
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethod for extraction and analysis of per- and poly-fluoroalkyl substances in contaminated asphalt†
Prashant
Srivastava
 *a,
Mike
Williams
a,
Jun
Du
a,
Divina
Navarro
a,
Rai
Kookana
a,
Grant
Douglas
b,
Trevor
Bastow
b,
Greg
Davis
b and
Jason K.
Kirby
a
*a,
Mike
Williams
a,
Jun
Du
a,
Divina
Navarro
a,
Rai
Kookana
a,
Grant
Douglas
b,
Trevor
Bastow
b,
Greg
Davis
b and
Jason K.
Kirby
a
aCommonwealth Scientific and Industrial Research Organisation (CSIRO), Land and Water, Waite Campus, Urrbrae, South Australia, Australia. E-mail: Prashant.Srivastava@csiro.au
bCommonwealth Scientific and Industrial Research Organisation (CSIRO), Land and Water, Floreat, Western Australia, Australia
First published on 13th April 2022
Abstract
The legacy use of aqueous film-forming foam (AFFF) has led to the generation of large volumes of per- and poly-fluoroalkyl substances (PFAS)-contaminated asphalt materials, especially at airports and fire training areas. The management of such PFAS-contaminated asphalt materials requires an understanding of PFAS concentrations in these materials. This study, therefore, aimed to develop a suitable extraction methodology for the analysis of 22 target PFAS (i.e., carboxylic acids, sulfonic acids and fluorotelomers) in asphalt materials. A series of experiments was conducted to optimise extraction solvent composition, as well as to assess the performance of the chosen method under various conditions (i.e., sonication temperature, PFAS contamination level, asphalt core composition and timing of stable isotope addition used as internal standard). The methanol-based extractants performed best due to their accuracy and precision, which were within the acceptable range (extraction efficiency between 70 and 130% and RSD < 20%). The method which involved three successive extractions with methanol/1% NH3 by ultrasonication at 25 °C was selected due to its performance and ease of operation. The mean recovery of a vast majority of PFAS was found to be in the acceptable range. Tests on the timing of addition of stable isotope (SI)-labelled PFAS internal standards indicate that the recoveries obtained, regardless of when the stable isotopes were added, were within the acceptable range for PFAS. The accuracy and precision of PFAS recoveries were not affected by PFAS spike level (2 μg kg−1 and 200 μg kg−1), as well as sample composition (based on the location of asphalt material in the field). Low RSDs were achieved for asphalt cores collected from a contaminated site covering a wide range of concentrations (from LOQ to 2135 mg kg−1), demonstrating the suitability of the sample preparation method for real-world samples. The results from the interlaboratory testing were also in good agreement and validated the proposed PFAS extraction and analytical approach.
1. Introduction
Per- and poly-fluoroalkyl substances (PFAS) are a diverse group of man-made fluorinated chemicals with fluorine atoms attached to an alkyl chain with the moiety CnF2n+1.1 The carbon–fluorine bond, being one of the strongest bonds known in organic chemistry, imparts thermal and chemical stability, which makes PFAS highly resistant to degradation.2 Due to these properties, these chemicals have historically been used in aqueous film-forming foams (AFFF) for firefighting, aviation hydraulic fluids, non-stick cookware, fabrics, furniture, carpeting, and food packaging.3 PFAS are also often called “forever chemicals” due to their persistence in the environment,4 while their wide range of applications has resulted in a worldwide problem of contamination in the environment.3 In particular, the extensive historical use of AFFF-containing PFAS (precursors and transformation chemicals) at firefighting stations, large fuel storage facilities and military bases/airfields has led to significant PFAS contamination within related infrastructure materials (i.e., concrete and asphalt) and the surrounding environments (i.e., waters, soils, and sediments).5 The high mobility of PFAS has often resulted in large contamination zones from the original source zone due to surface water and groundwater transport.6 For example, in Australia, an estimated 90 sites (including airports, defence airforce bases, firefighting training areas, etc.) were reported to be under investigation for hosting elevated PFAS concentrations (https://pfas.australianmap.net/). In the United States, an estimated 2854 sites in 50 states and 2 territories are suspected to be contaminated with PFAS (https://www.ewg.org/interactive-maps/pfas_contamination/); whereas in Europe, more than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 sites are suspected as potentially PFAS contaminated sites.7
000 sites are suspected as potentially PFAS contaminated sites.7
The legacy and current use of AFFF as part of training activities and emergency responses at fire stations, fuel storage facilities and airports has resulted in often large volumes of PFAS contaminated asphalt (e.g., driveway, taxiway and runways) and concrete (e.g., training pads).5,8 These PFAS contaminated asphalt and concrete materials need to be managed, e.g., reused, remediated or disposed of, from these sites to prevent the spread of contamination and minimize potential impacts on human health and the environment. Given the lack of guidance on the management of PFAS-contaminated materials, decommissioning of sites or removal of surficial layers of asphalt or concrete during tarmac resurfacing or training pad removal have resulted in a considerable volume of waste materials potentially contaminated with PFAS. A better understanding of PFAS contamination levels in these materials is necessary to determine the most appropriate management strategy (i.e., reuse, remediation, or disposal).9 For instance, according to the Stockholm Convention,10 the quantification of total and leachable concentrations of the perfluoroalkyl acids (PFAAs) e.g. perfluorooctanoic acid (PFOA), and perfluorooctane sulfonate (PFOS) is required for disposal of waste materials; whereas, the Australian PFAS National Environmental Management Plan (NEMP)11 recommends assessing perfluorohexane sulfonate (PFHxS) in addition to PFOA and PFOS while disposing of PFAS-contaminated waste materials.
The measurement, management and mitigation of PFAS contaminated concrete and asphalt have received limited attention in the literature. Currently, there is no analytical method available to extract PFAS from asphalt, although similar work has been conducted to extract PFAS from concrete. The work by Baduel et al.8 and Thai et al.12 on the spatial and vertical distribution of PFAS in concrete were the only relevant study found in the literature. Indeed, results from these studies highlighted how fire-fighting training pads could be a source of PFAS for many decades.8,12 Crucial to studies like these is the development of analytical methodologies that can accurately measure PFAS from complex matrices. Currently, most researchers have used methanol for extraction of PFAS in matrices such as soil,6,13–17 biosolids,18,19 and concrete8,12 followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) for analysis.9,13,15,18,20–23 Given the lack of studies on asphalt, it is unclear if the same approach applies to these materials. Compared to concrete which comprises cement, sand and aggregates,24,25 asphalt is a much more complex matrix, hence, “extracting” PFAS in a form that can be analysed may prove to be more difficult. Asphalt mainly consists of bitumen (a black viscous petroleum product remaining after the distillation/refining of crude oil comprising of high molecular weight alkanes, aromatics, polar hydrocarbons and asphaltenes) and mineral-based particles (such as aggregate).26–28 The presence of these highly hydrophobic components may reduce the overall extractability of PFAS from the asphalt matrix using the commonly used extraction methodologies, as well as contribute to potential matrix effects that affect the response of PFAS in the mass spectrometer either by suppression or enhancement. In this regard, a method that can effectively extract PFAS from the solid phase without releasing the matrix that can interfere with LC-MS/MS analysis is critical for their quantitation.
The PFAS extraction methodologies using methanol are often modified with an acid or base to improve the extraction efficiencies of charged PFAS.20 For example, modifying methanol with acids, such as formic, acetic, or hydrochloric acid, has been found to increase the extraction efficiency (>80% recovery) of PFAS containing anionic functional groups, including perfluoroalkyl carboxylic acids (PFCAs), perfluoroalkyl sulfonic acids (PFSAs) and perfluorooctane sulfonamide acids (FOSAAs) from soils and sediments.14,29–31 Conversely, the modification of methanol with bases such as ammonia (NH3) or sodium hydroxide (NaOH) has been used to improve the extraction efficiency of not only anionic PFCAs, PFSAs and fluorotelomer sulfonates (FTSs) but also of zwitterionic fluorotelomer betaines, containing both acidic and basic functional groups, from contaminated soils.16 Sequential extraction is another approach to improve PFAS extractability, as reported by Higgins et al.29 using acetic acid and methanol/1% acetic acid mixture to successfully measure anionic PFAS in sediments and sludge. These approaches could provide an excellent starting point to develop an extraction method applicable to asphalt.
LC-MS/MS is the most common analytical technique used for the measurement of PFAS in environmental samples.20,30,32–34 Solid-phase extraction (SPE) is used following extraction for cleanup to remove matrix bias on quantification and preconcentration to improve detection limits.22,23,30,35–37 Yu et al.37 used a hydrophilic–lipophilic-balanced (HLB) SPE cartridge (Waters) to remove matrix interferences in wastewater and sludge extracts to improve recoveries (PFOS = 92% (wastewater) and 84% (sludge); PFOA = 82% (wastewater) and 70% (sludge)) and quantification limits (PFOS = 0.25 ng L−1 and PFOA = 1 ng L−1). Isotope dilution analysis is applied to improve the accuracy and precision of PFAS quantification in complex matrices (for example, matrix effect bias that results in enhanced or suppressed analyte measurement).15,20,36,38
To our knowledge, this is the first research attempted to extract PFAS from asphalt. This study aimed at developing an extraction method that can provide a robust and reliable quantification of 22 PFAS (short- and long-chain PFCAs and PFSAs and fluorotelomers), including PFHxS, PFOS and PFOA, present in asphalt collected from military bases. Herein, we compared 6 extraction solvents that were selected based on their use and performance in the literature. The effects of other variables such as sonication temperature, PFAS spike level, and sample composition (based on sample location in the asphalt core) on the extraction efficiency were also tested. In addition, we examined whether the timing of the addition of isotopically labelled internal standards during the PFAS extraction process had an impact on PFAS recovery and QA/QC. The preferred extraction method was then applied to the measurement of PFOA, PFOS and PFHxS concentrations in several historically contaminated asphalt cores collected from an operational airbase in Australia (i.e., runway apron, runway, taxiway near aircraft hangar, driveway and near a fire-fighting training area).
2. Materials and methods
2.1. Chemicals and standards
Twenty-two PFAS were assessed for extraction efficiency from asphalt, including PFCAs, PFSAs and FTS. Native (non-labelled) PFAS and stable isotope (SI) (13Cx and/or 18Ox) labelled PFAS (available and used as internal standards for 15 PFAS), viz., PFCAs (perfluorobutanoic acid (PFBA), perfluoropentanoic acid (PFPeA), perfluorohexanoic acid (PFHxA), perfluoroheptanoic acid (PFHpA), perfluorooctanoic acid (PFOA), perfluorononanoic acid (PFNA), perfluorodecanoic acid (PFDA), perfluoroundecanoic acid (PFUdA), perfluorododecanoic acid (PFDoA), perfluorotridecanoic acid (PFTrDA), perfluorotetradecanoic acid (PFTeDA)), PFSAs (perfluorobutane sulfonic acid (PFBS), perfluoropentane sulfonic acid (PFPeS), perfluorohexane sulfonic acid (PFHxS), perfluoroheptane sulfonic acid (PFHpS), perfluorooctane sulfonic acid (PFOS) linear, perfluorononane sulfonic acid (PFNS), and perfluorodecane sulfonic acid (PFDS)), and FTS (4:2 fluorotelomer sulfonate (4:2 FTS), 6:2 fluorotelomer sulfonate (6:2 FTS), 8:2 fluorotelomer sulfonate (8:2 FTS), and 10:2 fluorotelomer sulfonate (10:2 FTS)) were purchased from Wellington Laboratories (Canada) (Table S1†). The native and SI-labelled PFAS stock solutions (500 μg L−1) were prepared in methanol and stored in polypropylene tubes at 4 °C.
The chemicals used to extract PFAS from asphalt were methanol (LC-MS grade, >99.9%, Fisher Chemical), ammonia solution (NH3, reagent grade, 28%; Scharlau), hydrochloric acid (HCl, reagent grade, 37%; Fisher Scientific), ammonium acetate (NH4CH3COO, reagent grade, 98%; Sigma-Aldrich), acetonitrile (ACN, LC-MS grade, >99.9%, Fisher Chemical), and acetone (HPLC grade, >99.8%, Fisher Scientific). Ultrapure deionised water (18 MΩ, Milli-Q, Millipore) was used for the preparation of extraction solutions and standards.
2.2. Asphalt sample collection and preparation
The asphalt materials were sourced from cores collected at four locations at an operational airbase in Australia: runway apron (taxiway) (Core 1, Core 2), run-way (Core 3, Core 4), taxiway near aircraft hangar (Core 5), driveway (Core 6) and near a fire-fighting training area (Core 7) (Table 1). Asphalt cores (10 cm diameter and up to 40 cm long) were sampled at these locations by drilling cores vertically into the asphalt using a small mobile drilling rig using PFAS-free potable water. The core samples were placed in high-density polythene bags, sealed to prevent contamination, and refrigerated for transport to the laboratory. In the laboratory, asphalt cores were oven-dried at 60 °C for 12 h and cut into 2 to 5 vertical sections using a circular saw with a diamond cutting blade cleaned with methanol. The number and location of vertical sections were decided by visual inspection of individual cores that identified easily recognisable sections/layers of different compositions (for example, colour, aggregate sizes) (Fig. S1†). The vertical core sections were further cut in half using the circular saw with a diamond cutting blade cleaned with methanol.| Location | No. | Depth mm | PFOA mg kg−1 | PFHxS mg kg−1 | PFOS mg kg−1 | Sum of PFHxS and PFOS mg kg−1 |
|---|---|---|---|---|---|---|
| a LOQ = limit of quantitation. b Concentrations without standard deviation had non-detect values for other replicates. c Concentration exceeded landfill acceptance criteria1 of 50 mg kg−1. | ||||||
| LOQa | 0.60 | 1.00 | 0.70 | |||
| Runway apron | Core 1 | 0–50 | <LOQ | <LOQ | <LOQ | <LOQ |
| 190–240 | <LOQ | <LOQ | <LOQ | <LOQ | ||
| Core 2 | 0–50 | <LOQ | <LOQ | <LOQ | <LOQ | |
| 180–230 | <LOQ | <LOQ | <LOQ | <LOQ | ||
| Runway | Core 3 | 0–50 | <LOQ | <LOQ | 1.40 ± 0.30 | 1.40 ± 0.30 |
| 50–80 | <LOQ | <LOQ | 1.39 ± 0.12 | 1.39 ± 0.12 | ||
| 80–110 | <LOQ | <LOQ | 3.12b | 3.12b | ||
| 110–140 | <LOQ | <LOQ | <LOQ | <LOQ | ||
| 140–170 | <LOQ | <LOQ | <LOQ | <LOQ | ||
| 170–220 | <LOQ | <LOQ | <LOQ | <LOQ | ||
| 220–250 | <LOQ | <LOQ | <LOQ | <LOQ | ||
| Core 4 | 0–50 | <LOQ | 3.10 ± 0.04 | 9.90 ± 0.40 | 13.00 ± 0.80 | |
| 50–70 | < LOQ | 2.20 ± 0.27 | 22.00 ± 2.70 | 24.20 ± 2.97 | ||
| 70–110 | 0.82 | 3.56b | 61.00b,c | 64.56b,c | ||
| 110–140 | 0.65 | 3.46b | 13.30b | 16.76b | ||
| 140–180 | < LOQ | 2.70 ± 0.20 | 1.00 ± 0.10 | 3.70 ± 0.30 | ||
| Taxiway near aircraft hangar | Core 5 | 0–50 | <LOQ | <LOQ | 1.70 ± 0.50 | 1.70 ± 0.50 |
| 50–80 | <LOQ | <LOQ | 3.20 ± 0.34 | 3.20 ± 0.34 | ||
| 80–110 | <LOQ | <LOQ | 2.50b | 2.50b | ||
| 110–140 | <LOQ | 3.45b | 18.40b | 21.85b | ||
| 140–170 | 13.50 ± 1.30 | 148.00 ± 9.70c | 1201.00 ± 159.00c | 1349.00 ± 168.70c | ||
| Driveway | Core 6 | 0–50 | 5.50 ± 0.40 | 68.20 ± 1.50c | 487.00 ± 44.00c | 555.20 ± 45.50c |
| Near a fire-fighting training pad | Core 7 | 0–50 | 3.60 ± 0.20 | 183.00 ± 16.00c | 1952.00 ± 965.00c | 2135.00 ± 981.00c |
| 50–100 | 0.92 ± 0.02 | <LOQ | 2.30 ± 0.40 | 2.30 ± 0.40 | ||
The extraction method comparison (Core 1) and influence of method parameters (e.g., sonication temperature, sample composition and spike level) studies (Core 1 and Core 2) for measurement of PFAS in asphalt occurred on bulk material prepared from cores collected on the runway apron (taxiway). A bulk material was prepared using individual vertical half sections or by combining two vertical half sections (top and bottom) from individual cores. The individual vertical sections or combined vertical sections were crushed into small particles/aggregates using a stainless-steel hammer cleaned with methanol and passed through a 2 mm stainless steel sieve with approximately half of the <2 mm homogenized material ground to a powder using a stainless-steel mill.
The preferred extraction method was used to determine PFAS concentrations in cores collected at different locations at an operational airbase in Australia (Table 1). Powdered asphalt samples were collected by drilling (2–3 cm depth) into the internal face of vertical half core sections using a stainless-steel drill bit cleaned with methanol (Fig. S1†). A powder sample was collected using a 10 mm drill bit from 15 drilling points encompassing both aggregate and the organic matrix selected using a 3 × 5 matrix on the internal face of vertical half core sections (Fig. S1†). The individual powders from the holes were combined in a polypropylene tube to make a bulk powder for extraction.
2.3. Comparison of PFAS extraction methods from asphalt
Asphalt matrices are likely to pose significant challenges to the extraction and detection of PFAS due to the diverse nature of PFAS that may be present in asphalt samples (i.e., polar/nonpolar, nonvolatile/semi-volatile/volatile, and neutral/anionic/cationic/zwitterionic species, chemicals), the potential interactions these PFAS may have with the components of asphalt, such as bitumen (a high-molecular-weight petroleum comprising alkanes, aromatics, polar hydrocarbons and asphaltenes) and mineral particles (mainly aggregate) and the release of components that may interfere with PFAS extraction recoveries.A summary of common extraction methods used for the measurement of PFAS in environmental samples such as soils, sediments and biosolids/wastes is presented in Table S2.† Methanol is the preferred solvent used often with the addition of a modifying acid or base for the extraction of a broad range of PFAS.14,16,18,29–31,39,40 Other polar solvents, such as acetonitrile and acetone have been used to a much lesser extent.31,41
In this study, methanol, acidic and basic modifications of methanol, and basic acetonitrile:acetone ((1:1 v/v)/1% NH3) mixture were selected to compare performance during the extraction of PFAS from asphalt (Table 2). These extractants were selected based on the reported good performance in the extraction of PFAS from complex environmental matrices (e.g., soils, sediments and biosolids/wastes), simplicity/usability, and less aggressive behaviour to the solid phase (reducing the mobilisation of matrix constituents that may cause matrix effect bias during mass spectrometry analysis). A commonly used ultrasonic method was employed for all extraction methods.6,8,14–16,18,29–31,40,42
| Variables | Treatments |
|---|---|
| a NH3 = ammonia, NH4CH3COO = ammonium acetate, HCl = hydrochloric acid, SPE = solid-phase extraction. | |
| Extractant | Extractant 1: methanol |
| Extractant 2: methanol/1% NH3 | |
| Extractant 3: methanol, methanol/1% NH3 and methanol/1% NH3 | |
| Extractant 4: methanol, methanol/1% NH3, and methanol/0.4 M HCl | |
| Extractant 5: methanol/1% NH4CH3COO | |
| Extractant 6: acetonitrile/acetone (1:1 v/v)/1% NH3 |
|
| Timing of PFAS stable isotope-labelled internal standard addition | To asphalt before extraction |
| To extract solution prior to SPE | |
| To cleaned up solutions after SPE | |
| Sonication temperature | 25 and 50 °C |
| Spiking concentration | 2 μg kg−1 and 200 μg kg−1 |
| Sample location (asphalt composition) | Top and bottom |
For each extraction treatment, homogenised ∼0.5 g of asphalt powder in triplicate was weighed into 15 mL polypropylene (PP) tubes and spiked with 100 μL of a mixed PFAS non-labelled standard solution (10 μg L−1 or 2 μg kg−1 for each individual PFAS in ultra-pure deionised water). The spiked asphalt samples were vortex-mixed for 10 seconds and dried at 25 °C for 48 h. Prior to the extraction, 100 μL of the SI-labelled PFAS mixture (10 μg L−1 in ultra-pure deionised water) was added to spiked asphalt samples.
Four mL of the different extractants (Table 2) were added to the polypropylene (PP) tube containing native PFAS-spiked asphalt samples mixed with SI-labelled PFAS and the tube was sonicated in an ultrasonic bath at 25 °C for 20 min. Following sonication, the suspensions were centrifuged for 15 min at 2500g and the supernatants were transferred to another 15 mL PP tube. The extractions were carried out using three separate sequential steps (Table 2), pooling each extract in the same container. In methods that undertook multiple extraction steps, the extracts were combined prior to preconcentration and clean up. Methods 1 and 2 consisted of three separate extractions with a single extractant (i.e., methanol or methanol/1% NH3). For method 3, the first extraction was undertaken using methanol, followed by two separate extractions using methanol/1% NH3. For method 4, there were three different extractants used in succession, i.e., methanol, then methanol/1% NH3, followed by methanol/0.4 M hydrochloric acid. Methods 5 (methanol/1% NH4CH3COO) and 6 (acetonitrile/acetone (1:1 v/v)/1% NH3) were similar to method 1 which consisted of three separate extractions with a single extractant (Table 2).
The pooled extracts were concentrated to 1 mL under a gentle stream of high purity nitrogen and neutralised (if required) using CH3COOH in case of methanol/NH3 based extractant (e.g., methods 2, 3 and 5) or NH3 in case of HCl based extractant (e.g., method 4). The concentrated solutions were passed through a 3 mL Bond Elut carbon SPE cartridge (Agilent) collecting the cleaned-up samples in a clean PP tube. After sample elution, the SPE cartridge was rinsed with 1 mL of methanol, with the washings combined with the cleaned-up sample. The samples were then concentrated to under 1 mL under a gentle stream of high purity nitrogen and transferred into a PP HPLC vial making the final volume to 1 mL with methanol for LC-MS/MS analysis.
As part of the QA/QC, several blanks and controls were prepared alongside each batch of extractions. Tubes containing ultrapure water used to prepare the PFAS spiking solutions were used to test background concentrations of PFAS from the solvents and consumables. Unspiked asphalt core samples were used to check for background concentrations of PFAS in asphalt. All extractions were performed in triplicates unless specified otherwise.
2.4. Testing method performance: influence of other variables on PFAS extraction
The influence of other variables on PFAS extraction was evaluated using the chosen extractant.2.5. Liquid chromatography-mass spectrometry-mass spectrometry (LC-MS/MS)
Analysis by LC-MS/MS was conducted at CSIRO laboratories using a Thermo Altis Triple Quadrupole Mass Spectrometer equipped with Thermo Scientific UltiMate 3000 HPLC system (Thermo Fisher Scientific, Waltham, MA, USA). Chromatographic separation was performed using a Phenomenex Kinetex C18 column (100 × 2.1 mm, particle size = 2.6 μm) and a binary mobile phase introduced at an initial flow rate of 250 μL min−1, which was increased to 300 μL min−1, and then brought back to 250 μL min−1 to equilibrate the column. The low flow rate, in the beginning, was to keep polar PFAS (e.g., PFBA) retained. Other PFAS are retained well on the column; therefore, the flow rate was subsequently increased to reduce the run time. The binary mobile phase consisted of A: 5 mM ammonium acetate and B: methanol. The gradient program was as follows: 0–3.5 min: 5% B (flow rate = 250 μL min−1), increased to 20% B at 4 min (flow rate = 250 μL min−1), increased to 95% B at 13.5 min and held for 4.5 min (flow rate = 300 μL min−1), and then back to 5% B (flow rate = 250 μL min−1). The total run was 25 min. The column oven and autosampler temperature were set at 35 °C and 15 °C, respectively. The sample volume injected was 10 μL. A delay column (Phenomenex Luna C18, 30 × 5 mm) was installed between the mobile phase mixer and the sample injector to ensure the delay of the system-related interference for accurate measurement of PFAS in the samples. A guard column was installed between the injector and the analytical column to remove the impurities and suspended solids from reaching the analytical column.Analytes coming out of the column were introduced to the mass spectrometer coupled with negative electrospray ionization (ESI). To minimise contamination with matrix components to the mass spectrometer ion source, the initial 3 min of sample run were directed to waste via a 6 port-2-position valve installed post-column. The MS source parameters were optimized as spray voltage −2.5 kV, sheath gas pressure 50 arbitrary units, auxiliary gas pressure 5 arbitrary units, ion transfer tube temperature 350 °C, and collision gas pressure 1.5 mTorr. High purity nitrogen (>98%) was used as a desolvation and nebulizer gas. Argon was used as the collision gas. Optimization of multiple reaction monitoring (MRM) parameters (Table S3†) was performed by direct infusion of 200 μg L−1 of a mixed PFAS analytical standard. Collision energy and tube lens voltages (Table S3†) were optimised for PFAS MRM transitions, respectively. Data were acquired and processed using the TraceFinder 4.1 software.
For quantitative analysis, a range of calibration standards consisting of native and SI-labelled PFAS (where available) was prepared: 0.05–20 μg L−1 native PFAS with 1 μg L−1 SI-labelled PFAS. Internal standard calibration was used for PFAS quantitation (i.e., isotope dilution), particularly for the 15 PFAS with available SI-labelled counterparts. The other 7 PFAS (PFTrDA, PFBS, PFPeS, PFHpS, PFNS, PFDS and 10:2 FTS) were quantified by external calibration. Concentrations obtained were used to then calculate PFAS recoveries to compare method performance.
As part of the analytical QA/QC, separate calibration standard (5 μg L−1) and blank (unspiked methanol) samples were injected every 5–8 samples of every sequence to assess the stability of the response of analytical standards (compared with the initial calibration response) and carry-over of PFAS within the analytical system.
2.6. Method validation
The instrumental parameters were validated by determining linearity, the instrumental limit of detection (LOD) and limit of quantification (LOQ), and intraday (repeatability) and interday (reproducibility) precision. The linearity was evaluated using 50 μL of different concentrations (from 25 to 10000 ng L−1) of PFAS standard solutions in methanol. The correlation coefficients (R2) were close to 0.99 for each PFAS. The instrumental LOD was determined as the concentration of PFAS required to produce a signal-to-noise ratio (S/N) of 3:1, where the noise was calculated as three times the standard deviation of the background signal. The LOQ was also determined as the concentration of PFAS required to produce an S/N of 10:1. The LOD ranged from 25 to 250 ng L−1 and the LOQ ranged from 83.25 to 832.50 ng L−1, whereas the method limit of quantitation (MLOQ) ranged from 0.15 to 1 mg kg−1 (Table S1†).
2.7. Interlaboratory comparison
A selected asphalt sample from the airbase driveway (Core 6) was sent to three external laboratories for comparison and confirmation of results. The laboratories had extensive experience in PFAS analysis in environmental samples, including biosolids and soils. The external laboratories used an extraction methodology similar to that developed in our laboratory for asphalt. Laboratory 1 (a research laboratory) optimised the extraction procedure for asphalt combining methanol/2% ammonia and methanol/2% formic acid with a Bond Elut SPE clean-up. Whereas, Laboratory 2, which is an internationally-recognised commercial laboratory, used methanol/0.01 M sodium hydroxide as an extraction solvent and agitation (rotating shaker) rather than ultrasonication. Both laboratories used the same commercially available stable isotopes as in our methodology. In addition to these confirmatory PFAS analyses, the asphalt samples with quantifiable PFAS species were also analysed for total oxidisable precursor (TOP) concentrations to determine whether additional PFAS that were not included in the suite of PFAS may have been present in the asphalt samples. The TOP assay for a subset of asphalt samples was undertaken by another internationally-recognised commercial laboratory (Laboratory 3).3. Results and discussion
3.1. Comparison of PFAS extraction methods for asphalt
The recoveries as a percentage of PFAS from spiked asphalt with (15 PFAS) and without (22 PFAS) SI-correction are shown in Tables 3 and S4,† respectively. The performance of six extractants for PFAS from asphalt was compared based on their accuracy, i.e., closeness to 100% (within a recovery range of 70% to 130%) and precision (i.e., low variability, relative standard deviation or RSD ≤ 20%) as specified in Table B-15 of the United States Department of Defense Quality Systems Manual Version 5.3.48 Heatmaps demonstrating the accuracy and precision in PFAS recovery with SI-correction are shown in Fig. 1.| PFAS species | Extractant 1 | Extractant 2 | Extractant 3 | Extractant 4 | Extractant 5 | Extractant 6 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | % RSD | Mean | % RSD | Mean | % RSD | Mean | % RSD | Mean | % RSD | Mean | % RSD | ||
| a Denotes statistically significant difference in comparison to other extractants. | |||||||||||||
| Short-chain PFCA | PFBA | 115.3 | 4.6 | 109.8 | 5.1 | 108.7 | 2.5 | 37.7a | 18.6 | 113.6 | 5.7 | 119.4 | 2.4 |
| PFPeA | 112.4 | 4.3 | 112.3 | 4.4 | 111.9 | 2.9 | 40.4a | 32.2 | 116.8 | 9.8 | 121.9 | 2.9 | |
| PFHxA | 113.7 | 6.8 | 117.2 | 6.1 | 113.8 | 0.9 | 39.0a | 19.5 | 116.1 | 7.6 | 123.5 | 7.6 | |
| PFHpA | 116.2 | 4.9 | 111.2 | 8.9 | 121.0 | 6.3 | 46.5a | 18.8 | 123.1 | 8.0 | 128.4 | 5.8 | |
| Mean | 114.4 | 5.2 | 112.6 | 6.1 | 113.9 | 3.1 | 40.9 | 22.3 | 117.4 | 7.8 | 123.3 | 4.7 | |
| Long-chain PFCA | PFOA | 113.6 | 10.2 | 110.5 | 8.7 | 112.2 | 8.6 | 45.8a | 26.1 | 122.7 | 3.1 | 125.4 | 2.5 |
| PFNA | 118.3 | 11.6 | 113.8 | 13.2 | 112.8 | 7.9 | 38.3a | 28.3 | 118.4 | 3.6 | 106.0 | 9.8 | |
| PFDA | 162.9 | 16.6 | 130.5 | 30.7 | 97.0 | 10.3 | 35.1a | 34.4 | 115.9 | 30.7 | 97.2 | 10.1 | |
| PFUdA | 89.9 | 26.2 | 90.8 | 12.2 | 118.0 | 7.9 | 37.2a | 40.5 | 106.8 | 19.9 | 112.4 | 6.2 | |
| PFDoA | 102.9 | 19.0 | 115.8 | 8.2 | 115.5 | 4.6 | 34.7a | 30.1 | 109.9 | 5.4 | 126.0 | 0.2 | |
| PFTeDA | 150.9 | 0.7 | 146.5 | 7.5 | 162.2 | 6.4 | 42.8a | 53.6 | 172.2 | 7.2 | 236.7 | 3.4 | |
| Mean | 123.1 | 14.0 | 118.0 | 13.4 | 119.6 | 7.6 | 39.0 | 35.5 | 124.3 | 11.7 | 134.0 | 5.4 | |
| PFCA | Mean | 119.5 | 10.3 | 115.8 | 10.4 | 117.2 | 5.8 | 39.8 | 30.0 | 121.4 | 10.0 | 129.5 | 5.1 |
| PFSA | PFHxS | 106.2 | 6.8 | 112.4 | 9.2 | 105.7 | 4.5 | 115.3 | 6.9 | 114.8 | 5.9 | 119.3 | 4.5 |
| PFOS | 142.9 | 4.0 | 131.0 | 11.8 | 125.3 | 11.2 | 117.2 | 18.8 | 124.6 | 11.6 | 101.3 | 14.1 | |
| Mean | 124.5 | 5.2 | 121.7 | 10.6 | 115.5 | 8.1 | 116.2 | 12.9 | 119.7 | 8.9 | 110.3 | 8.9 | |
| FTS | 4:2 FTS |
112.3 | 6.5 | 109.0 | 2.7 | 109.4 | 4.9 | 128.9 | 3.7 | 126.2 | 15.1 | 131.5 | 5.3 |
| 6:2 FTS |
106.7 | 7.9 | 113.5 | 7.8 | 113.5 | 6.4 | 115.8 | 19.3 | 120.1 | 11.3 | 125.2 | 4.5 | |
| 8:2 FTS |
128.6 | 3.2 | 117.4 | 15.3 | 82.2 | 16.6 | 40.2a | 18.5 | 105.8 | 56.3 | 110.5 | 34.9 | |
| Mean | 115.9 | 5.7 | 113.3 | 8.7 | 101.7 | 8.6 | 95.0 | 12.2 | 117.4 | 26.2 | 122.4 | 13.9 | |
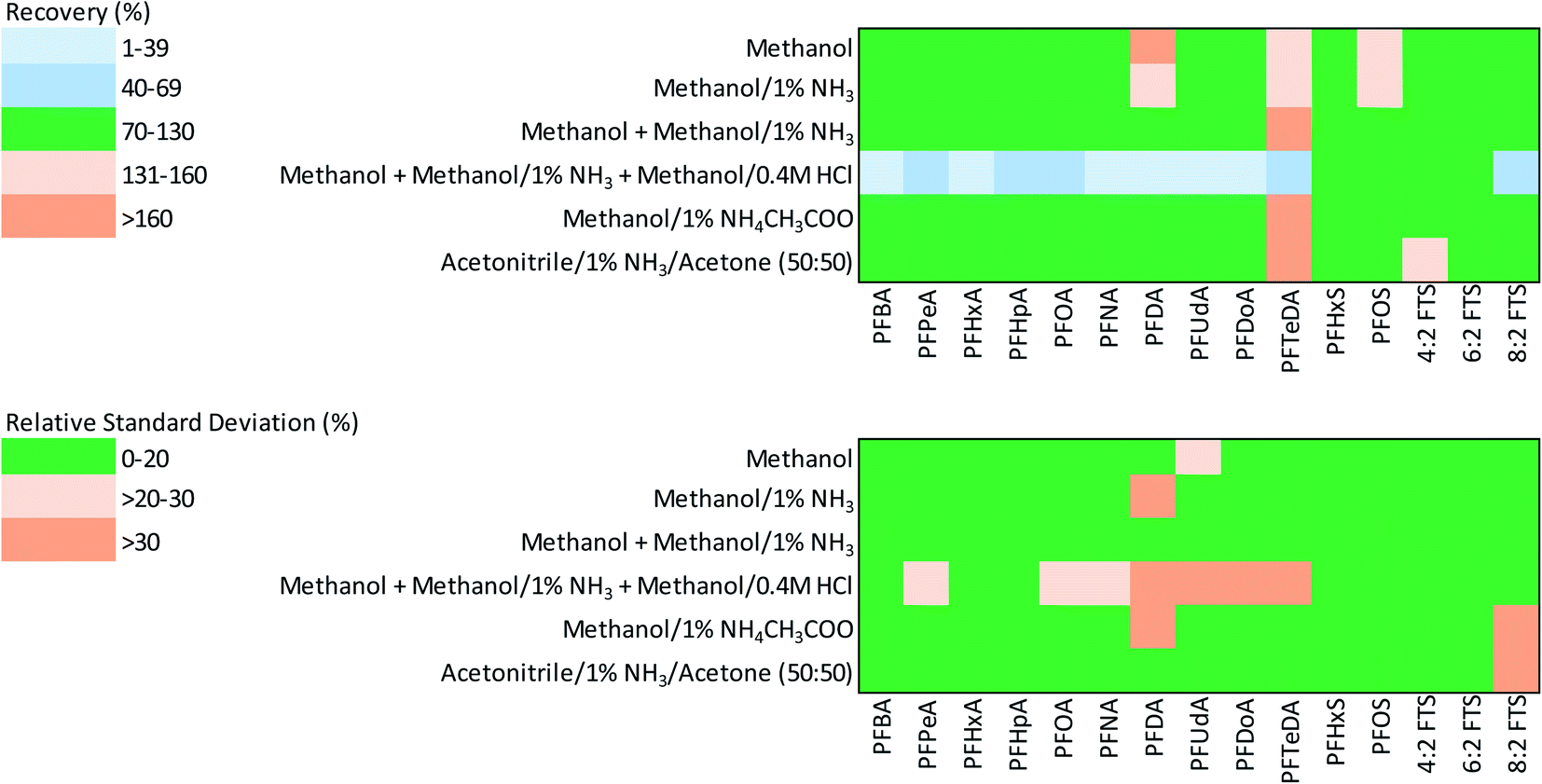 | ||
| Fig. 1 Heatmaps showing the accuracy (% recovery) and precision (% RSD) of stable isotope-corrected PFAS recovery from spiked asphalt using six extractants. | ||
The findings from the extraction recovery data for SI-corrected PFAS show that moderately polar solvents (e.g., methanol with and without additives) were efficient in the extraction of the majority of PFAS examined (Table 3 and Fig. 1). The mean recoveries for four of the six extractants tested (extractants 1, 2, 3 and 5) were within the acceptable range for PFCAs (70–130%); whereas the mean recovery for PFCAs by extractant 6 was slightly higher than the acceptable range (>130%). Recoveries of PFCAs using extractant 4 were below the lower acceptable range for PFAS examined (<70%). In general, the recovery of long-chain PFCAs was higher than that of short-chain PFCAs by most extractants examined except for extractant 4. The mean recoveries of PFSAs and FTSs by all extractants were within the acceptable range (70–130%).
The reproducibility or precision for recovery of PFAS (measured by RSD) was within the acceptable range (≤20%) for all extractants examined, except extractant 4 due to low recoveries found for some of the long-chain PFCAs (Table 3 and Fig. 1). In general, extractants 1, 2, 3 and 6 produced the most precise results with mean RSD ranging from 6.8 to 10.1.
The recoveries of PFAS for which SI correction could not be applied are shown in Table S4.† While the recoveries of short-chain PFSAs (PFBS and PFPeS) with all 6 extractants were within the acceptable range, only methanol-based extractants (1–3) gave acceptable recoveries for the long-chain compounds (PFCAs, PFSAs and FTS). The recoveries of PFNS and 10:2 FTS exceeded the acceptable range with most extractants. In general, the reproducibility of recovery for short-chain PFAS (i.e., PFCAs < C8 and PFSAs < C6) was better than for long-chain PFAS, whereas the recovery of PFSAs, was, in general, slightly more precise than that of PFCAs and fluorotelomers (Table S4†).
In this study, we found that the addition of acidified methanol (extractant 4) caused a poorer recovery range for a number of PFAS examined (Table 2). Previous studies on soils and sediments have also reported enhanced or suppressed recovery of PFAS due to HCl additive. For example, the recovery of 6:2 FTB from soil increased by 4-5-fold with the addition of HCl and NH4CH3COO to methanol compared to pure methanol extraction.49 The lower recoveries for some PFAS found in this study with the addition of an acidified methanol step may be due to the release of asphalt matrix constituents that can remove PFAS from samples following addition to a combined extract (e.g., flocculation and precipitation reactions) or interfere with PFAS measurement by mass spectrometry techniques (matrix effect bias that results in suppressed analyte measurement).
As evident from the heat maps (Fig. 1), the methanol-based extractants, especially extractants 1, 2, and 3, yielded more accurate and precise PFAS recoveries from asphalt. Particularly, from the perspective of PFAAs of current regulatory interest (i.e., PFOA, PFOS, PFHxS), the alkaline methanol extractants 2 and 3 provided the best results. In general, there was no substantial difference in PFAS recoveries from asphalt between extractants 2 and 3. Methanol based extractants have commonly been used for the extraction of PFAS from other complex environmental matrices such as soils and sediments.14,30,31 Therefore, extractant 2 was selected over extractant 3 as the preferred method for measurement of 22 PFAS from asphalt, especially as it required only a single extractant (methanol/1% NH3). The limits of detection and quantitation for the instrument and the method limit of quantitation method are given in Table S1.† The instrument LOD of 25 ng L−1 was achieved for most PFAS, whereas LOD of 100 ng L−1 was obtained for PFNS and LOD of 250 ng L−1 was achieved for PFOS, PFDS, PFUdA, PFDoA, PFTrDA, PFTeDA and 10:2 FTS. Accordingly, the instrumental LOD ranged from 83.25 to 832.50 ng L−1. The LOQ for the chosen method (based on seven samples) ranged from 0.15 mg kg−1 for PFBA to 1.00 mg kg−1 for PFHxS, PFHpS, PFNS, PFDS, 6:2 FTS and 10:2 FTS.
3.2. Influence of SI-labelled PFAS internal standard addition, sonication temperature, spike level and sample composition on PFAS extraction
The recovery of various PFAS (SI-corrected) from two different asphalt core materials (Core 1 and Core 2) with different stages of addition of SI-labelled PFAS internal standards are presented as boxplots in Fig. 2a. The addition of the SI-labelled PFAS internal standards prior to extraction represents the combined effects of extraction, SPE step, and instrument response; hence, recoveries obtained have been corrected for losses that may occur throughout the procedure. The respective data are reflective of the realistic scenario of the chosen method employing a cleanup step (via SPE). The addition of SI-labelled PFAS internal standards before the clean-up step only accounts for the effects of the SPE step and instrument response, hence, comparison with the “spike before extraction test” helps assess losses from the extraction step. To assess losses from the clean-up step, SI-labelled PFAS internal standards were added after the clean-up step; recoveries obtained would only be corrected for instrument response.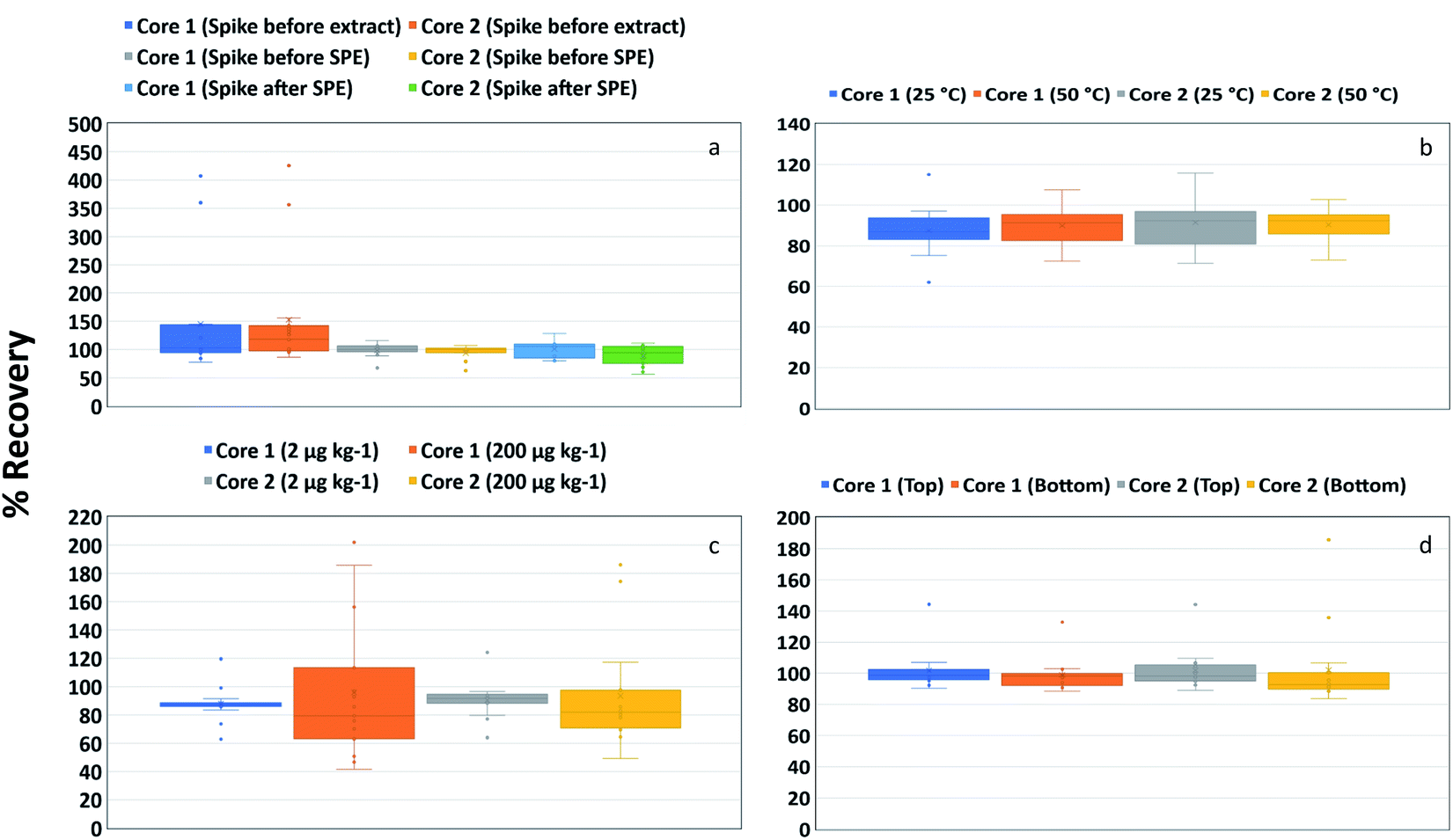 | ||
| Fig. 2 Box plot showing distribution of recovery of stable isotope-corrected PFAS in respective cores as a function of (a) PFAS stable isotope-labelled internal standard addition timing, (b) temperature, (c) spike concentration and (d) sample location (asphalt composition). Box plots showing distribution of individual PFAS compound in different cores are presented in Fig. S2.† | ||
Overall, the recoveries obtained, regardless of when the stable isotopes were added, were within the acceptable range for PFAS. While a comparison of “spike before extraction” and “spike before SPE” shows reduced recovery for the latter, the data are still within the acceptable range of recoveries. This would indicate that PFAS were effectively extracted from the asphalt, which agrees with the results in Table S4.† Recoveries appeared to be lower when the SI-labelled PFAS internal standards were added after SPE, suggesting some losses during the clean-up step. Nevertheless, the spread of the data points was small when SI-labelled PFAS internal standards were spiked before and after SPE compared to when SI-labelled PFAS internal standards were spiked before extraction (Fig. 2a). The individual PFAS compound-wise spread of the data points showed an acceptable recovery for most PFAS, except for some long-chain PFAS (such as PFUdA, PFDoA and 8:2 FTS) (Fig. S2a†).
The results of PFAS recoveries (SI-corrected) from the two cores (Core 1 and Core 2), loaded with 200 μg kg−1 PFAS concentrations, as extracted with extractant 2 at two sonication temperatures are presented as a box plot (Fig. 2b and S2b†). It is evident from the distribution of data that the recovery of PFAAs and FTS was generally slightly more accurate and precise at 50 °C. However, the overall difference was small (statistically not significant) and extraction at the higher temperature was not warranted. A comparison of the chain length of PFAS (Fig. S2b†) reveals that the distribution of recovery data for some long-chain PFAS was relatively wider than for other PFAS, although the recoveries were within the acceptable range. Previous work suggested that the association of PFAS with concrete aggregates might be weak,8 but there is no data on asphalt available in the literature at the moment. Therefore, it is not clear whether the association of PFAS was with the organic-rich bitumen phase within the asphalt core or the aggregate phase.50
A comparison of PFAS spike concentrations (Fig. 2c and S2c†) showed that the recoveries of most PFAS (SI-corrected) were neither affected by the PFAS loading nor the two different asphalt materials, although the distribution of the recovery data was much tighter and within the accepted limit at lower spiking level (2 μg kg−1) in both cores. Contrariwise, relatively large variability in the recovery was observed for Core 1 (200 μg kg−1), which was primarily due to the high recovery of PFUdA and PFDoA (Fig. S2c†). A comparison of the chain length of PFAS revealed that the recovery of some longer chain PFAAs was outside the acceptable range (Fig. S2c†). Minor differences (statistically not significant) in overall recoveries and their precision were noted with the two concentrations (2 μg kg−1 and 200 μg kg−1). This demonstrated that the chosen method was robust enough to give reliable recoveries for most of the PFAS tested here over a wide concentration range in the two different asphalt materials.
The recovery of PFAS (SI-corrected) from asphalt samples was not affected by sample composition. Similar recoveries were obtained for samples collected from different locations (top and bottom of a core) that were assumed to have different compositions (Fig. 2d and S2d†). The data, however, showed that the precision for short-chain PFAAs was much better than for long-chain PFAAs. Similarly, the recovery of short-chain FTS (4:2 and 6:2 FTS) was more precise than for the long-chain 8:2 FTS. Overall, the sample location did not have any impact on PFAS recovery in asphalt.
3.3. PFAS in asphalt samples collected from an operational airbase
The chosen extraction method 2 (three extractions with methanol/1% NH3) was applied to the determination of PFOA, PFOS and PFHxS concentrations in asphalt cores collected from an operational airbase in Australia (Table 1). The PFAS were selected based on interim landfill acceptance criteria (PFOA = 50 mg kg−1; ∑PFOS & PFHxS = 50 mg kg−1) and human health investigation levels for soil (industrial/commercial) (PFOA = 50 mg kg−1; ∑PFOS + PFHxS = 20 mg kg−1) outlined in the Australian PFAS NEMP (version 2).11Asphalt core samples collected from the runway apron (Core 1, Core 2) had PFOS and the sum of PFOS and PFHxS concentrations in vertical sections below the LOQ (Table 1). Six of the 24 asphalt vertical core sections were found to contain PFOA concentrations (0.65–13.5 mg kg−1) higher than the LOQ (0.6 mg kg−1). The highest PFOA concentration (13.5 ± 1.3 mg kg−1) was found in the deepest asphalt vertical core section (140–170 mm) from Core 5 collected from the taxiway near aircraft hangers (Table 1). Sixteen of the 24 asphalt vertical core sections were found to contain PFOS (1–1952 mg kg−1), whereas nine of the 24 asphalt vertical core sections had PFHxS (2.2–183 mg kg−1) – these concentrations were higher than the LOQ for PFOS and PFHxS (0.7 mg kg−1 and 1 mg kg−1, respectively). The highest sum of PFOS and PFHxS concentrations were found at the surface (0–50 mm) of Core 7 (2135 mg kg−1) (near a firefighting training pad) and the deepest vertical section (140–170 mm) of Core 5 (1349 mg kg−1) (taxiway) (Table 1). There were 4 vertical core sections (Core 4 (runway), Core 5 (taxiway near aircraft hangar), Core 6 (driveway) and Core 7 (near fire-fighting training pad)) that contained the sum of PFOS and PFHxS concentrations exceeding the interim landfill acceptance criteria or human health investigation levels for soil (industrial/commercial) (Table 1).
The three vertical sections of Core 3 (0–110 mm) were found to contain approximately 2–5 times higher concentrations than the LOQ of PFOS (Table 1). PFOS in the vertical core sections deeper than 110 mm were all less than the LOQ. There was a substantially higher PFOS concentration in the 80–110 mm vertical section compared to the 0–50 mm and 50–80 mm vertical core sections. This finding for Core 3 suggests that PFAS surface exposure (current or historical) had resulted in the migration of PFOS and accumulation in slightly deeper layers but that it had not penetrated the whole depth of the asphalt profile.
A substantially increasing concentration gradient for the sum of PFOS and PFHxS was found in Core 4 to a depth of 110 mm, which then decreased to the base of the asphalt core (>110–180 mm) (Table 1). Similar to Core 3, the finding for Core 4 suggests that PFAS surface exposure (current or historical) had resulted in the migration of PFOS, PFOA and PFHxS and accumulation in deeper asphalt vertical layers. The highest sum of PFOS and PFHxS concentration, similar to Core 3 on the runway, was in the 70–110 mm vertical asphalt core section, suggesting increased retention or reduction in penetration at this layer that may be due to a change in asphalt core geophysical or geochemical features.
The three vertical sections from Core 5 (0–110 mm) from the taxiway were found to contain approximately 2 to 5 times higher PFOS concentrations than the LOQ (Table 1). The bottom two core sections from Core 5 (110–170 mm) contained substantially higher PFOS and PFHxS concentrations than the sections above. This finding suggests that PFAS surface exposure (current or historical) had resulted in the migration of PFOA, PFOS, and PFHxS through the profile into deeper asphalt layers. The presence of substantially lower concentrations (<LOQ) in shallow vertical sections in this asphalt core may be due to their remobilisation/removal, for example, during weather events and cleaning, or removal during asphalt resurfacing and reworking.
The core collected from the driveway (Core 6) was found to contain PFOA, PFOS and PFHxS substantially above the LOQ with PFOS and PFHxS concentrations exceeding the interim landfill acceptance criteria or human health investigation levels for soil (industrial/commercial) (Table 1). This suggests that the driveway close to the fire-fighting training area was heavily contaminated with PFAS, as has been reported for concrete at other similar sites in Australia.8,12
The shallow vertical section from Core 7 (0–50 mm) near the firefighting training pad was found to contain PFOS and PFHxS concentrations exceeding the interim landfill acceptance criteria and human health investigation levels for soil (industrial/commercial) (Table 1). The sum of PFOS and PFHxS concentration in the bottom vertical core section from Core 7 was substantially lower than the shallow zone and only exceeded interim human health investigation levels for soil (industrial/commercial). This finding suggests that PFAS surface exposure (current or historical) had resulted in the migration of PFOS and PFHxS deeper than 50 mm into the asphalt profile at Core 7, albeit at reduced concentrations. The elevated PFAS concentrations in Core 7 are consistent with findings from concrete at firefighting training pads.8,12
3.4. Interlaboratory comparison
The driveway (Core 6) asphalt sample was extracted and analysed in the CSIRO laboratory and at Laboratory 1 and Laboratory 2, respectively. There was generally good agreement in the results obtained by all the laboratories (Fig. 3). In some cases, where concentrations were considerably greater than the respective LOD, the reported values were similar between laboratories. With respect to the TOP assay (Laboratory 3), it is expected that concentrations of only PFCAs are likely to increase due to the conditions of the assay, especially the shorter chain PFCAs (PFBA, PFPeA and PFHxA). Precursors that are known to oxidise to PFCAs include FOSAs, FTSs, FTABs, FTUCAs and PAPs.51,52 The concentration of PFCAs was typically greater than that measured pre-TOP assay, with PFOA apparently the main exception. In addition, although 8:2 FTS was measured pre-TOP, it was not quantifiable post-TOP. Further investigation would be required to determine the reason for these differences, such as historical fire suppression activities at the driveway sites or through transport from areas of fire suppression activities.
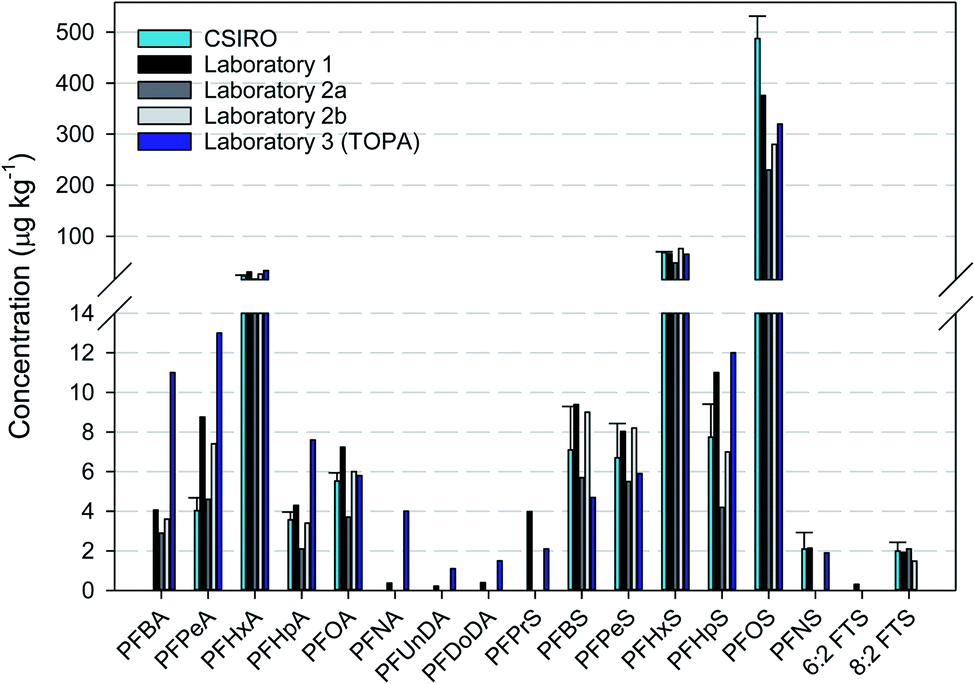 | ||
| Fig. 3 Interlaboratory comparison of PFAS concentrations in an asphalt core (Core 6 top 50 mm). CSIRO, Laboratory 1 and Laboratory 2a conducted the sample extraction and analysis independently. The extracts at CSIRO were also analysed by Laboratory 2 (represented as Laboratory 2b). Laboratory 3 only conducted total oxidisable precursor assay (TOPA). | ||
4. Conclusions
A series of experiments were conducted to develop an extraction methodology suitable for analysis of a wide range of PFAS (covering PFCAs, PFSAs, FTS) present at realistic environmental concentrations in asphalt. The method based on three successive extractions with methanol/1% NH3 was chosen as the most suitable method for extracting PFAS-contaminated asphalt because it gave the most accurate and precise results. Method performance was not affected by sonication temperature, PFAS spike level, and sample composition (based on the location of asphalt material in the field). The chosen method performed well with low RSD associated with the PFAS concentration of asphalt cores collected from the field covering a wide range of concentrations (from LOQ to 2135 mg kg−1) in the field-contaminated asphalt materials. The results from the interlaboratory testing were generally in good agreement and validated the proposed PFAS extraction and analytical approach.This PFAS extraction methodology provides confidence in investigating the presence and leachability of PFAS in asphalt, and in devising management options based on the estimated risk profile for PFAS in asphalt at impacted sites. Future investigations should consider the approaches to reducing the variations in the recoveries of different PFAS types (e.g., carboxylates, sulfonates, fluorotelomers, sulfonamides, sulfonamido ethanols, sulfonamido acetic acids, fluorotelomer carboxylic acid, zwitterions) and chain length, matrix effects in the asphalt samples. Orthogonal experiments should be conducted to examine factors affecting PFAS extractability from asphalt.
Author contributions
Prashant Srivastava: writing – original draft, data curation; Mike Williams: conceptualization, methodology, resources, formal analysis, validation, writing – review & editing; Jun Du: methodology, resources, formal analysis, writing – review & editing, validation; Divina Navarro: writing – review & editing; Rai Kookana: writing – review & editing; Grant Douglas: writing – review & editing; Trevor Bastow: writing – review & editing; Greg Davis: writing – review & editing; Jason K. Kirby: conceptualization, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Australian Department of Defence is gratefully acknowledged for funding this project. Special thanks are due to Mr Darren Skuse and Dr Karl Bowles for providing operational and logistical support for sample collection.References
- R. C. Buck, J. Franklin, U. Berger, J. M. Conder, I. T. Cousins, P. de Voogt, A. A. Jensen, K. Kannan, S. A. Mabury and S. P. van Leeuwen, Integr. Environ. Assess. Manage., 2011, 7, 513–541 CrossRef CAS PubMed.
- I. T. Cousins, J. C. DeWitt, J. Glüge, G. Goldenman, D. Herzke, R. Lohmann, C. A. Ng, M. Scheringer and Z. Wang, Environ. Sci.: Processes Impacts, 2020, 22, 2307–2312 RSC.
- J. Gluge, M. Scheringer, I. T. Cousins, J. C. DeWitt, G. Goldenman, D. Herzke, R. Lohmann, C. A. Ng, X. Trier and Z. Y. Wang, Environ. Sci.: Processes Impacts, 2020, 22, 2345–2373 RSC.
- K. R. Miner, H. Clifford, T. Taruscio, M. Potocki, G. Solomon, M. Ritari, I. E. Napper, A. P. Gajurel and P. A. Mayewski, Sci. Total Environ., 2021, 759, 7 CrossRef PubMed.
- C. Baduel, J. F. Mueller, A. Rotander, J. Corfield and M. J. Gomez-Ramos, Chemosphere, 2017, 185, 1030–1038 CrossRef CAS PubMed.
- E. F. Houtz, C. P. Higgins, J. A. Field and D. L. Sedlak, Environ. Sci. Technol., 2013, 47, 8187–8195 CrossRef CAS PubMed.
- The-Nordic-Council-of-Ministers, The Cost of Inaction: A Socioeconomic Analysis of Environmental and Health Impacts Linked to Exposure to PFAS, The-Nordic-Council-of-Ministers, Denmark, 2019 Search PubMed.
- C. Baduel, C. J. Paxman and J. F. Mueller, J. Hazard. Mater., 2015, 296, 46–53 CrossRef CAS PubMed.
- H. M. Solo-Gabriele, A. S. Jones, A. B. Lindstrom and J. R. Lang, Waste Manage., 2020, 107, 191–200 CrossRef CAS PubMed.
- UNEP, All POPs Listed in the Stockholm Convention, https://chm.pops.int/TheConvention/ThePOPs/ListingofPOPs/tabid/2509/Default.aspx Search PubMed.
- HEPA, PFAS National Environmental Management Plan Version 2.0, https://www.environment.gov.au/system/files/resources/2fadf1bc-b0b6-44cb-a192-78c522d5ec3f/files/pfas-nemp-2.pdf Search PubMed.
- P. K. Thai, J. T. McDonough, T. A. Key, J. Thompson, P. Prasad, S. Porman and J. F. Mueller, J. Hazard. Mater. Lett., 2022, 3, 100050 CrossRef.
- A. Ahmadireskety, B. F. Da Silva, T. G. Townsend, R. A. Yost, H. M. Solo-Gabriele and J. A. Bowden, Sci. Total Environ., 2021, 760, 143944 CrossRef CAS PubMed.
- K. A. Barzen-Hanson, S. E. Davis, M. Kleber and J. A. Field, Environ. Sci. Technol., 2017, 51, 12394–12404 CrossRef CAS PubMed.
- C. A. Huset and K. M. Barry, MethodsX, 2018, 5, 697–704 CrossRef PubMed.
- S. Mejia-Avendano, G. Munoz, S. Sauve and J. X. Liu, Anal. Chem., 2017, 89, 2539–2546 CrossRef CAS PubMed.
- A. Nickerson, A. C. Maizel, P. R. Kulkarni, D. T. Adamson, J. J. Kornuc and C. P. Higgins, Environ. Sci. Technol., 2020, 54, 4952–4962 CrossRef CAS PubMed.
- J. G. Sepulvado, A. C. Blaine, L. S. Hundal and C. P. Higgins, Environ. Sci. Technol., 2011, 45, 8106–8112 CrossRef CAS PubMed.
- H. N. Zhang, B. Wen, W. Wen, Y. B. Ma, X. Y. Hu, Y. L. Wu, L. Luo and S. Z. Zhang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1072, 25–33 CrossRef CAS PubMed.
- I. Navarro, P. Sanz and M. A. Martinez, Anal. Bioanal. Chem., 2011, 400, 1277–1286 CrossRef CAS PubMed.
- T. Anumol, S. Merel, B. O. Clarke and S. A. Snyder, Chem. Cent. J., 2013, 7, 104 CrossRef PubMed.
- K. Dasu, S. F. Nakayama, M. Yoshikane, M. A. Mills, J. M. Wright and S. Ehrlich, J. Chromatogr. A, 2017, 1494, 46–54 CrossRef CAS PubMed.
- J. Janda, K. Nodler, H. J. Brauch, C. Zwiener and F. T. Lange, Environ. Sci. Pollut. Res., 2019, 26, 7326–7336 CrossRef CAS PubMed.
- M. M. A. Elahi, M. M. Hossain, M. R. Karim, M. F. M. Zain and C. Shearer, Constr. Build. Mater., 2020, 260, 119788 CrossRef CAS.
- S. Shinkhede, V. Katare, S. Joglekar, M. Madurwar and S. Mandavgane, Int. J. Sustain. Eng., 2021, 14, 2048–2059 CrossRef.
- S. Ho and L. Zanzotto, J. Test. Eval., 2009, 37, 387–395 CAS.
- M. L. Greenfield, Int. J. Pavement Eng., 2011, 12, 325–341 CrossRef CAS.
- T. Zhou, L. P. Cao, E. H. Fini, L. W. Li, Z. Y. Liu and Z. J. Dong, Constr. Build. Mater., 2020, 249, 118748 CrossRef CAS.
- C. P. Higgins, J. A. Field, C. S. Criddle and R. G. Luthy, Environ. Sci. Technol., 2005, 39, 3946–3956 CrossRef CAS PubMed.
- M. Lorenzo, J. Campo and Y. Pico, Anal. Bioanal. Chem., 2015, 407, 5767–5781 CrossRef CAS PubMed.
- G. Munoz, P. Ray, S. Mejia-Avendano, S. V. Duy, D. T. Do, J. X. Liu and S. Sauve, Anal. Chim. Acta, 2018, 1034, 74–84 CrossRef CAS PubMed.
- M. I. Beser, O. Pardo, J. Beltran and V. Yusa, J. Chromatogr. A, 2011, 1218, 4847–4855 CrossRef CAS PubMed.
- F. Li, C. J. Zhang, Y. Qu, J. Chen, X. Hu and Q. Zhou, Int. J. Environ. Anal. Chem., 2011, 91, 1117–1134 CrossRef CAS.
- S. T. Wolf and W. K. Reagen, Anal. Methods, 2013, 5, 2444–2454 RSC.
- M. Fernandez-Sanjuan, J. Meyer, J. Damasio, M. Faria, C. Barata and S. Lacorte, Anal. Bioanal. Chem., 2010, 398, 1447–1456 CrossRef CAS PubMed.
- M. Bartolome, A. Gallego-Pico, O. Huetos, M. A. Lucena and A. Castano, Anal. Bioanal. Chem., 2016, 408, 2159–2170 CrossRef CAS PubMed.
- J. Yu, J. Y. Hu, S. Tanaka and S. Fujii, Water Res., 2009, 43, 2399–2408 CrossRef CAS PubMed.
- L. M. Harrington, Anal. Methods, 2017, 9, 473–481 RSC.
- E. F. Houtz and D. L. Sedlak, Environ. Sci. Technol., 2012, 46, 9342–9349 CrossRef CAS PubMed.
- Y. M. Lee, J. Y. Lee, M. K. Kim, H. Yang, J. E. Lee, Y. Son, Y. Kho, K. Choi and K. D. Zoh, J. Hazard. Mater., 2020, 381, 120909 CrossRef CAS PubMed.
- T. Ruan, Y. F. Lin, T. Wang, R. Z. Liu and G. B. Jiang, Environ. Sci. Technol., 2015, 49, 6519–6527 CrossRef CAS PubMed.
- S. Q. Chen, Y. Q. Zhou, J. Meng and T. Y. Wang, Environ. Pollut., 2018, 242, 2059–2067 CrossRef CAS PubMed.
- J. Meng, T. Y. Wang, P. Wang, Y. Q. Zhang, Q. F. Li, Y. L. Lu and J. P. Giesy, Environ. Pollut., 2015, 199, 102–109 CrossRef CAS PubMed.
- Draft Method 1633 - Analysis of Per- and Polyfluoroalkyl Substances (PFAS) in Aqueous, Solid, Biosolids, and Tissue Samples by LC-MS/MS (August 2021), Office of Science and Technology, U.S. Environmental Protection Agency, https://www.epa.gov/system/files/documents/2021-09/method_1633_draft_aug-2021.pdf?msclkid=18391ba2b48c11ec9295f25ed092b1e8 Search PubMed.
- J. R. Dean and G. H. Xiong, TrAC, Trends Anal. Chem., 2000, 19, 553–564 CrossRef CAS.
- M. J. Laranja, R. C. J. da Silva, M. C. Bisinoti, A. B. Moreira, M. Boscolo, O. P. Ferreira and C. de Almeida Melo, Biomass Convers. Biorefin., 2022, 12, 81–90 CrossRef CAS.
- A. Antony and M. Farid, Appl. Sci., 2022, 12, 2107 CrossRef CAS.
- USDOD/USDOE, Department of Defense (DoD)/Department of Energy (DOE) Consolidated Quality Systems Manual (QSM) for Environmental Laboratories – DoD Quality Systems Manual Version 5.3, https://denix.osd.mil/edqw/documents/manuals/qsm-version-5-3-final/ Search PubMed.
- G. Munoz, S. V. Duy, H. Budzinski, P. Labadie, J. X. Liu and S. Sauve, Anal. Chim. Acta, 2015, 881, 98–106 CrossRef CAS PubMed.
- Y. S. Li, D. P. Oliver and R. S. Kookana, Sci. Total Environ., 2018, 628–629, 110–120 CrossRef CAS PubMed.
- S. Hutchinson, T. Rieck and X. L. Wu, Environ. Chem., 2020, 17, 558–567 CrossRef CAS.
- D. Martin, G. Munoz, S. Mejia-Avendano, S. V. Duy, Y. Yao, K. Volchek, C. E. Brown, J. X. Liu and S. Sauve, Talanta, 2019, 195, 533–542 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ay00221c |
| This journal is © The Royal Society of Chemistry 2022 |