
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid, energy-efficient and pseudomorphic microwave-induced-metal-plasma (MIMP) synthesis of Mg2Si and Mg2Ge†
Zhen
Fan
a,
Hsi-Nien
Ho
b,
Robert
Szczęsny
c,
Wei-Ren
Liu
 b and
Duncan H.
Gregory
*a
b and
Duncan H.
Gregory
*a
aWestCHEM, School of Chemistry, University of Glasgow, Joseph Black Building, Glasgow, G12 8QQ, UK. E-mail: Duncan.Gregory@glasgow.ac.uk; Tel: +44 (0)141 330 8128
bDepartment of Chemical Engineering, R&D Center for Membrane Technology, Research Center for Circular Economy, Chung Yuan Christian University, 32023, No. 200, Chun Pei Rd., Chung Li District, Taoyuan City 32023, Taiwan
cFaculty of Chemistry, Nicolaus Copernicus University in Toruń, ul. Gagarina 7, 87-100 Toruń, Poland
First published on 25th July 2022
Abstract
Polycrystalline magnesium silicide, Mg2Si and magnesium germanide, Mg2Ge were synthesised from the elemental powders via the microwave-induced-metal-plasma (MIMP) approach at 200 W within 1 min in vacuo for the first time. The formation of reactive Mg plasma facilitated by the high-frequency electromagnetic field (2.45 GHz) is at the origin of the ultrafast reaction kinetics in these preparations. Powder X-ray diffraction (PXRD), Scanning Electron Microscopy (SEM) combined with Energy Dispersive X-ray Spectroscopy (EDX) and X-ray Photoelectron Spectroscopy (XPS) attest to the high purity of the products. Both SEM and Transmission Electron Microscopy (with Selected Area Electron Diffraction) (TEM/SAED) demonstrate the pseudomophic nature of the metal plasma reactions such that use of nanoporous Ge starting material leads to the production of nanoporous germanide, Mg2Ge. Covalent Mg–Si and Mg–Ge bonds with partial ionic character are suggested by XPS, while the refined crystal structures are consistent with Mg–Mg interactions within the cubane-like clusters in Mg2X antifluorite unit cells. The MIMP method unlocks not only the sustainable synthesis of Mg2X materials but also the wider production of intermetallics and Zintl phases of prescribed morphology.
Introduction
The societal benefits of a low-carbon future demand the means of harvesting, producing, converting and storing renewable energy efficiently. Energy demand and utilisation must also be efficiently managed. The sustainable green synthesis and fabrication of functional solid-state materials and devices is an integral part of this process; energy consumption and environmental impact must be minimised as a priority.1–3 For several decades now, the physics of microwaves (MWs) has inspired the creativity of synthetic chemists.4,5 Correspondingly, MW synthesis routes have produced a myriad of inorganic solid materials with various structures and morphologies. Simultaneously, the major benefits of time/energy savings; rapid heating rates; fast throughput; precise, selective heating, and reduction of waste heat/hazardous chemicals have been delivered.2–11 Nevertheless, MW synthesis still faces many challenges before its implementation as a primary route to advanced materials manufacturing could be considered.2–7,10The Mg2X (X = Si, Ge, Sn) family crystallises with the face-centered cubic, antifluorite structure (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) in which Mg atoms occupy the 8c (1/4, 1/4, 1/4) interstitial sites within a CCP lattice of X atoms (4a sites; Fig. 1b).5,12 These Zintl phase compounds are intrinsically n-type semiconductors with narrow indirect band gaps (e.g. ca. 0.73, 0.72 and 0.31 eV experimentally for Mg2Si, Mg2Ge and Mg2Sn, respectively).13,14 Since the 1960s, members of the Mg2X family (often forming solid-solutions) have continuously emerged as promising energy materials given excellent mid-temperature (ca. 500–800 K) thermoelectric (TE) performance combined with simple structures and flexible compositions of abundant, non-toxic environmentally-friendly elements.5,12 Common traits of large Seebeck coefficients, high electrical conductivity and unusually low lattice thermal conductivities permit the dimensionless figure of merit (zT) values to exceed unity comfortably for n-type Mg2X materials.15–18 More recently, Mg2X compounds have been proposed as candidate anode materials for rechargeable Mg-ion batteries,19–21 and these Mg2X materials (especially Mg2Si) have also demonstrated promise as the basis for the large-scale fabrication of nanoporous X anodes for Li- and Na-ion secondary batteries.5,22–26 Beside applications in energy conversion and storage, narrow-gap Mg2X compounds are considered as good candidates for infrared optoelectronic devices,27 whereas the mechanical and anti-corrosive properties of Mg2Si prove it an effective additive in metallurgy and for biodegradable implants.28–30 Presciently, the high-profile medical value of non-toxic Mg2Si nanoparticles as an outstanding deoxygenation agent for cancer starvation therapy has been discovered recently.31
m) in which Mg atoms occupy the 8c (1/4, 1/4, 1/4) interstitial sites within a CCP lattice of X atoms (4a sites; Fig. 1b).5,12 These Zintl phase compounds are intrinsically n-type semiconductors with narrow indirect band gaps (e.g. ca. 0.73, 0.72 and 0.31 eV experimentally for Mg2Si, Mg2Ge and Mg2Sn, respectively).13,14 Since the 1960s, members of the Mg2X family (often forming solid-solutions) have continuously emerged as promising energy materials given excellent mid-temperature (ca. 500–800 K) thermoelectric (TE) performance combined with simple structures and flexible compositions of abundant, non-toxic environmentally-friendly elements.5,12 Common traits of large Seebeck coefficients, high electrical conductivity and unusually low lattice thermal conductivities permit the dimensionless figure of merit (zT) values to exceed unity comfortably for n-type Mg2X materials.15–18 More recently, Mg2X compounds have been proposed as candidate anode materials for rechargeable Mg-ion batteries,19–21 and these Mg2X materials (especially Mg2Si) have also demonstrated promise as the basis for the large-scale fabrication of nanoporous X anodes for Li- and Na-ion secondary batteries.5,22–26 Beside applications in energy conversion and storage, narrow-gap Mg2X compounds are considered as good candidates for infrared optoelectronic devices,27 whereas the mechanical and anti-corrosive properties of Mg2Si prove it an effective additive in metallurgy and for biodegradable implants.28–30 Presciently, the high-profile medical value of non-toxic Mg2Si nanoparticles as an outstanding deoxygenation agent for cancer starvation therapy has been discovered recently.31
 | ||
| Fig. 1 Results from PXRD and electron microscopy for the MIMP-synthesised Mg2Si powders, showing (a and b) profile plot of the Rietveld refinement against PXD data and the corresponding crystal structure; (c and d) SEM images, (e–g) TEM images, and (h) SAED pattern taken from the area shown by the image in (g). | ||
Among several well-established synthesis routes to Mg2X materials, conventional high-temperature solid-state methods and reactive ball-milling synthesis typically require long durations, whereas newer spark plasma sintering (SPS) and self-propagating high-temperature synthesis methods still require high temperatures and special equipment requirements.5,12,15–17,20 Careful and/or multiple-step treatments are often needed. These synthesis approaches have to compromise the different physical and chemical properties of reactants and products, such as the solubility of Mg cations in solid-state X phases, boiling/melting points, volatility of Mg, mechanical hardness or ductility, and the ready tendency of Mg (and its products) to oxidise in air.5,12,15–17,32,33 Accordingly, the compositions of final products can be challenging to control, often involving impurities and potential safety issues (e.g. air exposure may cause the self-ignition of ball-milled products).5,17,20,32 A few early attempts to synthesise Mg2Si-based TE materials in the solid state using MWs demanded very high input powers (a few KW) for tens of minutes under inert conditions.5,34,35 It was the pioneering work of Savary et al. in 2010 that first led to the successful production of Mg2Si nanopowders, placing an encouraging milestone for the MW synthesis of Mg2X compounds (although control of purity could not then be mastered).36 In this case, pucks of Mg and Si powders were ball-milled for 2 h prior to heating using an incident MW power of only 175 W for 2 min under N2.36 Savary et al. applicator possessed the capability to deliver two modes of MW excitation separately to the sample; both magnetic and electric field components. Their experimental observations indicated that semiconducting Si coupled with the magnetic field whereas Mg coupled to neither mode and that it was the rapid heating of Si that principally drove the reaction towards Mg2Si.5,36 A similar instrument and methodology were then applied to prepare both n- and p-doped Mg2Si.37 This was perhaps unexpected given that both theory and experiment demonstrate that fine metal powders can couple with MWs (at 2.45 GHz) to reach high temperatures rapidly.2–5,10
We previously conceived a new microwave-induced-metal-plasma (MIMP) approach towards the synthesis of phase-pure Mg2Sn within 1 min, employing an incident power of 200 W in vacuo.3,5 In (sub)micron form, Mg and Sn powders coupled effectively with the MW field to form reactant metal plasmas (composed of ionised cations and electrons) in situ, such that the reaction kinetics were significantly promoted in the presence of the 2.45 GHz electromagnetic field.3,5 This new approach, where synthesis occurs via the reaction of species in the plasma phase, contrasts markedly with high-temperature solid-state reactions. The diffusion of atoms/ions in the solid state is relatively slow even at elevated temperatures and at the increased heating rates achievable by MW heating. Herein, we extend the MIMP method to the reaction of Mg with the metalloids Si and Ge in the rapid, energy-efficient synthesis of crystalline Mg2Si and Mg2Ge. Notably, the MIMP reaction between Mg and nanoporous Ge preserved the nanostructure of the latter in the Mg2Ge product, affirming that Mg plasma is the key reactive species in the Mg2X syntheses. In these respects, the MIMP method contrasts distinctively with MW syntheses in the solid state,36,37 showing great promise for Mg-based phases and for other alloys and compounds more broadly.
Experimental
Materials synthesis
The syntheses of Mg2Si and Mg2Ge followed similar procedures to that of Mg2Sn in our previous study.3,5 All manipulations and preparations were performed inside an N2-filled LABstar glovebox (mBraun; H2O, O2 < 0.5 ppm). Typically, either 70 mg (2.88 mmol) of Mg (99.8%, 325 mesh, Alfa Aesar) was combined with 35 mg (1.25 mmol) of Si (99.5%, 325 mesh, Sigma Aldrich) or 35 mg (1.44 mmol) of Mg (99.8%, 325 mesh, Alfa Aesar) with 45 mg (0.62 mmol) of nanoporous Ge (synthesised from Mg2Ge by de-alloying using literature methods38) and mixed thoroughly before the transfer of the respective powders into an alumina crucible (which can be considered MW-transparent). One control experiment using 35 mg of Mg and 45 mg of commercial bulk Ge powders (Trace metal basis, 99.999%, Acros Organics) was also performed (full details in ESI†). In each case, an excess of Mg powder (15 at%) was employed (equating to a 2.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) to compensate for the evaporation of Mg.3,5 The crucible was placed within a quartz tube, which was subsequently closed and connected to a vacuum line outside the glovebox.
:1 molar ratio) to compensate for the evaporation of Mg.3,5 The crucible was placed within a quartz tube, which was subsequently closed and connected to a vacuum line outside the glovebox.
A modified single-mode cavity MW reactor (CEM Discovery, 2.45 GHz) with the input power adjustable from 0–300 W was employed.3,5 An incident MW power of 200 W was applied to the powder mixture under a static vacuum of P < 10−6 mbar.5 The irradiation time was typically 60 s for both Mg2Si and Mg2Ge. In order to analyse the composition during the reaction, one Mg2Ge experiment was performed for 30 s. Following the irradiation, the quartz tube was allowed to cool naturally to room temperature. All samples were ground and stored in the glovebox for further characterisation.
Materials characterisation
Powder X-ray diffraction (PXRD) was performed using a PANalytical X'pert Pro MPD diffractometer in Bragg–Brentano geometry (Cu-Kα1, λ = 1.5406 Å; accelerating voltage of 40 kV; emission current of 40 mA). For phase identification purposes, PXRD patterns were collected at room temperature over a 2θ range of 15–85° with a step size of 0.0334° and 30 s per step. For structure determination/quantitative analysis by Rietveld refinement, PXRD datasets were collected over extended ranges of 15–110° and 15–100° (2θ) for Mg2Si and Mg2Ge respectively, with a step size of 0.01667° and 100 s per step. Previously published Mg2Si, Mg2Ge, Si, Ge, MgO and Mg structures were used for phase identification purposes and as initial models for Rietveld refinements.39–44 Rietveld refinements were performed by using GSAS via the EXPGUI interface.45 As-refined crystal structures were plotted using the VESTA package.46Scanning Electron Microscopy (SEM) and Energy Dispersive X-ray Spectroscopy (EDX) were performed using a Philips/FEI XL30 ESEM instrument (beam voltage 20 kV, maximum magnification 20 k) equipped with an INCA X-Act detector (Oxford Instruments Analytical, UK). The samples were coated with Pt plasma under vacuum to optimise the quality of SEM images. Transmission Electron Microscopy (TEM) and selected area electron diffraction (SAED) were performed using an FEI Tecnai G2 F20 X-Twin; 200 kV, FEG microscope. High-resolution Si2p, Ge3d, and Mg1s X-ray Photoelectron Spectroscopy (XPS) spectra were measured using a monochromatic Al-Kα Photoelectron Spectrometer (Thermo Scientific) under vacuum, both before and after etching with an Ar-ion beam (at 2.0 keV for 30 s).
Results and discussion
MIMP synthesis and characterisation of Mg2Si
MW syntheses of Mg2Si and Mg2Ge were conducted from elemental mixtures with an initial Mg/Si or Mg/Ge ratio of 2.3:1, under a static vacuum of P < 10−6 mbar with an incident power of 200 W, typically for a duration of 60 s. Plasma formed almost immediately (within ca. 2 s of MW irradiation), evolving initially from a pale purple colour to persistent green for both samples. These closely resembled the plasmas observed in the MIMP synthesis of Mg2Sn,5 suggesting the dominance of Mg plasma formation across the MIMP syntheses of the wider Mg2X family (X = Sn, Si, Ge).3,5 Fine dark blue powders were obtained from the reaction in each case, with no visible evidence of sintering or agglomeration to larger solid pieces.
Fig. 1a shows the profile plot obtained from the Rietveld refinement performed against laboratory PXRD data for the Mg2Si product synthesised from 60 s of irradiation. The silicide formed with the cubic antifluorite structure (space group Fmm) with a lattice parameter of a = 6.3525(1) Å (Fig. 1b; Table 1), in good agreement with previous reports in the literature.39 The refinement indicated that the sample was Mg-deficient with a site-occupancy-factor (SOF) of 0.976(4) for the Mg site (Table 2). Mg sub-stoichiometry and vacancies on the Mg site are not uncommon features in Mg2Si synthesized at high temperature (and/or reduced pressure) and occur even in the presence of excess magnesium due to a combination of evaporation and oxidation (to MgO).17,47,48 Interestingly, such Mg vacancies rarely lead to intrinsic p-doping due to the trapping of states within the band gap. Nevertheless, the PXRD results showed that the MIMP method yields crystalline Mg2Si of high purity (98.9(6) wt%), with the almost negligible amount of MgO (1.1(6) wt%) likely originating from the surface oxidation of Mg2Si during handling and the reaction of Mg plasma with trace amounts of oxygen in the closed quartz tube (Table 1).5,17,31
| Chemical formula | Mg2Si | MgO |
|---|---|---|
| Crystal system | Cubic | Cubic |
| Space group |
Fmm (no. 225) |
Fmm (no. 225) |
| Lattice parameter, a/Å | 6.3525(1) | 4.2176(3) |
| Cell volume/Å3 | 256.346(9) | 75.024(17) |
| Formula weight/g mol−1 | 302.032 | 161.216 |
| Formula units, Z | 4 | 4 |
| Calculated density/g cm−3 | 1.956 | 3.568 |
| Phase fraction/wt% | 98.9(6) | 1.1(6) |
| No. of variables | 35 | |
| No. of observations | 5628 | |
| wRp | 0.1576 | |
| R p | 0.1194 | |
| χ 2 | 5.611 | |
| Atom | Site | x | y | z | 100 × Uiso/Å2 | SOF |
|---|---|---|---|---|---|---|
| Mg | 8c | 0.25 | 0.25 | 0.25 | 2.85(4) | 0.976(4) |
| Si | 4a | 0 | 0 | 0 | 2.74(5) | 1 |
The SEM images show highly crystalline fine powders with a relatively uniform size distribution ranging from sub-microns to ∼1 μm across (Fig. 1c and d). In addition to this majority of particles, small clusters of much smaller (nanoscale) particles can be observed, which were potentially formed by the rapid reaction of Mg plasma with pulverised or existing Si nanoparticles and with the small amount of residual oxygen in the tube. Additional pulverisation of Mg2Si itself is also perhaps not surprising given the action of the plasma on the particles in the MW field. The observation of the nanoparticles herein is consistent with findings from our previous MIMP study on Mg2Sn.5 The EDX spectrum corresponding to the section of sample imaged in Fig. 1d led to an Mg/Si atomic ratio of 2.0 ± 0.1, in excellent agreement with the theoretical composition of Mg2Si (Fig. S1a, ESI†). The presence of a small concentration of O at the sample surface might be attributed largely to the brief air exposure during specimen transfer. The maps of Mg and Si confirm the uniform distribution of the elements across the surface of the MIMP-synthesised powder (Fig. S1b and c, ESI†).
The low-magnification TEM images in Fig. 1e showed that typical micron/sub-micron Mg2Si particles were rich in grain boundaries. At higher magnification (Fig. 1f), it was evident that the Mg2Si particles were composed of a multitude of crystalline nanograins, each ca. 5–10 nm across. On closer inspection (Fig. 1g), TEM images provided evidence of: (i) a disordered surface layer of ca. 1–2 nm in thickness, which potentially stemmed from a surface reaction when exposed to oxygen/water in air. This is consistent with the reactivity of Mg2Si, especially in powder form, and the formation of a passivation layer31 and (ii) highly crystalline regions beneath the surface layer, where lattice fringe spacings of 0.371 nm predominated, matching well with the (111) planes in Mg2Si (and consistent with PXRD results; Fig. 1a).39Fig. 1h shows a typical SAED pattern from the sample and as taken from the region shown in Fig. 1g. The pattern is typical of a polycrystalline material, with diffraction rings that can be indexed to Mg2Si. These data affirm that the crystalline material is bulk Mg2Si encapsulated with a thin layer of oxide that likely forms instantaneously when handled in air (Table S1, ESI†).
The oxidation states and binding energies of Mg and Si in the Mg2Si Zintl phase were further investigated by measuring high-resolution Si2p and Mg1s XPS spectra (Fig. 2). Compared to a reference spectrum taken for Si powder, which also featured distinct peaks for SiO2 in addition to those for Si(0), the spectrum from a sample of as-synthesised Mg2Si powder showed the co-existence of two broad peaks covering binding energies corresponding to SiO2 (SiOx), Si(0) and Si species that appeared more anionic in character as would befit Mg2Si (at 98.58 eV) (Fig. 2a).49,50 Impurity peaks might be expected from the surface oxidation of Mg2Si in air (just as the Si reference sample was surface-oxidised). The sample was subsequently etched with an Ar-ion beam and although the treatment could not completely remove all the oxidised surface species, the predominant Si peak was identified at a binding energy of 97.77 eV, which is significantly lower than that expected for elemental Si (99.29 eV), revealing the nominally negative oxidation state of Si in Mg2Si (Fig. 2a).49,50 One peak was discovered in the Mg1s XPS spectrum (Fig. 2b) and this shifted to lower binding energy post-etching. The initial observation suggests the presence of a passivating surface layer of MgO, as could be inferred from electron microscopy. Post etching, however, the Mg peak is located at a binding energy intermediate between MgO (1304.5 eV) and Mg (1303.0 eV), suggesting an oxidation state of Mg between formally +2, as in MgO, and of 0 in Mg metal.51 The XPS data indicated that the bonding in Mg2Si was by no means purely ionic (i.e. in which Mg valence electrons were entirely transferred to Si) but possessed significant covalent character.13,14,52,53 The spectra and inferences are entirely consistent with comprehensive studies of (oxidised) Mg2Si on Si (111) made more than 2 decades ago.54 Interestingly, the refined Mg–Mg distance in Mg2Si was only 3.1726(1) Å, which is rather shorter than the equivalent distance in Mg metal (3.1967(4) Å). This presents the possibility of “free” electrons within the cubic Mg8 sub-lattice in Fig. 1b.40,52 Our XPS and PXRD results concur with previous studies on Mg2Si when a charge transfer from Mg to Si was calculated as 0.16 units by applying a potential model to XPS data; this corresponds to an Mg–Si bond with an ionicity of 8%.50,55 Subsequent Bader charge analysis implied that Mg carries a charge of +1.45 and Si could be attributed a charge of −2.90.52 The vacant cavity at the body centre of the unit cell is proposed to form a concentration of electron density originating from the overlapping Mg 3p states, which in turn are primarily responsible for the conduction band minimum (CBM) (at the X point in the Brillouin zone) in the electronic structure of Mg2Si. This is a point that we will return to when discussing the Mg2X phases below.
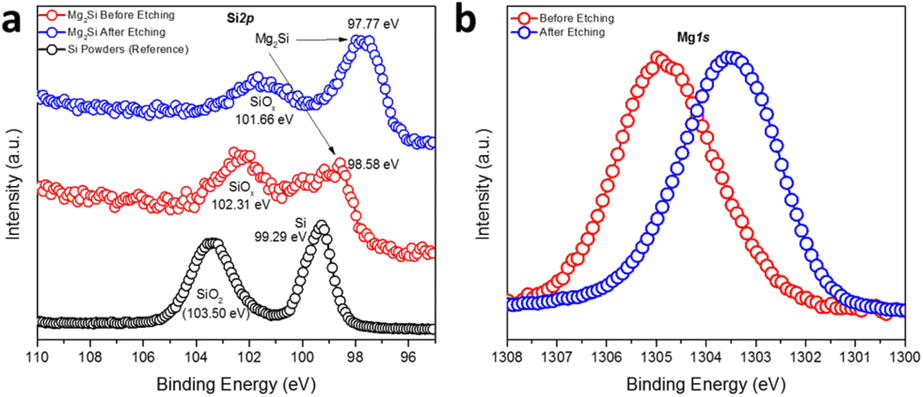 | ||
| Fig. 2 High-resolution (a) Si2p and (b) Mg1s XPS spectra of MIMP-synthesised Mg2Si powders (irradiated for 60 s). | ||
MIMP synthesis and characterisation of Mg2Ge
The MW exposure of Mg and Ge powders to an incident MW power of 200 W for 30 s was able to generate a product dominated by Mg2Ge (94.3(3) wt%). The remainder of the sample was composed of a small amount of unreacted starting materials (Mg: 4.1(3) wt% and Ge: 1.5(1) wt%), as indicated by the Rietveld refinement profile shown in Fig. 3a (see also ESI:† Tables S2 and S3). PXRD confirmed that irradiation of the elements at 200 W for double the duration (60 s) led to their complete reaction, with one very weak diffraction peak at 42.8° 2θ suggesting the presence of MgO.44 Correspondingly, the Rietveld refinement revealed a phase purity of 98.1(1) wt% for Mg2Ge with the remainder of the sample corresponding to MgO (1.9(1) wt%) (Fig. 3b; Table 3). As with the Mg2Si sample, the presence of MgO as a minor phase could originate from handling during sample transfer and measurement, with the additional caveat that the nanoporous Ge reactant is quite likely to also react with air to oxidise at its surface, presenting a potential source of oxygen at the outset.17 The Rietveld refinement confirmed Mg2Ge to be isostructural with Mg2Si, with a lattice parameter of a = 6.3940(1) Å and an Mg–Mg distance of 3.1970(1) Å, equivalent to that in Mg metal (3.1967(4) Å) within 1σ.40 In contrast to Mg2Si, we found no evidence of non-stoichiometry at the Mg 8c site; attempts to refine the SOF led to no significant departure from unity and did not improve the overall fit to the profile or the R factors (Table 4).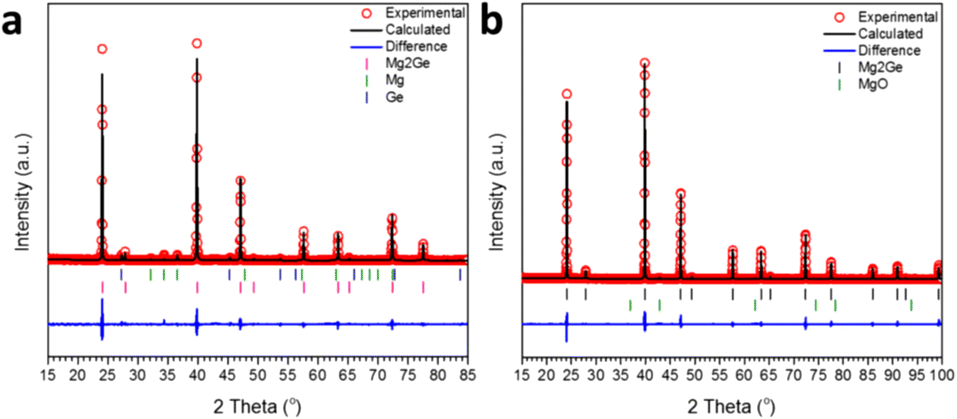 | ||
| Fig. 3 Profile plot of the Rietveld refinements against PXRD data for Mg2Ge samples synthesised at 200 W with a MW irradiation time of: (a) 30 s and (b) 60 s, respectively. | ||
| Chemical formula | Mg2Ge | MgO |
|---|---|---|
| Crystal system | Cubic | Cubic |
| Space group |
Fmm (no. 225) |
Fmm (no. 225) |
| Lattice parameter, a/Å | 6.3940(1) | 4.2236(4) |
| Cell volume/Å3 | 261.403(6) | 75.344(22) |
| Formula weight/g mol−1 | 484.800 | 161.216 |
| Formula units, Z | 4 | 4 |
| Calculated density/g cm−3 | 3.080 | 3.553 |
| Phase fraction/wt% | 98.1(1) | 1.9(1) |
| No. of variables | 34 | |
| No. of observations | 5085 | |
| wRp | 0.1626 | |
| R p | 0.1190 | |
| χ 2 | 2.382 | |
| Atom | Site | x | y | z | 100 × Uiso/Å2 | SOF |
|---|---|---|---|---|---|---|
| Mg | 8c | 0.25 | 0.25 | 0.25 | 2.52(6) | 1 |
| Ge | 4a | 0 | 0 | 0 | 2.10(4) | 1 |
Fig. 4a and b show SEM images taken for the Mg2Ge powder synthesised at 200 W for 60 s. For comparison, the size and morphology of the nanoporous Ge reactant powder are indicated in the SEM images in the ESI† (Fig. S2). The Mg2Ge powders take the appearance of individual micron-sized particles, which can be viewed as pieces of a nanoporous “bulk” matrix composed of nano ligaments ranging from ca. 85–300 nm across. An appraisal of the SEM images from both the nanoporous Ge reactant powder and the nanoporous Mg2Ge product demonstrates the close resemblance of the former and latter. This similarity signals the pseudomorphic nature of the MIMP reaction. In many ways, this is not an unreasonable result. For example, there are many examples of nanoporous matrices of p-block metals/metalloids possessing the capability for Li+, Na+ or Mg2+ ion insertion during electrochemical alloying reactions in rechargeable-ion batteries; such processes can have minimal effects on the nanostructure, extending cyclability over equivalent bulk materials.22–26 One can also further infer from this result that the MIMP process, at least in the case of Mg2Ge, is one that involves a solid-plasma phase reaction between Ge and Mg respectively, reinforcing the hypothesis made above concerning the MW reaction mechanism in the Mg–Si system.3,5 This premise was reinforced by conducting a supplementary MIMP synthesis of Mg2Ge from a mixture of Mg and commercial, bulk Ge powers (99.999%, Acros Organics). Revealingly, this experiment led to the production of non-porous, micron-sized Mg2Ge particles (ESI:† Fig. S3).
 | ||
| Fig. 4 SEM characterisation of Mg2Ge powders synthesised from Mg and Ge at 200 W with an irradiation time of 60 s, showing: (a and b) SEM images at different magnification; (c) a representative EDX spectrum taken from the area shown in the image in (b); and (d–f) elemental maps of Mg, Ge and O, respectively, taken from the same area shown in (b). | ||
Fig. 4c shows a representative EDX spectrum, yielding an atomic Mg:Ge:O ratio of 60.39:23.81:15.80. Fig. 4d–f show that elemental distributions of Mg, Ge and O were uniform across the surface of the specimen, demonstrating the homogeneity of the MIMP-synthesised product. In light of the known reactivity of Mg2Ge with water,5,31,56 the EDX data suggest that a surface passivation occurs following exposure to air as the sample was transferred to the SEM chamber:
| Mg2Ge(s) + 2H2O(g) → 2MgO(s) + GeH4(g) | (1) |
The surface composition and the oxidation states of Mg and Ge in Mg2Ge were further investigated by the measurement of high-resolution Ge3d and Mg1s XPS spectra (Fig. 5). Spectra confirmed the presence of the oxide passivation layer suggested by SEM/EDX results. The Ge3d XPS spectra (Fig. 5a) showed that Ar-ion beam etching can effectively remove this surface oxide layer (which likely formed during the transfer of the XPS specimen to the vacuum chamber). The experimental spectra for Mg2Ge were compared to those from a reference of Ge powder. Prior to Ar-etching, the Ge3d spectra for Mg2Ge showed little evidence for Ge(0) and only a relatively small peak corresponding to a binding energy typical for GeO2 (at 32.21 eV). This peak was effectively removed from the spectrum post-etching. As witnessed for Si in the equivalent Si2p XPS spectra taken for Mg2Si, the main Ge peak shifts significantly to lower binding energy as compared to that in the elemental spectrum (of Ge(0)). The shift of >1 eV (from 29.29 eV to 28.2 eV) indicates a nominal negative oxidation state for Ge in Mg2Ge.57 The Mg1s spectrum is rather similar to that seen for Mg2Si. Fig. 5b shows that, post-etching, the Mg1s peak is centred at ca. 1303.5 eV, which is located between the binding energies expected for MgO (1304.5 eV) and Mg (1303.0 eV), suggesting an oxidation state of Mg between +2 in MgO and 0 in Mg metal.51–53 In similarity to Mg2Si, therefore, XPS reveals that the bonding in the germanide is likely to be predominantly covalent.
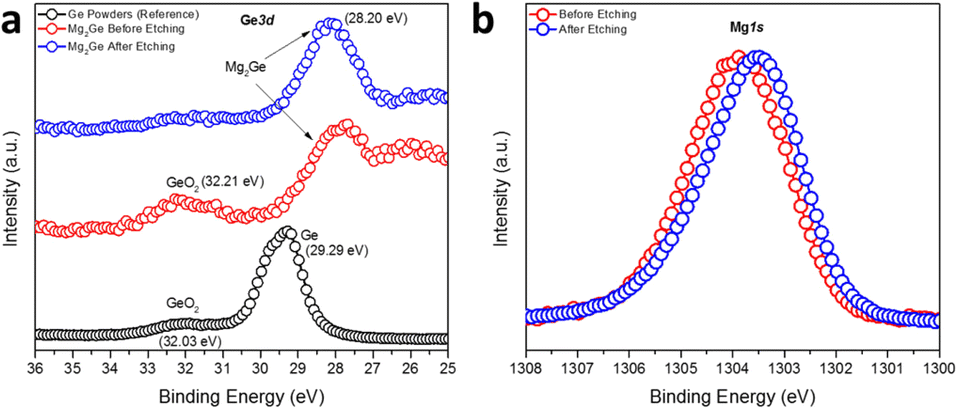 | ||
| Fig. 5 High-resolution (a) Ge3d and (b) Mg1s XPS spectra of the MIMP-synthesised Mg2Ge powders with an irradiation time of 60 s. | ||
Trends in the structures of MIMP-synthesised Mg2X (X = Si, Ge, Sn) and common features of the MIMP syntheses
Selected structural data for the Mg2X (X = Si, Ge, Sn) antifluorite compounds, as determined from Rietveld refinement, are presented in Fig. 6. As would be expected, Mg2Sn has a larger cubic lattice parameter (a = 6.7653(1) Å), Mg–X bond length (2.9295(1) Å) and Mg–Mg distance (3.3827(1) Å) when compared to Mg2Si and Mg2Ge.5 Mg2Ge exhibits a very similar cubic lattice parameter to Mg2Si, reflecting the rather similar covalent radii of the elements. Consequently, and given that both Mg and X are located on special positions (of (1/4, 1/4, 1/4) and (0,0,0), respectively), the Mg–X (X = Si, Ge) bond lengths and Mg–Mg distances vary by only ca. 0.02 Å. It should also be noted that the Mg–Mg distances (within the cubane-like Mg8 cluster) is only 3.1762(1) and 3.1970(1) Å for Mg2Si and Mg2Ge, respectively and therefore equal or less than the Mg–Mg distance in Mg metal (3.1967(4) Å).40,52,58 This implies potential metal–metal interactions and the presence of “free” electrons within the central cavity of the cubic Mg8 sub-lattice in both Mg2Si and Mg2Ge. The recent computational study that identified such electrons as being associated with the CBM (through both σ and π bonding Mg 3py states interacting with Si 3d states) suggested that these cavity electrons are likely mobile and associated with the intrinsic n-type semiconductivity of Mg2Si (and, in fact, these cavity states are deep lying enough to resemble those in inorganic electrides).52 The structural data would suggest that Mg2Ge is likely to behave in a similar way and that the transport properties of both compounds could be tuned by modifying the size of the central cavity. Interestingly, single crystal X-ray structural studies had previously suggested that Mg2Si could accommodate additional interstitial Mg in the vacant body centre (1/2,1/2,1/2) position, albeit at very low levels (<1%).59 Simultaneously, there was a tendency for the crystals to exhibit Mg vacancies of up to 1% at the 8c (1/4, 1/4, 1/4) position and that as these vacancies increased so the electrical conductivity decreased (and the Seebeck coefficient increased). Our structural data suggest Mg vacancy levels of ca. 2% at the 8c site in Mg2Si, but lab PXRD could not resolve the presence of electron density at the 1–2% level at the body centre. The presence of analogous vacancies in MIMP-synthesised Mg2Ge and Mg2Sn5 could not be so readily established. There is clearly a need for a still more comprehensive understanding of the links between structure, composition and electronic properties in Mg2Si and to explore to what extent these are replicated or otherwise in other Mg2X Zintl phases. The links between “cation” vacancies and the presence of either interstitial Mg or electrons could also have profound implications for ionic transport and the application of Mg2X as anodes in Mg-ion batteries.19–21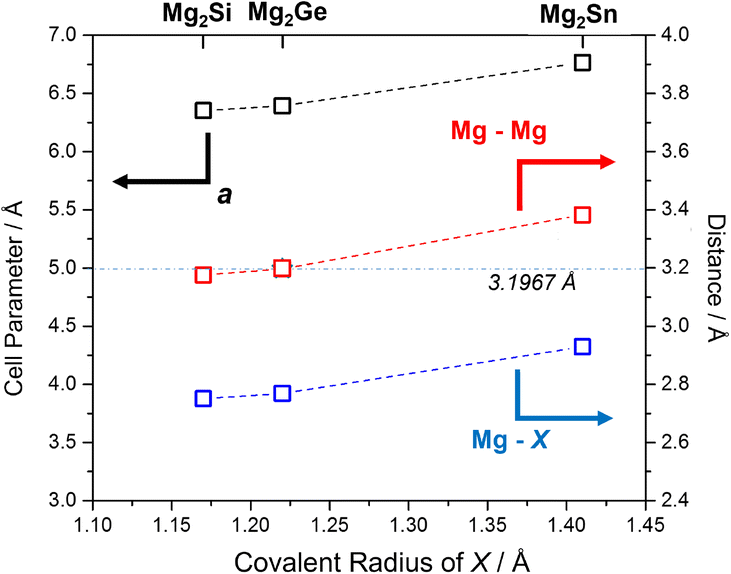 | ||
| Fig. 6 Plot of refined lattice parameter, a (black), Mg–X (blue) and Mg–Mg (red) distances5 for MIMP-synthesised Mg2X (X = Si, Ge, Sn) (200 W, 1 min) against covalent radius of X.60 The horizontal line and value represents the Mg–Mg distance in Mg metal.40 | ||
The observed plasma generation during our MIMP syntheses of Mg2X (X = Si, Ge) and for Mg2Sn5 was very similar, i.e. a transition from an initial purple coloured plasma (within ca. 10 s) to a steady green plasma. This is despite the very different physical and chemical properties of Sn, Si and Ge as reactants, where semiconducting Si and Ge have much higher melting points and hardness factors, for example, than metallic Sn.5,17 Given the volatility of Mg, it is not altogether surprising that Mg plasma formation is pivotal in the synthesis of Mg2X (X = Si, Ge, Sn) Zintl phases in a microwave field under vacuum.3,5 The pseudomorphic reaction of Mg with nanoporous Ge underlines the premise that Mg is in the plasma state during each of these MW syntheses. Given that plasma is essentially a gas formed from ions and electrons, the Mg2Si and Mg2Ge synthesis processes can be likened to many conventional high temperature solid–gas reactions, which are often similarly pseudomorphic.61,62 The principal difference in the MIMP case is that it is the metal starting material that reacts ostensibly in the gaseous phase. Any contrast, therefore, between the MIMP Mg2X reactions mainly lies in the state of X. For Si and Ge, with high melting points and vapour pressures, the non-metal remains in the solid state (Scheme 1). For X = Sn, which has very different physical properties and where Sn interacts considerably with the MW field itself, the situation is rather different and a range of possible reaction processes are possible, as discussed previously.5
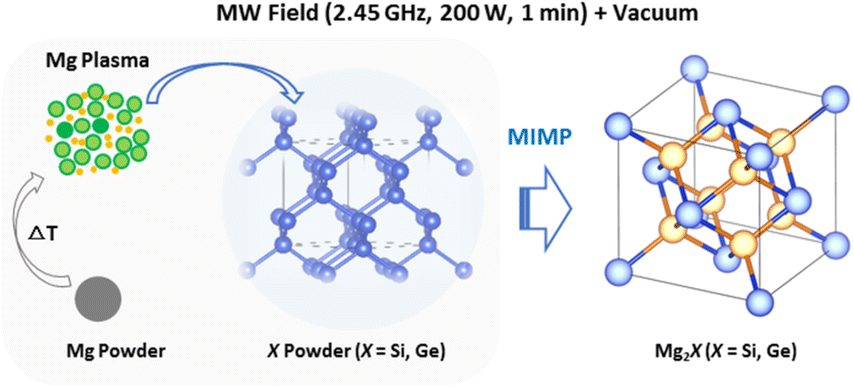 | ||
| Scheme 1 Schematic of the principal Mg plasma-enabled reaction route proposed for the MIMP synthesis of Mg2Si and Mg2Ge. | ||
Performing the Mg2X MW syntheses under vacuum has profound effects on not only the way in which the reaction proceeds, but also on reaction efficiency and product purity. Comparing the purity of MIMP-synthesised Mg2Si with that prepared using MW in the solid state in a nitrogen atmosphere under otherwise similar conditions previously (175 W, 120 s), the latter contained both Si and Mg as impurity phases among the products.36 Given that the coupling of solid Si with the magnetic component of the field was proposed as the primary heating mechanism towards reaction with Mg, it is perhaps not unexpected that the products lacked homogeneity. By contrast, the study noted that the fine powdered Mg starting material did not couple effectively with either component of the 2.45 GHz MW field.36
In the MIMP reactions, the coupling and direct heating of both of the Mg and X reactant powders with MWs is clearly important and this is especially true for Mg, as the initial heating stage is the vital precursor to Mg plasma formation. This can be observed in the opening seconds of the MIMP reactions when from an initial state of no plasma formation, the colour of the evolving plasma transforms from purple (residual trace gas in the tube) to green (magnesium). This almost immediate and steady formation of reactant metal plasma in vacuo contributes hugely to the rapid reaction kinetics, as the motions of the charged particles in the plasma phase are enhanced by the 2.45 GHz electromagnetic field.3,5 The possibility of producing Mg2X (X = Si, Ge, Sn) within 60 s contrasts enormously with equivalent conventional high-temperature solid-state approaches which typically require in excess of 10 h to complete.5,17 While the results also demonstrate the powerful potential to fabricate bespoke nanostructured intermetallic materials via the judicious selection of precursors, it will likely be the ability to scale up the MIMP method that will have the biggest impact on large scale materials production for applications such as rechargeable ion batteries and biomedical therapeutics.
Conclusions
In summary, the MIMP approach has proved highly effective in the synthesis of the high purity, polycrystalline Mg2Si and Mg2Ge Zintl phases. Application of an incident power of merely 200 W to the solid elemental reactants in vacuo, led to each product within 60 s. The reaction of Mg with Ge was revealed as pseudomorphic, with nanoporous Mg2Ge attainable from Mg powder and nanoporous Ge starting material. The experiments shed further light on the mechanisms of the MIMP process, demonstrating that the formation of metal (Mg) plasma is fundamental to its success. The plasma facilitates rapid reaction rates and the physical state of the other reactant, X, with which the plasma interacts, is pivotal in determining the microstructure of the products. The synthesis of high purity Mg2X has also enabled further investigations of the structure–composition relationships of the silicide, germanide and stannide Zintl phases. The implications of Mg–Mg interactions within the Mg8 sub-lattices and the (non)existence of vacancies at the 8c (1/4, 1/4, 1/4) site are manifestly likely to be pivotal in determining the electronic (and possibly ionic) transport properties. Further systematic studies (including neutron diffraction combined with electrical and thermal conductivity measurements, for example) should be able to test these hypotheses further. Meanwhile, further investigations should be conducted to extend the MIMP method towards the rapid energy-efficient synthesis of other Zintl phases, intermetallics and alloys.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DHG and ZF thank the University of Glasgow and the China Scholarship Council for the co-funding of a studentship for ZF. DHG thanks the Royal Society and EPSRC for associated funding under an International Exchange grant (IEC\R3\183040) and grant EP/N001982/1, respectively. W.-R. L. gratefully acknowledges the Ministry of Science and Technology, Taiwan, for support under project grants MOST 110-2923-E-006-011, 110-3116-F-011-002, 110-2622-E-033-009, 109-2911-I-033-502 and 108-E-033-MY3. ZF acknowledges the valuable experimental assistance and discussions from Dr. Mauro Davide Cappelluti (School of Chemistry, University of Glasgow). The authors acknowledge Mr. Cheng-Yi Lin (Department of Chemical Engineering, Chung Yuan Christian University) for assistance with SEM measurements.References
- S. Maldonado, ACS Energy Lett., 2020, 5(11), 3628–3632 CrossRef CAS.
- J. P. Siebert, C. M. Hamm and C. S. Birkel, Appl. Phys. Rev., 2019, 6(4), 041314 Search PubMed.
- Z. Fan, G. Baranovas, A. Y. Holly, R. Szczęsny, W. R. Liu and D. H. Gregory, Green Chem., 2021, 23(18), 6936–6944 RSC.
- H. J. Kitchen, S. R. Vallance, J. L. Kennedy, N. Tapia-Ruiz, L. Carassiti, A. Harrison, A. G. Whittaker, T. D. Drysdale, S. W. Kingman and D. H. Gregory, Chem. Rev., 2014, 114(2), 1170–1206 CrossRef CAS PubMed.
- Z. Fan, M. D. Cappelluti and D. H. Gregory, ACS Sustainable Chem. Eng., 2019, 7(24), 19686–19698 CrossRef CAS.
- S. Chahal, S. M. Kauzlarich and P. Kumar, ACS Mater. Lett., 2021, 3(5), 631–640 CrossRef CAS.
- S. Głowniak, B. Szczęśniak, J. Choma and M. Jaroniec, Adv. Mater., 2021, 33(48), 2103477 CrossRef PubMed.
- S. R. Vallance, S. Kingman and D. H. Gregory, Adv. Mater., 2007, 19(1), 138–142 CrossRef CAS.
- L. Carassiti, A. Jones, P. Harrison, P. S. Dobson, S. Kingman, I. MacLaren and D. H. Gregory, Energy Environ. Sci., 2011, 4(4), 1503–1510 RSC.
- S. Horikoshi, R. F. Schiffmann, J. Fukushima and N. Serpone, Microwave Chemical and Materials Processing, Springer, Singapore, 2018 Search PubMed.
- A. G. Whittaker and D. M. P. Mingos, J. Chem. Soc., Dalton Trans., 1995, 2073–2079 RSC.
- R. Santos, S. A. Yamini and S. X. Dou, J. Mater. Chem. A, 2018, 6(8), 3328–3341 RSC.
- J. Tejeda, M. Cardona, N. J. Shevchik, D. W. Langer and E. Schönherr, Phys. Status Solidi B, 1973, 58(1), 189–200 CrossRef CAS.
- B. Ryu, S. Park, E. Choi, J. D. Boor, P. Ziolkowski, J. Chung and S. D. Park, J. Korean Phys. Soc., 2019, 75(2), 144–152 CrossRef CAS.
- A. Sankhla, A. Patil, H. Kamila, M. Yasseri, N. Farahi, E. Mueller and J. D. Boor, ACS Appl. Energy Mater., 2018, 1(2), 531–542 CrossRef CAS.
- S. Yi, V. Attari, M. Jeong, J. Jian, S. Xue, H. Wang, R. Arroyave and C. Yu, J. Mater. Chem. A, 2018, 6(36), 17559–17570 RSC.
- L. Zhang, Synthesis and thermoelectric properties of Mg2Si-Mg2Sn solid solutions for waste heat recovery, Doctoral dissertation, The University of Texas at Austin, 2015 Search PubMed.
- W. Liu, X. Tan, K. Yin, H. Liu, X. Tang, J. Shi, Q. Zhang and C. Uher, Phys. Rev. Lett., 2012, 108(16), 166601 CrossRef PubMed.
- A. B. Ikhe, S. C. Han, S. J. R. Prabakar, W. B. Park, K. S. Sohn and M. Pyo, J. Mater. Chem. A, 2020, 8(28), 14277–14286 RSC.
- D. T. Nguyen and S. W. Song, J. Power Sources, 2017, 368, 11–17 CrossRef CAS.
- H. Yaghoobnejad Asl, J. Fu, H. Kumar, S. S. Welborn, V. B. Shenoy and E. Detsi, Chem. Mater., 2018, 30(5), 1815–1824 CrossRef CAS.
- W. An, B. Gao, S. Mei, B. Xiang, J. Fu, L. Wang, Q. Zhang, P. K. Chu and K. Huo, Nat. Commun., 2019, 10(1), 1–11 CrossRef CAS.
- Y. An, Y. Tian, C. Wei, Y. Tao, B. Xi, S. Xiong, J. Feng and Y. Qian, Nano Today, 2021, 37, 101094 CrossRef CAS.
- Y. An, H. Fei, G. Zeng, L. Ci, S. Xiong, J. Feng and Y. Qian, ACS Nano, 2018, 12(5), 4993–5002 CrossRef CAS PubMed.
- J. B. Cook, E. Detsi, Y. Liu, Y. L. Liang, H. S. Kim, X. Petrissans, B. Dunn and S. H. Tolbert, ACS Appl. Mater. Interfaces, 2017, 9(1), 293–303 CrossRef CAS PubMed.
- J. Niu, H. Gao, W. Ma, F. Luo, K. Yin, Z. Peng and Z. Zhang, Energy Storage Mater., 2018, 14, 351–360 CrossRef.
- H. Udono, H. Tajima, M. Uchikoshi and M. Itakura, Jpn. J. Appl. Phys., 2015, 54(7S2), 07JB06 CrossRef.
- M. Sikora-Jasinska, P. Chevallier, S. Turgeon, C. Paternoster, E. Mostaed, M. Vedani and D. Mantovani, RSC Adv., 2018, 8(18), 9627–9639 RSC.
- M. Ebrahimi, A. Zarei-Hanzaki, H. R. Abedi, M. Azimi and S. S. Mirjavadi, Tribol. Int., 2017, 115, 199–211 CrossRef CAS.
- M. Sikora-Jasinska, C. Paternoster, E. Mostaed, R. Tolouei, R. Casati, M. Vedani and D. Mantovani, Mater. Sci. Eng., C, 2017, 81, 511–521 CrossRef CAS PubMed.
- C. Zhang, D. Ni, Y. Liu, H. Yao, W. Bu and J. Shi, Nat. Nanotechnol., 2017, 12(4), 378–386 CrossRef CAS PubMed.
- I. H. Kim, J. Korean Phys. Soc., 2018, 72(10), 1095–1109 CrossRef CAS.
- C. R. Clark, C. Wright, C. Suryanarayana, E. G. Baburaj and F. H. Froes, Mater. Lett., 1997, 33(1–2), 71–75 CrossRef CAS.
- S. C. Zhou and C. G. Bai, Trans. Nonferrous Met. Soc. China, 2011, 21(8), 1785–1789 CrossRef CAS.
- S. Zhou and C. Bai, J. Cent. South Univ., 2012, 19(9), 2421–2424 CrossRef CAS.
- E. Savary, F. Gascoin and S. Marinel, Dalton Trans., 2010, 39(45), 11074–11080 RSC.
- D. Berthebaud and F. Gascoin, J. Solid State Chem., 2013, 202, 61–64 CrossRef CAS.
- Y. G. Zhang, N. Du, C. M. Xiao, S. L. Wu, Y. F. Chen, Y. F. Lin, J. W. Jiang, Y. H. He and D. R. Yang, RSC Adv., 2017, 7, 33837–33842 RSC.
- R. Saravanan and M. C. Robert, J. Alloys Compd., 2009, 479(1–2), 26–31 CrossRef CAS.
- H. E. Swanson, T. Eleanor and K. F. Ruth, Natl. Bur. Stand. Circ., 1953, 539, 1–95 Search PubMed.
- G. H. Grosch and K. J. Range, J. Alloys Compd., 1996, 235(2), 250–255 CrossRef CAS.
- A. S. Cooper, Acta Crystallogr., 1962, 15(6), 578–582 CrossRef CAS.
- D. M. Többens, N. Stüßer, K. Knorr, H. M. Mayer and G. Lampert, Mater. Sci. Forum, 2001, 378, 288–293 Search PubMed.
- S. Sasaki, K. Fujino and Y. Takéuchi, P. JPN Acad. B, 1979, 55(2), 43–48 CrossRef CAS.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34(2), 210–213 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44(6), 1272–1276 CrossRef CAS.
- T. Dasgupta, C. Stiewe, R. Hassdorf, A. J. Zhou, L. Boettcher and E. Mueller, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83(23), 235207 CrossRef.
- K. Yin, Q. Zhang, Y. Zheng, X. Su, X. Tang and C. Uher, J. Mater. Chem. C, 2015, 3(40), 10381–10387 RSC.
- J. Liang, X. Li, Z. Hou, C. Guo, Y. Zhu and Y. Qian, Chem. Commun., 2015, 51(33), 7230–7233 RSC.
- K. Sekino, M. Midonoya, H. Udono and Y. Yamada, Phys. Procedia, 2011, 11, 171–173 CrossRef CAS.
- L. Umaralikhan and M. Jamal Mohamed Jaffar, Iran. J. Sci. Technol., 2018, 42(2), 477–485 CrossRef.
- H. Mizoguchi, Y. Muraba, D. C. Fredrickson, S. Matsuishi, T. Kamiya and H. Hosono, Angew. Chem., Int. Ed., 2017, 56, 10135–10139 CrossRef CAS PubMed.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- M. Brause, B. Braun, D. Ochs, W. Maus-Friedrichs and V. Kempter, Surf. Sci., 1998, 398(1–2), 184–194 CrossRef CAS.
- M. R. J. V. Buuren, F. Voermans and H. V. Kempen, J. Phys. Chem., 1995, 99, 9519–9522 CrossRef.
- C. R. Whitsett, Electrical properties of magnesium silicide and magnesium germanide, Iowa State University, 1955 Search PubMed.
- Y. Zhang, N. Du, C. Xiao, S. Wu, Y. Chen, Y. Lin, J. Jiang, Y. He and D. Yang, RSC Adv., 2017, 7(54), 33837–33842 RSC.
- H. Hosono and M. Kitano, Chem. Rev., 2021, 121(5), 3121–3185 CrossRef CAS PubMed.
- M. Kubouchi, K. Hayashi and Y. Miyazaki, J. Alloys Compd., 2014, 617, 389–392 CrossRef CAS.
- S. S. Batsanov, Russ. Chem. Bull., 1995, 44(12), 2245–2250 CrossRef.
- J. Ahn, B. Kim, G. Jang and L. Moon, ChemElectroChem, 2018, 5(19), 2729–2733 CrossRef CAS.
- M. Saruyama, R. Sato and T. Teranishi, Acc. Chem. Res., 2021, 54, 765–775 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ce00721e |
| This journal is © The Royal Society of Chemistry 2022 |