mol−1).
O of the ester bonds in PLA disappeared, and instead new peaks at 1652 cm−1 (vCO, amide I), 1557 cm−1 (N–H bending, amide II) and 1179 cm−1 (vC–N, amide III) appeared. This result suggests that the ester bonds from PLA chains were completely cleaved by EA, and amide bonds were formed during the aminolysis. Moreover, the degradation product exhibited a strong and broad peak at ∼3400 cm−1, which was attributed to the stretching vibration of hydroxyl groups (vO–H). This is because the cleavage of ester bond released the hydroxyl group from the lactic acid, and the amide formed with EA introduced another new hydroxyl group. The 1H-NMR spectrum (Fig. 2b) and 13C-NMR spectrum (Fig. S2†) confirmed the degradation product is N-lactoyl ethanolamine (N-LEA). In the 1H-NMR spectrum, the chemical shift at ∼7.6 ppm is associated with the hydrogen of the amide group, and the peaks at ∼4.7 ppm and ∼5.5 ppm are attributed to the two hydroxyl groups. From the 13C-NMR spectrum, the chemical shift at ∼176 ppm is assigned to the carbon of the amide group. LC mass spectrometry was employed to further confirm the structure of N-LEA (Fig. S3†). There is a single, symmetric peak in the selected ion mode (Fig. S3a†), and the m/z of this peak mainly was 134.08 (Fig. S3b†), which is consistent with the molecular weight of N-LEA (Mw = 133.15 g mol−1).
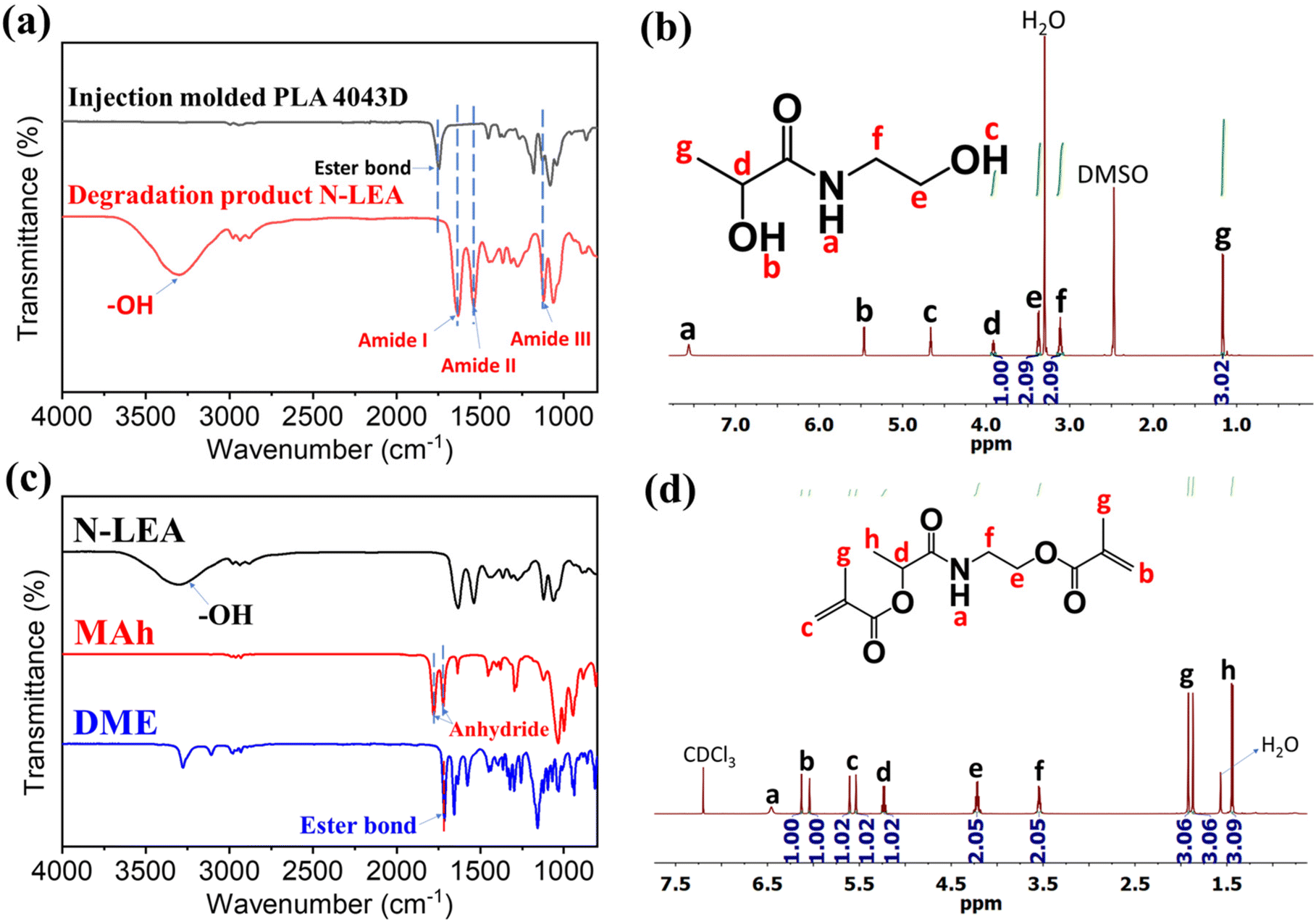 | ||
| Fig. 2 Characterizations of chemical structures of the monomeric degradation product and its methacrylated derivative. (a) FTIR spectra of injection molded PLA before and after degradation; (b) 1H-NMR spectra of N-LEA; (c) FTIR spectra of N-LEA, MA, and DME; and (d) 1H-NMR spectra of DME. | ||
3.3. Synthesis of dimethacrylate ester (DME) of N-LEA
Esterification of N-LEA and MAh resulted in a dimethacrylate compound, which has the functionality to crosslink with other acrylic monomers. After methacrylation, the broad peak of hydroxyl groups of N-LEA at around 3400 cm−1 in the FTIR spectra disappeared as seen in Fig. 2c, indicating the complete consumption of the hydroxyl groups of N-LEA by reacting with anhydride groups of MAh. Also, peaks at around 1780 cm−1 and 1720 cm−1 attributed to anhydride groups also disappeared. The ester bonds at around 1715 cm−1 were formed through the esterification. The product is a light yellowish powder, with a melting point of 94.5 °C by the DSC test (Fig. S4†). In the 1H-NMR spectrum (Fig. 2d), chemical shifts at ∼6.20 ppm (1H, b), ∼6.15 ppm (1H, b), ∼5.70 ppm (1H, c), and ∼5.60 ppm (1H, c) are assigned to –CH2CH2–, which possesses the same peak area by integration. From the 13C-NMR spectrum (Fig. S5†), the chemical shift at ∼173 ppm is assigned to the carbon of the amide group. The peaks at ∼166 and ∼168 ppm are attributed to the carbon of ester bonds. Moreover, carbons from –CC– can be seen at the chemical shift of ∼126 ppm and ∼136 ppm. The resulting product (DME) is a dimethacrylate compound with a structure that is clearly defined by the above analysis.
3.4. MSLA 3D printing and properties of the printed resins
Because the odorless, low viscosity ACMO is a good solvent for DME, it was selected as a reactive diluent to copolymerize with DME. Furthermore, ACMO is also known for its fast polymerization rate.31 Afterward, DME–ACMO photo-curable resin solutions with different DME/ACMO mass ratios were printed using a Masked Stereolithography (MSLA) 3D printer. Fig. 3a shows the FTIR spectra of test samples produced using DME–ACMO 3D printing resins. The intensity of the peaks attributed to the ester bond (∼1715 cm−1) increased with increasing DME content, while the intensities of peaks at ∼1627 cm−1 (CO, amide I), ∼1438 cm−1 (–C–N, amide III), ∼1111 cm−1 (C–O–C, ether) weakened due to the decrease in the ACMO content. There were no obvious peaks in the range of 750–810 cm−1, which indicates that almost all the –CC– were consumed during printing and post-curing. Tg's of 3D printed DME–ACMO resins increased from 151.2 to 173.0 °C with DME/ACMO mass ratio increasing from 10/90 to 30/70 (Fig. 3b). This is because DME with two double bonds acted as a crosslinker in the resin system and the crosslink density would increase with an increase in the DME content. In contrast, the 3D printed commercial photo-curable resins ANYCUBIC and MONOPRICE showed much lower Tg's, being 73.8 °C and 83.2 °C, respectively. This is probably because the DME–ACMO resins in this work had a higher crosslink density. Both DME–ACMO and commercial resins exhibited very similar density and gel content, but the latter showed a larger swelling ratio (Table S1†).
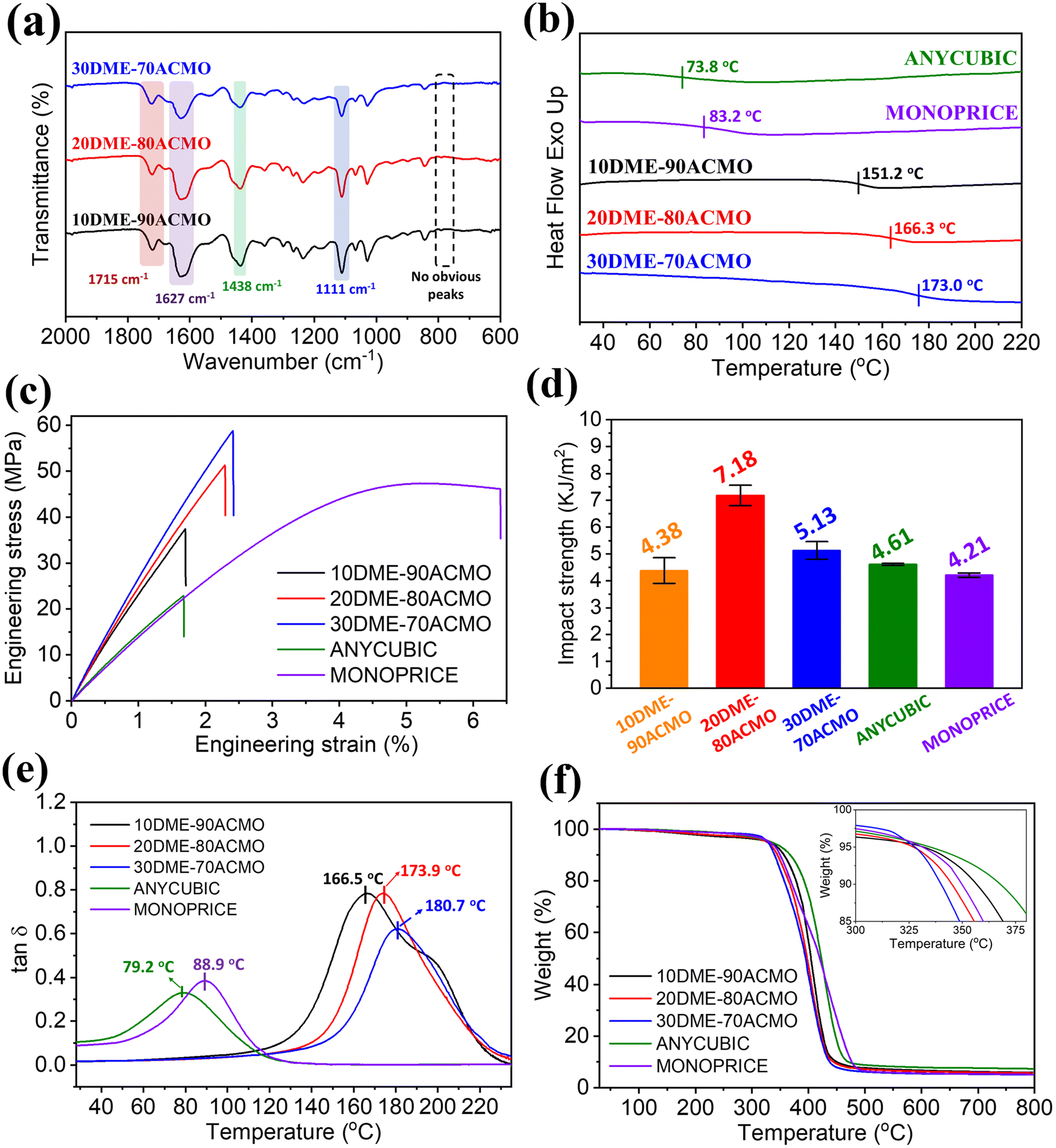 | ||
| Fig. 3 Chemical structure analysis, thermal and mechanical properties of 3D printed DME–ACMO resins and commercial resins. (a) FTIR spectra; (b) DSC thermograms; (c) stress–strain curves; (d) impact strengths; (e) tanδ; and (f) TGA curves. | ||
For the 3D printing specimens, the mechanical properties, including tensile strength, impact strength, and Young's modulus are critically important for practical use. Fig. 3c shows the comparison of tensile properties of 3D printed DME–ACMO resins and commercial resins. As the DME content increased from 10% to 30%, tensile strengths of DME–ACMO resins remarkably increased from 35.96 to 58.58 MPa. While the tensile strengths of ANYCUBIC and MONOPRICE were 21.59 and 47.54 MPa, respectively. Therefore, the tensile strengths of DME–ACMO resins derived from the decomposed PLA monomer (N-LEA) are comparable with commercial resins. Besides, 30DME–70ACMO even possesses a higher tensile strength value (∼58 MPa), which is 173% and 23.2% higher than those of ANYCUBIC and MONOPRICE, respectively. The Young's modulus of DME–ACMO resins increased from around 2.5 to 2.8 GPa with the increase of DME contents from 10 to 30 wt%, which was about 2 times as high as commercial resins (∼1.5 GPa). The impact strength increased from 4.18 to 7.18 kJ m−2, when the DME content was increased from 10 to 20 wt% (Fig. 3d). This is because the content of ACMO, which has a rigid ring structure was reduced. However, the impact strength decreased to 5.13 kJ m−2 at 30 wt% DME, which was probably due to the increased crosslink density. Similarly, the impact strengths of DME–ACMO resins were also comparable to those of commercial resins ANYCUBIC (4.61 kJ m−2) and MONOPRICE (4.21 kJ m−2). 3D printed DME–ACMO resins showed a single transition from the glassy state to the rubbery state at the temperature from 120 to 220 °C (Fig. S7†). Commercial resins, ANYCUBIC and MONOPRICE, also showed the same trend from the glassy state to rubber state with the temperature increasing from room temperature to 200 °C (Fig. S8†). The peaks of tanδ correspond to the glass transition temperature. Therefore, Fig. 3e indicates that Tg's of 3D printed DME–ACMO resins were 166.5, 173.9, and 180.7 °C, respectively. There was only one single apparent peak among DME–ACMO samples, which means there was only one Tg for each sample. However, there was a small shoulder peak present in the 10DME–90ACMO sample, because the low crosslink density induced thermal degradation. ANYCUBIC and MONOPRICE also possessed one single tanδ peak, but the Tg values for these resins were relatively low, which are 79.2 and 88.9 °C, respectively (Fig. 3e). Both 3D printed DME–ACMO and commercial resin samples possessed good thermostability from TGA tests (Fig. 3f). The temperatures corresponding to 5% weight loss (Td5) for all samples were above 325 °C (Table S1†). The solvent resistance of 3D printed DME–ACMO resins was investigated by testing the swelling ratio (SR) and sample weight remaining (wr) (Tables S2 & S3†). 3D printed DME–ACMO resins exhibited swelling behavior in a polar solvent. With the increase in the DME content, the SR of 3D printed DME–ACMO resins increased under immersion in distilled water, 5% acetic acid, methyl alcohol, and 10% NaOH solution, respectively. However, samples did not show apparent changes in ethyl acetate, which means they do not swell in non-polar solvent. All 3D printed DME–ACMO resins showed very high SR (>260%) when exposed to 10% NaOH solution, which indicates that solvent resistance in alkaline environments is limited. The wr values of all samples treated in all solvents is very high (>95%), suggesting that the crosslink networks of all samples are stable in these 5 solvents. The wr values of all samples are greater than 100% when they were treated in 10% sodium hydroxide solution. This is because the residual sodium hydroxide was deposited after drying.
3.5. General upcycling procedure
To demonstrate the application of the PLA aminolysis in solving the real-world problem, 3D printed PLA waste and support structures from our day-to-day prototyping were decomposed using this method. The major component of FDM 3D printed waste is PLA as revealed by FT-IR (Fig. S9†). Fig. 4 shows the general upcycling procedure from FDM 3D printed wastes to MSLA photo curable resins. Shredded FDM supporting base and wastes (Fig. 4a) were decomposed in EA (Fig. 4j) at 110 °C for 90 min, and the resulting black colored suspension (Fig. 4b) was filtered to separate the insolubles (black pigment + additives) (Fig. 4h) and a reaction mixture of the degradation product was dissolved in EA (Fig. 4c). EA was distilled under reduced pressure (Fig. 4i) to give a liquid degradation product. The 1H-NMR (Fig. S10†) spectra indicate that the recycled EA (rEA) had the same chemical structure as that of fresh EA. The only difference between EA and rEA is that rEA has a light-yellow hue due to the slight oxidation during PLA degradation and distillation. After EA was distilled, the collected degradation product (Fig. 4d) was found to have the same chemical structure as the degradation product obtained from aminolysis of injection molded PLA. Fig. S11† shows the FT-IR spectra of degradation products from aminolysis of FDM 3D printed waste and PLA injection molded waste, which are identical. 1H-NMR and 13C-NMR spectra of the two aminolysis products from the two different wastes (Fig. S12†) further proved that they are the same compound. Methacrylation of N-LEA from aminolysis of PLA 3D printed waste resulted in a light-yellow powder (Fig. 4e). Similarly, FT-IR and NMR analyses indicate that this methacrylation product was the same as that of DME derivatized from aminolysis product of injection molded PLA waste (Fig. S13 & S14†).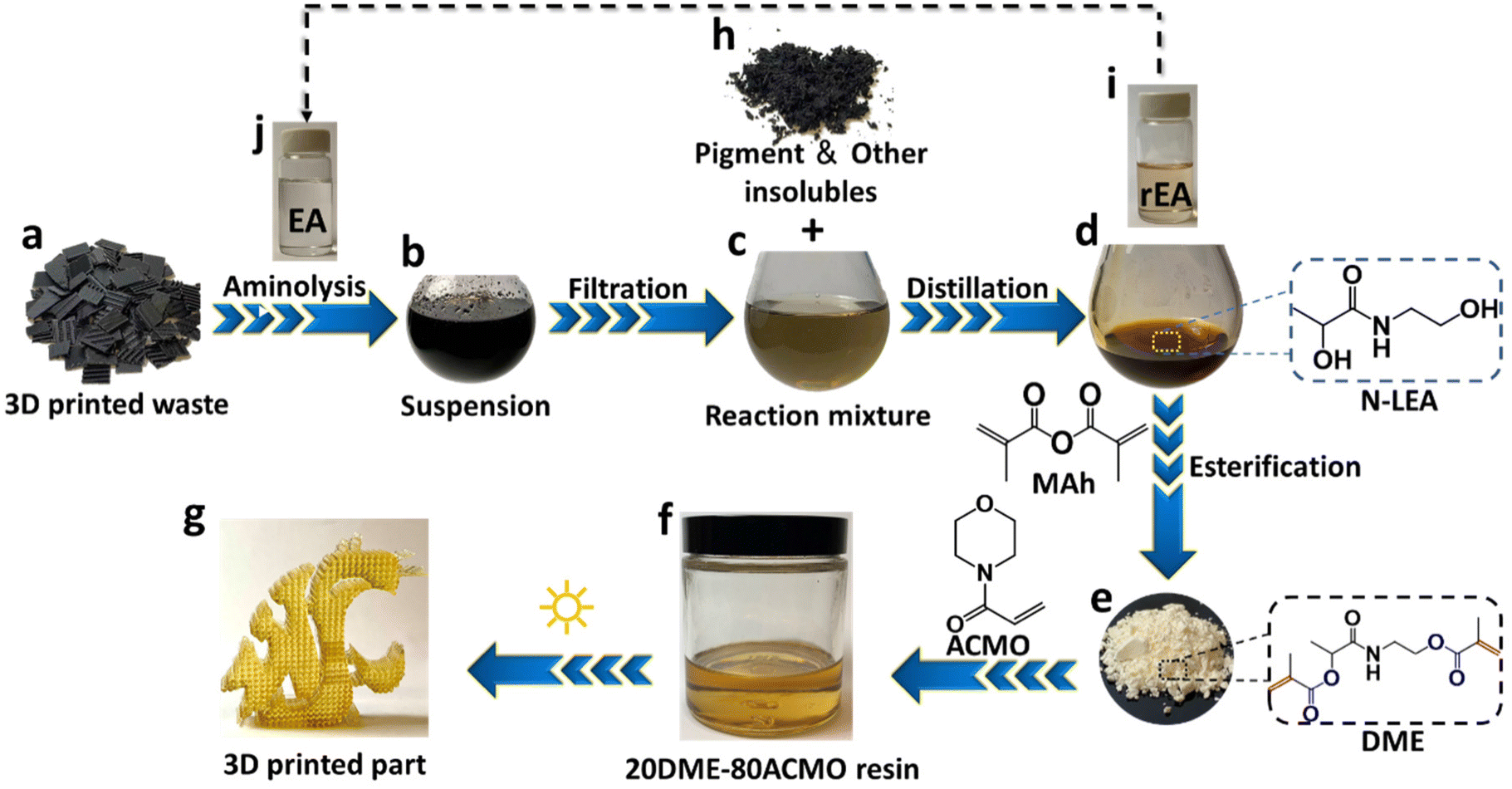 | ||
| Fig. 4 Schematic upcycling process from FDM 3D printed PLA waste to MSLA 3D printed photo-curable resins. | ||
ACMO was selected as a comonomer to copolymerize with DME to give a crosslinked polymer, because it serves as a good solvent and can dissolve up to 30 wt% of DME. Fig. 4g shows a fine lattice-like structure of a 3D printed photo cured 20DME–80ACMO object using a MSLA printer. Overall, this upcycling strategy realizes a route from FDM PLA 3D printed wastes to a new type of photo curable resin.
4. Conclusion
To cooperate with the future vision of biobased/biodegradable plastic like PLA, one must carefully consider the consequences of premature recycling and disposal strategies. In this work, an effective mild chemical recycling strategy from PLA waste to photo-curable 3D printing resins was developed. Injection molded and FDM 3D printed PLA waste was completely decomposed via aminolysis in ethanolamine (EA) with a PLA/EA mass ratio of 1:4 at 100 °C in 60 min. After aminolysis, a diol compound, N-lactoyl ethanolamine (N-LEA), was obtained as the sole degradation product. N-LEA was derivatized by reacting with methacrylic anhydride to give a dimethacrylate ester compound (DME), which was subsequently copolymerized with ACMO by photo curing to produce a crosslinked polymer. The DME/ACMO system could be 3D printed and exhibited mechanical and thermal properties comparable to or even higher than those of the commercial photo-curable resins, ANYBUBIC and MONOPRICE. The tensile modulus, in particular, is as high as 2.8 GPa, which is two times higher than that of commercial photo-curable resins.
Conflicts of interest
The authors declare the following competing interest: a coversheet provisional patent application has been filed for “A method of chemical recycling of poly(lactic acid) waste and preparation of new polymer materials”.Acknowledgements
Lin Shao and Ming-en Fei are grateful for the financial support from the China Scholarship Council (CSC, no. 201807565017). The authors acknowledge Dr Anna Berim and Dr David Gang for their support of LC Mass spectrometry testing. The authors also appreciate the financial support from the Department of Energy (DOE EERE), Office of Energy Efficiency and Renewable Energy (Award No. DE-EE0008931).References
- K. Masutani and Y. Kimura, Poly(lactic acid) Science and Technology, 2014. 10.1039/9781782624806-00001.
- L. Jiang, M. P. Wolcott and J. Zhang, Biomacromolecules, 2006, 7, 199–207 CrossRef CAS PubMed.
- Y.-C. Chang, Y. Chen, J. Ning, C. Hao, M. Rock, M. Amer, S. Feng, M. Falahati, L.-J. Wang and R. K. Chen, ACS Sustainable Chem. Eng., 2019, 7, 15304–15310 CrossRef CAS.
- G. Colomines, S. Domenek, V. Ducruet and A. Guinault, Int. J. Mater. Form., 2008, 1, 607–610 CrossRef.
- Y. Jiang, C. Yan, K. Wang, D. Shi, Z. Liu and M. Yang, Materials, 2019, 12, 1663 CrossRef CAS PubMed.
- B. Anneaux and E. Foley, A Novel Method for Chemical Recycling of PLA Under Mild Conditions, 2018 Search PubMed.
- F. M. Lamberti, L. A. Román-Ramírez and J. Wood, J. Polym. Environ., 2020, 28, 2551–2571 CrossRef CAS.
- A. R. Bagheri, C. Laforsch, A. Greiner and S. Agarwal, Global Challenges, 2017, 1, 1700048 CrossRef PubMed.
- I. Pillin, N. Montrelay, A. Bourmaud and Y. Grohens, Polym. Degrad. Stab., 2008, 93, 321–328 CrossRef CAS.
- F. Codari, S. Lazzari, M. Soos, G. Storti, M. Morbidelli and D. Moscatelli, Polym. Degrad. Stab., 2012, 97, 2460–2466 CrossRef CAS.
- V. Piemonte and F. Gironi, J. Polym. Environ., 2013, 21, 275–279 CrossRef CAS.
- V. Piemonte and F. Gironi, J. Polym. Environ., 2013, 21, 313–318 CrossRef CAS.
- D. Grewell, G. Srinivasan and E. Cochran, J. Renewable Mater., 2014, 2, 157–165 CrossRef.
- A. C. Sánchez and S. R. Collinson, Eur. Polym. J., 2011, 47, 1970–1976 CrossRef.
- H. Liu, X. Song, F. Liu, S. Liu and S. Yu, J. Polym. Res., 2015, 22, 1–7 CrossRef CAS.
- M. Liu, J. Guo, Y. Gu, J. Gao and F. Liu, ACS Sustainable Chem. Eng., 2018, 6, 15127–15134 CrossRef CAS.
- F. Liu, J. Guo, P. Zhao, Y. Gu, J. Gao and M. Liu, Polym. Degrad. Stab., 2019, 167, 124–129 CrossRef CAS.
- F. A. Leibfarth, N. Moreno, A. P. Hawker and J. D. Shand, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4814–4822 CrossRef CAS.
- F. Nederberg, E. F. Connor, T. Glausser and J. L. Hedrick, Chem. Commun., 2001, 2066–2067 RSC.
- S. Tian, Y. Jiao, Z. Gao, Y. Xu, L. Fu, H. Fu, W. Zhou, C. Hu, G. Liu, M. Wang and D. Ma, J. Am. Chem. Soc., 2021, 143, 16358–16363 CrossRef CAS PubMed.
- D. R. Merkel, W. Kuang, D. Malhotra, G. Petrossian, L. Zhong, K. L. Simmons, J. Zhang and L. Cosimbescu, ACS Sustainable Chem. Eng., 2020, 8, 5615–5625 CrossRef CAS.
- K. Fukushima, J. M. Lecuyer, D. S. Wei, H. W. Horn, G. O. Jones, H. A. Al-Megren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. McNeil and J. E. Rice, Polym. Chem., 2013, 4, 1610–1616 RSC.
- H. Liu, R. Zhao, X. Song, F. Liu, S. Yu, S. Liu and X. Ge, Catal. Lett., 2017, 147, 2298–2305 CrossRef CAS.
- X. Song, X. Zhang, H. Wang, F. Liu, S. Yu and S. Liu, Polym. Degrad. Stab., 2013, 98, 2760–2764 CrossRef CAS.
- F. M. Lamberti, L. A. Román-Ramírez, P. Mckeown, M. D. Jones and J. Wood, Processes, 2020, 8, 738 CrossRef CAS.
- L. A. Román-Ramírez, P. McKeown, C. Shah, J. Abraham, M. D. Jones and J. Wood, Ind. Eng. Chem. Res., 2020, 59, 11149–11156 CrossRef PubMed.
- R. Petrus, D. Bykowski and P. Sobota, ACS Catal., 2016, 6, 5222–5235 CrossRef CAS.
- C. Alberti, N. Damps, R. R. Meißner and S. Enthaler, ChemistrySelect, 2019, 4, 6845–6848 CrossRef CAS.
- L. Wan, C. Li, C. Sun, S. Zhou and Y. Zhang, Compos. Sci. Technol., 2019, 181, 107675 CrossRef.
- C. Shih, J. Controlled Release, 1995, 34, 9–15 CrossRef CAS.
- S. Deng, J. Wu, M. D. Dickey, Q. Zhao and T. Xie, Adv. Mater., 2019, 31, 1903970 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc01745h |
| This journal is © The Royal Society of Chemistry 2022 |