
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evidence for the encounter complex in frustrated Lewis pair chemistry
Andrew R.
Jupp

School of Chemistry, University of Birmingham, Edgbaston, Birmingham, West Midlands B15 2TT, UK. E-mail: a.jupp@bham.ac.uk
First published on 6th April 2022
Abstract
Frustrated Lewis Pairs (FLPs) are combinations of bulky Lewis acids and bases that can carry out small-molecule activation and catalysis. Mechanistically, the reaction of the acid, base and substrate involves the collision of three distinct molecules, and so the pre-association of the acid and base to form an encounter complex has been proposed. This article will examine the evidence for the formation of this encounter complex, focusing on the archetypal main-group combinations P(tBu)3/B(C6F5)3 and PMes3/B(C6F5)3 (Mes = mesityl), and includes quantum chemical calculations, molecular dynamics simulations, NMR spectroscopic measurements and neutron scattering. Furthermore, the recent discovery that the associated acid and base can absorb a photon to promote single-electron transfer has enabled the encounter complex to also be studied by UV-Vis spectroscopy, EPR spectroscopy, transient absorption spectroscopy, and resonance Raman spectroscopy. These data all support the notion that the encounter complex is only weakly held together and in low concentration in solution. The insights that these studies provide underpin the exciting transformations that can be promoted by FLPs. Finally, some observations and unanswered questions are provided to prompt further study in this field.
 Andrew R. Jupp | Andy obtained his Ph.D. (2012–2016) from the University of Oxford under the supervision of Prof. Jose Goicoechea, working on phosphorus analogues of the cyanate anion and urea. He subsequently carried out a Banting Postdoctoral Fellowship with Prof. Doug Stephan at the University of Toronto (2016–2018), working on frustrated Lewis pairs (FLPs) and the functionalisation of CO2. In 2018, he became an NWO VENI laureate at the University of Amsterdam, working with Prof. Chris Slootweg on the formation of main-group radicals in FLP systems. In 2020, he launched his independent career as a Birmingham Fellow at the University of Birmingham (UK), working on small-molecule activation and molecular photo-switches. He started a Royal Society University Research Fellowship in January 2021. |
Introduction
Combinations of a Lewis acid and a Lewis base typically form adducts via a dative bond, as described by Gilbert Lewis in his seminal work almost 100 years ago.1 Several exceptions to this general rule have been found in the intervening years. In 1942, Brown and co-workers observed that 2,6-lutidine and BMe3 do not form a Lewis adduct due to “steric interference”,2 and there are subsequent examples of non-quenching pairs of acids and bases reacting with unsaturated substrates.3,4 However, it was the discovery by Stephan and co-workers in 2006 of a molecule containing discrete Lewis acidic and basic sites that could reversibly activate dihydrogen that demonstrated the true potential of these systems.5 The following year, the term “Frustrated Lewis Pair” (FLP) was coined6 to describe this general concept of a combination of a bulky Lewis acid and Lewis base that is sterically precluded from forming a Lewis adduct.7 The unquenched reactive acidic and basic sites in FLPs have been exploited for a wide range of small-molecule activation and catalysis, and this concept has inspired research groups around the world to explore metal-free approaches to reactions that were once considered the preserve of transition metal complexes.8–13 These reactions include the capture of environmentally relevant small molecules such as CO2, N2O, and SO2;14–17 the catalytic hydrogenation of unsaturated organic substrates;18–20 and C–H activation.21Mechanism for FLP reactivity
The mechanism for the FLP activation of small molecules like dihydrogen has been widely discussed.22,23 The splitting of H2 by the cooperative action of a Lewis acid and Lewis base is a three-component reaction. Although termolecular reactivity is known in the literature,24,25 the probability of the acid, base and substrate colliding at the same time in the correct orientation for activation is low, and as such the pre-association of two of the three components has been proposed to explain the facile and rapid reactivity of FLPs (Scheme 1).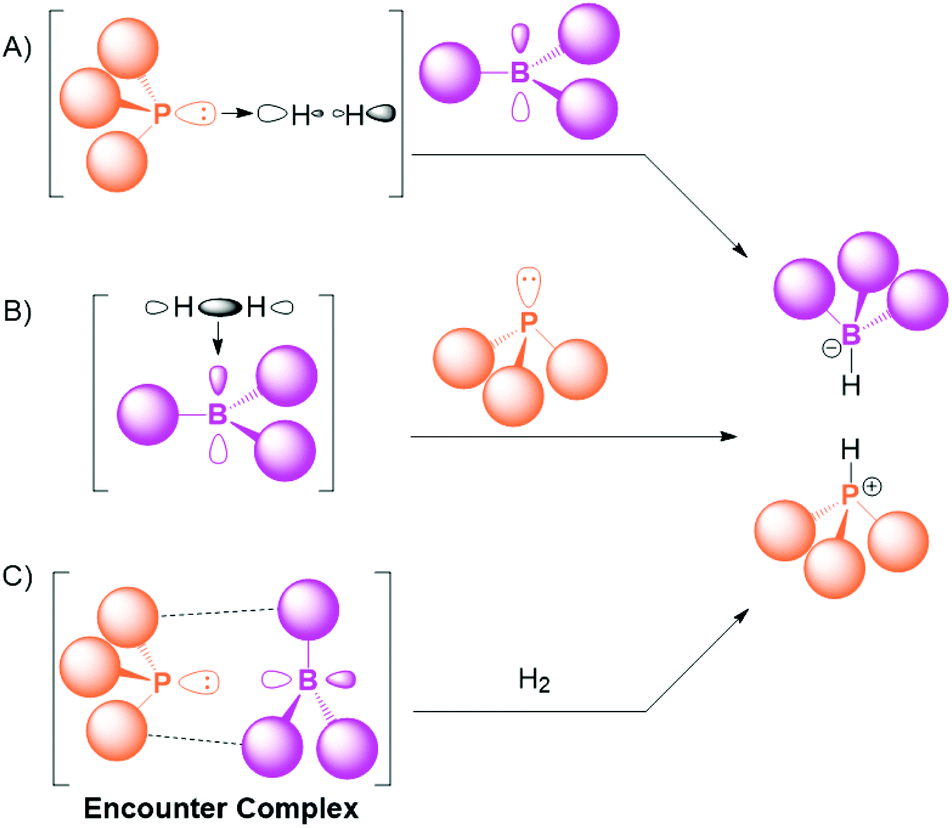 | ||
| Scheme 1 Mechanisms for the splitting of H2 by an FLP comprising a bulky phosphine and borane as alternatives to true termolecular reactivity. A: pre-association of the phosphine and H2; B: pre-association of the borane and H2; and C: pre-association of the phosphine and borane as an encounter complex. | ||
The Lewis base can theoretically interact with the substrate (typified by H2 in Scheme 1A), and phosphine⋯H2 interactions have been postulated in an argon matrix.26 However, for the case of P(tBu)3 with H2, the interaction is computed to be repulsive in the chemically relevant range.27 For certain small-molecule substrates like CO2, the initial base⋯substrate interaction is more of a possibility, and strong Lewis bases such as N-heterocyclic carbenes and imidazolin-2-ylidenamino-phosphines have been shown to bind CO2.28–31Scheme 1B shows the next alternative, where the substrate is initially bound to the Lewis acid. This pathway is well established for certain substrates; for example, the B(C6F5)3-catalysed hydrosilylation of aromatic ketones, aldehydes and esters by Piers and co-workers,32 where the borane activates the silane moiety,33 is regarded as an early example of FLP chemistry.34 Furthermore, an alkene⋯borane adduct has been observed at low temperature as an intermediate in FLP activation,35 and the zwitterionic adduct of B(C6F5)3 with an alkyne was structurally characterised recently.36 Regarding dihydrogen as the substrate, H2 has been shown to bind to BH3 in an argon matrix;37 HB(C6F5)2 can undergo σ-bond metathesis with H2;38 and antiaromatic boroles have been shown to activate H2.39,40 However, calculations suggest that the interaction of H2 with B(C6F5)3 is unfavourable due to Pauli repulsion.27
Therefore, particularly for the activation of H2 with the prototypical FLP combinations of P(tBu)3/B(C6F5)3 or PMes3/B(C6F5)3 (Mes = mesityl), the prevailing theory is that depicted in Scheme 1C, where there is a pre-association of the bulky acid and base. This adduct is commonly referred to as the encounter complex, and is held together by weak van der Waals interactions between the substituents on the phosphine and borane. Understanding the nature of the encounter complex and the factors that affect its formation are critical to rationalising and optimising subsequent FLP reactivity, and a great deal of work has gone into studying this ephemeral species. The fact that the adduct is only weakly held together limited the studies of the encounter complex in the early days of FLP chemistry to computational investigations, but recent developments have gleaned key information using a range of experimental techniques, including NMR spectroscopy, UV-Vis spectroscopy, EPR spectroscopy, transient absorption spectroscopy, resonance Raman spectroscopy, and neutron scattering. These experimental breakthroughs are the focus of this Frontier article.
It is worth noting at this stage that there are alternative strategies for enhancing the pre-organisation of the acid and base moieties in FLPs. The most widely employed approach is tethering the two components with a covalent linker, known as an intramolecular FLP. Erker's ethylene-bridged FLP, Mes2PCH2CH2B(C6F5)2, is a pioneering example of such a system.41 Many different covalent linkers have been employed in the ensuing years, and selected examples of intramolecular FLPs include a simple methylene-bridged P/B system;42 a dimethylxanthene-bridged system that enables the reversible capture of N2O;43 a phenylene bridged N/B system that could catalyse the selective Z-reduction of alkynes;44 a geminal S/B species that could be activated by light;45 and even chiral systems for asymmetric catalysis.46–48 An alternative approach is for the acid and base to interact in a classical manner; there are a number of systems that are capable of FLP-type reactivity where the Lewis acid and base interact via a dative bond. Examples of these systems include combinations of the Lewis acid B(C6F5)3 with 2,6-lutidine,49 Et2O,50 1,4-dioxane,51 or the proazaphosphatrane P(N(Me)CH2CH2)3N,52 where the classical adduct is in equilibrium with the dissociated acid and base.
However, this article will focus on the computational and experimental evidence for the presence of the encounter complex between discrete Lewis acids and bases to explain the termolecular reactivity of FLPs. There are a very large number of possible Lewis acids and bases; there have been interesting studies looking at N-heterocyclic carbene/borane combinations, although a large proportion of these systems either form a normal Lewis adduct or undergo a range of decomposition pathways via C–H or C–F activation, which limits the possibility of studying the encounter complexes in these systems experimentally.53–55 Recently, the trioxatriangulenium ion was explored as a carbon-centred Lewis acid in FLP chemistry, and the association with different phosphines was probed.56 To focus the discussion and explore the evidence for the encounter complexes in more detail, the FLPs in this article will be limited to P(tBu)3/B(C6F5)3 or PMes3/B(C6F5)3 (Fig. 1), as these are the most commonly used FLPs and have significance in a wide range of catalytic applications.
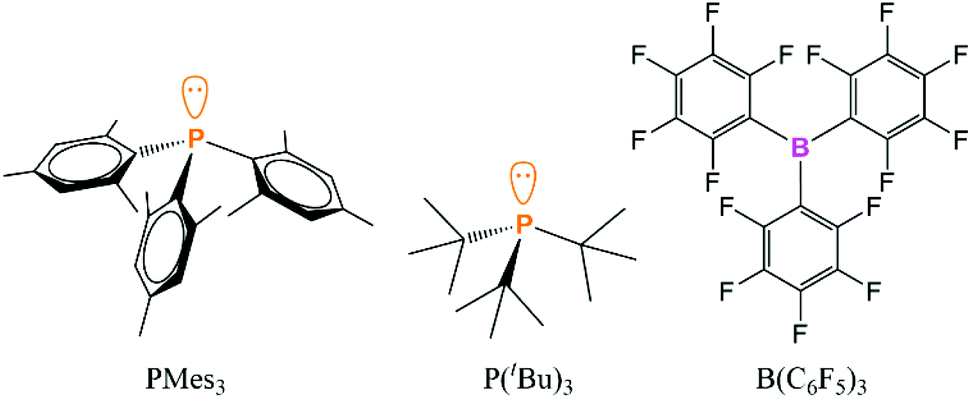 | ||
| Fig. 1 The phosphines and borane in the prototypical encounter complexes that are the focus of this article. | ||
Computational models of the encounter complex
The original research into the encounter complex was computational in nature. A seminal report from Pápai and co-workers first suggested the formation of the encounter complex in FLP chemistry.27 They identified a weakly associated [P(tBu)3]⋯[B(C6F5)3] adduct as a minimum on the potential energy surface, using both B3LYP and SCS-MP2 methods. There was no evidence of charge transfer from the phosphine lone pair into the vacant orbital on boron, as shown by the planarity of the central BC3 unit in B(C6F5)3. Furthermore, the P⋯B distance at the energetic minimum was 4.2 Å (for the SCS-MP2 calculation); for comparison, the sum of the covalent radii would predict a P–B bond length of 1.96 Å in a Lewis adduct.57 Instead, the adduct is stabilised by a combination of multiple C–H⋯F non-covalent interactions, and in the absence of any solvent modelling the association energy was calculated as ΔEassoc = −11.5 kcal mol−1. The nature of the encounter complex being stabilised by many weak dispersion interactions means that there is a degree of structural flexibility, and it was shown that large changes in the P⋯B distance can be achieved at a relatively small energetic cost.The significance of dispersion interactions to stabilise the encounter complex was also supported by a number of other studies.55,58,59 Note that there are contrasting theories for the mechanism of the activation of dihydrogen by the FLP.60 Pápai proposed an electron transfer mechanism based on synergistic interactions of the donor and acceptor orbitals on the base and acid with the acceptor and donor orbitals on dihydrogen, respectively,27,61 whereas Grimme has proposed an electric field model, where the H–H bond is polarised by a strong electric field generated between the donor/acceptor atoms.58,62 A more recent publication has sought to unify these two mechanisms,63 and very recently, Fernández and co-workers have explored the activation strain model-energy decomposition analysis as a tool to probe reactivity in FLPs.64,65 Crucial to these theories is the formation of the pre-organised encounter complex stabilised by weak dispersion interactions with a reactive “pocket” available for the small-molecule substrate to be activated.
A further computational study by Vankova and co-workers corroborated the formation of the encounter complex is energetically favourable, with an average association energy across a range of systems of ΔEassoc = −10 kcal mol−1.66 Incorporating solvent effects (toluene) using a polarisable continuum model led to only small changes in the association energy (less than 1 kcal mol−1). However, the favourable electronic interactions in the encounter complex are counterbalanced by the entropic cost of adduct formation. Entropy is the dominant factor at room temperature, and the formation of the encounter complex is endergonic (ΔGassoc = +5 ± 2 kcal mol−1), which is consistent with the difficulty in observing these species in the laboratory.66
To move beyond the static computational models used in quantum chemical calculations, Pápai and co-workers used molecular dynamics (MD) simulations to probe the encounter complex of P(tBu)3/B(C6F5)3 in toluene.67 The model system comprised one borane, one phosphine, and 1011 toluene molecules in a periodically repeated cell, which represents a relatively dilute solution compared to a typical experimental set-up. The Helmholtz free energy curve (see F(r) in Fig. 2) showed that the formation of the encounter complex is disfavoured; the structures with a P⋯B distance in the chemically useful range of 4.2–5.6 Å were approximately 1.2 kcal mol−1 higher in energy than the dissociation limit. The probability of finding a configuration with a P⋯B distance of less than 6 Å (see C(r) curve in Fig. 2 for cumulative probability of P(r)) was calculated to be roughly 2%, and those configurations featuring the optimal P⋯B distance of 4.5 Å were only present 0.5% of the time.
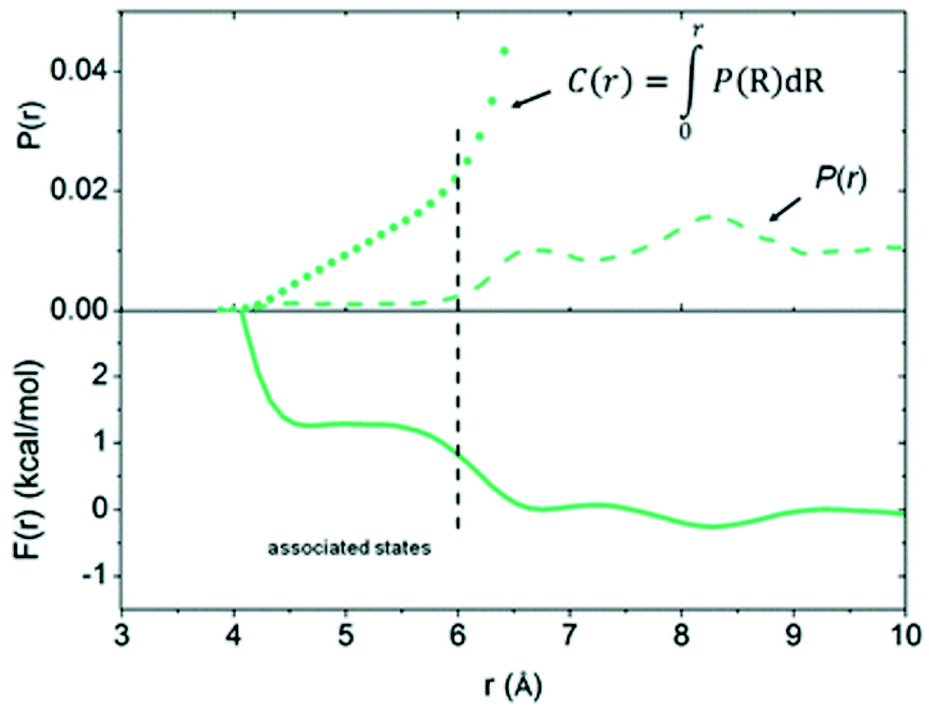 | ||
| Fig. 2 Free energy curve, F(r), and probability distribution, P(r), computed from MD simulations of P(tBu)3/B(C6F5)3 in toluene. Reproduced from ref. 67 with permission from the Royal Society of Chemistry. | ||
Experimental evidence for the encounter complex
Initial observation attempts
The weak stabilisation of the encounter complex in solution hampered early attempts to observe this species experimentally. It was reported that equimolar benzene or toluene solutions of P(tBu)3/B(C6F5)3 or PMes3/B(C6F5)3 gave no indication of any interactions by 1H, 11B, 19F, or 31P NMR spectroscopy, and in each case the NMR spectra for the FLP looked the same as those of the individual components.7,68 Furthermore, isothermal reaction calorimetry performed by Houghton and Autrey revealed that no appreciable heat was produced on mixing PMes3 and B(C6F5)3 in dichloromethane.69 This detailed calorimetric study concluded that the activation of H2 by this FLP is best modelled as a termolecular reaction with a rate-determining step of assembling the reactants into the solvent cage in the correct configuration.A further complication for exploring the encounter complex experimentally is that for one of the FLP combinations that we are discussing, P(tBu)3/B(C6F5)3, there is a competing side-reaction between the two components to form the ion pair [HP(tBu)3][FB(C6F5)3] and the intramolecular species (tBu)2P(C6F4)B(C6F5)2 with elimination of isobutylene (Scheme 2).70 However, despite these hurdles, there have been some recent breakthroughs in the characterisation of the encounter complex that will be explored below.
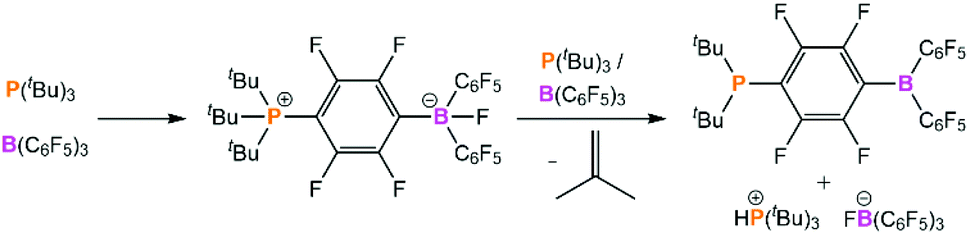 | ||
| Scheme 2 Possible side-reaction between P(tBu)3 and B(C6F5)3. | ||
NMR spectroscopy
The first report with unequivocal experimental evidence in support of the encounter complex was a comprehensive NMR spectroscopic study by Rocchigiani et al.68 They performed 19F,1H HOESY (Heteronuclear Overhauser Enhancement Spectroscopy) experiments on concentrated (220–230 mM) samples of P(tBu)3/B(C6F5)3 or PMes3/B(C6F5)3 in benzene or toluene, and clear cross-peaks corresponding to H/F interactions could be observed (Fig. 3). Furthermore, addition of a substoichiometric amount of B(C6F5)3 to a concentrated solution of PMes3 (relative ratio phosphine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :borane of 57:1) resulted in marginal shifts of the 19F resonances relative to the free borane, and substantial line-broadening of the para-F. The temperature-dependent broadening of the 19F NMR resonances of B(C6F5)3 in the presence of excess PMes3 at low temperature but not as the free borane is also consistent with aggregation of the phosphine and borane in solution.
:borane of 57:1) resulted in marginal shifts of the 19F resonances relative to the free borane, and substantial line-broadening of the para-F. The temperature-dependent broadening of the 19F NMR resonances of B(C6F5)3 in the presence of excess PMes3 at low temperature but not as the free borane is also consistent with aggregation of the phosphine and borane in solution.
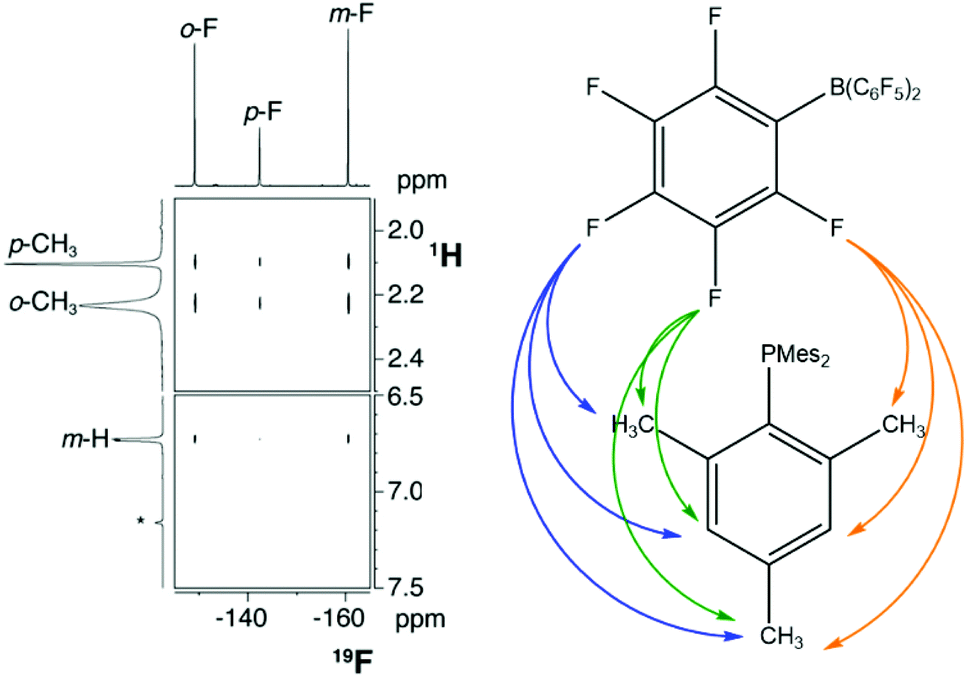 | ||
| Fig. 3 19F,1H HOESY NMR spectrum of PMes3/B(C6F5)3 in benzene-d6, showing cross-peaks arising from all fluorine and proton environments. Adapted with permission from ref. 68. Copyright 2014 American Chemical Society. | ||
The study also explored the relative orientation of the phosphine and borane in this aggregate to ascertain if there was any preferred directionality. The kinetics of NOE build-up and a comparison of the relative strengths of the NOEs within PMes3/B(C6F5)3 with computational predictions strongly indicate that the two components have a random relative orientation. This result suggests that the aggregation of the acid and base in solution is dominated by intramolecular H/F interactions, and not due to donation of the phosphine lone pair into the vacant p orbital on the borane. This hypothesis was corroborated with computations that revealed there was only 1 kcal mol−1 between the two limiting structures of PMes3/B(C6F5)3 shown in Fig. 4, and the two extremes were roughly equally likely to exist in solution. Grimme and co-workers carried out a comprehensive computational investigation to explore this further using state-of-the-art quantum chemistry methods, building on the NMR spectroscopic data, and extended the study to P(tBu)3/B(C6F5)3.71 The authors highlighted the importance of accurately modelling the dispersion interactions, and showed that for PMes3/B(C6F5)3 there is little energetic difference between the two extreme orientations depicted in Fig. 4 across a range of different methods, in agreement with experiment. However, for P(tBu)3/B(C6F5)3, the orientation labelled geometry a in Fig. 4 is energetically favoured by 3–5 kcal mol−1 (or 1–2 kcal mol−1 in free energies) compared to geometry b, indicating that the association is less driven by dispersion interactions, and that there is a small amount of P⋯B bonding present. This non-negligible P⋯B interaction in P(tBu)3/B(C6F5)3 has previously been discussed in terms of the frontier orbitals.72
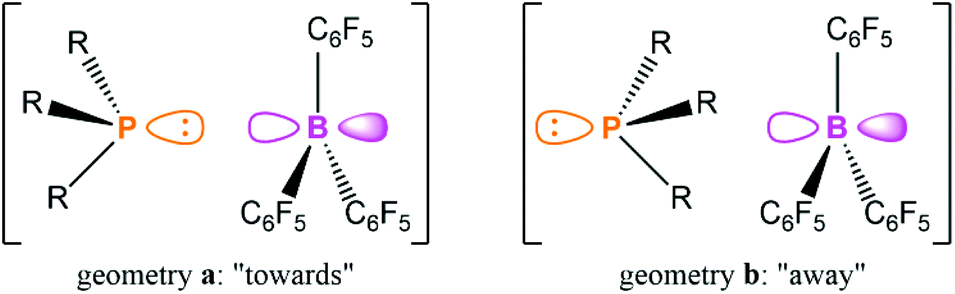 | ||
| Fig. 4 The two limiting geometries for the association of a phosphine with B(C6F5)3, with the P lone pair oriented “towards” or “away” from the p orbital on B. | ||
Rocchigiani and co-workers quantified the propensity of PMes3/B(C6F5)3 to associate in solution using diffusion 19F and 1H NMR spectroscopy; the average association constant was determined to be K = 0.5 ± 0.2 M−1, meaning that formation of the encounter complex is slightly endergonic (ΔG° = +0.4 ± 0.2 kcal mol−1). These results are consistent with the previously discussed data from molecular dynamics simulations.67
More recently, Swadźba-Kwaśny and co-workers carried out further NMR experiments on the FLP combination P(tBu)3/B(C6F5)3.73 Interestingly, their data show a clear change in the chemical shifts of the 19F NMR resonances of free B(C6F5)3 in benzene-d6versus the same resonances in a 1:1 mixture of P(tBu)3/B(C6F5)3 in benzene-d6, consistent with a small amount of P⋯B interaction. In a bid to determine whether ionic liquids can increase the concentration of the encounter complex in solution, they also carried out the same analysis of P(tBu)3/B(C6F5)3 using the ionic liquid [C10mim][NTf2] as the solvent (Fig. 5). The 19F NMR chemical shifts of free B(C6F5)3 are significantly different when dissolved in [C10mim][NTf2] compared to benzene-d6, consistent with interaction of the Lewis acidic borane with one of the components of the ionic liquid. There are undoubtedly new 19F and 31P NMR resonances that appear when P(tBu)3/B(C6F5)3 is dissolved in [C10mim][NTf2] compared to the individual components; according to the study, 24% of the B(C6F5)3 is no longer “free” (and this 24% is split across at least three different environments), while 78% of the P(tBu)3 is also in a new environment. The authors state that although it is not possible to make definitive assignments for these new resonances, they could be attributed to the interaction between the FLP components in the encounter complex, which is stabilised to a greater extent in the ionic liquid compared to benzene-d6. This notion was explored computationally in a further MD study by Liu and co-workers, comparing the association of P(tBu)3/B(C6F5)3 in toluene and a range of ionic liquids.74 They showed that in general the ionic liquids led to an enhanced probability of the phosphine and borane being associated with each other; the computed probability of finding P(tBu)3/B(C6F5)3 with a P⋯B distance of ≤6 Å in toluene was 2.32% (similar to the previously discussed value of 2% (ref. 67)), whereas this probability increased to 4.75–5.15% in the majority of the ionic liquids probed. For one of the ionic liquids, specifically [C6mim][CTf3], the same probability actually decreased to 1.08%, reflecting a decreased stability of the encounter complex in this case, so careful consideration of the nature of the ionic liquid is required. The authors propose that the ionic liquids pack together and leave large cavities that the encounter complex can accommodate, whereas toluene molecules move in between and separate the acid and base. These articles highlight the potential of ionic liquids as a tool to better study the encounter complex in FLP chemistry, although further work is required to unambiguously identify the encounter complex in these systems.
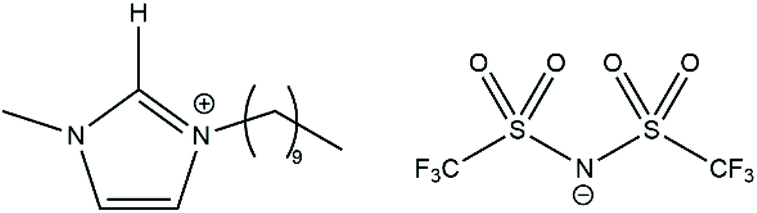 | ||
| Fig. 5 Structure of ionic liquid [C10mim][NTf2]. | ||
Neutron scattering
Swadźba-Kwaśny and co-workers also explored the nature of the association of P(tBu)3/B(C6F5)3 using neutron scattering experiments.73 Neutron diffraction has previously been used to study structure and solvation,75 and when combined with empirical potential structure refinement (EPSR) models can provide insight into complex systems. The measurements were carried out on equimolar solutions in benzene at 160 mmol concentration, which is relatively low for standard neutron scattering experiments, and resulted in poor resolution for the specific P⋯B interactions and significant variability between different refinement runs. Nevertheless, data could be obtained that provided evidence for association of the phosphine and borane molecules in solution. There was <1% chance of finding the P(tBu)3/B(C6F5)3 with a P⋯B distance of 5.7 Å, but this increased to 4.9% chance at P⋯B separations of <8 Å. These data are consistent with the weak association of the acid and base in solution, and highlight the utility of neutron diffraction for directly observing the encounter complex.Equivalence with the electron donor–acceptor complex
Recent experimental evidence for the encounter complex has arisen from the blossoming field of frustrated radical pairs.76–79 The single-electron transfer (SET) from the Lewis base to the Lewis acid to form the radical pair was first postulated by Piers and co-workers, although it was discounted as a viable mechanism in FLP chemistry due to the large discrepancy in redox potentials between P(tBu)3 and B(C6F5)3.80 A breakthrough from Stephan and co-workers was published in 2017, where they studied the FLP combination PMes3/B(C6F5)3 and saw evidence of the radical cation [PMes3]˙+, and postulated that the radical ion pair is in thermal equilibrium with the phosphine and borane.76 The [PMes3]˙+ could be observed by EPR spectroscopy, although the corresponding [B(C6F5)3]˙− radical was not observed, and this was attributed to the known and rapid decomposition of this radical anion via solvolytic pathways.81,82In light of the contradictory evidence above, Slootweg and co-workers sought to better understand the SET process in FLPs.83,84 Mulliken theory describes the interaction of an electron-rich donor (D) and an electron-poor acceptor (A) to form an electron donor–acceptor complex [D,A], which can subsequently absorb a photon of the correct energy to promote SET and afford the charge-transfer state [D˙+,A˙−] (Fig. 6).85–87 Relating these concepts to FLPs, Lewis acids are acceptors, and Lewis bases are electron donors, and therefore the encounter complex that we have been discussing is simply another name for the electron donor–acceptor complex.
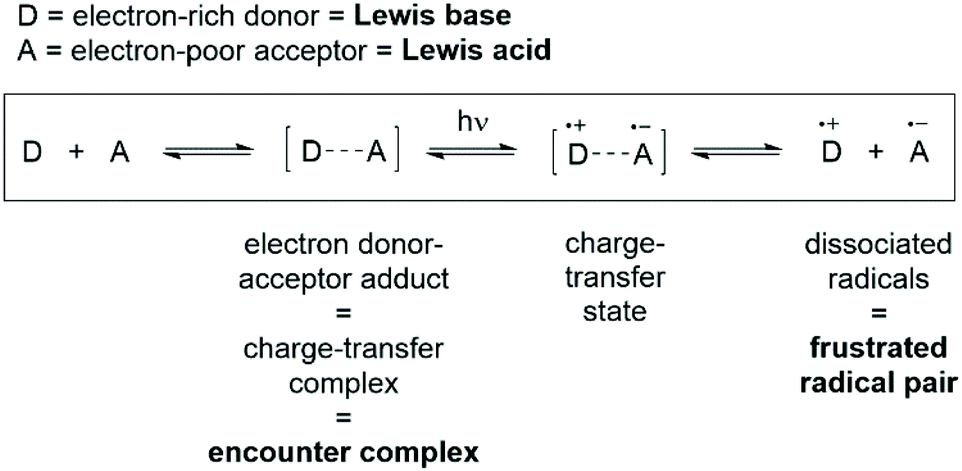 | ||
| Fig. 6 Mulliken theory for electron transfer in electron donor–acceptor adducts, with analogous terms relevant to FLP chemistry highlighted in bold. | ||
A toluene solution of PMes3/B(C6F5)3 is violet in colour, despite the individual components each being colourless in solution. This was noted in 2007 by Stephan, where they postulated that the colour arose from π-stacking of the aryl rings on the phosphine and borane.7 Then in 2017, it was postulated that the colour is due to a low concentration of the [PMes3]˙+ radical cation in equilibrium with the FLP.76 In 2020, Slootweg and co-workers proposed the violet colour is actually due to a charge-transfer band, where the electron donor–acceptor complex (i.e. encounter complex) can absorb a photon in the visible region to promote SET from the phosphine to the borane.83 This theory was supported computationally, as time-dependent density functional theory on the PMes3/B(C6F5)3 encounter complex revealed a band corresponding to this electronic transition. UV-Vis spectroscopy confirmed the presence of an absorption band at λ = 534 nm (Fig. 7a). Experimental verification that absorption of this band led to radical formation was obtained by EPR spectroscopy and transient absorption spectroscopy (Fig. 7b and c). Irradiation of the sample at the appropriate wavelength gave characteristic signals of the radical pair, while the same signals were not present in the analogous experiments performed in the dark. The back-electron transfer to regenerate the neutral phosphine and borane was rapid, and the lifetime of this radical pair was only 237 ps. These same analyses were also carried out on the FLP P(tBu)3/B(C6F5)3, which is pale yellow in toluene, and showed the presence of a new absorption band at λ = 372 nm. The P(tBu)3/B(C6F5)3 samples were always freshly prepared and quickly analysed to mitigate the complication in this particular FLP system of the previously discussed side-reaction that occurs between the acid and base as much as possible (Scheme 2); Piers and co-workers have shown that the (tBu)2P(C6F4)B(C6F5)2 product is also yellow.70 Irradiation of this new absorption band also showed diagnostic signals corresponding to the radical pair in the EPR spectrum, and transient absorption spectroscopy revealed a lifetime of only 6 ps.
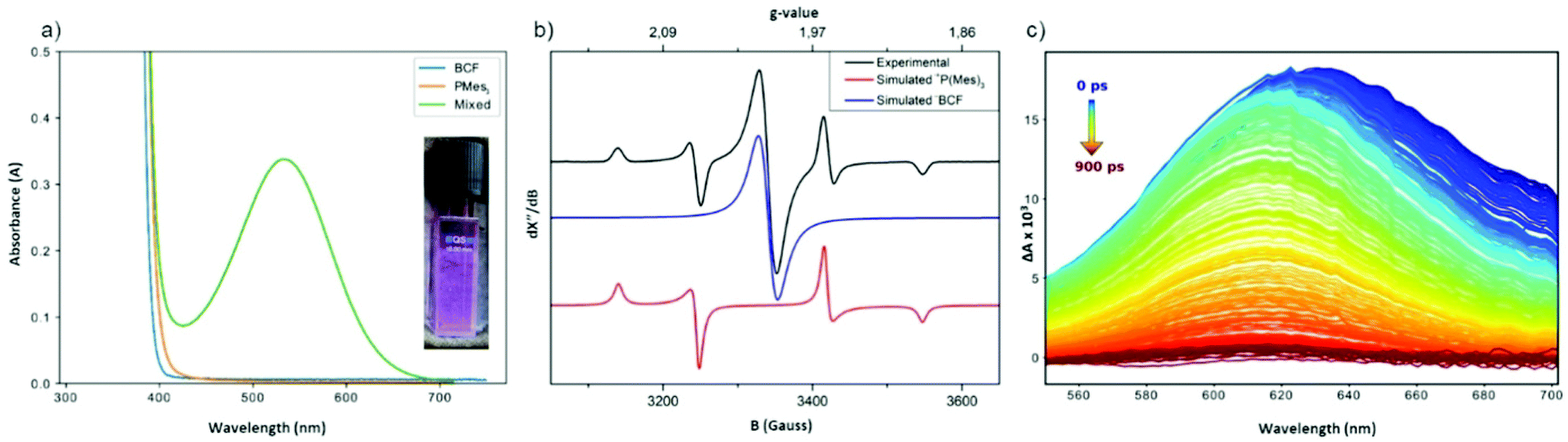 | ||
| Fig. 7 Evidence for encounter complex of PMes3/B(C6F5)3 due to SET processes (B(C6F5)3 is abbreviated as BCF in the figure above, as this is how it appears in the original article): (a) UV-Vis spectrum of toluene solution of PMes3/B(C6F5)3 compared to spectra of individual components; (b) experimental EPR spectrum of toluene solution of PMes3/B(C6F5)3 measured at 30 K during irradiation with visible light (390–500 nm) and simulated spectra of [PMes3]˙+ and [B(C6F5)3]˙−; (c) transient absorption spectra measured after pulsed excitation of PMes3/B(C6F5)3 with 530 nm light. Figure from ref. 83 used with permission from John Wiley and Sons. Copyright 2020 Wiley. | ||
The charge transfer in PMes3/B(C6F5)3 was further studied by resonance Raman spectroscopy by Ando and co-workers.88 Resonance Raman spectroscopy can provide information on vibrational modes that are associated with a particular electronic transition. The authors showed that the Raman spectrum of the FLP was the same as the superposition of the spectra of the individual components, consistent with the previous spectroscopic evidence that there is very limited interaction between the acid and base in solution. The resonance Raman spectrum of the FLP in CH2Cl2 (irradiated at λ = 457 nm) did show some enhancement of certain bands compared to the normal Raman spectrum, and as these vibrational modes were associated with both the borane and the phosphine, it was concluded that there must be some association of the two components in solution.
These spectroscopic measurements, namely UV-Vis, EPR, transient absorption, and resonance Raman spectroscopy, all arise from the charge transfer between the Lewis base and the Lewis acid. These analytical methods all provide direct evidence of the encounter complex in solution, as SET is only possible when the acid and base are in close proximity with a suitable orbital arrangement. This finding could unlock more ways for researchers to probe and understand the encounter complex in the future.
Conclusions and outlook
The encounter complex is a key concept for rationalising the three-component reactivity of FLPs with substrates such as dihydrogen. The initial studies were limited to computational models,27,58,60,66,67,71,72 which showed that the association of the bulky Lewis acid and base is driven by dispersion interactions between the large substituents on each molecule. For example, the encounter complexes of PMes3/B(C6F5)3 and P(tBu)3/B(C6F5)3 are stabilised by multiple C–H⋯F non-covalent interactions, although some studies have also shown a small P⋯B interaction in the latter. The electronic stabilisation of the encounter complex is opposed by the decrease in entropy, leading to the formation of this adduct being unfavourable according to free energy calculations. The encounter complex therefore has a low concentration in solution, which hampered early attempts to characterise this species experimentally.The first unambiguous evidence for the encounter complex in solution came from NMR measurements,68 and showed that the association of PMes3/B(C6F5)3 had no preferred orientation, which is consistent with the association being driven by dispersion interactions. An experimental value for the association constant of this FLP combination was obtained from the data (K = 0.5 ± 0.2 M−1), which supported the endergonic nature of the encounter complex. The concept of using ionic liquids as the solvent to better stabilise the encounter complex has been explored, which could open up new avenues for aiding characterisation of the encounter complex and promoting FLP reactivity.73 Neutron scattering measurements have also provided direct evidence for the association of P(tBu)3/B(C6F5)3 in benzene that is consistent with previous studies.73
More recently, it was discovered that the encounter complex of PMes3/B(C6F5)3 can absorb a photon to promote SET and afford a short-lived frustrated radical pair.83,84 This charge-transfer process was confirmed by EPR spectroscopy and transient absorption spectroscopy, and has enabled the encounter complex to be directly studied using UV-Vis spectroscopy and resonance Raman spectroscopy.88
The studies discussed in this article have provided evidence for the encounter complex in FLP chemistry. However, there are still questions to be answered to expand knowledge and application in this area of chemistry:
• Can the encounter complex be observed crystallographically?
• Can we experimentally determine the effects of concentration and temperature on encounter complex formation?
• Can we find an experimental probe to measure the concentration of “active” encounter complex in solution, i.e. those combinations that are oriented for small-molecule activation, instead of including the non-directional and non-productive orientations?
• Can the substituents around the Lewis acidic and Lewis basic centres be tuned to increase the concentration of the encounter complex in solution?
• Can we correlate concentration of encounter complex with catalytic activity for different FLP combinations, and therefore design FLP systems that are more active?
FLPs have unlocked reactivity and catalysis that was unthinkable by main-group compounds only twenty years ago, and will undoubtedly continue to provide new and sustainable routes to fundamental transformations. A thorough understanding of how the Lewis acids and bases associate and interact with small molecules in solution will be essential to driving this area of chemistry forward.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. R. J. would like to thank his mentors and colleagues for all their support in his career so far, and particularly to Prof. Doug Stephan, Dr Tim Johnstone, and Prof. Chris Slootweg for the stimulating discussions about FLPs and their mode of reactivity that have prompted this article. A. R. J. would also like to thank the Royal Society (URF\R1\201636) and the School of Chemistry at the University of Birmingham for funding.References
- G. N. Lewis, Valence and the Structure of Atoms and Molecules, Chemical Catalogue Company, Inc., New York, 1923 Search PubMed.
- H. C. Brown, H. I. Schlesinger and S. Z. Cardon, J. Am. Chem. Soc., 1942, 64, 325–329 CrossRef CAS.
- G. Wittig and E. Benz, Chem. Ber., 1959, 92, 1999–2013 CrossRef CAS.
- W. Tochtermann, Angew. Chem., Int. Ed. Engl., 1966, 5, 351–371 CrossRef CAS.
- G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- G. C. Welch, L. Cabrera, P. A. Chase, E. Hollink, J. D. Masuda, P. Wei and D. W. Stephan, Dalton Trans., 2007, 3407–3414 RSC.
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS PubMed.
- G. Erker and D. W. Stephan, Frustrated Lewis Pairs I: Uncovering and Understanding, Springer, Berlin, Heidelberg, 2013 Search PubMed.
- G. Erker and D. W. Stephan, Frustrated Lewis Pairs II: Expanding the Scope, Springer, Berlin, Heidelberg, 2013 Search PubMed.
- J. C. Slootweg and A. R. Jupp, Frustrated Lewis Pairs, Springer, Cham, 2021 Search PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- A. R. Jupp and D. W. Stephan, Trends Chem., 2019, 1, 35–48 CrossRef CAS.
- C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 CrossRef PubMed.
- E. Otten, R. C. Neu and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 9918–9919 CrossRef CAS PubMed.
- M. Sajid, A. Klose, B. Birkmann, L. Liang, B. Schirmer, T. Wiegand, H. Eckert, A. J. Lough, R. Fröhlich, C. G. Daniliuc, S. Grimme, D. W. Stephan, G. Kehr and G. Erker, Chem. Sci., 2013, 4, 213–219 RSC.
- D. W. Stephan and G. Erker, Chem. Sci., 2014, 5, 2625–2641 RSC.
- P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050–8053 CrossRef CAS PubMed.
- J. Lam, K. M. Szkop, E. Mosaferi and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3592–3612 RSC.
- D. W. Stephan, J. Am. Chem. Soc., 2021, 143, 20002–20014 CrossRef CAS PubMed.
- M. A. Légaré, M. A. Courtemanche, E. Rochette and F.-G. Fontaine, Science, 2015, 349, 513–516 CrossRef PubMed.
- J. Paradies, Eur. J. Org. Chem., 2019, 283–294 CrossRef CAS.
- L. Liu, B. Lukose, P. Jaque and B. Ensing, Green Energy Environ., 2019, 4, 20–28 CrossRef.
- M. P. Burke and S. J. Klippenstein, Nat. Chem., 2017, 9, 1078–1082 CrossRef CAS PubMed.
- R. T. Skodje, Nat. Chem., 2017, 9, 1038–1039 CrossRef CAS PubMed.
- A. Moroz, R. L. Sweany and S. L. Whittenburg, J. Phys. Chem., 1990, 94, 1352–1357 CrossRef CAS.
- T. A. Rokob, A. Hamza, A. Stirling, T. Soós and I. Pápai, Angew. Chem., Int. Ed., 2008, 47, 2435–2438 CrossRef CAS PubMed.
- H. Zhou and X. Lu, Sci. China: Chem., 2017, 60, 904–911 CrossRef CAS.
- H. A. Duong, T. N. Tekavec, A. M. Arif and J. Louie, Chem. Commun., 2004, 112–113 RSC.
- H. Zhou, W. Z. Zhang, C. H. Liu, J. P. Qu and X. B. Lu, J. Org. Chem., 2008, 73, 8039–8044 CrossRef CAS PubMed.
- F. Buß, P. Mehlmann, C. Mück-Lichtenfeld, K. Bergander and F. Dielmann, J. Am. Chem. Soc., 2016, 138, 1840–1843 CrossRef PubMed.
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983–988 CrossRef CAS PubMed.
- F.-G. Fontaine and D. W. Stephan, Philos. Trans. R. Soc., A, 2017, 375, 20170004 CrossRef PubMed.
- X. Zhao and D. W. Stephan, J. Am. Chem. Soc., 2011, 133, 12448–12450 CrossRef CAS PubMed.
- A. Bismuto, G. S. Nichol, F. Duarte, M. J. Cowley and S. P. Thomas, Angew. Chem., Int. Ed., 2020, 59, 12731–12735 CrossRef CAS PubMed.
- T. J. Tague and L. Andrews, J. Am. Chem. Soc., 1994, 116, 4970–4976 CrossRef CAS.
- G. I. Nikonov, S. F. Vyboishchikov and O. G. Shirobokov, J. Am. Chem. Soc., 2012, 134, 5488–5491 CrossRef CAS PubMed.
- C. Fan, L. G. Mercier, W. E. Piers, H. M. Tuononen and M. Parvez, J. Am. Chem. Soc., 2010, 132, 9604–9606 CrossRef CAS PubMed.
- A. Y. Houghton, V. A. Karttunen, C. Fan, W. E. Piers and H. M. Tuononen, J. Am. Chem. Soc., 2013, 135, 941–947 CrossRef CAS PubMed.
- P. Spies, G. Erker, G. Kehr, K. Bergander, R. Fröhlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072–5074 RSC.
- F. Bertini, V. Lyaskovskyy, B. J. Timmer, F. J. de Kanter, M. Lutz, A. W. Ehlers, J. C. Slootweg and K. Lammertsma, J. Am. Chem. Soc., 2012, 134, 201–204 CrossRef CAS PubMed.
- Z. Mo, E. L. Kolychev, A. Rit, J. Campos, H. Niu and S. Aldridge, J. Am. Chem. Soc., 2015, 137, 12227–12230 CrossRef CAS PubMed.
- K. Chernichenko, A. Madarasz, I. Pápai, M. Nieger, M. Leskela and T. Repo, Nat. Chem., 2013, 5, 718–723 CrossRef CAS PubMed.
- L. Fan, A. R. Jupp and D. W. Stephan, J. Am. Chem. Soc., 2018, 140, 8119–8123 CrossRef CAS PubMed.
- G. Ghattas, D. Chen, F. Pan and J. Klankermayer, Dalton Trans., 2012, 41, 9026–9028 RSC.
- M. Lindqvist, K. Borre, K. Axenov, B. Kotai, M. Nieger, M. Leskela, I. Pápai and T. Repo, J. Am. Chem. Soc., 2015, 137, 4038–4041 CrossRef CAS PubMed.
- X. Feng, W. Meng and H. Du, in Frustrated Lewis Pairs, Molecular Catalysis Vol. 2, ed. J. C. Slootweg and A. R. Jupp, Springer, Cham, 2021, pp. 29–86 Search PubMed.
- S. J. Geier and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 3476–3477 CrossRef CAS PubMed.
- T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809–15812 CrossRef CAS PubMed.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS PubMed.
- T. C. Johnstone, G. Wee and D. W. Stephan, Angew. Chem., Int. Ed., 2018, 57, 5881–5884 CrossRef CAS PubMed.
- D. Holschumacher, T. Bannenberg, C. G. Hrib, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2008, 47, 7428–7432 CrossRef CAS PubMed.
- S. Kronig, E. Theuergarten, D. Holschumacher, T. Bannenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, Inorg. Chem., 2011, 50, 7344–7359 CrossRef CAS PubMed.
- G. Bistoni, A. A. Auer and F. Neese, Chem. – Eur. J., 2017, 23, 865–873 CrossRef CAS PubMed.
- A. C. Shaikh, J. M. Veleta, J. Moutet and T. L. Gianetti, Chem. Sci., 2021, 12, 4841–4849 RSC.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef PubMed.
- S. Grimme, H. Kruse, L. Goerigk and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 1402–1405 CrossRef CAS PubMed.
- G. Skara, B. Pinter, J. Top, P. Geerlings, F. De Proft and F. De Vleeschouwer, Chem. – Eur. J., 2015, 21, 5510–5519 CrossRef CAS PubMed.
- T. A. Rokob, I. Bakó, A. Stirling, A. Hamza and I. Pápai, J. Am. Chem. Soc., 2013, 135, 4425–4437 CrossRef CAS PubMed.
- A. Hamza, A. Stirling, T. A. Rokob and I. Pápai, Int. J. Quantum Chem., 2009, 109, 2416–2425 CrossRef CAS.
- B. Schirmer and S. Grimme, Chem. Commun., 2010, 46, 7942–7944 RSC.
- G. Skara, F. De Vleeschouwer, P. Geerlings, F. De Proft and B. Pinter, Sci. Rep., 2017, 7, 16024 CrossRef PubMed.
- J. J. Cabrera-Trujillo and I. Fernández, Inorg. Chem., 2019, 58, 7828–7836 CrossRef PubMed.
- I. Fernández, Chem. Commun., 2022 10.1039/d2cc00233g.
- L. L. Zeonjuk, N. Vankova, A. Mavrandonakis, T. Heine, G. V. Röschenthaler and J. Eicher, Chem. – Eur. J., 2013, 19, 17413–17424 CrossRef CAS PubMed.
- I. Bakó, A. Stirling, S. Bálint and I. Pápai, Dalton Trans., 2012, 41, 9023–9025 RSC.
- L. Rocchigiani, G. Ciancaleoni, C. Zuccaccia and A. Macchioni, J. Am. Chem. Soc., 2014, 136, 112–115 CrossRef CAS PubMed.
- A. Y. Houghton and T. Autrey, J. Phys. Chem. A, 2017, 121, 8785–8790 CrossRef CAS PubMed.
- A. J. Marwitz, J. L. Dutton, L. G. Mercier and W. E. Piers, J. Am. Chem. Soc., 2011, 133, 10026–10029 CrossRef CAS PubMed.
- C. Bannwarth, A. Hansen and S. Grimme, Isr. J. Chem., 2015, 55, 235–242 CrossRef CAS.
- H. W. Kim and Y. M. Rhee, Chem. – Eur. J., 2009, 15, 13348–13355 CrossRef CAS PubMed.
- L. C. Brown, J. M. Hogg, M. Gilmore, L. Moura, S. Imberti, S. Gärtner, H. Q. N. Gunaratne, R. J. O'Donnell, N. Artioli, J. D. Holbrey and M. Swadźba-Kwaśny, Chem. Commun., 2018, 54, 8689–8692 RSC.
- X. Liu, X. Wang, Y. Li, T. Yu, W. Zhao and L. Liu, Phys. Chem. Chem. Phys., 2021, 23, 12541–12548 RSC.
- T. F. Headen, C. A. Howard, N. T. Skipper, M. A. Wilkinson, D. T. Bowron and A. K. Soper, J. Am. Chem. Soc., 2010, 132, 5735–5742 CrossRef CAS PubMed.
- L. Liu, L. L. Cao, Y. Shao, G. Ménard and D. W. Stephan, Chem, 2017, 3, 259–267 CAS.
- L. L. Liu and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3454–3463 RSC.
- A. Dasgupta, E. Richards and R. L. Melen, Angew. Chem., Int. Ed., 2021, 60, 53–65 CrossRef CAS PubMed.
- F. Holtrop, A. R. Jupp and J. C. Slootweg, in Frustrated Lewis Pairs, Molecular Catalysis Vol. 2, ed. J. C. Slootweg and A. R. Jupp, Springer, Cham, 2021, pp. 361–385 Search PubMed.
- W. E. Piers, A. J. Marwitz and L. G. Mercier, Inorg. Chem., 2011, 50, 12252–12262 CrossRef CAS PubMed.
- R. J. Kwaan, C. J. Harlan and J. R. Norton, Organometallics, 2001, 20, 3818–3820 CrossRef CAS.
- E. J. Lawrence, V. S. Oganesyan, G. G. Wildgoose and A. E. Ashley, Dalton Trans., 2013, 42, 782–789 RSC.
- F. Holtrop, A. R. Jupp, N. P. van Leest, M. Paradiz Dominguez, R. M. Williams, A. M. Brouwer, B. de Bruin, A. W. Ehlers and J. C. Slootweg, Chem. – Eur. J., 2020, 26, 9005–9011 CrossRef CAS PubMed.
- F. Holtrop, A. R. Jupp, B. J. Kooij, N. P. van Leest, B. de Bruin and J. C. Slootweg, Angew. Chem., Int. Ed., 2020, 59, 22210–22216 CrossRef CAS PubMed.
- R. S. Mulliken, J. Am. Chem. Soc., 1952, 74, 811–824 CrossRef CAS.
- R. Foster, J. Phys. Chem., 1980, 84, 2135–2141 CrossRef CAS.
- S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641–653 CrossRef CAS PubMed.
- L. R. Marques and R. A. Ando, ChemPhysChem, 2021, 22, 522–525 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |