
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssembling photoactive materials from polycyclic aromatic hydrocarbons (PAHs): room temperature phosphorescence and excimer-emission in co-crystals with 1,4-diiodotetrafluorobenzene†
Alessandra
Azzali
 a,
Simone
d'Agostino
a,
Mattia
Capacci
a,
Floriana
Spinelli
a,
Barbara
Ventura
*b and
Fabrizia
Grepioni
*a
a,
Simone
d'Agostino
a,
Mattia
Capacci
a,
Floriana
Spinelli
a,
Barbara
Ventura
*b and
Fabrizia
Grepioni
*a
aDipartimento di Chimica “Giacomo Ciamician”, Università di Bologna, Via F. Selmi, 2, 40126 Bologna, Italy. E-mail: fabrizia.grepioni@unibo.it
bIstituto ISOF-CNR, Via P. Gobetti, 101, 40219 Bologna, Italy. E-mail: barbara.ventura@isof.cnr.it
First published on 15th July 2022
Abstract
Co-crystallization between the polycyclic aromatic hydrocarbons anthracene (A) and 9-methylanthracene (MA) with 1,4-diiodotetrafluorobenzene (I2F4) afforded three novel co-crystals, viz. A·(I2F4)2 and an unexpectedly complex system with two distinct compositions, namely MA·I2F4 and (MA)4·I2F4, which can be mechanochemically interconverted by a change in the stoichiometry of the reactive mixtures. Interestingly, all co-crystals are dual-emissive materials and exhibit different mechanisms of emission. A·(I2F4)2 and MA·I2F4 fluoresce from isolated molecules, whereas the luminescence of (MA)4·I2F4 is dominated by excimer emission. In all cases, phosphorescence at RT (RTP) is observed and interpreted as a direct consequence of the interactions between the iodine atoms of the I2F4 co-former and the π-electron density of the anthracene aromatic rings. Furthermore, [4 + 4] photoactivity within (MA)4·I2F4 was also investigated by means of FTIR/NMR spectroscopy and PXRD. The photophysical and photochemical behaviors of all solids are discussed and rationalized based on their structural features.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are a vast class of organic compounds that are composed of two or more fused aromatic rings, sharing pairs of C-atoms.1–3PAHs can be the result either of natural processes, e.g., the transformation of organic sediments into fossil fuels, volcanic eruptions and natural forest fires,4,5 or, as a significant source, from anthropogenic activities, such as partial combustion of fossil fuels in industrial processes, wood-burning, tobacco-smoking, and exhaust from vehicles.4,6 PAHs are found everywhere, from aquatic and terrestrial systems to the atmosphere,7 and are considered highly polluting since they persist in soil and water once released into the environment.8–12 They also tend to accumulate in food like meat, vegetables, cereals, edible oils, and seafood13–17 and are categorized as highly toxic, mutagenic, and carcinogenic to various forms of life, including humans.18–22 For all these reasons their emissions and levels in the environment are being regulated by government agencies, and various remediation treatments have been proposed.4,23–25 Since the discovery of this family of compounds, scientists have proposed new applications for these pollutants, taking advantage of peculiar features such as their remarkable and unique electrical and optical properties.26–31 By using crystal engineering principles,32–35 valuable applications for PAHs have been developed in recent years, ranging from optical waveguides,36 OFET-devices,37 and additives for improving photovoltaic cell performances,38 to bright solids, including fluorescent,39–43 room temperature phosphorescent44–51 and ultralong phosphorescent materials.52–55
In this context, co-crystallization of luminescent molecules proved to be a powerful tool for developing new materials and achieving emission type and color fine-tuning.48,56–61 The type of co-formers to be used in co-crystallization with PAHs is rather limited, as hydrocarbons do not possess typical hydrogen bonding groups. Suitable systems are perfluoropolyhalogenated molecules, since they can drive co-crystallization by establishing halogen bonding interactions of the type X⋯π with the electron density of the aromatic rings in PAH molecules.62–68 These X⋯π intermolecular interactions can be seen as a particular case of directional halogen bonding where the electron depleted polar regions of the polarized halogens approach the electron-rich π-orbitals of an aromatic compound. Moreover, halogen atoms in close proximity to the photoactive PAH components may also promote interesting changes in their optical properties.
Within the frame of our studies on PAH co-crystals as solid luminescent materials,53 we report here the preparation of bright crystalline materials based on the co-crystallization of anthracene (A) and its derivative 9-methylanthracene (MA), as photoactive components, and 1,4-diiodo-tetrafluorobenzene (I2F4), as a co-former (Chart 1).
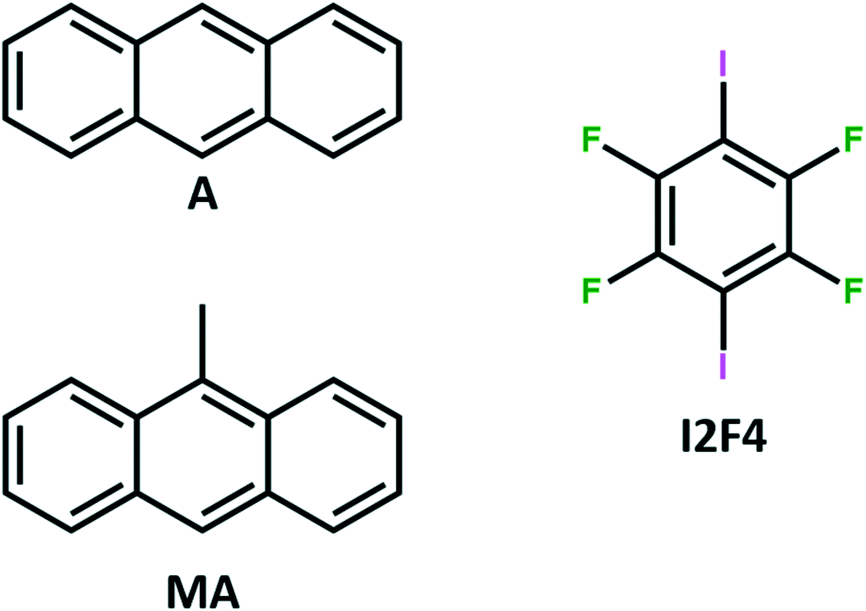 | ||
| Chart 1 Molecular structure of the building blocks used in this work: the polycyclic aromatic hydrocarbons, namely, anthracene (A) and 9-methylanthracene (MA), and the co-former 1,4-diiodotetrafluorobenzene (I2F4). | ||
New applications for these pollutants are actively being sought and are of paramount importance, especially if PAHs can be turned into valuable materials through green synthetic methods. To this end, in our co-crystallization experiments we reacted A and MA with I2F4 directly in the solid state, via mechanochemistry, testing different stoichiometric ratios of the pure components. Three novel co-crystals, A·(I2F4)2, MA·I2F4, and (MA)4·I2F4, have been obtained and characterized via a combination of X-ray diffraction (XRD) techniques and solid-state luminescence spectroscopy, the latter highlighting different optical features among the co-crystals and between the co-crystals and the pure aromatic compounds. Single crystals analysis was essential to rationalize the structure–property relationship for these solids. All co-crystals are dual-emissive materials: in A·(I2F4)2 and MA·I2F4 both fluorescence and phosphorescence from isolated A or MA molecules are observed, whereas emission from (MA)4·I2F4 is characterized by concomitant fluorescence from MA excimers and phosphorescence from the isolated molecules. Crystalline (MA)4·I2F4 is also photoactive, and its [4 + 4] photocycloaddition reaction was observed by means of FTIR spectroscopy. Finally, the stoichiometry mediated solid-state interconversion between MA·I2F4 and (MA)4·I2F4 was detected and investigated.
Experimental
Materials
All reactants were purchased from Sigma-Aldrich and used after purification. Two PAHs, namely, anthracene (A) and 9-methylanthracene (MA), and a co-former, 1,4-diiodotetrafluorobenzene (I2F4), were recrystallized from ethanol. All solid materials were preliminarily screened for chemical and crystallographic purity via powder XRD analyses (see Fig. ESI-2†) and identified as anthracene form I (ANTCEN),69 9-methylanthracene (MANTHR01),70 and 1,4-diiodotetrafluorobenzene form I (ZZZAVM02).71 Reagent grade solvents were used without further purification.Synthesis
For co-crystals A·(I2F4)2, MA·I2F4, and (MA)4·I2F4, the co-former I2F4 and the two polycyclic aromatic hydrocarbons, A or MA, were weighed in different stoichiometric ratios for a total of ca. 200 mg (see Table ESI-1†), transferred into 5 mL agate jars, each containing two 5 mm balls, and ground together for 2 h with a ball-milling apparatus operated at 15 Hz. The solid product was dissolved in CHCl3:EtOH 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, transferred into a conical flask and kept in the dark. Slow evaporation at room temperature afforded diffraction quality single crystals.
:1, transferred into a conical flask and kept in the dark. Slow evaporation at room temperature afforded diffraction quality single crystals.
Single-crystal XRD
Single crystal data for A·(I2F4)2, MA·I2F4, and (MA)4·I2F4 were collected at RT with an Oxford XCalibur S CCD diffractometer equipped with a graphite monochromator (Mo-Kα radiation, λ = 0.71073 Å). The crystals of MA·I2F4 were affected by twinning, for the best sample found and analyzed, two twin unit cells were indexed, and the reflection data were integrated with the default configuration for twinned crystals of the CrysAlisPro software. Subsequent structure solution and refinement were performed using the HKLF4 file containing non-overlapped reflections. The structures were solved by intrinsic phasing with SHELXT72 and refined on F2 by full-matrix least squares refinement with the program SHELXL,73 implemented in the Olex2 software.74,75 All non-hydrogen atoms were refined anisotropically, applying the rigid-body RIGU restraint.76 HCH atoms for all compounds were added in calculated positions and refined riding on their respective carbon atoms. Data collection and refinement details are listed in Table 1 and Fig. ESI-1† for the Ortep plots. The programs Mercury77 and Platon78 were used for molecular graphics and packing index analysis, respectively. The magnitude of the main intermolecular interactions detected with co-crystals was evaluated with the Crystal Explorer Package and using CE-HF/3-21G (k_ele = 1.019; k_pol = 0.651; k_disp = 0.901; k_rep = 0.811) as energy model and basis set.79–81 CCDC numbers 2166101–2166103.| A·(I2F4)2 | MA·I2F4 | (MA)4·I2F4 | |
|---|---|---|---|
| Formula | C26H10F8I4 | C21H12F4I2 | C66H48F4I2 |
| FW/g mol−1 | 981.94 | 594.11 | 1170.84 |
| Crystal system | Triclinic | Monoclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pn |
P |
| a/Å | 5.9997(4) | 15.2587(8) | 8.1105(5) |
| b/Å | 9.7209(10) | 8.2100(3) | 10.3429(5) |
| c/Å | 12.2681(10) | 16.2077(9) | 16.2211(8) |
| α/° | 73.604(8) | 90 | 97.450(4) |
| β/° | 89.751(6) | 104.852(5) | 103.319(5) |
| γ/° | 81.821(7) | 90 | 98.140(5) |
| Volume/Å3 | 678.94(10) | 1962.57(17) | 1292.05(12) |
| Packing coefficient | 0.68 | 0.67 | 0.67 |
| ρ calc/g cm−3 | 2.402 | 2.011 | 1.505 |
| μ/mm−1 | 4.660 | 3.244 | 1.274 |
| λ/Å | 0.71073 | 0.71073 | 0.71073 |
| Measd rflns | 4495 | 9172 | 8611 |
| Indep rflns | 2655 | 5933 | 4551 |
| R 1 | 0.0325 | 0.0289 | 0.0529 |
| wR 2 | 0.0703 | 0.0910 | 0.1030 |
Powder XRD
For phase identification diffractograms were recorded on a PANalytical X'Pert Pro automated diffractometer equipped with an X'Celerator detector in Bragg–Brentano geometry, using Cu-Kα radiation (λ = 1.5418 Å) without a monochromator in the 2θ range between 3° and 30° (continuous scan mode, step size 0.0167°, counting time 19.685 s, Soller slit 0.04 rad, antiscatter slit 1/2, divergence slit 1/4, 40 mA × 40 kV). The program Mercury77 was used for the calculation of the powder patterns, on the basis of single-crystal data retrieved from the Cambridge Structural Database (CSD) or collected in this work.Thermogravimetric analysis
TGA measurements on all samples were performed with a PerkinElmer TGA7 thermogravimetric analyser, in the 30–300 °C temperature range, under an N2 gas flow and at the heating rate of 5.00 °C min−1 (see ESI†).Irradiation experiments
Single crystals and polycrystalline samples of (MA)4·I2F4 were irradiated for 5 hours using a UV-LED (Led EnginLZ1-10UV00-0000) with λ = 365 nm and placed at 2 cm.FTIR spectroscopy
Attenuated total reflectance Fourier transform IR (ATR-FTIR) spectra were obtained using a Bruker Alpha FT-IR spectrometer.Optical spectroscopy
Room temperature measurements were performed on powder samples placed between two quartz slides. Reflectance spectra were acquired with a PerkinElmer Lambda 950 UV/vis/NIR spectrophotometer equipped with a 100 mm integrating sphere and converted into absorption spectra using the Kubelka–Munk function.82 A thin layer of powder was used (absorbance values below 0.2) for luminescence measurements. Emission spectra were collected in the front-face mode with an Edinburgh FLS920 fluorimeter equipped with a Peltier-cooled Hamamatsu R928 PMT (280–850 nm) and corrected for the wavelength dependent phototube response. Gated emission spectra were acquired in the front-face mode with the same fluorimeter by using a time-gated spectral scanning mode and a μF920H Xenon flash lamp (pulse width <2 μs, repetition rate between 0.1 and 100 Hz) as an excitation source. As gating parameters, a delay of 0.1 ms and a gate of 0.5 ms were used. The spectra were corrected for the wavelength dependent phototube response. Excitation spectra were collected with the same fluorimeter operating in both modes. Emission quantum yields were determined in the solid state with the absolute method, according to Würth et al.,83 by using a Labsphere barium sulfate-coated integrating sphere (diameter of 3 inches) placed in the same fluorimeter and elaborating the corrected emission spectra. The limit of detection is of the order of 1%.Fluorescence and phosphorescence lifetimes were measured by using an IBH time-correlated single-photon counting (TCSPC) apparatus with a pulsed NanoLED excitation source at 368 nm for fluorescence and a SpectraLED at 370 nm for phosphorescence. Analysis of the luminescence decay profiles against time was accomplished with the Decay Analysis Software DAS6 provided by the manufacturer. The reported lifetimes are averaged values from 3–4 measurements. For A and A·(I2F4)2 a time-resolved luminescence analysis has been performed with the same TCSPC apparatus, and the decay associated spectra (DAS) have been obtained by global analysis of the kinetic data by using the global fitting module of the DAS v6.5 software. The wavelength dependences of the amplitudes of the individual kinetic components were plotted as decay associated spectra. Estimated errors are 10% on lifetimes and quantum yields and 2 nm on emission and absorption peaks.
Solution NMR spectroscopy
1H- and 19F-NMR spectra (Fig. ESI-8 and ESI-9†) were recorded on a Varian Mercury 400, and chemical shifts (δ) of NMR signals were expressed in parts per million (ppm) using an internal standard, TMS (δH = 0.00). CDCl3 was bought from Cambridge Isotope Lab.1H-NMR (400 MHz, CDCl3, TMS): (MA)4·I2F4 before irradiation δ (ppm) 8.33 (s, 1H), 8.29–8.26 (d, 2H, CH), 8.00–7.98 (d, 2H, CH), 7.52–7.43 (m, 4H), 7.24 (CDCl3), 3.09 (s, 3H, CH3). (MA)4·I2F4 after irradiation 1H-NMR (400 MHz, CDCl3, TMS): δ (ppm): 8.33 (s, 1H), 8.29–8.26 (d, 2H, CH), 8.00–7.98 (d, 2H, CH), 7.52–7.43 (m, 4H), 7.24 (CDCl3), 7.13–7.11 (d, 2H, CH), 6.90–6.79 (m, 4H, CH), 3.99 (s,1H, CH), 3.09 (s, 3H, CH3), 2.16 (s, 3H, CH3). (MA)4·I2F4 before and after irradiation 19F NMR (377 MHz, CDCl3) δ −116.89.
Results and discussion
Co-crystal synthesis and mechanochemical interconversion
Co-crystallization was carried out via ball-milling of the PHAs anthracene (A) or 9-methylanthracene (MA) with the co-former 1,4-diiodotetrafluorobenzene (I2F4) in several stoichiometric ratios (see Table ESI-1†).For the pair A:I2F4, we identified a unique and novel crystalline form only when reacted in a 1:2 ratio, whereas for all the other stoichiometries tested, we detected unreacted materials. On the other hand, the system MA:I2F4 showed a more complex and interesting behavior, and two different and unique new phases were obtained only with the 1:1 and 4:1 ratios, whereas for all the other stoichiometries tested, we noticed also the concomitant presence of unreacted MA crystals.
The TGA measurements for all co-crystals (see ESI-4–ESI-6†) did not evidence any water or solvent loss.
The three solid products recrystallized from a CHCl3:EtOH 1:1 mixture and were then analyzed via single-crystal XRD (see Table 1).
Structural analysis allowed the formulation of the co-crystals as A·(I2F4)2, MA·I2F4, and (MA)4·I2F4 (vide infra for structural description), while the comparison between the experimental and calculated patterns allowed the verification of the purity of the polycrystalline samples (see Fig. ESI-3†).
As the co-crystals MA·I2F4 and (MA)4·I2F4 differ for the amount of MA in the unit cell, we tried to convert a polycrystalline sample of MA·I2F4 into (MA)4·I2F4 and vice versa through mechanochemistry; pure (MA)4·I2F4 was obtained by milling MA·I2F4 powders with three additional equivalents of MA. Analogously, conversion of (MA)4·I2F4 into MA·I2F4 was achieved by adding three equivalents of co-former to powders of (MA)4·I2F4. Scheme 1 depicts the overall process.
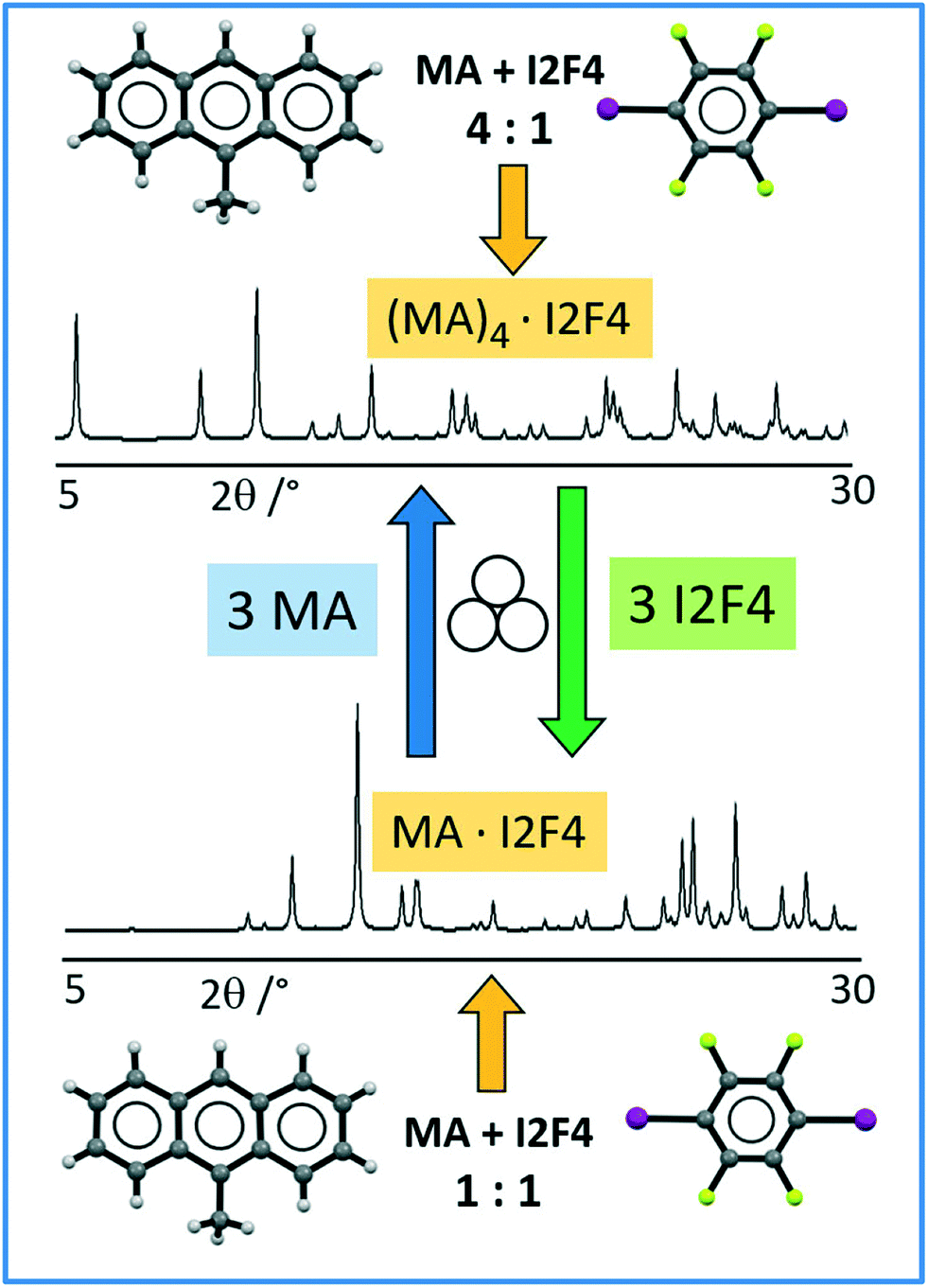 | ||
| Scheme 1 Representation of mechanochemical synthesis and interconversion of crystalline (MA)4·I2F4 (top) and MA·I2F4 (bottom). | ||
Structural features of A·(I2F4)2, (MA)4·I2F4 and MA·I2F4
The reaction of anthracene with I2F4 in the 1:2 stoichiometric ratio yields the co-crystal A·(I2F4)2 (see Table 1). The I2F4 molecules are arranged in a ladder-like fashion (Fig. 1), with π-stacked pairs of I2F4 molecules inserted almost perpendicularly (angle between the planes ca. 81°) between the anthracene aromatic rings. This is reminiscent of the packing in solid anthracene; as it can be appreciated in Fig. 1c, a layer of I2F4 molecules takes the place of an analogous layer of anthracene molecules, depicted in orange in Fig. 1d. Intermolecular interactions of the X⋯π type66,67 between the iodine atoms on I2F4 and the anthracene aromatic rings are also present [I⋯π = 3.465(6) − 3.588(4) Å], see Fig. 1d.
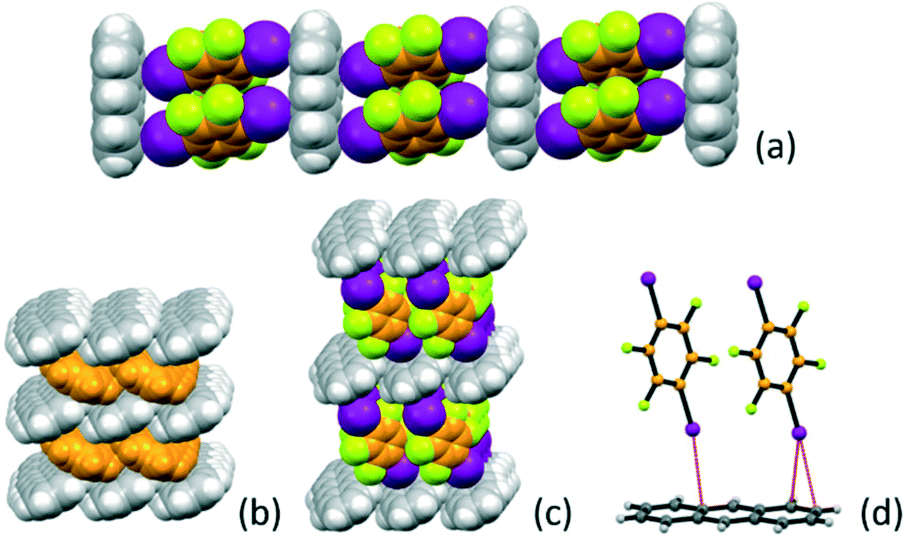 | ||
| Fig. 1 (a) Space-filling representation of crystalline A·(I2F4)2 showing the ladder-like alternation of anthracene and I2F4 molecules. Comparison of the layered structures in (b) solid anthracene (CSD refcode: ANTCEN) and (c) A·(I2F4)2, and (d) a depiction of the X⋯π interactions at work between iodine atoms and the anthracene aromatic rings. | ||
Alternation of the flat co-formers and 9-methylanthracene molecules is also evident in crystalline MA·I2F4. Fig. 2a shows the π-stacking (distance between the aromatic rings ca. 3.5 Å) of MA and I2F4 molecules along the crystallographic b-axis. As a consequence of this arrangement the crystal can also benefit from the stabilization effect of inter-layer weak X⋯π66,67 halogen bonds, as depicted in Fig. 2b [I⋯π = 3.57(9) − 3.88(1) Å].
 | ||
| Fig. 2 (a) Columnar stacking of MA and I2F4 molecules along the b-axis in crystalline MA·I2F4, and (b) geometry of the X⋯π interactions at work between stackings. | ||
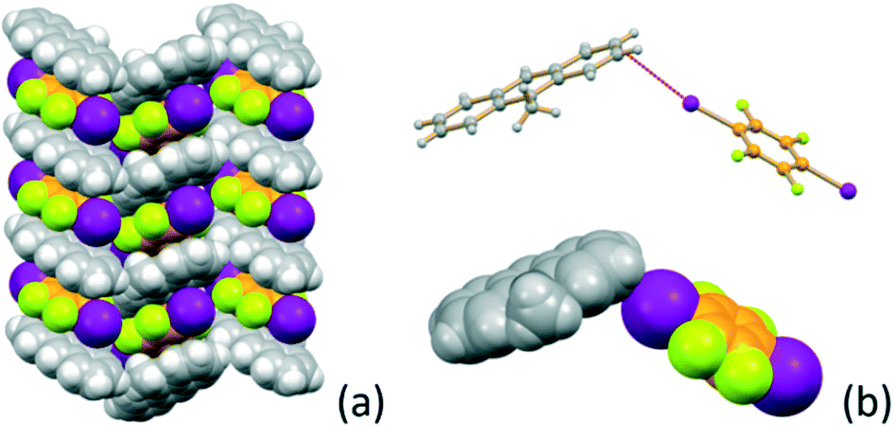
If the MA:I2F4 stoichiometric ratio is increased to 4:1, the co-crystal (MA)4·I2F4 is obtained, which presents some interesting features. The packing in this crystal can be seen to be formed from layers of the type shown in Fig. 3a. The I2F4 molecules are sandwiched (lime rectangle) between two symmetry equivalent MA molecules referred by symmetry (distance between the aromatic planes ca. 3.5 Å), while the MA not involved in the “sandwich” are arranged in pairs (blue rectangle) and parallel and in a head-to-tail fashion, as observed in pure MA crystals;70 this feature allows strong π–π stacking interactions, with the MA molecules at a distance of ca. 3.5 Å and shifted, transversally and longitudinally, by ca. 1.3 and 1.1 Å, respectively (Fig. 3b). The I2F4 co-former interacts laterally with these MA pairs via weak I⋯π halogen bonds66,67 [I⋯π = 3.413(5), 3.413(5) Å] (see Fig. 3c).
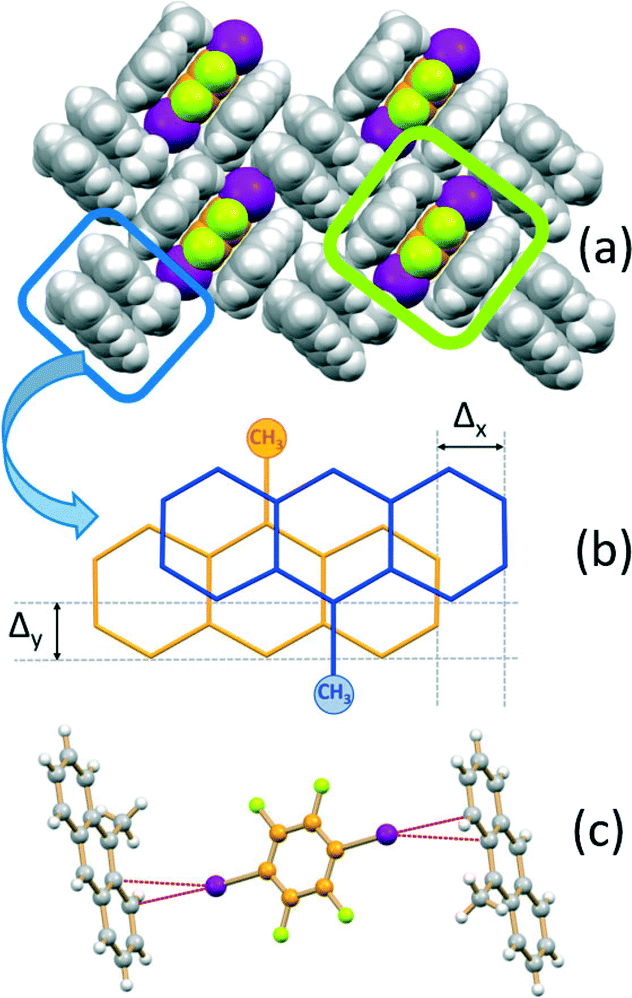 | ||
| Fig. 3 Packing features of (MA)4·I2F4 showing (a) the I2F4 molecules “sandwiched” between MA molecules (lime rectangle) and the π-stacked MA pairs (blue rectangle), (b) details of the head-to-tail pair arrangement (Δy and Δx indicate the transversal and longitudinal offsets, respectively), and (c) the geometry of the I⋯π interactions. | ||
It is worth noting that, in both A·(I2F4)2 and MA·I2F4, close interaction between PAHs molecules is hindered by the presence of the I2F4 co-former, which acts as a solid “dilutant”; on the other hand, head-to-tail π-stacking of 9-methylanthracene is observed in (MA)4·I2F4, analogously to what was observed in pure MA crystals.
The structural features of the compounds described above have also been examined, with the help of the program CrystalExplorer,79–81 in terms of intermolecular interaction energies, resulting from the sum of electrostatic, polarization, dispersion and repulsion contributions. In all solids the most relevant contribution to the total potential energy comes from π-stacking arrangements, both between the I2F4 molecules, as observed in A·(I2F4)2, and between MA and I2F4 or MA and MA, as can be seen for MA·I2F4 and (MA)4·I2F4. I⋯π interactions also contribute in a significant manner to the total energy (see Fig. ESI-14 to ESI-16 and Tables ESI-4 to ESI-6†).
All these features play an essential role in the rationalization of the photochemical and photophysical behaviors of the three co-crystalline materials (see below).
Photoreactivity
Pairs of double bonds can behave as reactive centers and, upon irradiation, take part in a [2 + 2] or [4 + 4] solid-state photocyclization process, depending on the system under consideration. According to the topochemical postulate developed by Schmidt and coworkers in the 1960s, when the distance between the potentially reactive centers is comprised between approximately 3.6 and 4.1 Å, photocyclization, following UV irradiation, is likely to occur.84–86 Since then, the photoreactivity of anthracene and its derivatives within molecular crystals has fascinated scientists and has been the subject of intense research over the last decades, both for theoretical purposes and potential applications.87–95The arrangement of MA pairs in crystalline (MA)4·I2F4 is promising from a purely geometrical point of view; therefore the possible [4 + 4] photoreactivity of the co-crystal was investigated. UV irradiation experiments on the co-crystal were first conducted on the same specimen used for single crystal XRD data collection and structural determination. Upon UV irradiation, however, the single crystals simply “crumbled”. This is not surprising, as the photocyclization processes taking place within solids are almost invariably accompanied by the accumulation of stress, and when this cannot be relieved, the internal pressure may cause the single crystal to collapse and the consequent formation of amorphous or polycrystalline material.96–98 The photoreactivity of the powder sample was also tested, and the PXRD pattern was measured after irradiation. Unfortunately, the loss of crystallinity made it impossible to identify the resulting product.
FTIR spectroscopy was therefore employed, and we could detect the presence of a photodimer within the amorphous powders. Fig. 4 shows the FTIR spectra recorded before and after irradiation. Compared to the original sample, the UV-exposed one shows a marked decrease in the band at ca. 885 cm−1, associated with the C–H out-of-plane bending vibrations of unreacted anthracene. In contrast, the rest of the spectrum remains unaltered, thus indicating that only a fraction of MA molecules has reacted. For comparison, a pure crystalline sample of MA was also irradiated; the FTIR spectrum of the product shows the absence of the diagnostic band at ca 885 cm−1, and the powder is then identified as a head-to-tail photodimer99 (CSD refcode QQQFES04, see Fig. ESI-7†).
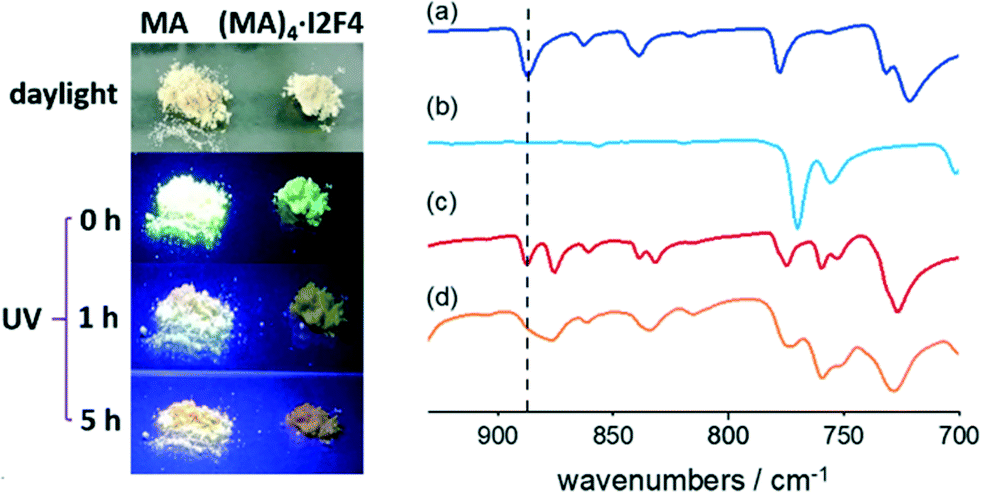 | ||
| Fig. 4 Left: pictures taken during the UV irradiation of the polycrystalline samples of MA and (MA)4·I2F4. Right: details of the FTIR spectra recorded on (a) crystalline MA, (b) the head-to-tail MA photodimer, (c) co-crystal (MA)4·I2F4, and (d) the resulting amorphous solid after UV irradiation (right). | ||
The identity of the photoproduct is also confirmed by the emergence of a new set of signals in the 1H-NMR spectrum (Fig. ESI-8†), which agrees with the reported one for the head-to-tail photodimer.100 Furthermore, 19F-NMR rules out the side reactions between the polyhalogenated co-former and 9-methylanthracene (Fig. ESI-9†).
The pictures of the MA sample, taken under UV irradiation (see Fig. 4), show that, as the irradiation proceeds, the green fluorescence characteristic of this PAH vanishes with time, due to the formation of photodimers. In contrast, when crystalline (MA)4·I2F4 is irradiated with UV, the color of its emission progressively changes from green to orange, likely due to the presence of unreacted MA molecules.
Luminescence spectroscopy
Photophysical features of A·(I2F4)2, MA·I2F4, and (MA)4·I2F4 were also investigated using solid-state absorption and luminescence spectroscopy, with the purpose of (i) characterizing the emission properties of the compounds resulting from the co-crystallization and (ii) highlighting the differences with those of parent crystalline materials, i.e., anthracene (A) and 9-methylanthracene (MA).For co-crystal A·(I2F4)2, the absorption and luminescence profiles are very similar to those of pure anthracene (see ESI†). Both compounds display fluorescence with a clear vibronic structure (see Fig. 5 and Fig. ESI-10;† main maxima at 424, 446 and 472 nm for A, blue-shift of 2 nm in A·(I2F4)2), as reported for anthracene crystals.101 In A·(I2F4)2, as mentioned above, the I2F4 co-former acts as a “dilutant” towards anthracene molecules, preventing the formation of strong intermolecular interactions of π-stacking or herring-bone type, and this accounts for a monomeric-type emission. The emission of the co-crystal is much weaker than that of pure A: quantum yields of 12.6% and 0.8% are measured for A and A·(I2F4)2, respectively. Moreover, close inspection of the emission spectrum of A·(I2F4)2 evidences the additional emission bands in the range 650–800 nm. By using a pulsed source and applying a gated detection we could assign these bands to the phosphorescence spectrum of anthracene in the co-crystal. Fig. 5 shows the isolated spectrum, with two clear vibronic bands at 676 nm and 746 nm. These results indicate that the presence of I⋯π interactions in the co-crystal promotes the intersystem crossing in the PAHs molecules, thus quenching fluorescence and enabling phosphorescence at RT in a completely organic material.44,53 The excitation spectra collected in both the fluorescence and phosphorescence modes are almost superimposable (see Fig. ESI-10†).
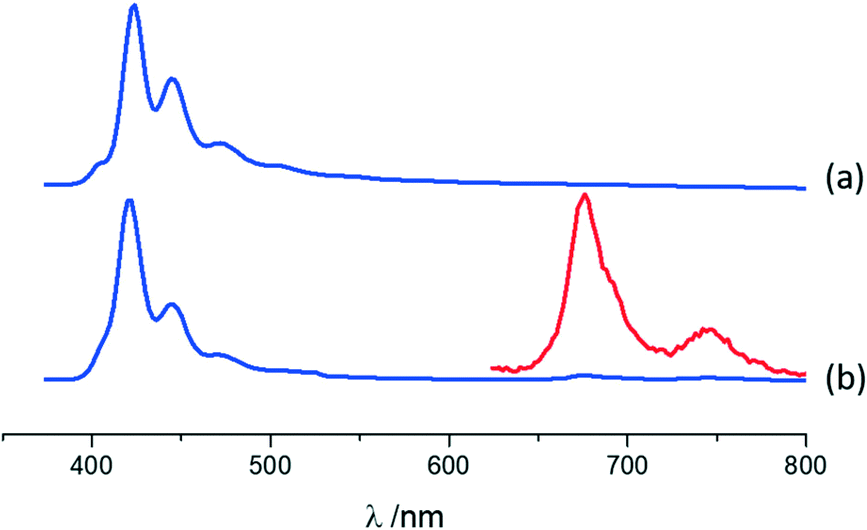 | ||
| Fig. 5 Normalized luminescence spectra in the solid state for (a) pure A and (b) A·(I2F4)2 (arbitrary units on the y-axis). λexc = 380 nm. The phosphorescence spectrum in the 650–800 nm region, isolated with a gated detection, is magnified in the inset for A·(I2F4)2. The phosphorescence emission is clearly evident in A·(I2F4)2 compared to solid anthracene, where no phosphorescence was detected. | ||
The fluorescence lifetime analysis for A and A·(I2F4)2 is reported in the ESI.†
A similar characterization technique was performed for pure MA and its co-crystals. The emission of MA is characterized by a broad band peaking at 522 nm (Fig. 6 and ESI-11†), typical of excimer fluorescence.102 Excimer emission in crystals of anthracene derivatives is not common and has been observed under high pressures101,103 or in particular crystal designs.104,105 The luminescence features of MA·I2F4 are markedly different from those of pure MA crystals: a weak and vibronically resolved fluorescence is observed, accompanied by a clear phosphorescence emission (Fig. 6 and ESI-11†). The latter spectrum, isolated by means of gated detection, shows bands at 700 nm and 774 nm. Such differences are a consequence of the two types of MA molecular arrangements. In the parent crystal, PAHs are suitably paired to produce excimer emission and photoreact.99 On the other hand, within MA·I2F4, the MA molecules are almost isolated and at a longer distance, hindering de-facto the formation of excimers. The “lighting-up” of phosphorescence in the range 650–850 nm is a result of the external heavy atom effect exerted by the co-former.
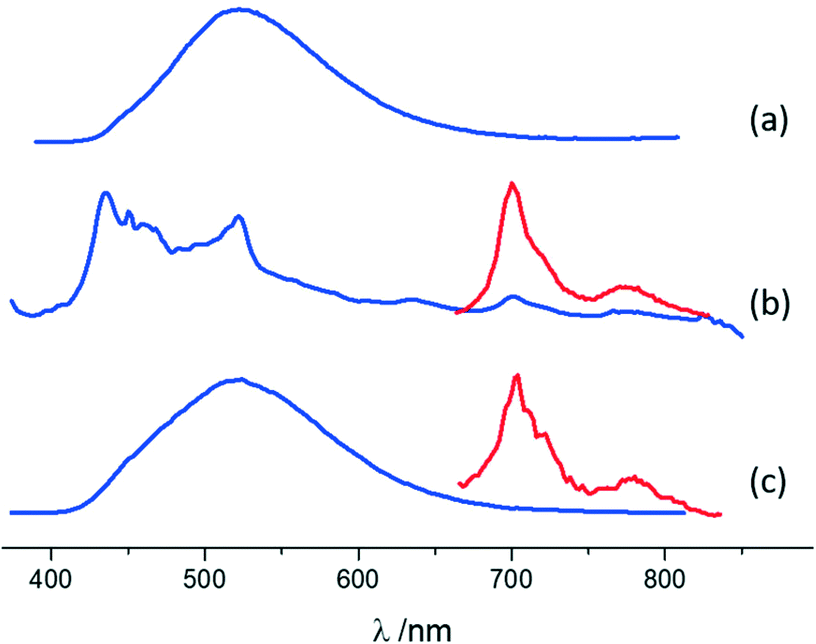 | ||
| Fig. 6 Normalized luminescence spectra in the solid state of (a) pure MA, (b) MA·I2F4, and (c) (MA)4·I2F4 (arbitrary units on the y-axis). λexc = 380 nm. The phosphorescence spectra in the 650–850 nm region, isolated with a gated detection, are magnified in the inset for MA·I2F4 and (MA)4·I2F4. In both co-crystals the phosphorescence emission is clearly evidenced compared to the parent PAH 9-methylanthracene, where no phosphorescence was detected at all. Moreover, for MA·I2F4 the spectrum is not dominated by excimer emission. | ||
Co-crystal (MA)4·I2F4 still shows different luminescence properties, since both excimer fluorescence (with maximum at 522 nm) and phosphorescence (with peaks at 702 nm and 778 nm) are observed (Fig. 6 and ESI-11†). The crystal arrangement of (MA)4·I2F4 features both paired and isolated MA molecules, leading to an emission pattern where the excimer contribution prevails. The presence of phosphorescence, even if weaker than in MA·I2F4, is particularly interesting in combination with excimer emission and can be ascribed to the isolated molecules interacting with the co-former.
Emission quantum yields of 9.0% and 13.4% are found for MA and (MA)4·I2F4, respectively, while for MA·I2F4, a low value of ca. 0.1% (below experimental resolution) is measured.
The fluorescence lifetimes measured for MA and its co-crystals agree with the proposed luminescence mechanisms. A lifetime of ca. 6 ns is detected for MA·I2F4, where a monomeric emission prevails, and a bi-exponential decay with longer lifetimes of ca. 27 ns and 62 ns is observed for pure MA, indicative of excimer emission (Table ESI-2†). In the case of (MA)4·I2F4, lifetimes of the order of 6 ns and 36 ns can be ascribed to the presence of both monomeric and excimer emission, even if the latter is dominant in the steady-state spectrum.
Phosphorescence lifetimes were measured for A·(I2F4)2, MA·I2F4 and (MA)4·I2F4. In all cases the decays could be fitted using bi-exponential functions, with lifetimes of the order of 30 μs and 200 μs for A·(I2F4)2 and MA·I2F4 and longer lifetimes of ca. 90 μs and 320 μs for (MA)4·I2F4 (Table ESI-3†). The increase of the phosphorescence lifetime in (MA)4·I2F4 can be due to the lower conformer/PAH ratio of this co-crystal, which leads to a reduced heavy atom effect in promoting both intersystem crossing and the phosphorescence rate.
Conclusions
In this work, we reported the mechanochemical synthesis of the three novel co-crystals, namely, A·(I2F4)2, MA·I2F4, and (MA)4·I2F4, containing the polycyclic aromatic hydrocarbons, anthracene (A) or methylanthracene (MA), and the co-former 1,4-diodotetrafluorobenzene (I2F4).The mainframe for this work was the potential re-use of pollutants as novel interesting materials. The choice of a polyhalogenated co-former was instrumental in obtaining solid-state products possessing remarkable and unprecedented luminescence features.
A peculiar aspect is that all co-crystals exhibit phosphorescence at room temperature, contrary to what observed for the parent crystalline anthracene and 9-methylanthracene, which show only fluorescence. The presence of excimer emission in (MA)4·I2F4 is also interesting, as this emission mechanism is combined with the phenomenon of phosphorescence within the same material.
The two co-crystals MA·I2F4 and (MA)4·I2F4 were shown to interconvert in the solid state, via mechanochemistry, upon the addition of an excess of the limiting component; finally, in the case of (MA)4·I2F4, containing pairs of MA molecules in a favourable relative orientation, the potential [4+4] photoreactivity was also investigated.
Work is in progress to further test the ability of polyhalogenated co-formers to direct the assembly of organic co-crystals with controllable luminescence, able to display phosphorescence at room temperature. This approach also offers the possibility to achieve photoreactive materials with intriguing photophysical properties.
Author contributions
AA, MC and FS: experimentation and data collection; AA and SD: conceptualization and manuscript preparation; SD: structural analysis and data validation; BV and FG: funding acquisition and supervision.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge the University of Bologna (RFO-scheme), MUR, project PRIN2020 “Nature inspired crystal engineering”, and CNR (Project PHEEL) for financial support.Notes and references
- J. C. Fetzer, Polycyclic Aromat. Compd., 2007, 27, 143–162 CrossRef CAS.
- A. T. Lawal, Cogent Environ. Sci., 2017, 3, 1339841 CrossRef.
- B. R. T. Simoneit, in PAHs and Related Compounds. The Handbook of Environmental Chemistry, ed. A. H. Neilson, Springer, Berlin, Heidelberg, 1998, pp. 175–221 Search PubMed.
- H. I. Abdel-Shafy and M. S. M. Mansour, Egypt. J. Pet., 2016, 25, 107–123 CrossRef.
- K. Ravindra, R. Sokhi and R. Van Grieken, Atmos. Environ., 2008, 42, 2895–2921 CrossRef CAS.
- M. Tobiszewski and J. Namieśnik, Environ. Pollut., 2012, 162, 110–119 CrossRef CAS PubMed.
- B. J. Finlayson-Pitts and J. N. Pitts, Science, 1997, 276, 1045–1051 CrossRef CAS PubMed.
- K. C. Jones, J. A. Stratford, K. S. Waterhouse, E. T. Furlong, W. Giger, R. A. Hites, C. Schaffner and A. E. Johnston, Environ. Sci. Technol., 1989, 23, 95–101 CrossRef CAS.
- C.-C. Lee, C. S. Chen, Z.-X. Wang and C.-J. Tien, Sci. Total Environ., 2021, 795, 148867 CrossRef CAS PubMed.
- A. O. Adeniji, O. O. Okoh and A. I. Okoh, Arch. Environ. Contam. Toxicol., 2019, 76, 657–669 CrossRef CAS PubMed.
- A. O. Adeniji, O. O. Okoh and A. I. Okoh, in Recent Insights in Petroleum Science and Engineering, ed. M. Zoveidavianpoor, IntechOpen, Rijeka, 2018, pp. 393–428 Search PubMed.
- A. Turaki Usman, H. O. Abugu and C. O. B. Okoye, J. Environ. Health Sci. Eng., 2021, 19, 1523–1534 CrossRef CAS PubMed.
- O. S. Olatunji, O. S. Fatoki, B. O. Opeolu and B. J. Ximba, Food Chem., 2014, 156, 296–300 CrossRef CAS PubMed.
- G. R. Sampaio, G. M. Guizellini, S. A. da Silva, A. P. de Almeida, A. C. C. Pinaffi-Langley, M. M. Rogero, A. C. de Camargo and E. A. F. S. Torres, Int. J. Mol. Sci., 2021, 22, 6010 CrossRef CAS PubMed.
- R. H. de Vos, W. van Dokkum, A. Schouten and P. de Jong-Berkhout, Food Chem. Toxicol., 1990, 28, 263–268 CrossRef CAS PubMed.
- H. Ekner, K. Dreij and I. Sadiktsis, Food Control, 2022, 132, 108528 CrossRef CAS.
- Y.-R. Ju, C.-F. Chen, M.-H. Wang, C.-W. Chen and C.-D. Dong, J. Hazard. Mater., 2022, 421, 126708 CrossRef CAS PubMed.
- R. Stading, G. Gastelum, C. Chu, W. Jiang and B. Moorthy, Semin. Cancer Biol., 2021, 76, 3–16 CrossRef CAS PubMed.
- C. Torres-Moreno, L. Puente-DelaCruz, G. Codling, A. L. Villa, M. Cobo, J. Klanova and B. Johnson-Restrepo, Environ. Res., 2022, 204, 111981 CrossRef CAS PubMed.
- Z. Nováková, J. Novák, M. Bittner, P. Čupr, P. Přibylová, P. Kukučka, M. Smutná, B. I. Escher, H. Demirtepe, A. Miralles-Marco and K. Hilscherová, J. Hazard. Mater., 2022, 424, 127306 CrossRef PubMed.
- L. Zhang, H. Wang, Z. Yang, B. Fang, H. Zeng, C. Meng, S. Rong and Q. Wang, Environ. Pollut., 2022, 293, 118493 CrossRef CAS PubMed.
- R. Tong, L. Cao, X. Yang and B. Zhang, Int. Arch. Occup. Environ. Health, 2021, 94, 1917–1929 CrossRef CAS PubMed.
- C. E. Cerniglia, Curr. Opin. Biotechnol., 1993, 4, 331–338 CrossRef CAS.
- A. B. Patel, S. Shaikh, K. R. Jain, C. Desai and D. Madamwar, Front. Microbiol., 2020, 11, 1–23 CrossRef PubMed.
- L. N. Ukiwe, U. U. Egereonu, P. C. Njoku, C. I. A. Nwoko and J. I. Allinor, Int. J. Chem., 2013, 5, 43–55 Search PubMed.
- P. Yu, Y. Li, H. Zhao, L. Zhu, Y. Wang, W. Xu, Y. Zhen, X. Wang, H. Dong, D. Zhu and W. Hu, Small, 2021, 17, 2006574 CrossRef CAS PubMed.
- L. H. Nguyen and T. N. Truong, ACS Omega, 2018, 3, 8913–8922 CrossRef CAS PubMed.
- C. Aumaitre and J.-F. Morin, Chem. Rec., 2019, 19, 1142–1154 CrossRef CAS PubMed.
- G. Malloci, G. Cappellini, G. Mulas and A. Mattoni, Chem. Phys., 2011, 384, 19–27 CrossRef CAS.
- I. Cherchneff, J. R. Barker and A. G. G. M. Tielens, Astrophys. J., 1991, 377, 541 CrossRef.
- G. M. Paternò, Goudappagouda, Q. Chen, G. Lanzani, F. Scotognella and A. Narita, Adv. Opt. Mater., 2021, 9, 2100508 CrossRef.
- D. Braga, F. Grepioni, L. Maini and S. D'Agostino, IUCrJ, 2017, 4, 369–379 CrossRef CAS PubMed.
- D. Braga, F. Grepioni, L. Maini and S. d'Agostino, Eur. J. Inorg. Chem., 2018, 2018, 3597–3605 CrossRef CAS.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- C. B. Aakeröy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397–407 RSC.
- D. Tian and Y. Chen, Adv. Opt. Mater., 2021, 9, 2002264 CrossRef CAS.
- B. L. Hu and Q. Zhang, Chem. Rec., 2021, 21, 116–132 CrossRef CAS PubMed.
- J. Lee, J. Shim, J. S. Lee, C.-J. Choi, S.-Y. Yim, Y. Jin, K. Yu, Y. J. Park, S. Ahn and D. I. Son, J. Mater. Chem. C, 2021, 9, 13081–13089 RSC.
- R. M. Hochstrasser and A. Malliaris, Mol. Cryst. Liq. Cryst., 1970, 11, 331–342 CrossRef CAS.
- Y. Ishii and A. Matsui, J. Phys. Soc. Jpn., 1967, 22, 926 CrossRef CAS.
- H.-D. Wu, H.-D. Peng and G.-B. Pan, RSC Adv., 2016, 6, 78979–78983 RSC.
- R. M. Hochstrasser, J. Chem. Phys., 1962, 36, 1099–1100 CrossRef CAS.
- H. Wu, M. Li, C. Sun, X. Wang and Z. Su, Dalton Trans., 2020, 49, 5087–5091 RSC.
- S. d'Agostino, F. Grepioni, D. Braga and B. Ventura, Cryst. Growth Des., 2015, 15, 2039–2045 CrossRef.
- L. Li, Z. F. Liu, W. X. Wu and W. J. Jin, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2018, 74, 610–617 CrossRef CAS.
- M. Villa, S. D'Agostino, P. Sabatino, R. Noel, J. Busto, M. Roy, M. Gingras and P. Ceroni, New J. Chem., 2020, 44, 3249–3254 RSC.
- B. Ventura, A. Bertocco, D. Braga, L. Catalano, S. d'Agostino, F. Grepioni and P. Taddei, J. Phys. Chem. C, 2014, 118, 18646–18658 CrossRef CAS.
- M. Singh, K. Liu, S. Qu, H. Ma, H. Shi, Z. An and W. Huang, Adv. Opt. Mater., 2021, 9, 1–24 Search PubMed.
- D. Li, F. Lu, J. Wang, W. Hu, X.-M. Cao, X. Ma and H. Tian, J. Am. Chem. Soc., 2018, 140, 1916–1923 CrossRef CAS PubMed.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 1–10 CrossRef PubMed.
- Y. Xie, Y. Ge, Q. Peng, C. Li, Q. Li and Z. Li, Adv. Mater., 2017, 29, 1606829 CrossRef PubMed.
- X.-N. Li, M. Yang, X.-L. Chen, J.-H. Jia, W.-W. Zhao, X.-Y. Wu, S.-S. Wang, L. Meng and C.-Z. Lu, Small, 2019, 15, 1903270 CrossRef CAS PubMed.
- S. D'Agostino, F. Spinelli, P. Taddei, B. Ventura and F. Grepioni, Cryst. Growth Des., 2019, 19, 336–346 CrossRef.
- R. Kabe and C. Adachi, Nature, 2017, 550, 384–387 CrossRef CAS PubMed.
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920–9940 CrossRef CAS PubMed.
- J. C. Christopherson, F. Topić, C. J. Barrett and T. Friščić, Cryst. Growth Des., 2018, 18, 1245–1259 CrossRef CAS.
- F. Grepioni, S. D'Agostino, D. Braga, A. Bertocco, L. Catalano and B. Ventura, J. Mater. Chem. C, 2015, 3, 9425–9434 RSC.
- B. Lu, X. Fang and D. Yan, ACS Appl. Mater. Interfaces, 2020, 12, 31940–31951 CrossRef CAS PubMed.
- X. G. Yang, W. J. Qin, J. R. Zhang, X. K. Tian, X. Fan, L. F. Ma and D. Yan, Front. Chem., 2021, 9, 851 Search PubMed.
- Q. Feng, W. Huan, J. Wang, F. Guo, J. Lu, G. Diao and Y. Shan, Crystals, 2018, 8, 392 CrossRef.
- W. Ji, B. Xue, S. Bera, S. Guerin, Y. Liu, H. Yuan, Q. Li, C. Yuan, L. J. W. Shimon, Q. Ma, E. Kiely, S. A. M. Tofail, M. Si, X. Yan, Y. Cao, W. Wang, R. Yang, D. Thompson, J. Li and E. Gazit, ACS Nano, 2020, 14, 10704–10715 CrossRef CAS PubMed.
- V. Kumar, P. Scilabra, P. Politzer, G. Terraneo, A. Daolio, F. Fernandez-Palacio, J. S. Murray and G. Resnati, Cryst. Growth Des., 2021, 21, 642–652 CrossRef.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed.
- C. B. Aakeröy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem. – Eur. J., 2013, 19, 16240–16247 CrossRef PubMed.
- L. Li, W. X. Wu, Z. F. Liu and W. J. Jin, New J. Chem., 2018, 42, 10633–10641 RSC.
- X. Pang, H. Wang, W. Wang and W. J. Jin, Cryst. Growth Des., 2015, 15, 4938–4945 CrossRef CAS.
- M. Arhangelskis, F. Topić, P. Hindle, R. Tran, A. J. Morris, D. Cinčić and T. Friščić, Chem. Commun., 2020, 56, 8293–8296 RSC.
- R. Mason, Acta Crystallogr., 1964, 17, 547–555 CrossRef CAS.
- J. C. J. Bart and G. M. J. Schmidt, Isr. J. Chem., 1971, 9, 429–448 CrossRef CAS.
- S. Y. Oh, C. W. Nickels, F. Garcia, W. Jones and T. Friščić, CrystEngComm, 2012, 14, 6110–6114 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. A. Sarjeant, 2015, 1–50.
- A. Thorn, B. Dittrich and G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2012, 68, 448–451 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- S. P. Thomas, P. R. Spackman, D. Jayatilaka and M. A. Spackman, J. Chem. Theory Comput., 2018, 14, 1614–1623 CrossRef CAS PubMed.
- K. Rurack and M. Spieles, Anal. Chem., 2011, 83, 1232–1242 CrossRef CAS PubMed.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protoc., 2013, 8, 1535–1550 CrossRef PubMed.
- G. M. J. Schmidt, J. Chem. Soc., 1964, 385 Search PubMed.
- M. D. Cohen, G. M. J. Schmidt and F. I. Sonntag, J. Chem. Soc., 1964, 2000–2013 RSC.
- E. Heller and G. M. J. Schmidt, Isr. J. Chem., 1971, 9, 449–462 CrossRef CAS.
- T. Salzillo, I. Bilotti, R. G. Della Valle, E. Venuti and A. Brillante, J. Am. Chem. Soc., 2012, 134, 17671–17679 CrossRef CAS PubMed.
- T. Salzillo, E. Venuti, C. Femoni, R. G. Della Valle, R. Tarroni and A. Brillante, Cryst. Growth Des., 2017, 17, 3361–3370 CrossRef CAS.
- L. Opilik, P. Payamyar, J. Szczerbiński, A. P. Schütz, M. Servalli, T. Hungerland, A. D. Schlüter and R. Zenobi, ACS Nano, 2015, 9, 4252–4259 CrossRef CAS PubMed.
- F. Spinelli, S. d'Agostino, P. Taddei, C. D. Jones, J. W. Steed and F. Grepioni, Dalton Trans., 2018, 47, 5725–5733 RSC.
- T. Salzillo, S. Zaccheroni, R. G. Della Valle, E. Venuti and A. Brillante, J. Phys. Chem. C, 2014, 118, 9628–9635 CrossRef CAS.
- T. Salzillo, E. Venuti, R. G. Della Valle and A. Brillante, J. Raman Spectrosc., 2017, 48, 271–277 CrossRef CAS.
- T. Salzillo and A. Brillante, CrystEngComm, 2019, 21, 3127–3136 RSC.
- G. Hofer, A. D. Schlüter and T. Weber, Macromolecules, 2022, 55, 568–583 CrossRef CAS.
- T. Kim, L. Zhu, L. J. Mueller and C. J. Bardeen, J. Am. Chem. Soc., 2014, 136, 6617–6625 CrossRef CAS PubMed.
- D. Braga, S. d'Agostino and F. Grepioni, Cryst. Growth Des., 2012, 12, 4880–4889 CrossRef CAS.
- G. Kaupp, J. Schmeyers, M. Kato, K. Tanaka and F. Toda, J. Phys. Org. Chem., 2002, 15, 148–153 CrossRef CAS.
- D. Bučar, T. D. Hamilton and L. R. MacGillivray, in Organic Nanostructures, ed. J. L. Atwood and J. W. Steed, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2008, pp. 305–311 Search PubMed.
- A. F. Mabied, M. Müller, R. E. Dinnebier, S. Nozawa, M. Hoshino, A. Tomita, T. Sato and S. Adachi, Acta Crystallogr., Sect. B: Struct. Sci., 2012, 68, 424–430 CrossRef CAS PubMed.
- D. Y. Wu, B. Chen, X. G. Fu, L. Z. Wu, L. P. Zhang and C. H. Tung, Org. Lett., 2003, 5, 1075–1077 CrossRef CAS PubMed.
- Z. A. Dreger, H. Lucas and Y. M. Gupta, J. Phys. Chem. B, 2003, 107, 9268–9274 CrossRef CAS.
- J. B. Birks and J. B. Aladekomo, Photochem. Photobiol., 1963, 2, 415–418 CrossRef CAS.
- Y. Dai, H. Liu, T. Geng, F. Ke, S. Niu, K. Wang, Y. Qi, B. Zou, B. Yang, W. L. Mao and Y. Lin, J. Mater. Chem. C, 2021, 9, 934–938 RSC.
- H. Liu, D. Cong, B. Li, L. Ye, Y. Ge, X. Tang, Y. Shen, Y. Wen, J. Wang, C. Zhou and B. Yang, Cryst. Growth Des., 2017, 17, 2945–2949 CrossRef CAS.
- Y. Mizobe, M. Miyata, I. Hisaki, Y. Hasegawa and N. Tohnai, Org. Lett., 2006, 8, 4295–4298 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Powder XRD patterns, TGA traces, interaction energies, NMR spectra, photophysical measurements. CCDC 2166101–2166103. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce00720g |
| This journal is © The Royal Society of Chemistry 2022 |