
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow do H2 oxidation molecular catalysts assemble onto carbon nanotube electrodes? A crosstalk between electrochemical and multi-physical characterization techniques†
Ahmed
Ghedjatti
abc,
Nathan
Coutard
 a,
Laura
Calvillo
d,
Gaetano
Granozzi
d,
Bertrand
Reuillard
a,
Vincent
Artero
a,
Laure
Guetaz
e,
Sandrine
Lyonnard
c,
Hanako
Okuno
b and
Pascale
Chenevier
*c
a,
Laura
Calvillo
d,
Gaetano
Granozzi
d,
Bertrand
Reuillard
a,
Vincent
Artero
a,
Laure
Guetaz
e,
Sandrine
Lyonnard
c,
Hanako
Okuno
b and
Pascale
Chenevier
*c
aUniv. Grenoble Alpes, CNRS, CEA, IRIG, Laboratoire de Chimie et Biologie des Métaux, 38000 Grenoble, France
bUniv. Grenoble Alpes, CEA, IRIG, MEM, LEMMA, 38000 Grenoble, France
cUniv. Grenoble Alpes, CEA, CNRS, IRIG, SYMMES, 38000 Grenoble, France. E-mail: pascale.chenevier@cea.fr
dDepartment of Chemical Sciences, University of Padova, Via F. Marzolo 1, Padova 35131, Italy
eUniv. Grenoble Alpes, CEA, LITEN, DTNM, 38000 Grenoble, France
First published on 19th November 2021
Abstract
Molecular catalysts show powerful catalytic efficiency and unsurpassed selectivity in many reactions of interest. As their implementation in electrocatalytic devices requires their immobilization onto a conductive support, controlling the grafting chemistry and its impact on their distribution at the surface of this support within the catalytic layer is key to enhancing and stabilizing the current they produce. This study focuses on molecular bioinspired nickel catalysts for hydrogen oxidation, bound to carbon nanotubes, a conductive support with high specific area. We couple advanced analysis by transmission electron microscopy (TEM), for direct imaging of the catalyst layer on individual nanotubes, and small angle neutron scattering (SANS), for indirect observation of structural features in a relevant aqueous medium. Low-dose TEM imaging shows a homogeneous, mobile coverage of catalysts, likely as a monolayer coating the nanotubes, while SANS unveils a regular nanostructure in the catalyst distribution on the surface with agglomerates that could be imaged by TEM upon aging. Together, electrochemistry, TEM and SANS analyses allowed drawing an unprecedented and intriguing picture with molecular catalysts evenly distributed at the nanoscale in two different populations required for optimal catalytic performance.
Introduction
While electrocatalysis is becoming a key enabling technology for renewable energy production, the anticipated shortage of an increasing number of critical elements, especially metals, has triggered a shift towards molecularly-defined materials development with lower metal loadings but enhanced performance. On the one hand, traditional industrial metal or metal-oxide catalysts are continuously downsized, from the nano shape1,2 to ultimately isolated metal atom active sites in so-called single-atom catalysts (SAC3) or surface confined molecular catalysts, in order to increase the share of atoms effectively involved in the catalytic process.4–7 SAC, generally stabilized within N-doped sp2 carbon matrices, first flourished through uncontrolled metal inclusions during pyrolysis,8 but synthetic methods recently emerged9–11 enabling high control and almost molecularly defined design of active centers to build efficient catalysts for the O2 and CO2 reduction reactions (ORR and CO2RR)5–7,12 or H2 evolution reaction (HER).9,10,13,14 On the other hand, molecular catalysts possibly inspired from enzymes' active sites, immobilized onto nanocarbons,15,16 can also yield to electrode materials capable of attaining industrially-relevant current densities for the hydrogen oxidation reaction (HOR),17–20 ORR8,12 or CO2RR under technologically-relevant conditions.4,21–23 Here, heterogenized molecular catalysis comes with a fine understanding of the catalytic mechanisms at stake15,24 and of the structural and electronic factors favoring catalytic efficiency or selectivity.25–28 However, maintaining full availability and reactivity of active molecules on solid support often remains a challenge, as well as the development of immobilization methods that are robust enough to enable long-term operation. So far, most success in the ORR or CO2RR arose from immobilization of planar macrocyclic complexes such as porphyrins and phthalocyanins, either incorporated into highly porous structures, such as covalent organic frameworks29 or polymers of catalytic subunits,23 or π-stacked onto graphitic materials such as carbon nanotubes.4,30 Regarding the HOR, the most prominent catalysts are the so-called DuBois' complexes25,26 that operate reversibly and with a catalytic capacity per atom close to that of platinum after immobilization onto carbon nanotubes.17 Two diaza-diphosphacyclooctane ligands complex the Ni redox center and position flexible amine functions so that they form frustrated Lewis pairs with the Ni center where H2 can be cleaved in a heterolytic manner allowing an efficient HOR.25,26 So far the most HOR-active catalyst in this series after immobilization is an arginine derivative [Ni(P2CyN2Arg)2]7+ (NiArg, Fig. 1) designed by Shaw and coworkers.31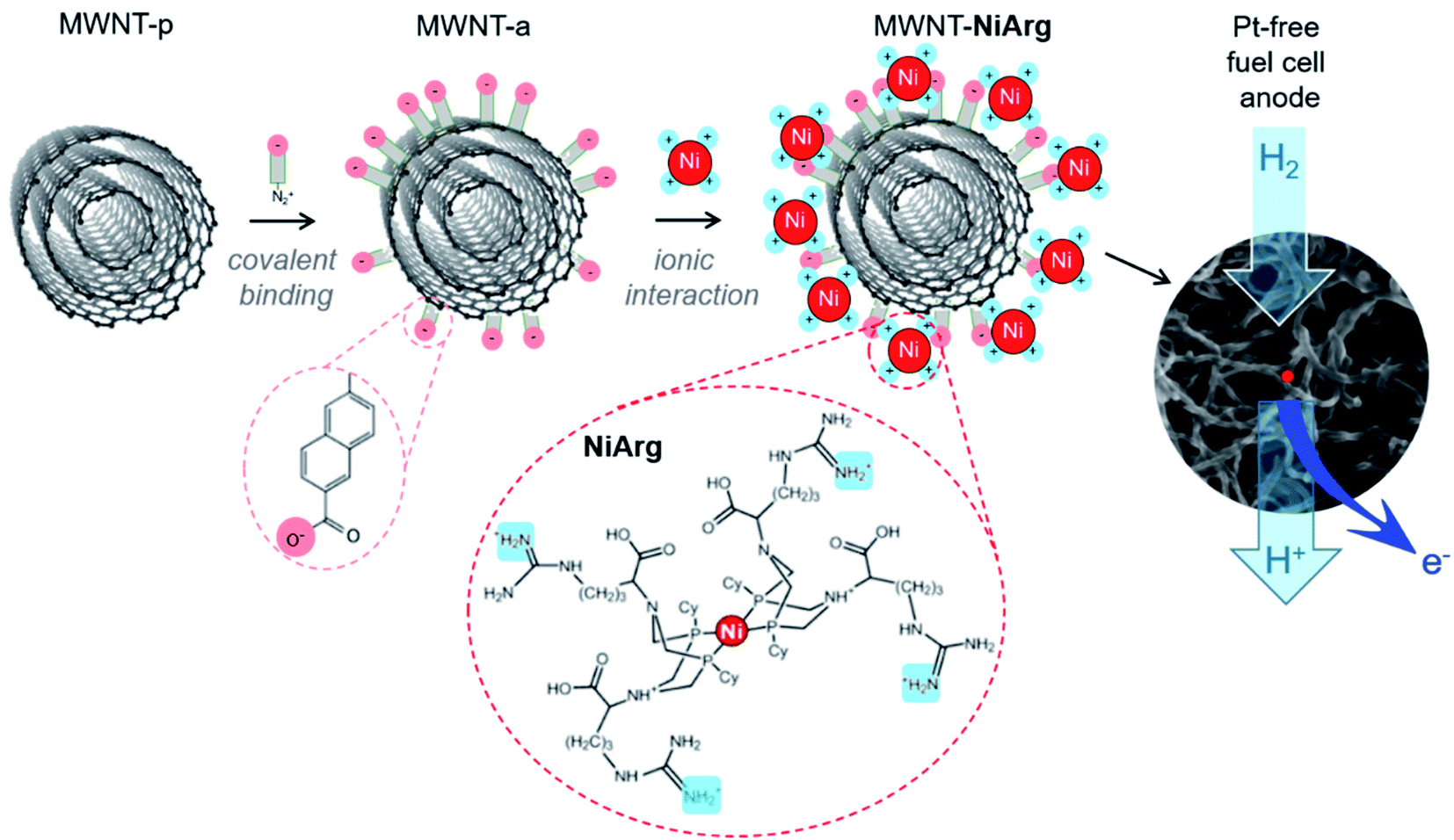 | ||
| Fig. 1 Scheme of the functionalization of pristine MWNTs (MWNT-p) with naphthoic acid moieties to yield the anionic MWNT-a, and subsequent electrostatic adsorption of NiArg catalyst to yield the functional MWNT-NiArg, as the catalytic material for platinum-group metal-free fuel cell anodes. | ||
Multiwalled carbon nanotubes (MWNT) form a well-adapted platform for electrode development as they provide both a large surface area to support the catalyst adsorption,17,18,20,32–34 and a high conductivity in film.35 Such molecularly-engineered nanomaterials proved operational once implemented in the platinum-group-metal-free (PGM-free) anode of a H2/O2 fuel cell device.18,36–38
The current density displayed by such electrode materials directly related to the rate of catalysis, which is limited, for a given immobilized catalyst, by the surface coverage and by the mass transport of the substrate (hydrogen) and products (protons) of the reaction. The understanding and optimization of mass transport effects in the catalytic layer (access to hydrogen gas and electrolyte, flooding effects) have been the subject of other recent papers by our groups.18,20 Here we focus on the structure of the catalytic material to optimize the grafting of catalysts to gain control on the distribution of catalytic sites on MWNTs. Such structural characterization of molecular-engineered carbon nanotubes are scarce.39
The NiArg catalyst contains a single metal atom in a 3D flexible structure. It can therefore adopt various conformations once immobilized on the MWNT. This feature is actually required to minimize reorganization energy when transitioning through the redox states, and to position pendent proton relays along the catalytic cycle. Thus, finding out where and how the catalyst distributes along the MWNT surface is a difficult task. In this study, we use two complementary techniques: transmission electron microscopy (TEM) for a direct imaging of the catalyst onto the carbon nanotube surface, and small angle neutron scattering (SANS) for an indirect observation of catalyst-loaded carbon nanotubes in a relevant solvent medium.
Results
Materials and electrocatalysis
Among the different grafting strategies to immobilize DuBois' catalysts on carbon nanotubes, adhesion through electrostatic interactions gave rise to the highest HOR currents even after implementation in functional fuel cells.18,20,37 Pristine MWNTs are first functionalized with diazonaphthoic acid to yield MWNT-a covered with carboxylate functions (a stands to indicate the anionic nature of the MWNT surface).37 Then MWNT-a are incubated for a few minutes with the cationic NiArg catalyst (Fig. 1) to yield MWNT-NiArg. The binding is strong thanks to the multiple positive charges of the guanidinium groups on the NiArg catalyst molecule interacting with the polyanionic surface. The material is later used to formulate a catalytic ink, in some cases along with Nafion ionomer, and can be deposited to form a catalytic layer on a conductive, porous support to act as the anode of a hydrogen fuel cell.Fig. 2 displays the voltammetric response of such anodes under argon in neutral pH conditions (Fig. 2a) and under hydrogen flow in acidic media (Fig. 2b). The voltammetric test under H2 in H2SO4 (0.5 M) electrolyte is representative of the acidic conditions found in proton-exchange membrane fuel cells. Importantly, the presence of Nafion does not drastically modify the shape of the catalytic response but lowers the catalytic current density by ∼30% at all potentials. Nafion's effect being low, advanced characterization was mainly led on the adsorption of the catalyst onto the MWNTs without Nafion.
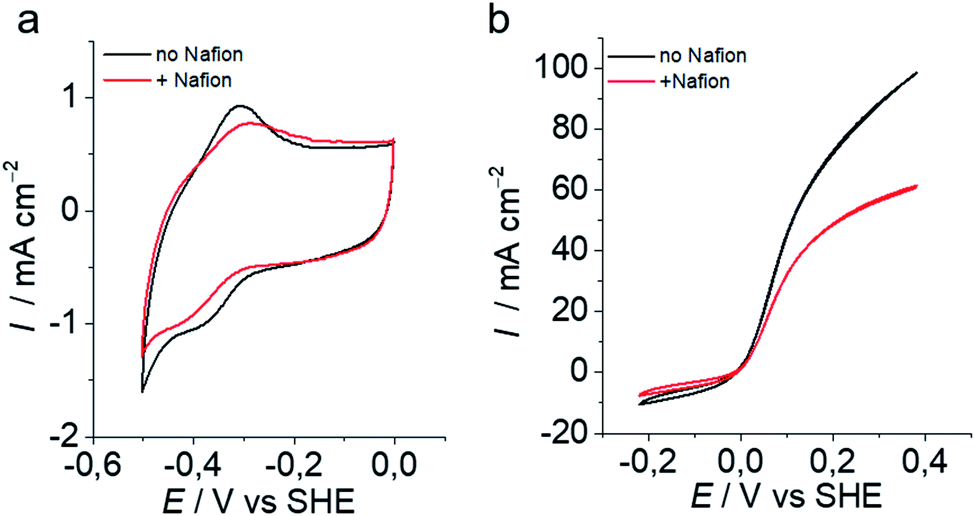 | ||
| Fig. 2 Electrochemical characterization of MWNT-NiArg anodes without (black) and with (red) Nafion (20 mV s−1) at room temperature. (a) Cyclic voltammetry (CV) traces in 0.2 M phosphate buffer pH 7 under argon. (b) CV traces at pH 0.3 (H2SO4 0.5 M) under H2. | ||
Structural characterization
The MWNT-NiArg composite was observed by scanning transmission electron microscopy (STEM) and compared with pristine MWNTs (MWNT-p) and anionic MWNT-a.As the materials are sensitive to the electron beam irradiation, all observations were performed at an acceleration voltage of 80 kV. The images were recorded simultaneously by two detectors, the bright field (BF) and the high-angle annular dark-field (HAADF) detectors. Structural details of carbon materials are more easily revealed in BF images whose contrast is close to the conventional TEM image, while HAADF images having a contrast depending on the thickness and density of the material highlight heavy atoms. Generally, the images were acquired at first exposure, after focusing in a nearby region in order to minimize the beam damage. The beam irradiation effect on structural modification was studied and used for specific analyses.
Fig. 3a–i show the STEM images in HAADF and BF modes of the 3 different states of MWNTs. MWNT-p have an average diameter of 10 nm, with a luminal core of 4 nm and 7 ± 4 graphene layers with a high crystallinity without any visible structural defect on the surface layer (Fig. 3a–c, Table 1). The anionic layer in MWNT-a is recognized in HAADF images as a homogeneous amorphous-like coverage distributed on all MWNTs (Fig. 3d), appearing as an increase in MWNT diameter and brightness. BF images also show a thin layer of curly structures all over the surface, indicating the presence of some added material, as expected from the grafting of naphthoic acid moieties. Numerous structural defects such as holes are recognized, in the 2–3 first graphitic layers, in the high resolution BF images (arrows on Fig. 3f), which may be due to covalent bonding formed during the diazonaphthoic acid grafting and subsequent local disruption of the Csp2 structure of the MWNT. Note that the inner surface of the MWNT-a is also partly functionalized, which indicates that the lumen of the MWNTs is at least partly accessible to the solvent and diazonium reagent.
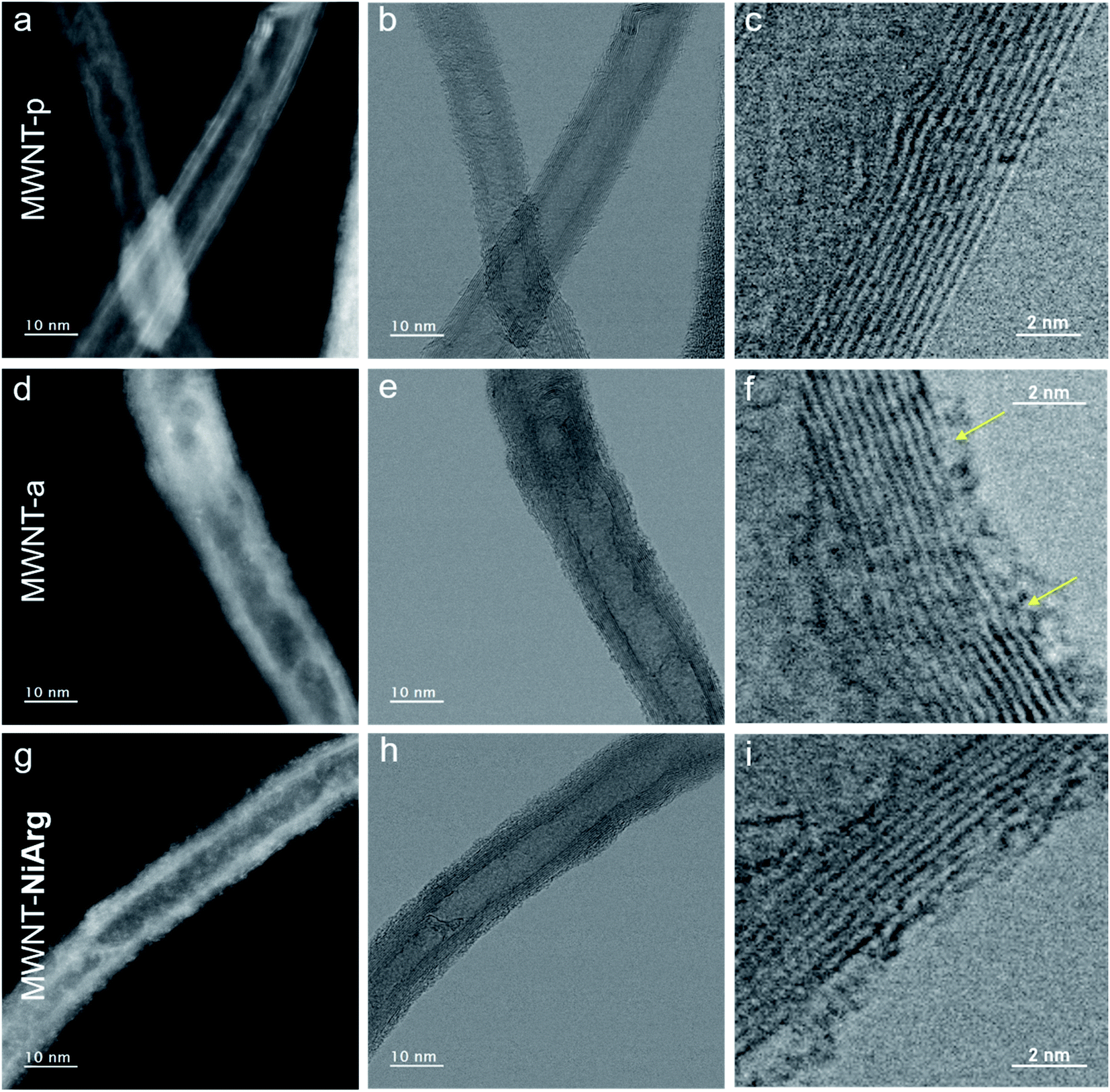 | ||
| Fig. 3 Structural characterization of NiArg adsorption on MWNT-a. (a–i) Simultaneous STEM images in HAADF (a, d, g) and BF (b, c, e, f, h, i) mode of MWNT-p (a–c), MWNT-a (d–f) and MWNT-NiArg (g–i). The images were acquired at first exposure corresponding to an electron dose of 2 × 106 e nm−2. | ||
| Core | Carbon | External diameter | ||||
|---|---|---|---|---|---|---|
| Diameter | Distribution | Thickness | Distribution | Diameter | Distribution | |
| a Averaged size from >600 measurements. b Size and size distribution from best fit with core–shell cylinder profile. | ||||||
| STEM of MWNT-p | 4.8 nma | 39%a | 2.5 nma | 45%a | 9.8 nma | 33%a |
| SANS of MWNT-a | 4.3 nmb | 46%b | 2.7 nmb | 30%b | 9.7 nm | 39% |
According to the BF image, the MWNT-NiArg present the same structural features as MWNT-a, but with thicker curly structures at the surface (Fig. 3h and i). The HAADF images of MWNT-NiArg also show a bright coverage of uniform thickness with a thin granular contrast (Fig. 3g). This coverage was found to be homogeneous all along the MWNTs, and on all MWNTs in reproduced MWNT-NiArg samples. The images do not show individual molecules: because of their flexible structure enabling catalytic activity,40,41NiArg catalysts probably cannot be imaged unlike other more rigid metal–organic molecules,42,43 where isolated metal atoms could be singly imaged.
Chemical analysis and beam damage effect
As expected for organic and metal–organic materials, the grafted layers are sensitive to electron irradiation. The blur coverage surrounding fresh MWNT-NiArg tends to agglomerate locally under the electron beam and stabilizes as bright clusters after several sequences of scanning (Fig. 4a). In the case of MWNT-a, the MWNT surface also evolves toward granular aspect under beam irradiation, but with much less brightness and contrast than for MWNT-NiArg (Fig. S2†). We thus hypothesize that the organic part of the NiArg molecule slowly fragments away, ultimately leaving a mostly inorganic Ni-rich residue. Indeed, arginine is known to undergo easy fragmentation under thermal or electronic irradiation degradation near or within its guanidine moiety.44,45 To confirm this assignment, chemical analysis was carried out using electron energy loss spectroscopy (EELS) in the STEM microscope. Measurements were recorded after irradiation-induced agglomeration, to ensure a stable signal. EELS elemental maps obtained for the C-K and Ni-L23 edges could undoubtedly identify the presence of Ni from NiArg in the bright spots observed on beam-irradiated MWNT-NiArg (Fig. 4b–d).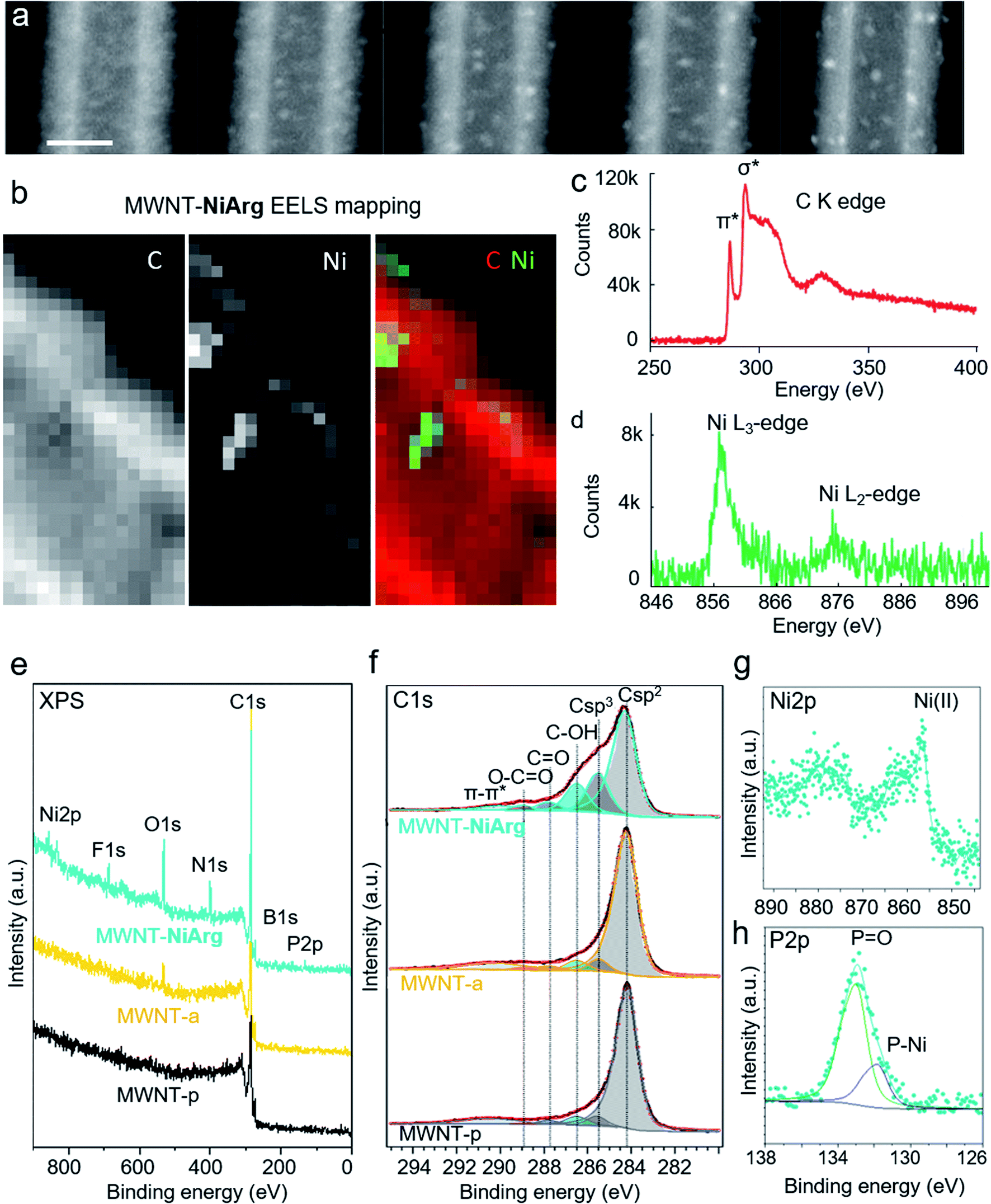 | ||
| Fig. 4 Chemical analysis by EELS and XPS. Series of HAADF-STEM images of MWNT-NiArg recorded sequentially (approximate dose per image 5 × 10−6 e nm−2, scale bar 10nm), showing the effect of beam damage (a). EELS mapping of C, Ni (pixel size 0.5 nm) and color-coded superimposed C and Ni map of a detail region of MWNT-NiArg after beam exposure (b), and corresponding EELS spectra at the carbon K edge (c) and nickel L edge (d). XPS spectra of MWNT-p (black line), MWNT-a (yellow line) and MWNT-NiArg (blue line): survey spectra (e) and detail spectra of the C 1s (f), Ni 2p (g) and P 2p (h) regions. See O 1s and N 1s spectra in Fig. S1.† | ||
As a complement, an analysis carried out by X-ray photoelectron spectroscopy (XPS) offered a quantitative determination of the elements at the surface of MWNTs, as well as their chemical state. We expect less pronounced but similar beam damage in the XPS analysis (long exposure to the high-energy X-ray beam in the vacuum chamber) as from the STEM electron beam. XPS spectra (Fig. 4e–h) showed a 4-fold increase in O content for MWNT-a, compared to the MWNT-p, and a small addition of N from the coupling of the diazo moiety (quantitative ratios in Tables S1 and S2†), as previously described.46 For MWNT-NiArg, XPS revealed the presence of additional Ni, P, N, B and F attributed to the incorporation of the Ni complex. The shape and the binding energy (BE) of the Ni 2p spectrum confirmed the presence of Ni2+ (Fig. 4g).33,47 The P 2p line (Fig. 4h) can be separated into two different chemical shifted components: one at 131.7 eV related to Ni–P interactions and another one at 133.1 eV that could be attributed to the oxidation of phosphine to phosphine oxide,19 which could also be a ligand for the Ni. The surface composition calculated from XPS showed a P/Ni atomic ratio of 4.1, very close to the stoichiometric one (4 P atoms per Ni center in NiArg). The presence of B and F suggests that some of the initial BF4− counter-ions adsorb to the MWNT-a surface with the NiArg cation: the B/Ni and F/Ni ratios indicate that about one out of two BF4− per NiArg is still present. Finally, comparing the recorded N/Ni and C/Ni ratios (taking into account all C 1s peaks except the sp2 carbons, present only in MWNT-a) to what is expected from the NiArg structure (Table S1†), it appears that at least 50% in N and 30% in C are lost. This loss of organic elements is in accordance with the observed beam damage of the above mentioned guanidinium moieties in NiArg, en route to the formation of Ni-rich clusters (Fig. 4a and b).
Nanostructure of the NiArg coverage in MWNT-NiArg
As a complement to STEM, SANS analysis was carried out to confirm the observed NiArg structure. Indeed, STEM analysis requires to place the material in a deep vacuum environment, while SANS analysis is directly performed on the aqueous catalytic ink.We thus recorded SANS data for MWNT-a and MWNT-NiArg to get structural insights of the catalyst/MWNT organization in a wet environment (Fig. 5a). MWNT-a and MWNT-NiArg were ground at a final concentration of 3% w/v of MWNTs in a water/ethanol mixture, i.e. the solvent in the catalytic ink used to prepare the electrodes characterized in Fig. 2. The obtained paste was shot by a neutron beam in an airtight quartz cell of thickness 100 μm, and the scattered intensity was converted to a SANS 1D profile. MWNT-a and MWNT-NiArg SANS profiles superimpose completely (Fig. 5a 100% H). Their profile is typical of a cylinder signal: a strong intensity decay with two oscillatory features that fits with a core–shell cylinder scattering model (Fig. S3†) as we reported previously.18 The average inner and outer diameters and the size distribution determined by curve fitting (Table 1) are in excellent accordance with the size measurements by STEM.
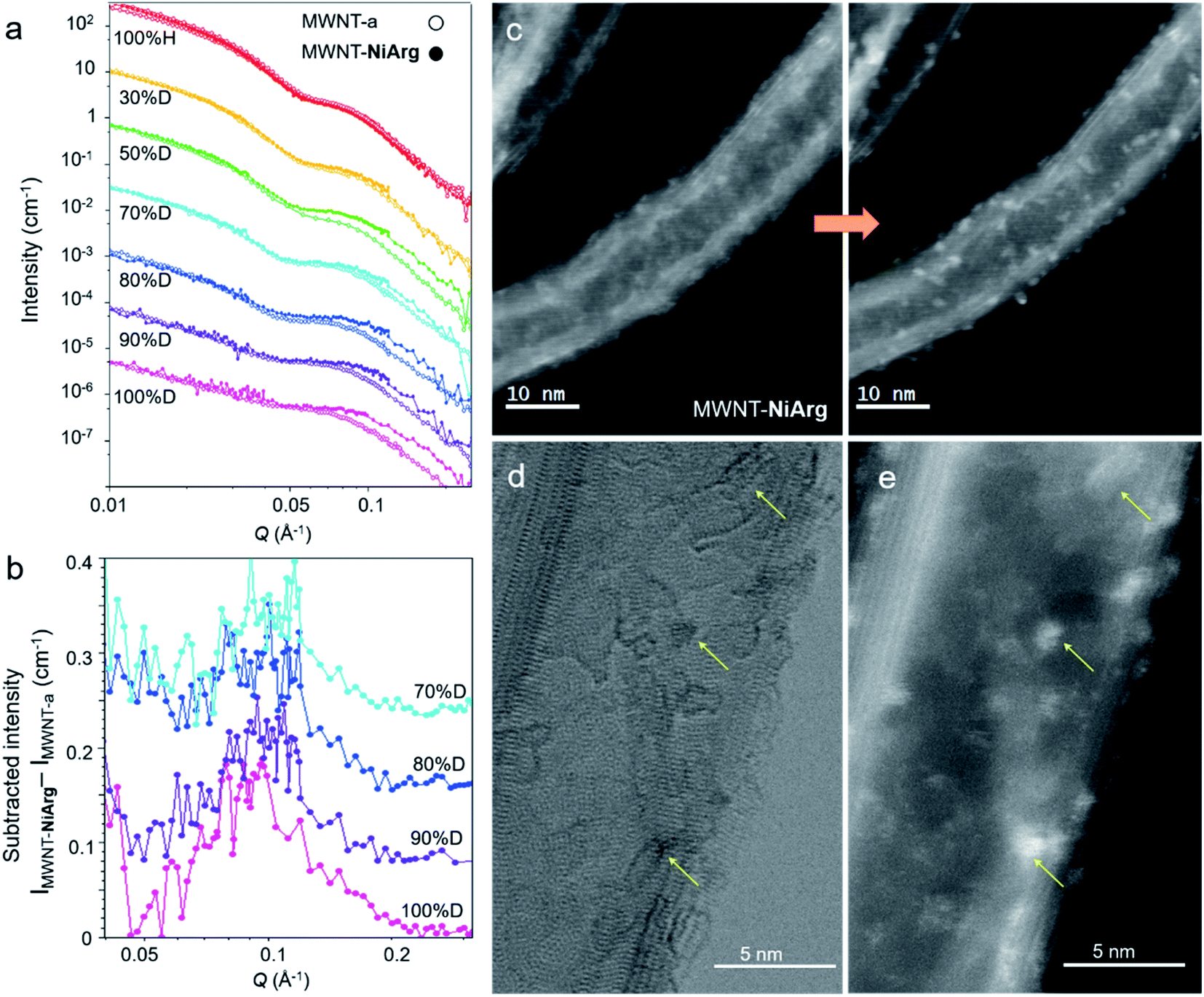 | ||
| Fig. 5 SANS and STEM evidence of an organized distribution of NiArg on MWNT. (a) SANS profiles of MWNT-a (circles) and MWNT-NiArg (dots) in solvents of % D 0, 30, 50 70, 80, 90 and 100%. Data at successive % D are down-shifted by a factor of 0.1 for clarity. (b) SANS profile of MWNT-NiArg subtracted by the corresponding SANS profile of MWNT-a for % D 70, 80, 90 and 100%, showing a repeatable peak centered at 0.096 Å−1. Successive plots are down-shifted by an increment of 0.08 for clarity. (c) Successive STEM-HAADF images of MWNT-NiArg at first scan and after exposure to electron beam. (d, e) Simultaneously recorded BF (d) and HAADF (e) STEM images of the same detail of MWNT-NiArg after beam exposure, showing co-localization (arrows) of MWNT structural defects (d) and Ni-rich clusters (e). | ||
Metal–organic compounds such as NiArg have a scattering efficiency in the neutron beam (known as scattering length density, SLD) with a low contrast to standard solvents, as both contain a high amount of hydrogen atoms. They are thus transparent under these recording conditions. To increase the contrast, we progressively replaced the hydrogenated solvent by its deuterated equivalent with a percentage of deuteration (% D) varying from 0 to 100. Below 30% D, the solvent SLD matches that of organic and metal–organic compounds, so that the naphthoate layer and NiArg give little signal and the MWNT carbon shell gives a strong signal. Above 70% D, the solvent SLD matches the SLD of the MWNT carbon shell, so that the MWNT signal is attenuated while organic and metal–organic compounds show a higher contrast. Any nanostructure made of NiArg should thus appear as a SANS signal in the 70–100% D media. The SANS profiles of MWNT-a and MWNT-NiArg in the series of deuterated solvents (Fig. 5a) superimpose in the low Q region, but discrepancies indeed appear at Q > 0.05 Å−1 for % D > 50.
A change in the core–shell cylinder form factor could not account for this change in SANS profile in deuterated solvents. Indeed, the addition of a layer around the MWNT should result in a larger outer diameter due to the added matter, and thus in a displacement of the SANS profile to the lower Q. We observe the reverse: the observed profile displacement at solvent % D > 70 can only fit with a core–shell cylinder with a 20% reduction in diameter, which is physically inconsistent and in disagreement with STEM data. Instead, taking advantage of the very good quality of the data, we could directly subtract the SANS profile of MWNT-a to that of MWNT-NiArg (Fig. 5b) to enlighten an additional signal. The subtracted intensity profiles show a consistent peak at Q = 0.095 Å−1, with a constant width at half maximum of ±15%. As this structure shows contrast only in highly deuterated solvents with SLD >4.2 × 10−6 Å−2, its SLD must be lower than 3 × 10−6 Å−2. This is in good accordance with the SLD of the NiArg catalyst estimated to 1 to 3 × 10−6 Å−2, depending on the possible protonation of its acid and amine moieties with H+/D+ from the solvent. This peak thus interprets as stemming from an organized structure made of NiArg with a repeating distance of 6.5 ± 1.5 nm.
Discussion
The MWNT-NiArg composite takes advantage of the very high specific area of MWNTs to integrate high amounts of NiArg while ensuring direct electrical contact of the catalyst molecule. A good balance of electrocatalytic HOR current versus catalyst loading were obtained for a loading of 220 nmolNiArg/mgMWNT in the catalytic ink (Fig. 2), while higher loadings resulted in unstable catalytic currents. Theoretically, considering the NiArg hydrodynamic radius measured by NMR of 0.87 nm, a single, close-packed monolayer of catalyst onto the MWNTs corresponds to a higher estimate of 130 nmolNiArg/mgMWNT (see experimental part for details). It thus appears that a significant amount of the introduced NiArg catalyst is in excess. Indeed, integration of the cyclic voltammograms shown in Fig. 2a allows to probe the directly connected NiArg, estimated to the even lower value of ∼40 nmolNiArg/mgMWNT.The excess catalyst can either leach out during electrochemical measurements or stay within the catalytic layer. The hypothesis of the excess of catalyst leaching out to solution is unlikely, as we found out that such an excess of catalyst contributes to the high current densities, sustained over several hours of continuous operation.18 We thus expect some accumulation of NiArg somewhere trapped in the composite material, although not directly electronically connected. The point of this study was thus to get more insights into the structural distribution of NiArg within the catalytic layers.
The present study first allows concluding on a regular distribution of the NiArg catalyst across the modified electrode, at the macroscale. No heterogeneity was observed at neither the micron nor the millimeter scales, and the NiArg coverage of MWNT-NiArg appears regular in thickness, similar and repeatable on all MWNTs by electron microscopy. It follows that excess NiArg distributes in a stable and homogeneous manner in the material. We can thus exclude the segregation of a NiArg phase as bulk deposits within the catalytic layer.
Still, SANS data indicate the presence of a nanostructure with a repeating distance of 6.5 ± 1.5 nm that could correspond to accumulation of NiArg at the surface of MWNTs. Actually, the Ni-rich clusters with a typical size of 0.5–1 nm observed by STEM on irradiated MWNT-NiArg (Fig. 5b) also scatter homogeneously on the surface with an apparent distance of 3–5 nm. Taking into account the cylindrical shape and the superimposed imaging of both sides of the MWNT, this apparent distance in a 2D projection corresponds to a real inter-cluster distance in 3D of 5–9 nm, congruent with the repeating distance of 6.5 ± 1.5 nm detected by SANS in the NiArg distribution. The SANS signal would then arise from a nanostructured layer made of excess catalyst as aggregates on MWNT-NiArg.
A closer look at the simultaneous BF and HAADF images of MWNT-NiArg after irradiation (Fig. 5d–e) allows observing an overlap between the position of bright clusters in HAADF and of carbon layer curls in BF, corresponding to defect sites in the nanotube structure. These defect sites stem from the diazonium functionalization, as they are identical to those observed on MWNT-a, but are absent on MWNT-p (Fig. 3). These MWNT-a are heavily functionalized, inducing structural defects in the structure of the MWNT not only in the outer layer, but across two to three carbon layers (Fig. 3f). This inhomogeneity in functionalization creates carboxylate-rich sites at the MWNT surface. The subsequent multi-anionic sites have a strong affinity for cations, which will attract the polycationic NiArg molecules. We suggest that NiArg concentrates on these densely functionalized sites through electrostatic interactions (Fig. 6), thus creating a soft structure of aggregated catalysts on top of a continuous monolayer of catalysts along the whole MWNT surface. These aggregation sites separate by a typical distance of about 6.5 nm, as observed by SANS. The typical distance between the NiArg aggregates in MWNT-NiArg would thus arise from the diazonium-induced defect distribution in MWNT-a.
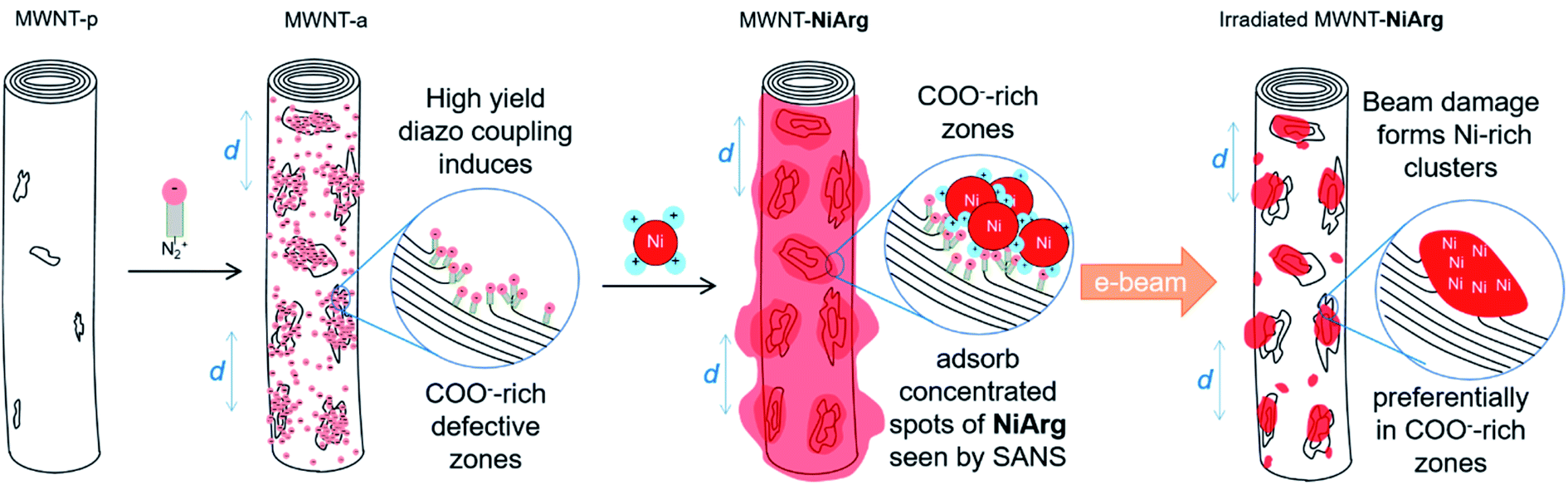 | ||
| Fig. 6 Scheme of the proposed model for the formation of a cluster distribution of NiArg in MWNT-NiArg over two steps: diazonaphthoate coupling onto MWNT-p, adsorption of NiArg onto MWNT-a. Transformation of the NiArg layer following electron-beam exposure is proposed in a third step. | ||
Under the electron beam irradiation of the STEM, the organic part of NiArg is damaged and partly evaporated, leaving a residue enriched in Ni and P. This residue is inhomogeneously but regularly distributed as clusters from the start, as it directly arises from the distribution of aggregated NiArg at the surface of the MWNT. In successive HAADF images of MWNT-NiArg, we observe that clusters grow but barely move on the MWNT surface. We thus suggest that the clusters mostly grow on the carboxylate-rich defect sites where NiArg molecules were initially aggregated, as schematically represented in Fig. 6. NiArg distributed in less dense adsorption areas (i.e. as monolayer) could lead to the growth of lower size clusters, as observed on Fig. 5f and indicated as smaller red spots in Fig. 6d.
Unfortunately, it was not possible to detect the spot structure in MWNT-NiArg in the presence of Nafion, as the Nafion self-assembled nanostructure gives a strong interfering STEM and SANS signal. In a previous study on grafting methods to immobilize DuBois's catalysts onto MWNTs,18 we showed that Nafion could interfere in the catalyst/MWNT interaction depending on the employed anchoring strategies, resulting in catalytic activity loss. NiArg proved the most resilient catalyst as it is not significantly displaced from the MWNT surface by Nafion.18 Actually, SANS data indicate that NiArg can displace Nafion from the MWNT-a surface (Fig. S6†) while integration of the cyclic voltammograms shown in Fig. 2a indicates that the presence of Nafion diminished by ∼25% the amount of electrically connected NiArg catalyst (∼30 vs. 40 nmolNiArg/mgMWNT without and with Nafion, Fig. 2a). This 25% lower amount of electrically connected catalyst in the presence of Nafion results in the same 29% drop in electrocatalytic current (32 vs. 45 mA cm−2 at 0.1 V vs. NHE, Fig. 2b), suggesting that the activity of each connected catalyst would be the same. These hints suggest that the spot structure in MWNT-NiArg would be maintained in the presence of Nafion in the fuel cell catalytic layer.
Conclusion
Although not covalent, the polyanion/polycation grafting method used to immobilize NiArg on MWNT-a thus proves to be a robust catalyst anchoring approach. Relying on a series of cutting-edge characterization techniques at the nanoscale, we could show that the molecular catalyst evenly distributes along the MWNT outer wall, generating a regular layer with a dense array of aggregates in addition to the expected monolayer of directly connected catalysts. These aggregates seem to more specifically attach to structural defects in the functionalized MWNT-a sidewalls, where a higher local concentration of carboxylate moieties can be found. Importantly, the identification of two populations of NiArg catalyst within the composite layer open new horizons for the comprehensive understanding of the catalytic performances. In particular, it would be of great importance to determine which of the NiArg catalysts, adsorbed as a (sub)monolayer or within the aggregates, are the most active ones and how the less active population is recruited to enhance the catalytic current. Understanding the dynamics of exchange between these two populations is also of utmost interest as it may allow designing systems with self-repair mechanisms. Alternatively, it may be possible to exploit the dual properties of non–connected NiArg catalysts to promote self-defense against oxygen crossing over from the cathode in an operating proton-exchange membrane fuel cell.48 These results thus pave the way to the further implementation of such molecular systems into appropriate composite electrode designs to be implemented into functional devices, which should soon allow reaching industrially relevant current and stability performances.20Experimental methods
Materials
All chemicals and solvents, including the Nafion perfluorinated resin solution (5 wt% in lower aliphatic alcohols and water), were purchased from Sigma-Aldrich and used without further purification unless otherwise stated. UP-NC7000WT MWNTs (purity > 90% in carbon) were obtained from Nanocyl (Belgium) and used as received (as MWNT-p). The gas diffusion layers (GDL w/o MPL) were purchased from Paxitech (Grenoble, France). [Ni(P2CyN2Arg)2](BF4)2 (NiArg)31 was synthesized according to published procedures. MilliQ water was systematically used.MWNT-a synthesis
MWNTs were treated with diazonaphthoic acid following a published procedure.18 Briefly, MWNTs (50 mg) are sonicated in 150 mL of argon-degassed DMF for 45 minutes. 1.5 g of 6-amino-2-naphtoic acid and 1.1 mL of isoamyl nitrite are added, to trigger the in situ formation of 2-naphthoic acid 6-diazonium and its subsequent reduction onto the MWNTs under argon at 80 °C for 12 hours. The resulting solid in suspension was filtered, washed thoroughly twice with DMF, then with ethanol and dried to yield around 50 mg of MWNT-a.SANS sample preparation
MWNT-a were dispersed into a MWNT-a stock paste using a certain percentage of deuterated solvent (X% D) by sonication and grinding in mixtures of deuterated ethanol/absolute ethanol at a ratio of X% vol: (100-X)% vol at a MWNT concentration of 4 mg mL−1. NiArgX% D stock solutions were obtained by dissolving NiArg in D2O/H2O in a X% vol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(100-X)% vol ratio at a concentration of 3 mg mL−1. For the preparation of MWNT-NiArgX% D samples, 67 μL of NiArgX% D stock was thoroughly mixed with 200 μL of MWNT-a X% D stock and incubated for 24 h. For samples containing no catalyst, in order to preserve proper solvent and deuteration ratios, MWNT-a X% D samples were prepared by mixing 67 μL of a X% D D2O/H2O mixture with 200 μL of MWNT-a X% D stock and incubated for 24 h. For samples with Nafion, 10 mg MWNT-a were mixed with 200 μL Nafion 5% solution, and residual solvents were evaporated. This solid was used in place of pure MWNT-a in the preparation of X% D samples as described above. 10-fold dilutions of MWNT-a 0% D and MWNT-NiArg 0% D in ethanol/water 3:1 v/v were used for STEM sample preparation.
:(100-X)% vol ratio at a concentration of 3 mg mL−1. For the preparation of MWNT-NiArgX% D samples, 67 μL of NiArgX% D stock was thoroughly mixed with 200 μL of MWNT-a X% D stock and incubated for 24 h. For samples containing no catalyst, in order to preserve proper solvent and deuteration ratios, MWNT-a X% D samples were prepared by mixing 67 μL of a X% D D2O/H2O mixture with 200 μL of MWNT-a X% D stock and incubated for 24 h. For samples with Nafion, 10 mg MWNT-a were mixed with 200 μL Nafion 5% solution, and residual solvents were evaporated. This solid was used in place of pure MWNT-a in the preparation of X% D samples as described above. 10-fold dilutions of MWNT-a 0% D and MWNT-NiArg 0% D in ethanol/water 3:1 v/v were used for STEM sample preparation.
SEM measurements
Scanning electron microscopy (SEM) images was acquired with a ZEISS Ultra 55+ electron microscope, using an electron beam at 5.0 kV.Electrode preparation
MWNT-NiArg electrodes were prepared as previously described.18 Briefly, MWNT-a inks were prepared by sonicating MWNT-a in EtOH (3 mg mL−1) with or without Nafion (in 1:1 ratio with MWNT-a) for 30 minutes in an ultrasonic bath. Then, the dispersion was mixed with a solution of NiArg in milliQ water (4 mM) in a 1:3 ratio (30 μL of MWNT-a with 10 μL of NiArg) and 20 μL of the resulting mixture were then deposited directly onto the GDL disk (0.125 cm2) and left to dry for 5 minutes (the drying time had a strong impact on the electrocatalytic performances as previously reported20). The as prepared disk was then directly used for electrochemical measurements in aqueous media.
Electrochemical measurements
Electrochemical experiments were carried out using a Bio-Logic SP300 potentiostat (equipped with an additional impedance module) and using a three-electrode electrochemical cell. The working electrode was the GDL-MWNT assembly in a homemade miniaturized half-cell setup,19,32 the counter-electrode was a platinum mesh and a Hg/Hg2SO4 (0.5 M H2SO4) was used as reference electrode. Gases were provided in the back of the GDL at atmospheric pressure (Patm) and 10 mL min−1 and atmospheric pressure with the use of Bronkhorst Mass-Flows through Swagelok fittings. Measurements in neutral conditions were performed in 0.2 M potassium phosphate buffer at pH7 degased with argon and with an argon feed at the back of the working electrode. Integration of the reversible wave at E1/2 = 0.04 V vs. RHE, attributed to the Ni(0)/Ni(II) couple, gives the density of electrically connected catalysts. Electrocatalytic HOR measurement were performed in acidic electrolyte (pH 0.3, 0.5 M H2SO4) degassed with argon and with an H2 feed at the back of the working electrode (10 mL min−1 and at atmospheric pressure using a Bronkhorst Mass-Flows controller). iR drop was measured prior to the electrocatalytic measurements using the ZIR technique of EC-lab. The compensated resistance (Rc) was set to 85% of the uncompensated resistance (Ru).Hydrodynamic radius estimation
The hydrodynamic radius of NiArg was estimated using previously reported calculations by Shaw and coworkers49 and using the Stokes–Einstein equation as follow:where r is the hydrodynamic radius (m), k is the Boltzmann constant (1.38 × 10−23 J K−1), T is the temperature (K), D is the diffusion coefficient of NiArg that was previously calculated using NMR pulsed field gradient methods49 (at 298 K) of 2.8 × 10−10 m2 s−1 and η is the viscosity of the medium at the given temperature (for water,50η = 0.89 × 10−3 Pa s at 298 K). This gave a hydrodynamic radius of rNiArg = 0.87 nm.
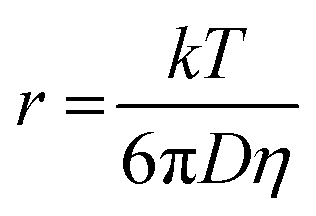
To estimate the amount of NiArg in a single layer covering the MWNTs, we assume that NiArg are close-packed solid spheres of radius rNiArg. The molar density dmonolayer of this NiArg layer is thus:
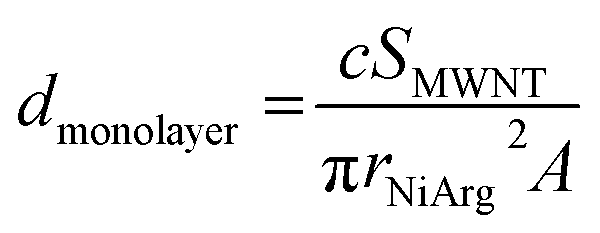
XPS
XPS measurements were taken at RT in a custom designed UHV system equipped with an Omicron electron analyser, working at a base pressure of 10−10 mbar. Core-level photoemission spectra were acquired in normal emission using a dual non-monochromatized Mg Kα X-ray source (1253.6 eV). Single spectral regions were collected using 0.1 eV steps, 0.5 s collection time and 20 or 50 eV pass energy. C 1s region was separated into chemically shifted components in order to determine the nature and amount of the oxygenated species. An asymmetrical shape was used for the sp2 component, whereas symmetrical Voigt functions were used for the sp3 component and the C–O functional groups. The π–π* transition at binding energy (BE) of 290.4 eV was also included in the fit. P 2s region was separated into two different chemical shifted components using symmetrical Voigt functions. The surface composition was calculated from the XPS peaks areas and the corresponding sensitivity factors.STEM and EELS analysis
The different types of MWNTs were dispersed into iso-propyl alcohol (IPA) and drop-casted onto a lacy carbon membrane on Cu TEM grid. STEM analyses were performed using a probe Cs corrected FEI Themis microscope operating at 80 kV. BF and HAADF-STEM images were acquired using a convergence semi-angle of 20 mrad. The images acquired at first exposure correspond to an electron dose of 2 × 106 e nm−2. EELS measurements were carried out using a Gatan imaging filter Quantum with 1 eV energy dispersion and the elemental maps was acquired using the Spectrum Imaging technique allowing a spectrum acquisition for each pixel of beam scanning in HAADF-STEM mode for the C-K and Ni-L23 edge.Small angle neutron scattering (SANS)
SANS measurements were performed on the D22 spectrometer at the Institut Laue Langevin (ILL, Grenoble, France) and on the PAXY spectrometer at the Laboratoire Léon Brillouin (LLB, Paris, France). Two configurations are used to cover an extended Q-range, from 4 × 10−3 to 0.6 Å−1 and 7 × 10−3 to 0.6 Å−1 (ILL and LLB experiments, respectively), Q being the scattering vector defined as Q = (4π/λ)sin(θ/2) where λ is the wavelength of the incident neutron beam, and θ the total scattering angle. The isotropic 2D patterns recorded in the two configurations are radially averaged to obtain the 1D scattered intensities I(Q), and further corrected using standard procedures (detector efficiency, background, and empty cell subtraction) using PASINET and GRASP data reduction in-house software. As data taken in the two configurations are perfectly overlapping in the intermediate Q-region, the I(Q) are merged to obtain a single profile for each sample. The sample pastes are shot by a neutron beam in an air-tight quartz cell of thickness 100 μm. Absolute intensities are obtained by normalizing I(Q) by the intensity of the incoherent scattering plateau at high Q (plateau due to solvent scattering), which allows to correct for experimental differences in thickness among quartz cells. A typical distance d is generally related to a peak at Q by Bragg's law d = 2π/Q.Spectra fitting is performed using SASView software. The best fits are obtained as previously described18 with the core–shell cylinder form factor and the hard-sphere structure factor. As in our previous study, the best fits give a constant shell SLD of 7 × 10−6 to 8 × 10−6 Å−2 (Fig. S4†), consistent with the graphite-like composition of the MWNTs, and constant shell and core diameters and size distribution for all % D. Notably, the SLD of the MWNT core increases linearly with the % D (Fig. S4†) and is equal to 55% of the solvent SLD, indicating a partial filling of the MWNT cores by the solvent. As SANS profiles for MWNT-a and MWNT-NiArg at the same % D superimpose (except for % D >70% and Q > 0.05 Å−1), the fits give the same SLD and size parameters for MWNT-a and MWNT-NiArg (Fig. S5†).
Data availability
Data can be made available upon request to the corresponding author.Author contributions
Investigations and data curation: A. G., N. C., L. C., B. R., L. G., H. O., S. L., P. C.; writing – original draft: P. C.; formal analysis, writing – review & editing: B. R., L. G., H. O., V. A., S. L., P. C.; funding acquisition, conceptualization: V. A., S. L.; supervision: G. G., P. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. G. thanks for funding the CEA PTC Program on Materials and Processes (Bio-PAC project). P. C., V. A. and B. R. acknowledge support from the French Agence Nationale de la Recherche (Labex ARCANE, CBH-EUR-GS, and ANR-17-EURE-0003). The research leading to these results has also received funding from the Fuel Cells and Hydrogen 2 Joint Undertaking (FCH-JU, GAN, under Grant Agreement No. 779366). FCH-JU receives support from the European Union's Horizon 2020 research and innovation programme, Hydrogen Europe and Hydrogen Europe Research. A. G., S. L. and P. C. acknowledge ILL and LLB for beam-time allocation and Alexis Chennevière, Jacques Jestin and Lionel Porcar for their help as local contacts.References
- M. Li, Z. Zhao, T. Cheng, A. Fortunelli, C.-Y. Chen, R. Yu, Q. Zhang, L. Gu, B. V. Merinov, Z. Lin, E. Zhu, T. Yu, Q. Jia, J. Guo, L. Zhang, W. A. Goddard, Y. Huang and X. Duan, Ultrafine Jagged Platinum Nanowires Enable Ultrahigh Mass Activity for the Oxygen Reduction Reaction, Science, 2016, 354(6318), 1414–1419, DOI:10.1126/science.aaf9050.
- K. Kodama, T. Nagai, A. Kuwaki, R. Jinnouchi and Y. Morimoto, Challenges in Applying Highly Active Pt-Based Nanostructured Catalysts for Oxygen Reduction Reactions to Fuel Cell Vehicles, Nat. Nanotechnol., 2021, 16(2), 140–147, DOI:10.1038/s41565-020-00824-w.
- H. Zhang, W. Cheng, D. Luan and X. W. (David) Lou, Atomically Dispersed Reactive Centers for Electrocatalytic CO2 Reduction and Water Splitting, Angew. Chem., Int. Ed., 2021, 60(24), 13177–13196, DOI:10.1002/anie.202014112.
- X. Zhang, Y. Wang, M. Gu, M. Wang, Z. Zhang, W. Pan, Z. Jiang, H. Zheng, M. Lucero, H. Wang, G. E. Sterbinsky, Q. Ma, Y.-G. Wang, Z. Feng, J. Li, H. Dai and Y. Liang, Molecular Engineering of Dispersed Nickel Phthalocyanines on Carbon Nanotubes for Selective CO2 Reduction, Nat. Energy, 2020, 5(9), 684–692, DOI:10.1038/s41560-020-0667-9.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Single-Atom Electrocatalysts, Angew. Chem., Int. Ed., 2017, 56(45), 13944–13960, DOI:10.1002/anie.201703864.
- X.-L. Lu, X. Rong, C. Zhang and T.-B. Lu, Carbon-Based Single-Atom Catalysts for CO2 Electroreduction: Progress and Optimization Strategies, J. Mater. Chem. A, 2020, 8(21), 10695–10708, 10.1039/d0ta01955k.
- J. Zhang, W. Cai, F. X. Hu, H. Yang and B. Liu, Recent Advances in Single Atom Catalysts for the Electrochemical Carbon Dioxide Reduction Reaction, Chem. Sci., 2021, 12(20), 6800–6819, 10.1039/d1sc01375k.
- A. Morozan, B. Jousselme and S. Palacin, Low-Platinum and Platinum-Free Catalysts for the Oxygen Reduction Reaction at Fuel Cell Cathodes, Energy Environ. Sci., 2011, 4(4), 1238, 10.1039/c0ee00601g.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Single-Atom Catalysts: Synthetic Strategies and Electrochemical Applications, Joule, 2018, 2(7), 1242–1264, DOI:10.1016/j.joule.2018.06.019.
- Y. Peng, B. Lu and S. Chen, Carbon-Supported Single Atom Catalysts for Electrochemical Energy Conversion and Storage, Adv. Mater., 2018, 30(48), 1801995, DOI:10.1002/adma.201801995.
- C. Tang, L. Chen, H. Li, L. Li, Y. Jiao, Y. Zheng, H. Xu, K. Davey and S.-Z. Qiao, Tailoring Acidic Oxygen Reduction Selectivity on Single-Atom Catalysts via Modification of First and Second Coordination Spheres, J. Am. Chem. Soc., 2021, 143(20), 7819–7827, DOI:10.1021/jacs.1c03135.
- C. Wan, X. Duan and Y. Huang, Molecular Design of Single-Atom Catalysts for Oxygen Reduction Reaction, Adv. Energy Mater., 2020, 10(14), 1903815, DOI:10.1002/aenm.201903815.
- H. Liu, X. Peng and X. Liu, Single-Atom Catalysts for the Hydrogen Evolution Reaction, ChemElectroChem, 2018, 5(20), 2963–2974, DOI:10.1002/celc.201800507.
- C. Zhu, Q. Shi, S. Feng, D. Du and Y. Lin, Single-Atom Catalysts for Electrochemical Water Splitting, ACS Energy Lett., 2018, 3(7), 1713–1721, DOI:10.1021/acsenergylett.8b00640.
- N. Coutard, N. Kaeffer and V. Artero, Molecular Engineered Nanomaterials for Catalytic Hydrogen Evolution and Oxidation, Chem. Commun., 2016, 52(95), 13728–13748, 10.1039/c6cc06311j.
- V. Artero, Bioinspired Catalytic Materials for Energy-Relevant Conversions, Nat. Energy, 2017, 2(9), 1–6, DOI:10.1038/nenergy.2017.131.
- T. N. Huan, R. T. Jane, A. Benayad, L. Guetaz, P. D. Tran and V. Artero, Bio-Inspired Noble Metal-Free Nanomaterials Approaching Platinum Performances for H2 Evolution and Uptake, Energy Environ. Sci., 2016, 9(3), 940–947, 10.1039/c5ee02739j.
- N. Coutard, B. Reuillard, T. N. Huan, F. Valentino, R. T. Jane, S. Gentil, E. S. Andreiadis, A. Le Goff, T. Asset, F. Maillard, B. Jousselme, A. Morozan, S. Lyonnard, V. Artero and P. Chenevier, Impact of Ionomer Structuration on the Performance of Bio-Inspired Noble-Metal-Free Fuel Cell Anodes, Chem Catalysis, 2021, 1, 1, DOI:10.1016/j.checat.2021.01.001.
- B. Reuillard, M. Blanco, L. Calvillo, N. Coutard, A. Ghedjatti, P. Chenevier, S. Agnoli, M. Otyepka, G. Granozzi and V. Artero, Noncovalent Integration of a Bioinspired Ni Catalyst to Graphene Acid for Reversible Electrocatalytic Hydrogen Oxidation, ACS Appl. Mater. Interfaces, 2020, 12(5), 5805–5811, DOI:10.1021/acsami.9b18922.
- J. Schild, B. Reuillard, A. Morozan, P. Chenevier, E. Gravel, E. Doris and V. Artero, Approaching Industrially Relevant Current Densities for Hydrogen Oxidation with a Bio-Inspired Molecular Catalytic Material, J. Am. Chem. Soc., 2021, 143(43), 18150–18158, DOI:10.1021/jacs.1c07093.
- M. E. Ahmed, S. Adam, D. Saha, J. Fize, V. Artero, A. Dey and C. Duboc, Repurposing a Bio-Inspired NiFe Hydrogenase Model for CO2 Reduction with Selective Production of Methane as the Unique C-Based Product, ACS Energy Lett., 2020, 5(12), 3837–3842, DOI:10.1021/acsenergylett.0c02002.
- A. Maurin and M. Robert, Noncovalent Immobilization of a Molecular Iron-Based Electrocatalyst on Carbon Electrodes for Selective, Efficient CO2-to-CO Conversion in Water, J. Am. Chem. Soc., 2016, 138(8), 2492–2495, DOI:10.1021/jacs.5b12652.
- N. Han, Y. Wang, L. Ma, J. Wen, J. Li, H. Zheng, K. Nie, X. Wang, F. Zhao, Y. Li, J. Fan, J. Zhong, T. Wu, D. J. Miller, J. Lu, S.-T. Lee and Y. Li, Supported Cobalt Polyphthalocyanine for High-Performance Electrocatalytic CO2 Reduction, Chem, 2017, 3(4), 652–664, DOI:10.1016/j.chempr.2017.08.002.
- Z. Thammavongsy, I. P. Mercer and J. Y. Yang, Promoting Proton Coupled Electron Transfer in Redox Catalysts through Molecular Design, Chem. Commun., 2019, 55(70), 10342–10358, 10.1039/C9CC05139B.
- D. L. DuBois, Development of Molecular Electrocatalysts for Energy Storage, Inorg. Chem., 2014, 53(8), 3935–3960, DOI:10.1021/ic4026969.
- A. Dutta, A. M. Appel and W. J. Shaw, Designing Electrochemically Reversible H2 Oxidation and Production Catalysts, Nat. Rev. Chem., 2018, 2(9), 244–252, DOI:10.1038/s41570-018-0032-8.
- U. J. Kilgore, J. A. S. Roberts, D. H. Pool, A. M. Appel, M. P. Stewart, M. R. DuBois, W. G. Dougherty, W. S. Kassel, R. M. Bullock and D. L. DuBois, [Ni(PPh2NC6H4X2)2]2+ Complexes as Electrocatalysts for H2 Production: Effect of Substituents, Acids, and Water on Catalytic Rates, J. Am. Chem. Soc., 2011, 133(15), 5861–5872, DOI:10.1021/ja109755f.
- B. Ginovska-Pangovska, A. Dutta, M. L. Reback, J. C. Linehan and W. J. Shaw, Beyond the Active Site: The Impact of the Outer Coordination Sphere on Electrocatalysts for Hydrogen Production and Oxidation, Acc. Chem. Res., 2014, 47(8), 2621–2630, DOI:10.1021/ar5001742.
- C. S. Diercks, S. Lin, N. Kornienko, E. A. Kapustin, E. M. Nichols, C. Zhu, Y. Zhao, C. J. Chang and O. M. Yaghi, Reticular Electronic Tuning of Porphyrin Active Sites in Covalent Organic Frameworks for Electrocatalytic Carbon Dioxide Reduction, J. Am. Chem. Soc., 2018, 140(3), 1116–1122, DOI:10.1021/jacs.7b11940.
- S. Campidelli, B. Ballesteros, A. Filoramo, D. D. Diaz, G. de la Torre, T. Torres, G. M. A. Rahman, C. Ehli, D. Kiessling, F. Werner, V. Sgobba, D. M. Guldi, C. Cioffi, M. Prato and J. P. Bourgoin, Facile Decoration of Functionalized Single-Wall Carbon Nanotubes with Phthalocyanines via “Click Chemistry, J. Am. Chem. Soc., 2008, 130(34), 11503–11509 CrossRef CAS PubMed.
- A. Dutta, J. A. S. Roberts and W. J. Shaw, Arginine-Containing Ligands Enhance H2 Oxidation Catalyst Performance, Angew. Chem., Int. Ed., 2014, 53(25), 6487–6491, DOI:10.1002/anie.201402304.
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Metaye, A. Fihri, S. Palacin and M. Fontecave, From Hydrogenases to Noble Metal-Free Catalytic Nanomaterials for H-2 Production and Uptake, Science, 2009, 326(5958), 1384–1387 CrossRef CAS PubMed.
- P. D. Tran, A. Le Goff, J. Heidkamp, B. Jousselme, N. Guillet, S. Palacin, H. Dau, M. Fontecave and V. Artero, Noncovalent Modification of Carbon Nanotubes with Pyrene-Functionalized Nickel Complexes: Carbon Monoxide Tolerant Catalysts for Hydrogen Evolution and Uptake, Angew. Chem., Int. Ed., 2011, 50(6), 1371–1374, DOI:10.1002/anie.201005427.
- P. Chenevier, L. Mugherli, S. Darbe, L. Darchy, S. Dimanno, P. D. Tran, F. Valentino, M. Iannello, A. Volbeda, C. Cavazza and V. Artero, Hydrogenase Enzymes: Application in Biofuel Cells and Inspiration for the Design of Noble-Metal Free Catalysts for H2 Oxidation, C. R. Chim., 2013, 16(5), 491–505 CrossRef CAS.
- M. F. De Volder, S. H. Tawfick, R. H. Baughman and A. Hart, J. Carbon Nanotubes: Present and Future Commercial Applications, Science, 2013, 339(6119), 535–539, DOI:10.1126/science.1222453.
- P. D. Tran, A. Morozan, S. Archambault, J. Heidkamp, P. Chenevier, H. Dau, M. Fontecave, A. Martinent, B. Jousselme and V. Artero, A Noble Metal-Free Proton-Exchange Membrane Fuel Cell Based on Bio-Inspired Molecular Catalysts, Chem. Sci., 2015, 6, 2050, 10.1039/c4sc03774j.
- S. Gentil, N. Lalaoui, A. Dutta, Y. Nedellec, S. Cosnier, W. J. Shaw, V. Artero and A. Le Goff, Carbon-Nanotube-Supported Bio-Inspired Nickel Catalyst and Its Integration in Hybrid Hydrogen/Air Fuel Cells, Angew. Chem., Int. Ed., 2017, 56(7), 1845–1849, DOI:10.1002/anie.201611532.
- S. Gentil, J. K. Molloy, M. Carrière, A. Hobballah, A. Dutta, S. Cosnier, W. J. Shaw, G. Gellon, C. Belle, V. Artero, F. Thomas and A. Le Goff, A Nanotube-Supported Dicopper Complex Enhances Pt-Free Molecular H2/Air Fuel Cells, Joule, 2019, 3(8), 2020–2029, DOI:10.1016/j.joule.2019.07.001.
- F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. Maccato, S. Rapino, B. R. Gonzalez, H. Amenitsch, T. Da Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Efficient Water Oxidation at Carbon Nanotube-Polyoxometalate Electrocatalytic Interfaces, Nat. Chem., 2010, 2(10), 826–831, DOI:10.1038/nchem.761.
- M. R. DuBois and D. L. DuBois, The Roles of the First and Second Coordination Spheres in the Design of Molecular Catalysts for H-2 Production and Oxidation, Chem. Soc. Rev., 2009, 38(1), 62–72, 10.1039/B801197b.
- A. D. Wilson, R. K. Shoemaker, A. Miedaner, J. T. Muckerman, D. L. DuBois and M. R. DuBois, Nature of Hydrogen Interactions with Ni(II) Complexes Containing Cyclic Phosphine Ligands with Pendant Nitrogen Bases, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(17), 6951–6956 CrossRef CAS PubMed.
- D. Zhang, W. Chen, Z. Li, Y. Chen, L. Zheng, Y. Gong, Q. Li, R. Shen, Y. Han, W. C. Cheong, L. Gu and Y. Li, Isolated Fe and Co Dual Active Sites on Nitrogen-Doped Carbon for a Highly Efficient Oxygen Reduction Reaction, Chem. Commun., 2018, 54(34), 4274–4277, 10.1039/c8cc00988k.
- Y. Min, X. Zhou, J.-J. Chen, W. Chen, F. Zhou, Z. Wang, J. Yang, C. Xiong, Y. Wang, F. Li, H.-Q. Yu and Y. Wu, Integrating Single-Cobalt-Site and Electric Field of Boron Nitride in Dechlorination Electrocatalysts by Bioinspired Design, Nat. Commun., 2021, 12(1), 303, DOI:10.1038/s41467-020-20619-w.
- M. Gu, R. Guo, J. Zhang, Y. Yao and L. Yang, Thermal Dissociation of the Singly Protonated Arginine: Competition between Side-Chain and Backbone Fragmentation, Chem. Phys., 2020, 538, 110890, DOI:10.1016/j.chemphys.2020.110890.
- P. M. Gehrig, K. Nowak, C. Panse, M. Leutert, J. Grossmann, R. Schlapbach and M. O. Hottiger, Gas-Phase Fragmentation of ADP-Ribosylated Peptides: Arginine-Specific Side-Chain Losses and Their Implication in Database Searches, J. Am. Soc. Mass Spectrom., 2021, 32(1), 157–168, DOI:10.1021/jasms.0c00040.
- P. Doppelt, G. Hallais, J. Pinson, F. Podvorica and S. Verneyre, Surface Modification of Conducting Substrates. Existence of Azo Bonds in the Structure of Organic Layers Obtained from Diazonium Salts, Chem. Mater., 2007, 19(18), 4570–4575, DOI:10.1021/cm0700551.
- F. Petraki, V. Papaefthimiou and S. Kennou, A Study of the Ni-Phthalocyanine/Gold Interface Using x-Ray and Ultraviolet Photoelectron Spectroscopies, J. Phys.: Conf. Ser., 2005, 10, 135–138, DOI:10.1088/1742-6596/10/1/033.
- A. A. Oughli, A. Ruff, N. P. Boralugodage, P. Rodríguez-Maciá, N. Plumeré, W. Lubitz, W. J. Shaw, W. Schuhmann and O. Rüdiger, Dual Properties of a Hydrogen Oxidation Ni-Catalyst Entrapped within a Polymer Promote Self-Defense against Oxygen, Nat. Commun., 2018, 9(1), 864, DOI:10.1038/s41467-018-03011-7.
- A. Dutta, D. L. DuBois, J. A. S. Roberts and W. J. Shaw, Amino Acid Modified Ni Catalyst Exhibits Reversible H2 Oxidation/Production over a Broad PH Range at Elevated Temperatures, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(46), 16286–16291 CrossRef CAS PubMed.
- L. Korson, W. Drost-Hansen and F. J. Millero, Viscosity of Water at Various Temperatures, J. Phys. Chem., 1969, 73(1), 34–39, DOI:10.1021/j100721a006.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05168g |
| This journal is © The Royal Society of Chemistry 2021 |