
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reaction of a bis(pentafulvene)titanium complex with an N-heterocyclic olefin: C–H-activation leads to resonance between a titanium vinyl and titanium alkylidene complex†
Malte
Fischer
 *ab,
Matthew M. D.
Roy
c,
Sascha
Hüller
b,
Marc
Schmidtmann
b and
Rüdiger
Beckhaus
*b
*ab,
Matthew M. D.
Roy
c,
Sascha
Hüller
b,
Marc
Schmidtmann
b and
Rüdiger
Beckhaus
*b
aInorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: malte.fischer@chem.ox.ac.uk
bInstitut für Chemie, Fakultät für Mathematik und Naturwissenschaften, Carl von Ossietzky Universität Oldenburg, Postfach 2503, D-26111 Oldenburg, Germany. E-mail: ruediger.beckhaus@uni-oldenburg.de
cDepartment of Chemistry, Catalysis Research Center and Institute for Silicon Chemistry, Technische Universität München, Germany
First published on 7th February 2022
Abstract
The N-heterocyclic olefin (NHO) ImMe4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 (2) (ImMe4CH2 = (MeCNMe)2CCH2) was employed for the synthesis of the titanium complex 3 derived from an NHO ligand precursor. By reacting 2 with the bis(π–η5:σ–η1-pentafulvene)titanium complex 1a, the terminal ylidic methylene unit of 2 is deprotonated by the quaternary exocyclic carbon atom of one pentafulvene ligand of 1a yielding the titanium complex 3 which bears an anionic NHO ligand (ImMe4CH−). 3 was characterized by NMR spectroscopy, single crystal X-ray diffraction and quantum chemical calculations. The latter highlight that 3 is best described as a titanium vinyl complex with significant contribution of the titanium alkylidene resonance structure.
CH2 (2) (ImMe4CH2 = (MeCNMe)2CCH2) was employed for the synthesis of the titanium complex 3 derived from an NHO ligand precursor. By reacting 2 with the bis(π–η5:σ–η1-pentafulvene)titanium complex 1a, the terminal ylidic methylene unit of 2 is deprotonated by the quaternary exocyclic carbon atom of one pentafulvene ligand of 1a yielding the titanium complex 3 which bears an anionic NHO ligand (ImMe4CH−). 3 was characterized by NMR spectroscopy, single crystal X-ray diffraction and quantum chemical calculations. The latter highlight that 3 is best described as a titanium vinyl complex with significant contribution of the titanium alkylidene resonance structure.
Introduction
N-Heterocyclic olefins (NHOs)1 formally consist of a terminal alkylidene unit bound to an N-heterocyclic carbene (NHC) backbone.2 This molecular composition leads to remarkable nucleophilic character of the exocyclic methylene group, especially when compared to more traditional olefins. The pendent N-heterocycle causes significant polarization of the exocyclic carbon–carbon double bond, leading to increased nucleophilicity of the ylidic carbon atom. As a result, this ligand class has been characterized as relatively strong σ-donors without the potential to act as π-acceptors. This can be illustrated by the zwitterionic resonance structure B shown in Scheme 1a.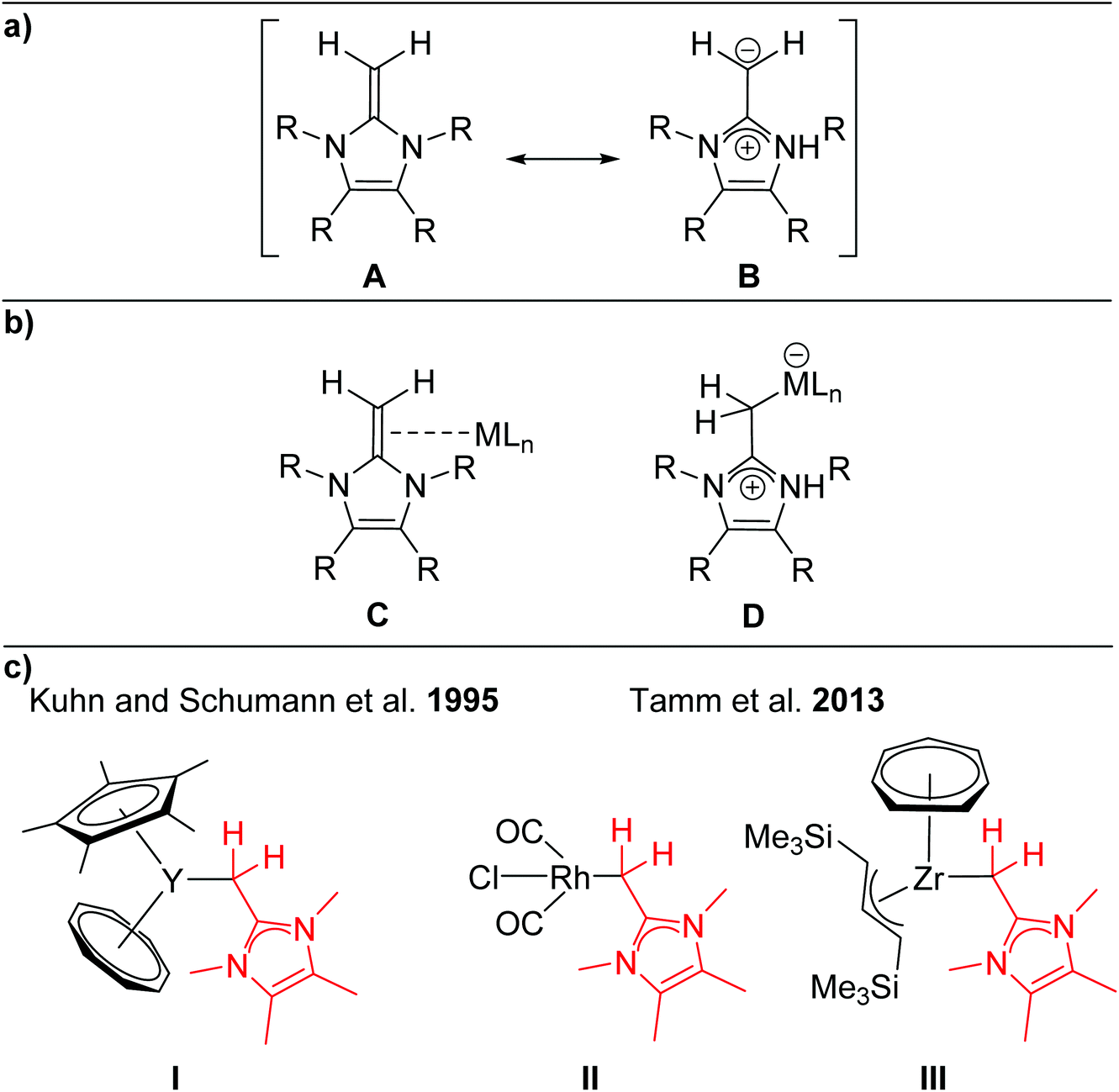 | ||
| Scheme 1 a) Neutral olefinic (A) and zwitterionic (B) resonance forms of an NHO; (b) coordination modes of NHOs to a metal centre (C and D) (R = H, alkyl, aryl); (c) selected examples of literature known transition metal complexes with NHO ligands (I–III). | ||
Early reports by Kuhn and coworkers in 1993/1994 demonstrated the nucleophilicity of NHOs by employing ImMe4CH2 for the formation of stable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 donor acceptor adducts with the Lewis acidic metal pentacarbonyl species of molybdenum and tungsten.3 Despite these examples in the 1990s, it took nearly twenty years for the renaissance of NHO chemistry when Beller et al. synthesized the more sterically encumbered NHO IPrCH2 (IPrCH2 = (HCNDipp)2CCH2; Dipp = 2,6-iPr2C6H3) by an in situ synthetic protocol and used it for subsequent transformations to cationic phosphine ligand precursor compounds.4 The corresponding palladium complexes bearing these phosphine ligands are active catalysts in C–E (E = C, N, O) bond formations.4
:1 donor acceptor adducts with the Lewis acidic metal pentacarbonyl species of molybdenum and tungsten.3 Despite these examples in the 1990s, it took nearly twenty years for the renaissance of NHO chemistry when Beller et al. synthesized the more sterically encumbered NHO IPrCH2 (IPrCH2 = (HCNDipp)2CCH2; Dipp = 2,6-iPr2C6H3) by an in situ synthetic protocol and used it for subsequent transformations to cationic phosphine ligand precursor compounds.4 The corresponding palladium complexes bearing these phosphine ligands are active catalysts in C–E (E = C, N, O) bond formations.4
Since these preliminary studies by Kuhn and Beller, NHOs have found their entry into the literature as stabilizing ligands for reactive low-valent main group compounds.2a,b As representative examples regarding this topic, NHOs have been employed for the synthesis of group 14 EH2 complexes by the Rivard group (E = Si, Ge, Sn).5 Further highly noteworthy applications of NHOs are their use as organocatalysts, as well as their use as ligands for transition metal complexes.2 But in contrast to the ubiquity of NHCs as ligands,2b,6 NHOs remain a fairly underrepresented ligand class for transition metals, which is underlined by the small number of reported transition metal complexes of Sc,7 Zr,8 W,3,9 Mo,3 Rh,10 Ir,11 Pd,12 Pt13 and Au14 (Scheme 1c, selected examples I–III), in which the NHO (subunit) adopts consistently an end-on coordination mode.
Given the shown underestimated advantages of NHOs in main group chemistry, intensification of research in connection with transition metals is desirable. In the first place, the reactive site in NHOs is less shielded and easier to address for further investigations compared to related NHCs. Secondly, the strong NHC–element interaction causes diminished nucleophilicity of the corresponding complexes which could be circumvented by employing NHOs due to them being milder donors.
As can be seen from the above overview of transition metal NHO complexes, NHOs have not yet been investigated in realm of titanium chemistry. We questioned whether NHOs might be potential ligands for the comprehensively studied bis(π–η5:σ–η1-pentafulvene)titanium complexes, whose reactivity is mainly driven by the π–η5:σ–η1 bonding mode of both pentafulvene ligands.15 In this context, the synthesis of a series of 1:1 titanium–NHC adducts has already been reported in which the NHC simply coordinates end-on to the metal centre, accompanied by a haptotropic shift of both pentafulvene ligands in direction to a π–η4 coordination mode.16 Motivated by these results, we were interested in the reactivity of an NHO towards bis(π–η5:σ–η1-pentafulvene)titanium complexes.
Results and discussion
Accordingly, the less sterically demanding NHO Ime4CH2 (2) was targeted and synthesized by employing a reported four step procedure starting from acetaldehyde and butane-2,3-dione (1H NMR spectrum of obtained 2 see Fig. S2†).17 The bis(π–η5:σ–η1-pentafulvene)titanium complexes 1a (R = Ad) and 1b (R = p-tolyl) were also prepared according to literature protocols.18 The stoichiometric combination of 1a and 2 in n-hexane at room temperature is accompanied by a color change from blue to magenta within 15 minutes of vigorous stirring (Fig. S1†), and after workup a magenta solid is obtained in a good isolated yield of 94%. Analysis of the NMR data revealed that instead of forming the supposed 1:1 adduct comparable to all reported transition metal NHO complexes so far (cf.Scheme 1), the product proved to be the result of deprotonation at the terminal methylene group of 2. Hence, the exocyclic quaternary carbon atom (Cq,exo) of one pentafulvene ligand is protonated and the corresponding anionic N-heterocyclic vinyl unit (ImMe4CH−) is added to the metal center to cleanly give complex 3 (Scheme 2).
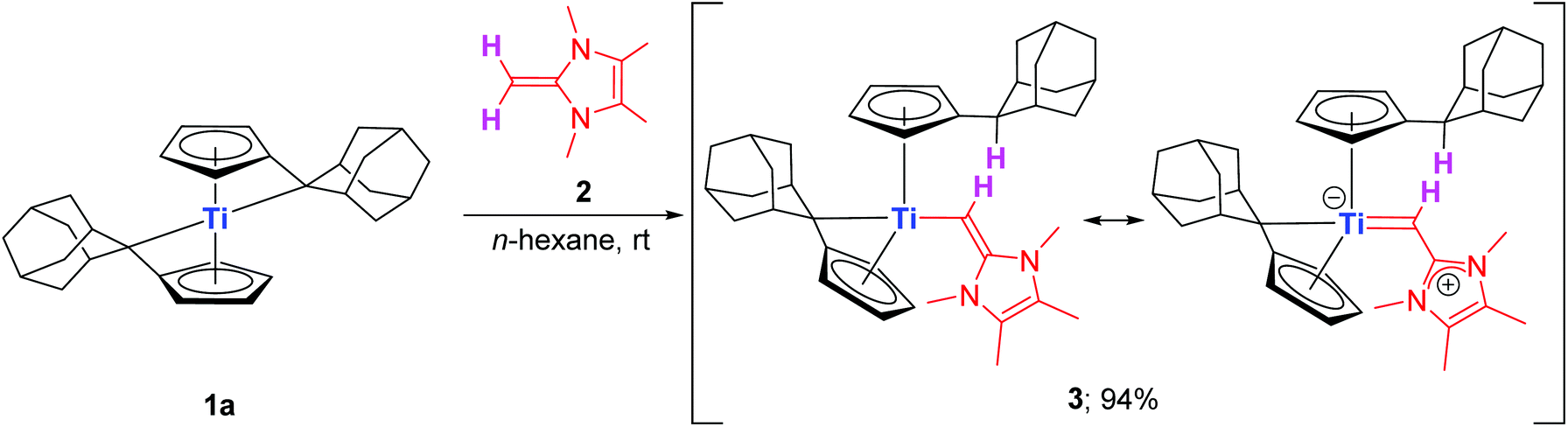 | ||
| Scheme 2 Synthesis of complex 3 by reaction of 1a with 2. | ||
Characteristic of the asymmetric environment at the central titanium atom are the eight signals in the 1H NMR spectrum for the four remaining pentafulvene hydrogen atoms and the four hydrogen atoms of the newly formed substituted cyclopentadienyl ligand between 4.30 and 6.47 ppm. The protonation of one Cq,exo of the former pentafulvene ligand is clearly verified by the 1H NMR signal localized at δ1H = 3.02 ppm with the corresponding 13C resonance at δ13C{1H} = 45.6 ppm, which is in good agreement to other complexes bearing the CpAdH ligand.19 The signals of the Cq,exo and Cq,ipso atoms of the remaining pentafulvene ligand are localized at δ13C{1H} = 111.1 and 124.7 ppm respectively, and consequently only marginally shifted compared to the starting material 1a (δ13C{1H} = 113.3 and 131.4 ppm).18 Most striking is the downfield shifted singlet signal at δ1H = 7.17 ppm correlating to the most downfield shifted signal in the 13C NMR at δ13C{1H} = 166.7 ppm of the CH functionality in position α to the titanium. The gated-decoupling 13C NMR experiment (Fig. S5†) reveals the corresponding 1JC,H coupling constant of 1JC,H = 118.5 Hz, indicative for sp2-hybridization of the respective carbon atom and that no Ti–Cα agostic interactions are present. The 1H/15N HMBC NMR spectrum (Fig. S6†) shows one resonance at δ15N = 123.4 ppm with cross peaks to the aforementioned CH unit and to both signals of the methyl groups (δ1H = 1.45 (2 × CqCH3) and 2.81 (2 × NCH3) ppm) of the N-heterocyclic backbone.
To the best of our knowledge, 3 is the first early transition metal complex with a formal N-heterocyclic vinyl ligand whereas this bonding motif is more commonly encountered in main group chemistry. Generally, these anionic ligands are strongly electron releasing. This has been exploited first by Rivard and coworkers, and later by Ghadwal et al., and more recently again the Rivard group reported stable and base-free divinylgermanes, divinylgermylenes, and expanded this protocol to a series of homoleptic tetrylenes and a zinc complex.20 To date, all reported examples of compounds bearing the anionic N-heterocyclic vinyl ligand require either stoichiometric amounts of an external base (e.g. a second equivalent of the NHO used), proceed via direct halosilane elimination of a preformed silylated NHO or via salt metathesis whereas in the herein reported example no byproducts are formed.20,21 Here we demonstrate the direct formation of this ligand class in the coordination sphere of titanium by formal intramolecular proton transfer to one pentafulvene ligand. Although in principal there is a second pentafulvene ligand to potentially react with, no second C–H activation occurs regardless of the stoichiometry used for this reaction. Even heating of a toluene solution of 3 to 100 °C for several days only resulted in the reisolation of 3.
Of particular note, the reaction of the aryl substituted bis(π–η5:σ–η1-pentafulvene)titanium complex 1b with 2 does not lead to a similar outcome. The increased propensity of the adamantyl-substituted derivative 1a to exhibit C–H activation chemistry can be attributed to the stronger basicity of an alkyl substituted Cq,exo atom when compared to a diaryl substitution pattern.15 Independent of the chosen stoichiometry, solvent, and reaction temperature, the reaction of 1b with 2 is always accompanied by release of the respective pentafulvene ligand (Fig. S7†).
Crystals of 3 suitable for single crystal X-ray diffraction were obtained from a saturated n-hexane/toluene (10:1) solution of 3 at −26 °C and the molecular structure is shown in Fig. 1. Complex 3 crystallizes in the orthorhombic space group Pccn with the titanium atom being best described as trigonal pyramidal according to the τ4/τd values of τ4 = 0.76 (τd = 0.65).22 The π–η5:σ–η1 coordination mode of the remaining pentafulvene ligand is unambiguously verified by the established parameters,23 and, as expected, the values deviate only marginal from the starting material 1 and other complexes of group 4 metals with π–η5:σ–η1 bonded pentafulvene ligands.18,24 The transition from the other former pentafulvene ligand to a substituted cyclopentadienyl ligand is proved by the C16–C21 bond lengths of 1.5130(19) Å, being typical for C(sp2)–C(sp3) single bonds.25 Both carbon atoms C31 and C32 are sp2-hybridized (in agreement to the 1JC,H coupling constant of C31) as verified by the sum of angles around both atoms (360°; the hydrogen atom H31 was located in the electron density map and refined isotropically). The presence of Ti–Cα agostic interactions can be excluded as shown by the Ti1–C31–H31 angle of 138.78(16)°. The respective C31–C32 bond length of 1.415(3) Å is significantly elongated in comparison to NHO 2 (1.357(3) Å),3a and for the dichlorogermylene (IDippCH)2GeCl2 similar values are found (1.371(4) Å and 1.359(4) Å).20b These short C–C distances in are in good agreement to C(sp2)–C(sp2) double bonds (1.32 Å (ref. 25)). This is further supported by the fact that the mentioned carbon atoms and the substituents (hydrogen atom and both nitrogen atoms of the imidazoline moiety) in (IDippCH)2GeCl2 are in-plane, which is not applicable for 3 (Fig. 1, right).20b In contrast, H31 in complex 3 is significantly more coplanar to the plane defined by the titanium atom and both centroids of the pentafulvene and cyclopentadienyl ligands (fold angle between the planes defined by Ct1, Ti1, Ct2 and H31, C31, Ti1: 26.9°). This supports the formulation of a titanium–carbon double bond. To classify the Ti1–C31 bond length of 2.0371(13) Å in 3, Table 1 summarizes selected titanium–carbon bond lengths of titanium alkyl complexes (IV and V), titanium vinyl complexes (VI and VII), and titanium alkylidene complexes (VIII, IX, X, and XI).26–32
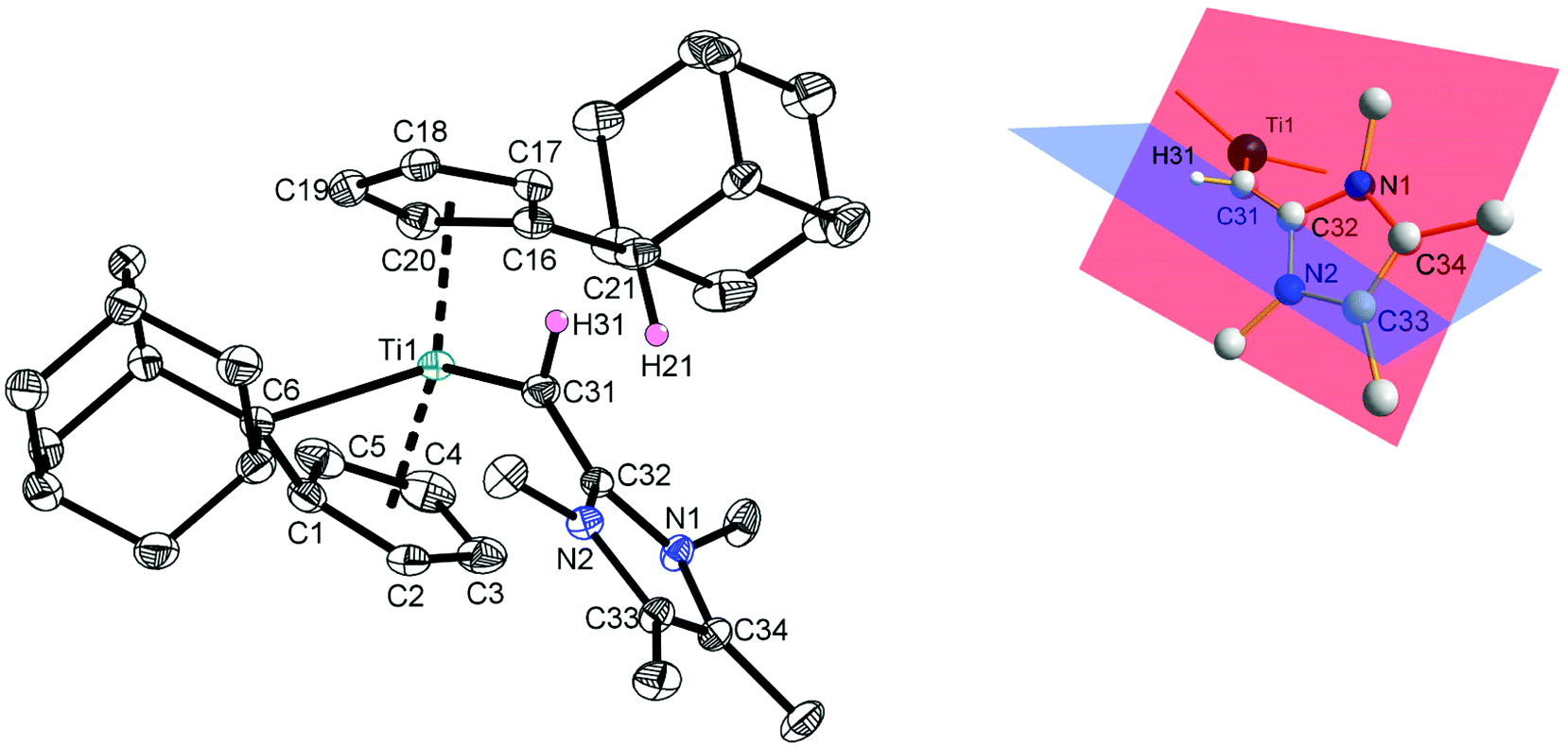 | ||
| Fig. 1 Left: Molecular structure of 3 in the crystal. Thermal ellipsoids are drawn at the 50% probability level (hydrogen atoms except H21, H31 have been omitted for clarity). Selected bond lengths (Å) and angles (°): Ti1–C6 2.378(2), Ti1–C31 2.0371(13), C1–C6 1.454(3), C16–C21 1.5130(19), C31–C32 1.415(3), N1–C32 1.375(2), N1–C34 1.404(2), N2–C32 1.376(3), N2–C33 1.394(2), C33–C34 1.352(3), Ti1–C31–C32 138.69(14), Ct1–Ti1–Ct2 136.0, ∑∠C31 360.0, ∑∠C32 360.0 (Ct1 = centroid of C1–C5; Ct2 = centroid of C16–C20); right: excerpt of the molecular structure of 3 in the crystal. Red plane defined by N1, C32, N2; blue plane defined by C32, C31, H31; fold angle between both planes: 61°; fold angle between the planes defined by Ct1, Ti1, Ct2 and H31, C31, Ti1: 26.9°. | ||
| Compound | Ti–Cα bond length | Compound | Ti–Cα bond length |
|---|---|---|---|
|
a Nacnac = ArNC(Me)C–C(Me)NAr, Ar = 2,6-iPr2C6H3.
b PNP = N[2-P(iPr2)2-4-MeC6H3]2.
c PN = N[2-P(iPr2)-4-MeC6H3]-2,4,6-MeC6H2.
|
|||
| Cp2Ti(CH3)2 (IV)26 | 2.170(2) Å | [(Nacnac)TiC(H)tBu][OTf]a (VIII)30 |
1.840(8) Å |
| 2.181(2) Å | |||
| Cp2Ti(CH2Ph)2 (V)27 | 2.239(6) Å | [(Nacnac)TiC(H)tBu][Cl]a (IX)31 |
1.830(3) Å |
| 2.210(5) Å | |||
| Cp2Ti(CHCH2)F (VI)28 |
2.098(6) Å | [(PNP)TiC(H)tBu(OTf)b (X)31 |
1.883(7) Å |
| Cp*2Ti(CHCH2)OC(Ph)CH2 (VII)29 |
2.141(2) Å | (PN)2TiCH2c (XI)32 |
1.939(3) Å |
Based on the Ti–Cα bond lengths shown in Table 1, the Ti1–C31 bond is best described as an elongated double bond. The elongation compared to classic titanium alkylidene complexes (VIII–XI) is attributed to the different chemical environment at the Cα atom and the formal negative charge in 3, whose impact is also critical for the significant downfield shifted 13C NMR signal compared to VIII–XI.
To further probe the nature of the vinylic ligand to metal interaction in 3, gas phase computations were carried out at the M06-2X-D3/def2-TZVP level of theory.33–38 Notably, the HOMO contains a polarized titanium–carbon π-bond, delocalized about the C32–C31–Ti1 motif (cf.Fig. 1 and 2 top left). The polarity of this interaction is also manifest in the natural atomic charges, with the titanium-bound carbon carrying a significant negative charge (−0.77) along with a highly charged titanium center (+0.70) (Fig. 2 top right). Selected NBOs of the Ti1–C31–C32 unit of 3 are shown in Table S2.† Natural resonance theory (NRT) analysis further supports a delocalized C32–C31–Ti1 π-bonding description, as can be seen by the six leading resonance structures (Fig. 2 bottom). Resonance structures containing either a C32C31 bond (A, 43%) or a Ti1C31 bond (B, E, F, 38%) account for a majority of the overall structure. Similarly, both C32–C31 and Ti1–C31 Wiberg bond indices indicate partial π-bond character (1.49 and 1.02, respectively; Fig. 2 top right).
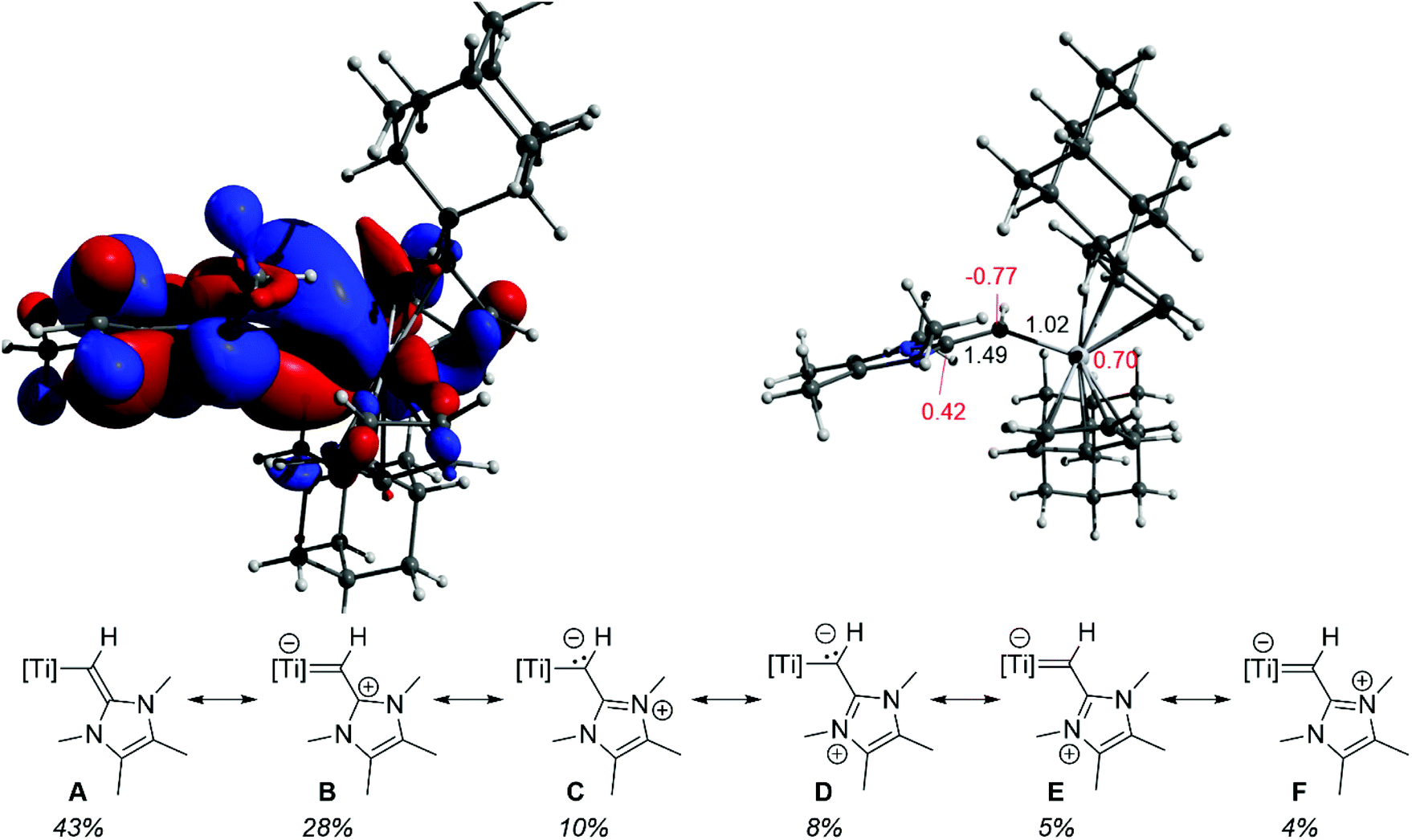 | ||
| Fig. 2 Top left: HOMO of 3; top right: optimized geometry of 3 computed at the M06-2X-D3/def2-TZVP level of theory with selected natural charges (red) and Wiberg bond indices (black); bottom: natural resonance theory computed leading resonance structures of 3 with their % weighting. | ||
Conclusion
In conclusion, the reaction of the NHO 2 with the bis(π–η5:σ–η1pentafulvene)titanium complex 1a selectively proceeds via formal deprotonation of the terminal ylidic methylene group by the Cq,exo atom of one pentafulvene ligand and addition of the formal ImMe4CH− unit to the titanium center to give complex 3, which has been characterized by means of multinuclear NMR spectroscopy, single crystal X-ray diffraction and quantum chemical analyses. These data suggest that the bonding situation in 3 is best described by the resonance structures shown in Scheme 2 with delocalized π bonding along the Ti–C–C axis (cf.Fig. 1).
Conflicts of interest
There are no conflict to declare.Acknowledgements
M. F. is thankful to the Alexander von Humboldt Foundation for funding and support (Feodor Lynen postdoctoral research fellowship). M. M. D. R. is thankful to the Natural Sciences and Engineering Research Council of Canada (NSERC) for a Postdoctoral Fellowship and thanks Dr Emanuel Hupf for access to computational resources.Notes and references
- The term of N-hterocyclic olefines (NHOs) was introduced in 2011: S. M. I. Al-Rafia, A. C. Malcolm, S. K. Liew, M. J. Ferguson, R. McDonald and E. Rivard, Chem. Commun., 2011, 47, 6987–6989 RSC.
- Articles and reviews on recent developments in the chemistry of NHOs: (a) A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed; (b) M. M. D. Roy and E. Rivard, Acc. Chem. Res., 2017, 50, 2017–2025 CrossRef CAS PubMed; (c) R. S. Ghadwal, Dalton Trans., 2016, 45, 16081–16095 RSC; (d) R. D. Crocker and T. V. Nguyen, Chem. – Eur. J., 2016, 22, 2208–2213 CrossRef CAS PubMed.
- (a) N. Kuhn, H. Bohnen, J. Kreutzberg, D. Bläser and R. Boese, J. Chem. Soc., Chem. Commun., 1993, 1136–1137 RSC; (b) N. Kuhn, H. Bohnen, D. Bläser and R. Boese, Chem. Ber., 1994, 127, 1405–1407 CrossRef CAS.
- A. Dumrath, X.-F. Wu, H. Neumann, A. Spannenberg, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8988–8992 ( Angew. Chem. , 2010 , 122 , 9172–9176 ) CrossRef CAS PubMed.
- (a) K. C. Thimer, S. M. I. Al-Rafia, M. J. Ferguson, R. McDonald and E. Rivard, Chem. Commun., 2009, 7119–7221 RSC; (b) S. M. I. Al-Rafia, A. C. Malcolm, S. K. Liew, M. J. Ferguson and E. Rivard, J. Am. Chem. Soc., 2011, 133, 777–779 CrossRef CAS PubMed; (c) S. M. I. Al-Rafia, A. C. Malcolm, R. McDonald, M. J. Ferguson and E. Rivard, Chem. Commun., 2012, 48, 1308–1310 RSC.
- Selected recent reviews: (a) P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem., 2021, 5, 711–725 CrossRef CAS; (b) D. Munz, Organometallics, 2018, 37, 275–289 CrossRef CAS; (c) F. E. Hahn, Chem. Rev., 2018, 118, 9455–9456 CrossRef CAS PubMed.
- H. Schumann, M. Glanz, J. Winterfeld, H. Hemling, N. Kuhn, H. Bohnen, D. Bläser and R. Boese, J. Organomet. Chem., 1995, 493, C14–C18 CrossRef CAS.
- A. Glöckner, S. Kronig, T. Bannenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, J. Organomet. Chem., 2013, 723, 181–187 CrossRef.
- (a) D. A. Imbrich, W. Frey, S. Naumann and M. R. Buchmeiser, Chem. Commun., 2016, 52, 6099–6102 RSC; (b) D. A. Imbrich, W. Frey, S. Naumann and M. R. Buchmeiser, Chem. Commun., 2016, 52, 6099–6102 RSC.
- (a) S. Kronig, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2013, 2013, 2301–2314 CrossRef CAS; (b) K. Powers, C. Hering-Junghans, R. McDonald, M. J. Ferguson and E. Rivard, Polyhedron, 2016, 108, 8–14 CrossRef CAS.
- (a) A. Iturmendi, N. Garcia, E. A. Jaseer, J. Munarriz, P. J. Sanz Miguel, V. Polo, M. Iglesias and L. A. Oro, Dalton Trans., 2016, 45, 12835–12845 RSC; (b) M. Iglesias, A. Iturmendi, P. J. Sanz Miguel, V. Polo, J. J. Pérez-Torrente and L. A. Oro, Chem. Commun., 2015, 51, 12431–12434 RSC; (c) M. Sriram, Y. Zhu, A. M. Camp, C. S. Day and A. C. Jones, Organometallics, 2014, 33, 4157–4164 CrossRef CAS; (d) M. Viciano, M. Feliz, R. Corberan, J. A. Mata, E. Clot and E. Peris, Organometallics, 2007, 26, 5304–5314 CrossRef CAS.
- (a) I. C. Watson, A. Schumann, H. Yu, E. C. Davy, R. McDonald, M. J. Ferguson, C. Hering-Junghans and E. Rivard, Chem. – Eur. J., 2019, 25, 9678–9690 CrossRef CAS PubMed; (b) O. Esposito, D. E. Roberts, F. G. N. Cloke, S. Caddick, J. C. Green, N. Hazari and P. B. Hitchcock, Eur. J. Inorg. Chem., 2009, 2009, 1844–1850 CrossRef.
- P. P. Ponti, J. C. Baldwin and W. C. Kaska, Inorg. Chem., 1979, 18, 873–875 CrossRef CAS.
- A. Fürstner, M. Alcarazo, R. Goddard and C. W. Lehmann, Angew. Chem., Int. Ed., 2008, 47, 3210–3214 ( Angew. Chem. , 2008 , 120 , 3254–3258 ) CrossRef PubMed.
- (a) R. Beckhaus, Coord. Chem. Rev., 2018, 376, 467–477 CrossRef CAS; (b) P. Preethalayam, K. S. Krishnan, S. Thulasi, S. S. Chand, J. Joseph, V. Nair, F. Jaroschik and K. V. Radhakrishnan, Chem. Rev., 2017, 117, 3930–3989 CrossRef CAS PubMed.
- M. Manßen, C. Adler and R. Beckhaus, Chem. – Eur. J., 2016, 22, 4405–4407 CrossRef PubMed.
- (a) N. Kuhn, H. Bohnen, G. Henkel and J. Kreutzberg, Z. Naturforsch., B: J. Chem. Sci., 1996, 51, 1267–1278 CrossRef; (b) N. Kuhn, G. Henkel and J. Kreutzberg, Z. Naturforsch., B: J. Chem. Sci., 1991, 46, 1706–1712 CrossRef CAS; (c) T. Peppel, C. Roth, K. Fumino, D. Paschek, M. Köckerling and R. Ludwig, Angew. Chem., Int. Ed., 2011, 50, 6661–6665 ( Angew. Chem. , 2011 , 123 , 6791–6795 ) CrossRef CAS PubMed.
- M. Diekmann, G. Bockstiegel, A. Lützen, M. Friedemann, W. Saak, D. Haase and R. Beckhaus, Organometallics, 2006, 35, 339–348 CrossRef.
- Selected most recent examples of titanium complexes bearing the CpAdH functionality: (a) M. Fischer, M. Manßen, M. Schmidtmann, T. Klüner and R. Beckhaus, Chem. Sci., 2021, 12, 13711–13718 RSC; (b) M. Fischer, L. Vincent-Heldt, M. Hillje, M. Schmidtmann and R. Beckhaus, Dalton Trans., 2020, 49, 2068–2072 RSC.
- (a) M. M. D. Roy, S. R. Baird, E. Dornsiepen, L. A. Paul, L. Miao, M. J. Ferguson, Y. Zhou, I. Siewert and E. Rivard, Chem. – Eur. J., 2021, 27, 8572–8579 CrossRef CAS PubMed; (b) C. Hering-Junghans, P. Andreiuk, M. J. Ferguson, R. McDonald and E. Rivard, Angew. Chem., Int. Ed., 2017, 56, 6272–6275 ( Angew. Chem. , 2017 , 129 , 6368–6372 ) CrossRef CAS PubMed; (c) R. S. Ghadwal, S. O. Reichmann, F. Engelhardt, D. M. Andrada and G. Frenking, Chem. Commun., 2013, 49, 9440–9442 RSC; (d) S. M. I. Al-Rafia, M. J. Ferguson and E. Rivard, Inorg. Chem., 2011, 50, 10543–10545 CrossRef CAS PubMed; (e) I. C. Watson, M. J. Ferguson and E. Rivard, Inorg. Chem., 2021, 60, 18347–18359 CrossRef CAS PubMed.
- (a) N. Kuhn, M. Göhner and M. Steimann, Z. Anorg. Allg. Chem., 2002, 628, 1108–1115 CrossRef CAS; (b) M. K. Sharma, S. Blomeyer, B. Neumann, H.-G. Stammler and R. S. Ghadwal, Chem. – Eur. J., 2019, 25, 8249–8253 CrossRef CAS PubMed; (c) M. M. D. Roy, M. J. Ferguson, R. McDonald, Y. Zhou and E. Rivard, Chem. Sci., 2019, 10, 6476–6481 RSC; (d) P. Gupta, J.-E. Siewert, T. Wellnitz, M. Fischer, W. Baumann, T. Beweries and C. Hering-Junghans, Dalton Trans., 2021, 50, 1838–1844 RSC; (e) A. Causero, H. Elsen, J. Pahl and S. Harder, Angew. Chem., Int. Ed., 2017, 56, 6906–6910 ( Angew. Chem. , 2017 , 129 , 7010–7014 ) CrossRef CAS PubMed; (f) C. C. Chong, B. Rao, R. Ganguly, Y. Li and R. Kinjo, Inorg. Chem., 2017, 56, 8608–8614 CrossRef CAS PubMed.
- (a) M. H. Reineke, M. D. Sampson, A. L. Rheingold and C. P. Kubiak, Inorg. Chem., 2019, 54, 3211–3217 CrossRef PubMed; (b) L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- Parameters for the π–η5:σ–η1 coordination mode of the pentafulvene ligand: angle θ of the C1–C6 bond out of the five-membered ring plane: θ = 31.7°; ring slippage Δ of the titanium atom toward the Cq,ipso atom of the five-membered ring: Δ = 0.27 Å; Ti1–C6 bond length of 2.378(2) Å significantly elongated compared to titanium–carbon single bonds; C1–C6 bond length of 1.441(2) Å constitutes an elongated C(sp2)–C(sp3) double bond.22.
- M. Fischer, T. Oswald, H. Ebert, M. Schmidtmann and R. Beckhaus, Organometallics, 2018, 37, 415–421 CrossRef CAS.
- M. B. Smith and J. March, Advanced Organic Chemistry, Wiley, New York, 6th edn, 2007 Search PubMed.
- U. Thewalt and T. Wöhrle, J. Organomet. Chem., 1994, 464, C17–C19 CrossRef CAS.
- J. Scholz, F. Rehbaum, K.-H. Thiele, R. Goddard, P. Betz and C. Krüger, J. Organomet. Chem., 1993, 443, 93–99 CrossRef CAS.
- R. Beckhaus, J. Sang, J. Oster and T. Wagner, J. Organomet. Chem., 1994, 484, 179–190 CrossRef CAS.
- R. Beckhaus, I. Strauß and T. Wagner, J. Organomet. Chem., 1994, 464, 155–161 CrossRef CAS.
- F. Basuli, B. C. Bailey, L. A. Watson, J. Tomaszewski, J. C. Huffman and D. J. Mindiola, Organometallics, 2005, 24, 1886–1906 CrossRef CAS.
- B. C. Bailey, J. C. Huffman, D. J. Mindiola, W. Weng and O. V. Ozerov, Organometallics, 2005, 24, 1390–1393 CrossRef CAS.
- L. N. Grant, S. Ahn, B. C. Manor, M.-H. Baik and D. J. Mindiola, Chem. Commun., 2017, 53, 3415–3417 RSC.
- All calculations were carried out using the Gaussian 16 software package.30 The initial
guess geometry of the titanium complex 3 was taken from the molecular structure obtained from X-ray crystallography and optimized in the gas phase using at the M06-2X/def2-TZVP31,32 level of theory using a Grimme D3 dispersion correction.33 The structure was confirmed to be a minimum on the potential energy surface using frequency analysis. Natural bond orbital analysis (NBO) and natural resonance theory (NRT) were carried out using the NBO 6.0 program.34 In the NRT analysis only resonance delocalization involving the N2CC–Ti atoms of the molecule were considered.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Phys. Chem. Chem. Phys., 2008, 120, 215–241 CAS.
- F. Weigand and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Anthony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2126498. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2dt00014h |
| This journal is © The Royal Society of Chemistry 2022 |