
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomic origins of the strong metal–support interaction in silica supported catalysts†
Feng
Yang‡
 ab,
Haofei
Zhao‡
c,
Wu
Wang‡
d,
Lei
Wang
b,
Lei
Zhang
b,
Tianhui
Liu
b,
Jian
Sheng
a,
Sheng
Zhu
a,
Dongsheng
He
e,
Lili
Lin
f,
Jiaqing
He
*d,
Rongming
Wang
*c and
Yan
Li
*a
ab,
Haofei
Zhao‡
c,
Wu
Wang‡
d,
Lei
Wang
b,
Lei
Zhang
b,
Tianhui
Liu
b,
Jian
Sheng
a,
Sheng
Zhu
a,
Dongsheng
He
e,
Lili
Lin
f,
Jiaqing
He
*d,
Rongming
Wang
*c and
Yan
Li
*a
aBeijing National Laboratory for Molecular Science, Key Laboratory for the Physics and Chemistry of Nanodevices, State Key Laboratory of Rare Earth Materials Chemistry and Applications, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: yanli@pku.edu.cn
bDepartment of Chemistry, Southern University of Science and Technology, Shenzhen, Guangdong 518055, China
cBeijing Advanced Innovation Center for Materials Genome Engineering, Center for Green Innovation, Beijing Key Laboratory for Magneto-Photoelectrical Composite and Interface Science, School of Mathematics and Physics, University of Science and Technology Beijing, Beijing 100083, China. E-mail: rmwang@ustb.edu.cn
dDepartment of Physics, Southern University of Science and Technology, Shenzhen, Guangdong 518055, China. E-mail: hejq@sustech.edu.cn
eCore Research Facilities, Southern University of Science and Technology, Shenzhen, Guangdong 518055, China
fState Key Laboratory of Green Chemistry Synthesis Technology, Zhejiang University of Technology, Hangzhou 310032, China
First published on 18th August 2021
Abstract
Silica supported metal catalysts are most widely used in the modern chemical industry because of the high stability and tunable reactivity. The strong metal–support interaction (SMSI), which has been widely observed in metal oxide supported catalysts and significantly affects the catalytic behavior, has been speculated to rarely happen in silica supported catalysts since silica is hard to reduce. Here we revealed at the atomic scale the interfacial reaction induced SMSI in silica supported Co and Pt catalysts under reductive conditions at high temperature using aberration-corrected environmental transmission electron microscopy coupled with in situ electron energy loss spectroscopy. In a Co/SiO2 system, the amorphous SiO2 migrated onto the Co surface to form a crystallized quartz-SiO2 overlayer, and simultaneously an interlayer of Si was generated in-between. The metastable crystalline SiO2 overlayer subsequently underwent an order-to-disorder transition due to the continuous dissociation of SiO2 and the interfacial alloying of Si with the underlying Co. The SMSI in the Pt–SiO2 system was found to remarkably boost the catalytic hydrogenation. These findings demonstrate the universality of the SMSI in oxide supported catalysts, which is of general importance for designing catalysts and understanding catalytic mechanisms.
Introduction
Oxide supported metal catalysts are of fundamental importance in a broad range of applications such as petrochemical engineering, energy conversion, and heterogeneous catalysis,1–5 for both organic and inorganic synthesis.6 Oxide supports were previously considered to be inert, and their primary role is to disperse and stabilize the metal catalysts. However, later it was demonstrated that oxide supports can manipulate the physicochemical properties of metal nanoparticles and influence the catalytic performance through so-called carrier effects.7,8 The so-called strong metal–support interaction (SMSI) was reported more than 40 years ago.9–11 It presents geometric, electronic, and compositional effects that can be used to control the catalyst structure, properties, and performance. SMSI in oxide-supported metal catalysts has been intensively studied.12–15Many efforts have been focused on addressing the SMSI effect using either reducible or hard-to-reduce oxides or inert non-oxides16,17 as supports. Among various supports, reducible oxide supports, such as TiO2,18–28 CeO2,29–32 FeOx,33–35 Ta2O5,36 and Nb2O5,36 were commonly adopted to establish the classical SMSI systems. These reducible oxide supports undergo redox-induced dynamic structural changes and then migrate onto the metal surface to encapsulate the metal nanoparticles, maximizing the metal-oxide contact surface and the interfacial energy.19 For example, in metal/TiO2 systems, SMSI resulted from the partial reduction of the oxide support, which induced the removal of the surface lattice oxygen of TiO2 and the migration of substoichiometric TiO2−x onto the metal surface to form an overlayer.18–20 It was hypothesized that bonding between the Ti atoms of the reducible TiO2 support and the metal catalyst surface made the migration of the oxide support onto the metal thermodynamically favorable.23,37 The chemical bonding between reducible TiO2 and the metal catalyst may also induce electron transfer from Ti3+ species to metal nanoparticles, and further migration to reactant molecules.21 Consequently, classical SMSI has been utilized to improve the catalyst stability20,38–40 and activity,41 and identify the mechanisms.18
It is generally accepted that encapsulation of metal nanoparticles hardly occurs on hard-to-reduce oxide-supports such as SiO2,10 MgO,42 and ZnO.23,43,44 Nonetheless, building an appropriate oxide–metal interface has always attracted great attention in industrial processes, in which SiO2 is of greater interest.45–48 For instance, the Fe/SiO2 catalyst has been shown to be promising for direct nonoxidative conversion of methane to ethylene, aromatics, and hydrogen. Iron species were found to interact extensively with SiO2 supports, becoming embedded in the silica matrix through bonding with Si after annealing at 1500 °C. Thus, these otherwise extremely reactive and coordinatively unsaturated iron atoms were stabilized and persisted under the very harsh reaction conditions.45 Recent reports proposed the construction of a Si–metal interface by depositing thin film SiO2 onto metal nanoparticles at high temperature.46,49 Owing to the interaction between SiO2 and metal, the catalyst was resistant to sintering and accelerated the activation of O2 (ref. 46) and the hydrogenation of CO2 to ethanol.49 Besides, a thin layer of Al2O3 between the metal catalysts and the silicon substrate was generally adopted to improve the growth efficiency of vertical single-walled carbon nanotube forests. It is believed that the presence of the Al2O3 layer is critical for the formation of high-density metal nanocatalysts of small size, which are stable throughout the high temperature (800–1000 °C) process.6,50–53 However, SiO2 was proven to be invalid in the same process. Though the mechanism is still not very clear, the interaction between silica and the metal catalyst may play a role. There have been only a few examples of metal–silica interaction revealed by X-ray photoelectron/absorption spectroscopy, diffraction or magnetic measurements.10,37,54–56 However, much less is known about the atomic-scale origins of the silica-based SMSI, silica–metal interface and their interfacial interaction under reactive conditions compared to that of titania–metal systems.
Here, we report the unequivocal finding of SMSI in an industrially relevant Co/SiO2 model catalyst where SiO2 migrated onto Co nanocrystals and the SMSI originated from the interfacial reduction induced SiO2–Si–Co structure, by employing the state-of-the-art aberration-corrected environmental transmission electron microscopy (ETEM)18,26,57–64 combined with in situ electron energy loss spectroscopy (EELS),65,66 thus providing direct visualization and chemical evolution at the atomic scale. The dynamic evolution of the interface accompanied by the interfacial Si–Co alloying was also investigated in this SMSI system. These findings greatly broaden the applicability of the SMSI concept on hard-to-reduce oxide supports and unlock new opportunities to design thermally stable, surface-alloyed metal catalysts.
Results and discussion
Sinter-resistant Co nanoparticles with an overlayer on the SiO2 substrate
The heating chips fitted in the Titan G2 80-300 ETEM micro-heater used in this work contain an amorphous SiNx membrane with a native oxide layer of SiO2, which was confirmed by energy dispersive X-ray (EDX) elemental mapping and quantification analysis (Fig. S1 and Table S1†). A Co thin film (thickness: ∼1 nm) was sputtered on the surface of the SiO2 film. Co nanoparticles formed on the substrate when annealing in 3% H2/Ar (50 Pa) at 750 °C with a size distribution of 5–10 nm. To avoid the effect of beam irradiation, we performed in situ heating with beam off and then captured the ETEM images with beam on under 80 kV. When annealing Co nanoparticles for 7294 s, surprisingly, we did not observe any significant aggregation or leaching of Co nanoparticles on the substrate (Fig. 1a and b), indicating the high thermal stability of these nanoparticles on SiO2/SiNx. The selected-area electron diffraction (SAED) patterns of the nanoparticles before and after annealing both show the diffraction rings of metallic Co (Fig. S2†). For comparison, we also prepared Co nanoparticles on amorphous carbon films and performed ETEM. It was clearly observed that some Co metal nanoparticles evaporated at 550 °C (Fig. 1c and d, marked by dashed circles, and Fig. S3†). The behaviors of Co nanocrystals on SiO2/SiNx and carbon substrates indicate different thermal stabilities. | ||
| Fig. 1 In situ ETEM characterization of the stability of Co nanocrystals. (a–d) Sequential ETEM images showing Co nanoparticles before and after annealing on SiO2/SiNx (a and b) and carbon film (c and d) in 3% H2/Ar, which were acquired at the same region of the sample. The circles are meant to help with comparison. (e–g) Typical high resolution ETEM images of semi-suspended Co nanoparticles with an overlayer on SiO2/SiNx acquired under different reactive conditions: CO (280 Pa) (e), 3% H2/Ar (50 Pa) (f), and CH4 (63 Pa) (g). Inset: FFT patterns of the particle region. | ||
To investigate the origin of the unexpected thermal stability of Co nanoparticles on SiO2/SiNx and achieve an atomic-scale observation, we chose the semi-suspended Co nanocrystals standing on the edge of the substrate to study the interaction between Co and SiO2/SiNx. We found that a crystallized single layer formed as a conformal coating on the surface of a Co nanocrystal from the ETEM image acquired in the H2 atmosphere (Fig. 1f) and Co nanoparticles of 4–20 nm could all be coated. The in situ high-angle annular dark field scanning TEM (HAADF-STEM) images also clearly demonstrated that the Co nanoparticles were partially covered by a layer which showed a similar contrast to the Co and SiO2/SiNx substrate (Fig. S4†), which excluded the overlayer consisting of light elements such as carbon. Furthermore, the crystallized overlayer was also observed when exposing Co nanoparticles to various reductive gases, such as CO (280 Pa), CH4 (63 Pa), and a mixture of CH4/H2 (v/v = 7/3, 50 Pa) (Fig. 1e and g, and S5†), and both face-centered-cubic (fcc) and hexagonal-close-packing (hcp) Co nanoparticles of 4–20 nm were coated with overlayers (Fig. S5†), suggesting the universality of the formation of the overlayer. Generally, there is only one atomic layer coated on each Co particle. In a few cases, there are 2 to 4 layers (Fig. S6†). It is also worth noting that all the overlayers started from the metal–substrate boundaries (see arrows in Fig. 1e–g and S5†), arguably through the diffusion of species from the substrate to the Co surface.
Atom-resolved structure and composition of the Co–SiO2 interface
To confirm the composition and structure of the overlayer on the Co particle surface, we performed in situ STEM-EELS characterization of Co nanoparticles on SiO2/SiNx under H2 at 750 °C. It was demonstrated by EELS elemental mapping that Si, O, and N were distributed continuously and uniformly on the substrate, again suggesting a homogeneous coating of SiO2 on SiNx (Fig. 2a–e). The EELS elemental mapping of an individual Co nanoparticle semi-suspended on the substrate confirmed that the overlayer on the Co nanoparticle was composed of both O and Si (Fig. 2d–f). It was consistently found from the line scan profile of EELS across the Co-overlayer that only Si and O coexist on the surface of Co (Fig. S7†). Carbon and SiNx were also excluded because there was no C or N signal on the Co particle (Fig. 2c, and S8a and b†). The study performed in a reductive H2 environment excluded the possibility of the presence of CoOx in the overlayer because CoOx can be reduced by H2 at 750 °C. For those Co nanoparticles annealed in CO and CH4, we also performed the EELS mapping of the sample under ETEM and did not find the C signal (Fig. S8c and d†). Hence, the overlayer is not a graphene layer.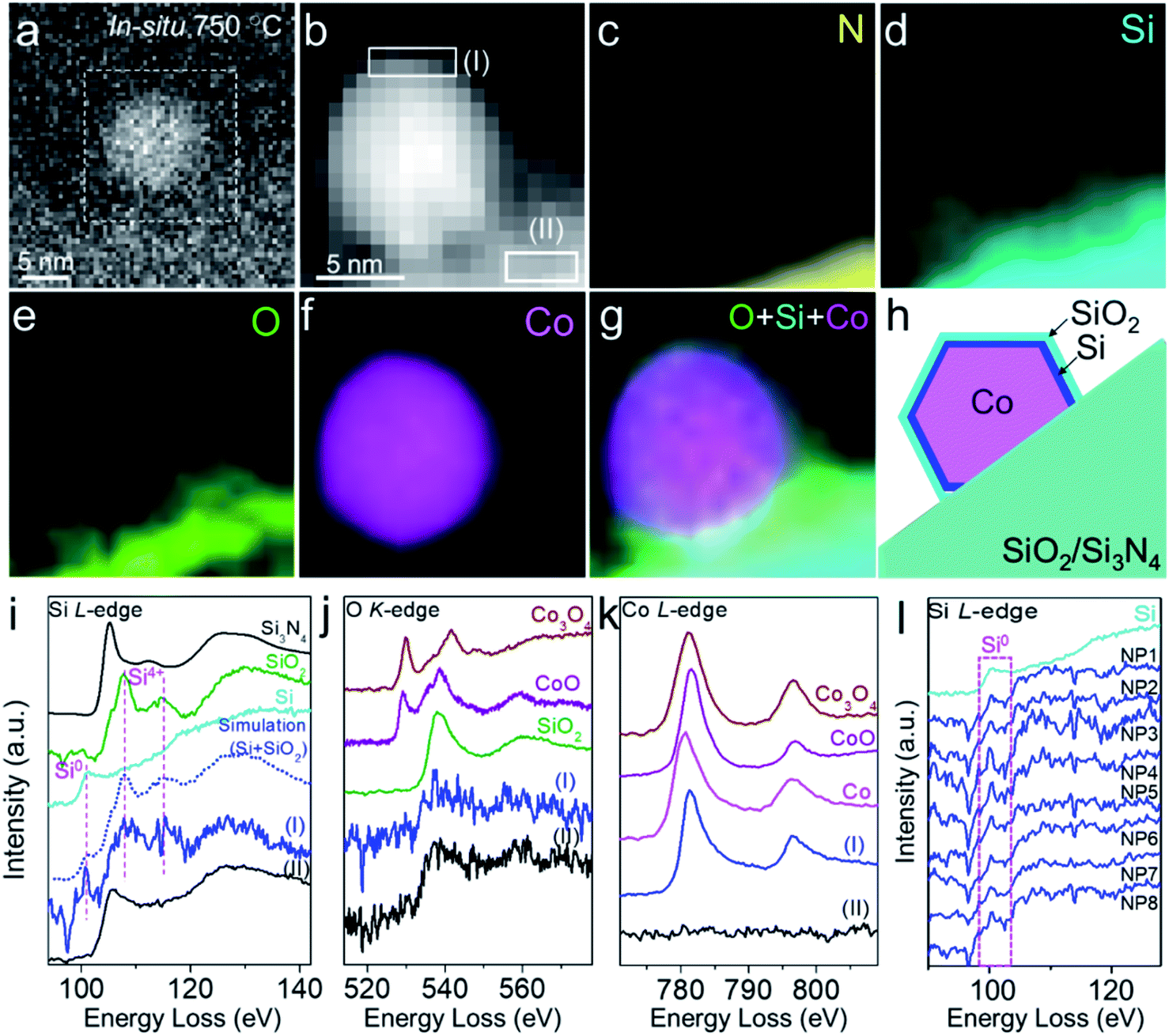 | ||
| Fig. 2 In situ EELS characterization of the crystalline SiO2 overlayer on the Co surface. (a and b) In situ HAADF-SETM (a) and EELS spectrum image (b) showing a Co nanoparticle on SiO2/SiNx in 3% H2/Ar (50 Pa) at 750 °C. (c–g) Corresponding EELS elemental mapping images of the N K-edge (c), Si L2,3-edge (d), O K-edge (e), and Co L2,3-edge (f), and the combined image (g). Mapping step size: 1.1 nm. (h) Schematic of the SiO2 overlayer on a Co nanoparticle with a Si interface. (i–k) EELS spectra extracted from regions I and II in (b), showing Si (i), O (j), and Co (k) signals from the near-surface of the nanoparticle and substrate. (l) Merged EELS spectra of the Si L-edge from different Co nanoparticles (NP1–8). Commercial Si and SiO2, Si3N4, Co, CoO, and Co3O4 from the EELS database were used as references. All the spectra were calibrated using the zero-loss peak (0 eV). | ||
In situ EELS spectra extracted from elemental mapping were also used to reveal the fine structure of the overlayer. The Si L2,3-edge spectra extracted from the near-surface (thickness: 1.6 nm) of a particle (Fig. 2b, region I) showed that its peak positions and fine structures were very similar to those of both quartz-structure α-SiO2 (107.7 and 115.0 eV) and Si (100.1 eV), while the Si signal from the substrate (Fig. 2b, region II, and Fig. 2i) exhibited a mixture of structures of Si3N4 and SiO2. We also performed EELS simulation with the combination of standard Si and SiO2 (molar ratio of Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SiO2 = 1:2). The simulated EELS spectrum matched well with the experimental result (Fig. 2i, dashed line), which further confirmed the existence of Si in the overlayer. The energy loss peaks of the O K-edge from the overlayer and substrate were both associated with SiO2 instead of Co3O4 or CoO (Fig. 2j). These data further verified that there was no cobalt oxide in the overlayer, and O was attributed to SiO2. In the Co regime, the energy loss peaks of the L2,3-edge (780.6 and 795.7 eV) and the intensity ratio of L2/L3 acquired from the near-surface were similar to those of metallic Co (Fig. 2k). The in situ EELS result clearly evidenced that the SiO2 overlayer migrated from SiO2/SiNx onto the Co surface with the formation of Si in-between and such interfaces existed in all observed nanoparticles (Fig. 2l). The configuration of the SiO2–Si–Co interface is schematically illustrated in Fig. 2h.
:SiO2 = 1:2). The simulated EELS spectrum matched well with the experimental result (Fig. 2i, dashed line), which further confirmed the existence of Si in the overlayer. The energy loss peaks of the O K-edge from the overlayer and substrate were both associated with SiO2 instead of Co3O4 or CoO (Fig. 2j). These data further verified that there was no cobalt oxide in the overlayer, and O was attributed to SiO2. In the Co regime, the energy loss peaks of the L2,3-edge (780.6 and 795.7 eV) and the intensity ratio of L2/L3 acquired from the near-surface were similar to those of metallic Co (Fig. 2k). The in situ EELS result clearly evidenced that the SiO2 overlayer migrated from SiO2/SiNx onto the Co surface with the formation of Si in-between and such interfaces existed in all observed nanoparticles (Fig. 2l). The configuration of the SiO2–Si–Co interface is schematically illustrated in Fig. 2h.
Based on EELS results, we combined the high-resolution ETEM images to resolve the atomic structure of the crystalline overlayer. Fig. 3a shows a close-up view of an overlayer stabilized on the surface of a Co nanocrystal. The fast Fourier transform (FFT) derived from the interfacial region between Co and the overlayer shows at least two groups of diffraction spots, indicating the inhomogeneous structure (Fig. 3b). Among them, one group of spots marked by circles corresponds to the fcc-Co along the [110] direction. Another group marked by boxes with spacings of 3.4 Å, 3.4 Å, and 4.3 Å could readily be attributed to quartz SiO2 along the [10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ] direction (ICSD 16331, space group: P3221) (Fig. S9†). Additionally, we also noted that a pair of diffraction spots at around 3.4 Å expanded (Fig. 3b, marked by dashed ellipses), which probably originated from different interlayer distances of SiO2–Si–Co as Si was evidenced by in situ EELS. An interface structure of SiO2–Si–Co was proposed, in which Si (ICSD 51688, space group Fd
] direction (ICSD 16331, space group: P3221) (Fig. S9†). Additionally, we also noted that a pair of diffraction spots at around 3.4 Å expanded (Fig. 3b, marked by dashed ellipses), which probably originated from different interlayer distances of SiO2–Si–Co as Si was evidenced by in situ EELS. An interface structure of SiO2–Si–Co was proposed, in which Si (ICSD 51688, space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) was orientated along the [001] direction. The simulated TEM image based on this proposed atomic interface structure matched well with the experimental image (Fig. 3c–e). These in situ ETEM and EELS results demonstrated that a crystallized SiO2 overlayer was formed on the Co surface with an interface structure of SiO2–Si–Co.
m) was orientated along the [001] direction. The simulated TEM image based on this proposed atomic interface structure matched well with the experimental image (Fig. 3c–e). These in situ ETEM and EELS results demonstrated that a crystallized SiO2 overlayer was formed on the Co surface with an interface structure of SiO2–Si–Co.
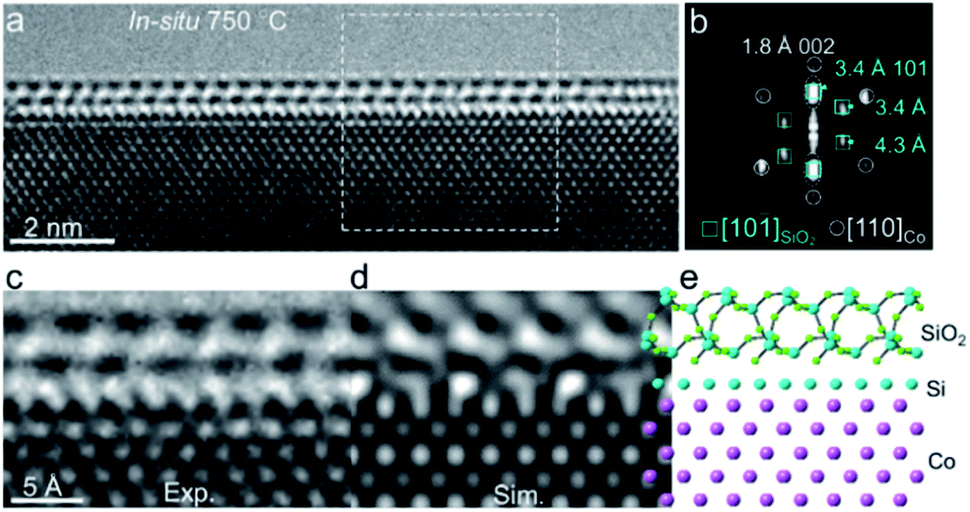 | ||
| Fig. 3 Atomic-resolution ETEM characterization of the crystalline SiO2 overlayer on the Co surface. (a) Close-up view of Co with a double overlayer acquired at 750 °C under 3% H2/Ar (50 Pa). (b) Corresponding FFT from the interface region marked in (a). (c–e) Experimental TEM image (c) and simulated TEM image (d) obtained from the proposed atomic model of the SiO2–Si–Co interface (e), along the orientations of α-SiO2 [10], Si [100], and fcc-Co [110], respectively. | ||
We also prepared supported Co nanoparticles on SiO2 spheres (∼400 nm in size). The powder sample was annealed in a tube furnace under 10% H2/Ar at 750 °C for 6 h. It was found from STEM-EDX elemental mapping that Si and O appeared on Co nanoparticles (Fig. S10†), demonstrating the general observation of diffusion of SiO2 onto Co.
SMSI of Co/SiO2 induced surface evolution of Co nanocrystals
Fig. 4a–d show a round-to-faceted Co particle transformation coinciding with the migration of the SiO2 overlayer on the Co surface, which was captured in situ under 3% H2/Ar (50 Pa) at 750 °C. At a moment designated as 0 s, the low index facets, {020} and {220}, and high index facet, {420}, were identifiable. The shape of Co appeared to be a truncated polyhedron with rounded corners. A crystallized SiO2 overlayer already formed on the (020) plane of the Co nanoparticle (Fig. 4a). When further heated, the SiO2 overlayer gradually grew from the (220) plane to the (200) plane of the Co particle accompanied by the transformation of SiO2 from an amorphous to a crystallized single layer. Meanwhile, the low index (220) and (200) facets gradually expanded, while the high index (420) facet shrank, as shown in the sequence of Fig. 4a–d. The FFT pattern derived from the particle region confirmed the orientation of the Co nanoparticle (Fig. 4d, inset). We further quantified the shape evolution by tracking the propagation of different Co facets. Changes in the length of each Co facet as a function of time were plotted in Fig. 4e, which clearly showed the transformation described above. Besides, we also noted that the high index (4![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0) facet, which is adjacent to the (200) facet, exhibited a clean surface without the SiO2 overlayer and did not significantly change during the process (Fig. 4a–e).
0) facet, which is adjacent to the (200) facet, exhibited a clean surface without the SiO2 overlayer and did not significantly change during the process (Fig. 4a–e).
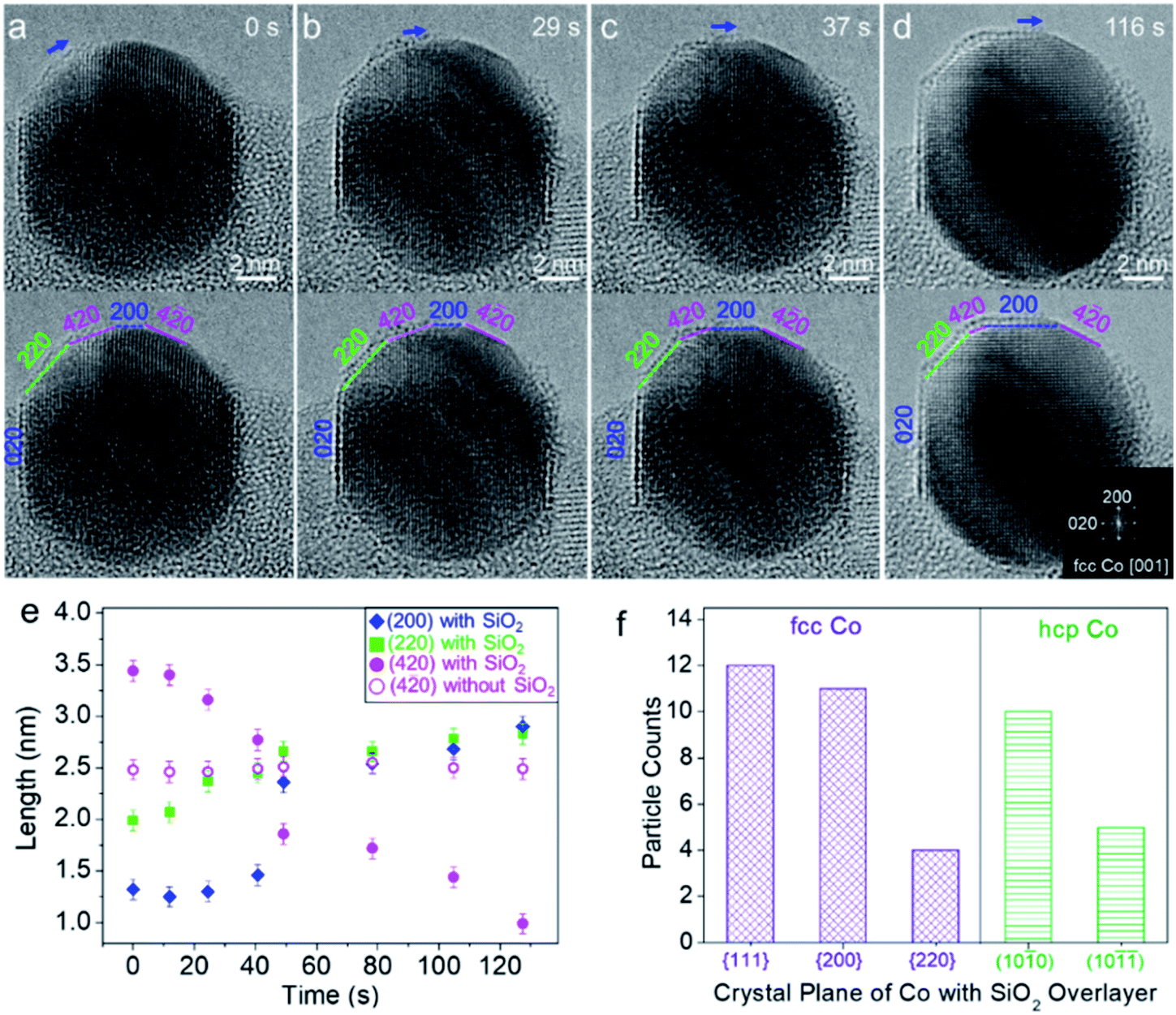 | ||
| Fig. 4 SMSI induced dynamic shape evolution of a Co nanocrystal with a SiO2 overlayer. (a–d) Time-sequenced ETEM images of a Co nanocrystal during SiO2 overlayer formation. The same TEM images were highlighted with facets of the nanocrystal. The growth direction of SiO2 was marked by arrows. Inset in (d): FFT pattern derived from the particle region. (e) Length of facets plotted against annealing time. (f) Statistic of the formation of the SiO2 overlayer on different crystal planes of fcc and hcp Co nanoparticles. | ||
To exclude the effect of electron beam irradiation induced facet evolution, we also performed in situ heating with beam off and then captured the ETEM images with beam on. It was found from single-shot ETEM images that the Co surface with a SiO2 overlayer exhibited a flat facet with a low index, such as fcc-Co (002), (111) and hcp-Co (100), while the surface without the SiO2 overlayer exposed the round surface consisting of high index crystal planes (Fig. S11†). The statistical TEM analysis based on these in situ observations further showed that the SiO2 overlayer preferentially stabilized on the low index facets of fcc-Co {111}, {200}, and {220} as well as hcp-Co (100) and (10) (Fig. 4f). The reconstruction and evolution of high index (420) facets to low index (200) facets during the SMSI process could be thermodynamically favourable. Because it was reported that adhesion energy between the metal and oxide overlayer is negative (γadh < 0 eV) and the extension energy of the two facets is inversely proportional to their surface energy, that is, the lower the surface energy, the more the facet is extended (A200/A420 ∝ γ420/γ200 > 1).4,19 Contact of the SiO2 overlayer with Co will result in a net increase of the ratio of the surface area A200/A420 (i.e., a net increase of ratio of surface energies (γ420 + γadh)/(γ200 + γadh)). A similar tendency has been reported in a TiO2–SMSI system where the decrease of the whole surface energy is generally considered to be the driving force for the encapsulation of reducible oxide TiO2.19,67 Besides the effect of SiO2 coverage, other factors such as alloying of two components68 and gas adsorption4,69 may also affect the surface energies and reconstruction of Co nanocrystal facets. From these results, it was believed that SMSI was initiated by the partial reduction of SiO2 at the SiO2–Co interface, which can be projected by the facet evolution of Co nanocrystals.
It was generally accepted that the ordered crystalline oxide was very effective in blocking access of gas molecules to the underlying metal surface, while the amorphous oxide overlayer was gas permeable.9,70 We found that most of the Co nanoparticles were coated by the amorphous SiO2 overlayer after heating at 750 °C for 8 h (Fig. 5a). The sample was annealed using an ETEM with the electron beam off. The order-to-disorder transition of the SiO2 overlayer could be attributed to continuous reduction of SiO2 at the interface, thus destroying the lattice of quartz SiO2. The as-reduced Si atoms gradually diffused into Co to probably form a silicide alloy, which was schematically shown in Fig. 5d and e. This deduced process was evidenced by in situ STEM-EELS measurement. The in situ EELS mapping showed that Si distributed in both the surface and body of Co nanoparticles while the O was surface-rich (Fig. 5b), indicating the diffusion of Si into the body of the Co nanoparticle. More Co/SiO2 particles displaying the similar behaviour of Si diffusion are shown in Fig. S12.† It was consistently found from in situ EELS spectra by comparing the intensity ratio of the Co L3/L2 edge that the Co signal was close to that of the Si–Co alloy instead of metallic Co after the order-to-disorder transition of the SiO2 overlayer (Fig. 5d, S13 and S14†).
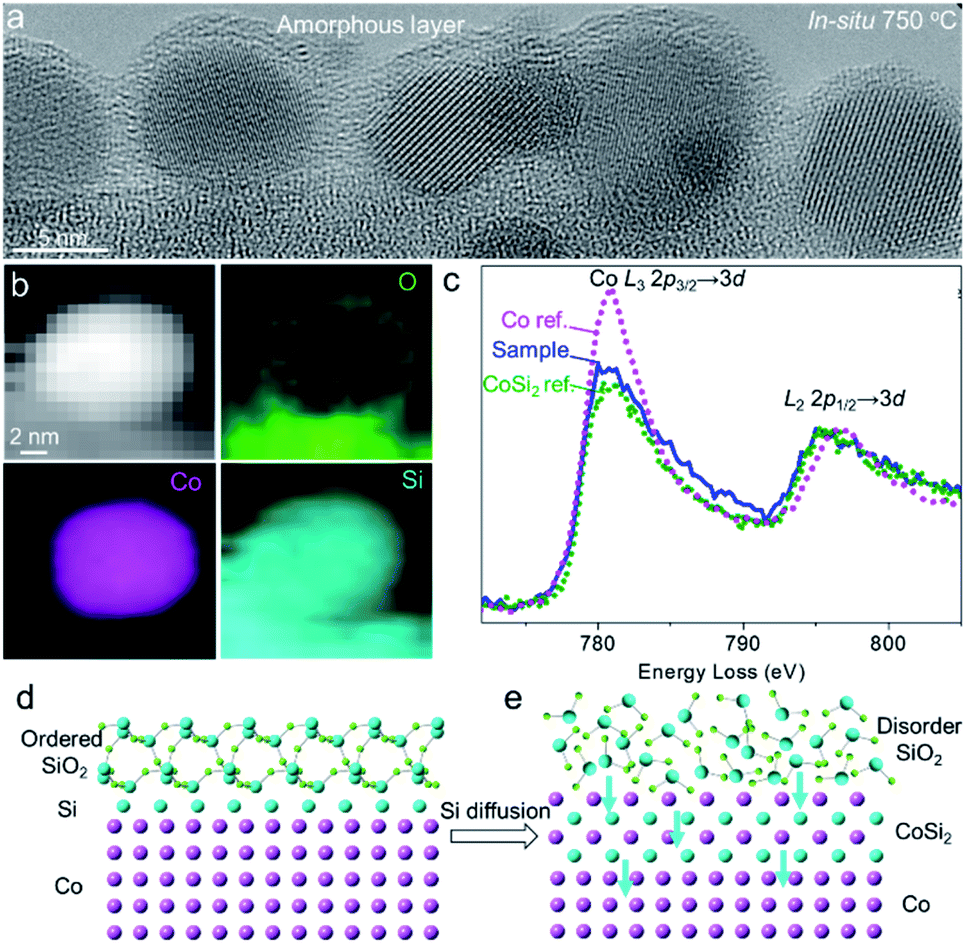 | ||
| Fig. 5 Order-to-disorder transition of the SiO2 overlayer on Co nanoparticles. (a) Single-shot ETEM image showing the amorphous SiO2 overlayer on Co nanoparticles. (b) In situ EELS mapping of a Co/SiO2 showing the distributions of O, Co, and Si. (c) In situ EELS Co L2,3 spectra after the SiO2 order-to-disorder transition. The metallic Co and CoSi2 were used as references. The spectra were calibrated by the zero-loss peak (0 eV) and then normalized by the energy loss peak of the Co L2-edge. (d and e) Proposed model of ordered to amorphous transition of the SiO2 overlayer coinciding with diffusion of Si in Co. | ||
These ETEM and in situ EELS results clearly reveal that SiO2 migrated to the Co surface and a quartz SiO2 overlayer was formed. The formation of Si most probably originated from a mechanism proposed by D. Duprez et al.,71 in which silica is reduced to form Si by hydrogen spillover from the metal to the oxide. In our in situ experiments, various reductive gases such as H2, CH4, and CO were used (Fig. 1e–g and S5†), which may undergo spillover from metallic Co to SiO2 and reduce SiO2 at 750 °C. The hydrogen spillover effect lowers the reduction temperature of SiO2 as follows:
| Co–Ointer–Si–O–Si–O → Co–Si–O–Si–O + ½O2 (g) | (i) |
| ½O2 (g) + H2 (g) → H2O (g) | (ii) |
Subsequently, the interfacial alloying of Co–Si promotes the interaction between Co and the SiO2 support and prevents the diffusion of Co atoms or the migration of Co nanoparticles, thus inhibiting the sintering of Co nanoparticles. The two-step reaction was dependent on the reaction temperature and the pressure of H2 (or other reducible gases) and O2. At lower temperature (<600 °C), we did not find the interfacial reaction induced SMSI effect. The vacuum could also promote the interfacial alloying of Co and Si, similar to what was proposed in early studies for the interaction of Cu/SiO2 (ref. 72) and Rh/SiO2.37 In the metal–silica (metal–O–Si–O) contact, oxygen was first transformed into a chemisorption state on the metal surface as shown in the above step (i), from where it is subsequently thermally desorbed and vacuumed. Additionally, the interaction between the metal and Si could further induce interfacial alloying to form a silicide alloy.
Boosting the catalysis of hydrogenation with SMSI induced Pt/SiO2 catalysts
Inspired by the ETEM results of SMSI in metal–silica, the special chemical properties derived from the strong interaction between supported metal and the silica substrates have been utilized in the catalytic hydrogenation reaction. We prepared Pt nanoparticles supported on SiO2 spheres (400 nm in size) (Fig. S16†). A series of high-resolution ETEM images of the same Pt nanoparticle on the SiO2 substrate annealed at different temperatures in 3% H2/Ar (50 Pa) are shown in Fig. 6a–d. The sample was annealed in ETEM with electron beam off. The SMSI induced by the migration of SiO2 on Pt and diffusion of Si in Pt nanoparticles also occurred when annealing the sample at temperatures higher than 600 °C, which was simultaneously evidenced by in situ ETEM and ex situ STEM-EDX elemental mapping (Fig. 6a–g). The ex situ high-resolution TEM images and corresponding FFT showed that the Pt nanoparticles were partially alloyed with Si to form Pt3Si (Fig. S16†), which show similar behavior to that of Co/SiO2. The thickness of the overlayer increased with increasing temperature from 600–750 °C (Fig. 6a–d).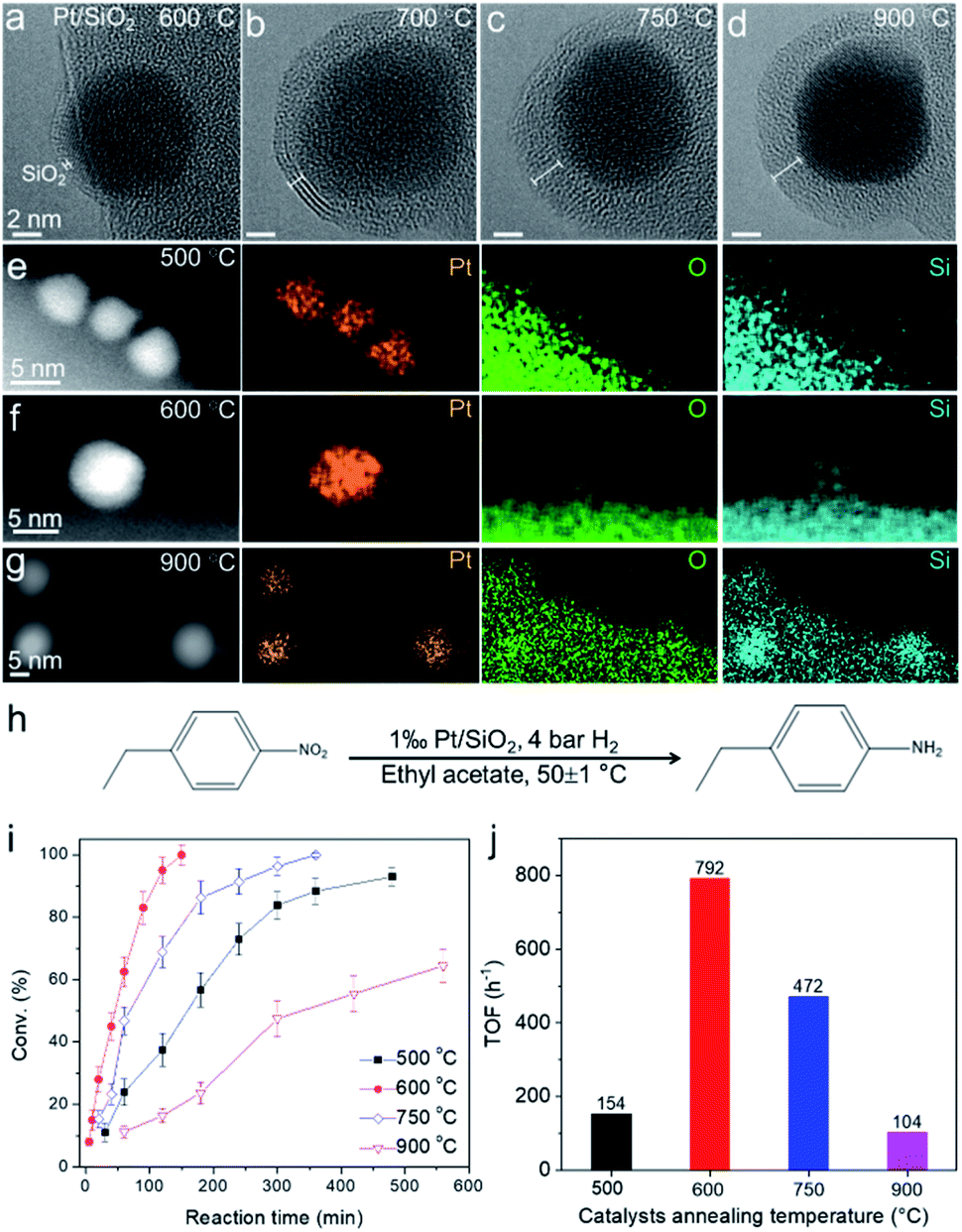 | ||
| Fig. 6 Pt/SiO2 catalysts for the hydrogenation reaction. (a–d) ETEM images of the same Pt/SiO2 particle acquired at 600–900 °C in H2 (50 Pa). (e–g) Ex situ STEM-EDX elemental mapping of Pt/SiO2 after annealing at 500, 600, and 900 °C. (h–j) Catalytic performance for the 4-nitroethylbenzene hydrogenation over Pt/SiO2 catalysts annealed at different temperatures (500, 600, 750, and 900 °C). The conversion values are the average of 3 parallel catalysis experiments. The TOF values were calculated under the conversion of <20% based on the total Pt loading in the catalysts. | ||
The Pt/SiO2 (0.1 at% Pt) powder samples annealed at 500–900 °C in H2 were tested for the catalytic hydrogenation of 4-nitroethylbenzene at 50 ± 1 ○C (Fig. 6h). The conversion of 4-nitroethylbenzene as a function of time was plotted in Fig. 6i for a series of catalysts annealed at 500–900 °C, and different catalytic behaviors were evident. The best performance with a turnover frequency (TOF) of 792 h−1 was observed for the 600 °C-annealed Pt/SiO2 catalyst (Fig. 6j). Since the formation of a SiO2 overlayer on Pt and diffusion of Si in Pt nanoparticles were observed in the sample annealed at 600 °C, we believe the strong interaction between Pt and diffused Si in the Pt/SiO2 catalyst played a great role in boosting the catalytic performance. Pt atoms became more electron-rich via the donation of electrons from Si because the electronegativity of Pt (2.28) is larger than that of Si (1.91). The electron-rich characteristics of Pt induced by the SMSI would lead to a higher hydrogen coverage and thus accelerate the hydrogenation reaction.73 However, the decreased activities of samples prepared at higher temperatures (750 °C and 900 °C) could be tentatively ascribed to two critical factors. One is the over-coated SiO2 layer on Pt, which may block the access of molecules to the underlying metal surface. This was evidenced by CO adsorption characterization where the active Pt surface area reaches the largest value of 4.15 m2 per g of catalyst (600 °C) and decreases sharply to 1.73 m2 per g of catalyst (900 °C) (Table S2†). The other is the catalyst particle aggregation, which may also result in decreased activities. We performed a statistical TEM analysis of the Pt nanoparticle size prepared at 500–900 °C. The average size of Pt nanoparticles increased from ∼3.6 nm to ∼5.7 nm when increasing the annealing temperature to 750–900 °C (Fig. S17†). The above results provide possibilities for controlling the electronic structure of the metal catalyst by accommodating diffusion of the Si via metal–silica SMSI. We performed a statistical analysis of size distribution of the Pt nanoparticles supported on SiO2 after the hydrogenation reaction and did not find the aggregation of catalysts (Fig. S18†). This indicated that the catalysts were also stable after the hydrogenation reaction.
Conclusion
The present observations illustrate spatially and chemically resolved evidence for SMSI induced structural and compositional evolution of SiO2 supported metal catalysts. Both atom and element resolved characterization clearly demonstrated that amorphous SiO2 migrated onto the Co nanoparticle surface to form a crystallized quartz SiO2 overlayer. We found that the SMSI is induced by the formation of Si at the Co–SiO2 interface at high temperature under various reductive atmospheres of H2, CO, and CH4. Upon subsequent exposure to high temperature, the metastable crystalline SiO2 transformed to an amorphous overlayer, with the diffusion of Si into Co and the formation of a silicide alloy at the surface of the Co nanoparticles. The SMSI was also validated in a Pt–SiO2 system, and it greatly boosted the catalytic hydrogenation. The metal nanocrystals with a Si-based intermetallic surface are stable and potentially open new possibilities for catalysis. Our research may also provide a route for the synthesis of nano-sized silicide intermetallic compounds that have recently become a central focus of research in magnetism.74Experimental section
Preparation of Co/SiO2 and ETEM experiments
The in situ experiments were conducted using an aberration-corrected Titan G2 80-300 ETEM operated at 80 kV equipped with a MEMS-based heating holder (NanoEx-i/v). The TEM images were acquired at an electron dose-rate of 107 e− Å−2 s−1. A thin film of Co (thickness: 1 nm) was sputtered onto the amorphous SiO2/SiNx thin film of the MEMS chip purchased from Thermo Fisher Scientific. The as-prepared specimen was heated to 750 °C under 3% H2/Ar (50 Pa). After that, TEM images and videos were captured. Other reducing gases, CH4 (99.99%, 63 Pa), CH4/H2 (v/v = 7/3, 50 Pa), and CO (99.999%, 280 Pa), were also used instead of H2/Ar to introduce to TEM chamber. Besides the sputtered Co film, we also used cobalt(II) acetate (Co(OAC)2) dissolved in ethanol (0.1 mmol L−1) as a precursor to prepare Co nanoparticles on the SiO2/SiNx thin film. The as-prepared Co(OAC)2 specimen was first heated to 500 °C in pure O2 (50 Pa) for 30 min to form Co3O4 and reduced in H2 (50 Pa) at 700 °C for 30 min to form metallic Co catalysts. The sizes of Co nanoparticles prepared from Co(OAC)2 are larger than those from the sputtering method. These larger Co nanoparticles (∼5–20 nm) were also used to perform ETEM, EELS, and EDX mapping to achieve high spatial resolution.In situ EELS experiments
In situ STEM-EELS data were collected in another, but otherwise the same, aberration-corrected Titan G2 80-300 ETEM. It was equipped with a Gatan image filter (Quantum 936) with an energy dispersion of 0.25 eV operated at an accelerating voltage of 300 kV. The zero-loss EELS was acquired immediately after obtaining the core-loss EELS on individual nanoparticles. The obtained EELS data were further analyzed in Digital Micrograph. The position of core-loss EELS was corrected with the corresponding zero-loss peak and the extrapolated background was then subtracted from the edge of interest. A Fourier-ratio deconvolution was further performed to remove the effect of plural scattering.Ex situ EDX mapping
STEM-EDX mapping was carried out on an FEI Talos F200X electron microscope with an HAADF detector operating at 200 kV. EDX was acquired using a Bruker Super-X detection system.TEM image simulation
The atomic coordination of the Co, Si, and SiO2 was obtained from the Inorganic Crystal Structure Database (ICSD). Structural models and simulated electron diffraction patterns were generated using CrystalMaker and SingleCrystal (CrystalMaker Software Ltd.). TEM image simulation was performed using the Quantitative STEM (QSTEM) simulation package.75 The atomic model of the Co–Si–SiO2 interface was constructed using the QSTEM Model Builder. The microscope parameters used for the simulations were similar to those for imaging.Preparation of SiO2 sphere supported Co and Pt powder samples
Typically, the SiO2/Co (weight ratio of Co to silica is 0.5) catalyst was prepared by an incipient wetness impregnation method. 150 mg cobalt acetate was dissolved in 3 mL distilled water, followed by the addition of 100 mg SiO2 spheres (diameter ∼ 500 nm). The solid-solution mixture was stirred at room temperature for 15 min and subsequently dried overnight at 80 °C in an oven. The obtained catalyst precursor was finally reduced in a quartz tube furnace under 10% H2/Ar at 750 °C for 4 h, resulting in a SiO2 supported Co powder catalyst. The preparation of Pt/SiO2 was carried out following the same procedures using H2PtCl6·6H2O as the precursor.Evaluation of catalytic performance of Pt/SiO2 catalysts
Pt/SiO2 was annealed at 500, 600, 750, and 900 °C under H2 (100 mL min−1) for 2 h, respectively. In a typical catalysis experiment, 0.5 mmol of 4-nitroethylbenzene, a certain amount of freshly prepared Pt/SiO2 catalyst, and 2 mL of ethyl acetate were added into a glass reaction vessel. Then the reaction vessel was placed into an autoclave reactor, subsequently purged using pure H2 feed 5 times, and pressurized to 4 bars. The catalytic reactions were carried out at 50 ± 1 °C with a stirring speed of 800 rpm. After reaction, the reactants and products were analyzed using a gas chromatograph (GC9720, Fuli Analytical Instruments Co., Ltd.) with a flame ionization detector and Agilent column (30 m × 0.320 mm×0.25 μm).Author contributions
F. Y. and Y. L. contributed to the idea and experimental design and wrote the manuscript. F. Y. and H. Z. prepared the samples, collected the ETEM data, and analysed the results. F. Y. and W. W. contributed to the EELS data. F. Y., L. W., L. Z., and T. L. prepared the powder sample, evaluated the catalysis, and performed ETEM. L. L. contributed to the CO adsorption characterization. All authors contributed to data analysis, interpreted the data, and approved the final manuscript. F. Y., H. Z., and W. W. contributed equally to this work.Conflicts of interest
The authors declare no financial interest.Acknowledgements
We gratefully acknowledge the Core Research Facilities of Southern University of Science and Technology for the TEM measurements. This work is financially supported by the National Key Research and Development Program of China (2016YFA0201904, 2018YFA0703700), National Natural Science Foundation of China (NSFC) (21631002, 12034002, 22120102004, 51971025), Beijing National Laboratory for Molecular Sciences (BNLMS-CXTD-202001), Shenzhen Basic Research Project (JCYJ20170817113121505), Shenzhen KQTD Project (KQTD20180411143400981) as well as Fundamental Research Funds for the Central Universities (FRFBD2013A). J. H. acknowledges the NSFC (11934007, 11874194, 51632005), the leading talents of Guangdong Province Program (00201517), the Science and Technology Innovation Committee Foundation of Shenzhen (KQTD2016022619565991, JCYJ20200109141205978, ZDSYS20141118160434515), High Level of Special Funds (G02206302). F. Y. acknowledges the NSFC (52002165), Beijing National Laboratory for Molecular Sciences (BNLMS202013), Guangdong Provincial Natural Science Foundation (2021A1515010229), Shenzhen Basic Research Project (JCYJ20210317150714001) Innovation Project for Guangdong Provincial Department of Education (2019KTSCX155), and a start-up fund from SUSTech.Notes and references
- C. Xie, Z. Niu, D. Kim, M. Li and P. Yang, Chem. Rev., 2019, 120, 1184–1249 CrossRef.
- G. Chen, Y. Zhao and N. F. Zheng, Science, 2014, 344, 495–499 CrossRef CAS.
- N. J. Divins, I. Angurell, C. Escudero, V. Perez-Dieste and J. Llorca, Science, 2014, 346, 620–623 CrossRef CAS PubMed.
- P. L. Hansen, J. B. Wagner, S. Helveg, J. R. Rostrup-Nielsen, B. S. Clausen and H. Topsoe, Science, 2002, 295, 2053–2055 CrossRef CAS PubMed.
- Y. He, J. C. Liu, L. Luo, Y. G. Wang, J. Zhu, Y. Du, J. Li, S. X. Mao and C. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7700–7705 CrossRef CAS.
- T. Tsuji, K. Hata, D. N. Futaba and S. Sakurai, J. Am. Chem. Soc., 2016, 138, 16608–16611 CrossRef CAS PubMed.
- T. W. van Deelen, C. H. Mejia and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- Q. Fu, F. Yang and X. H. Bao, Acc. Chem. Res., 2013, 46, 1692–1701 CrossRef CAS.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- H. Praliaud and G. A. Martin, J. Catal., 1981, 72, 394–396 CrossRef CAS.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef CAS.
- T. W. Hansen, A. T. Delariva, S. R. Challa and A. K. Datye, Acc. Chem. Res., 2013, 46, 1720–1730 CrossRef CAS.
- Z. Li, Y. Cui, Z. Wu, C. Milligan, L. Zhou, G. Mitchell, B. Xu, E. Shi, J. T. Miller, F. H. Ribeiro and Y. Wu, Nat. Catal., 2019, 1, 349–355 CrossRef.
- L. Wang, L. Wang, X. Meng and F. S. Xiao, Adv. Mater., 2019, 31, 1901905 CrossRef CAS.
- A. Parastaev, V. Muravev, E. Huertas Osta, A. J. F. van Hoof, T. F. Kimpel, N. Kosinov and E. J. M. Hensen, Nat. Catal., 2020, 3, 526–533 CrossRef CAS.
- J. Dong, Q. Fu, H. Li, J. Xiao, B. Yang, B. Zhang, Y. Bai, T. Song, R. Zhang, L. Gao, J. Cai, H. Zhang, Z. Liu and X. Bao, J. Am. Chem. Soc., 2020, 142, 17167–17174 CrossRef CAS.
- T. W. Hansen, J. B. Wagner, P. L. Hansen, S. Dahl, H. Topsoe and C. J. Jacobsen, Science, 2001, 294, 1508–1510 CrossRef CAS.
- J. C. Matsubu, S. Zhang, L. DeRita, N. S. Marinkovic, J. G. Chen, G. W. Graham, X. Pan and P. Christopher, Nat. Chem., 2017, 9, 120–127 CrossRef CAS PubMed.
- S. Zhang, P. N. Plessow, J. J. Willis, S. Dai, M. Xu, G. W. Graham, M. Cargnello, F. Abild-Pedersen and X. Pan, Nano Lett., 2016, 16, 4528–4534 CrossRef CAS.
- S. Liu, W. Xu, Y. Niu, B. Zhang, L. Zheng, W. Liu, L. Li and J. Wang, Nat. Commun., 2019, 10, 5790 CrossRef CAS.
- J. Huang, S. He, J. L. Goodsell, J. R. Mulcahy, W. Guo, A. Angerhofer and W. D. Wei, J. Am. Chem. Soc., 2020, 142, 6456–6460 CrossRef CAS.
- W. Yuan, D. Zhang, Y. Ou, K. Fang, B. Zhu, H. Yang, T. W. Hansen, J. B. Wagner, Z. Zhang, Y. Gao and Y. Wang, Angew. Chem., Int. Ed., 2018, 57, 16827–16831 CrossRef CAS.
- X. Y. Liu, M. H. Liu, Y. C. Luo, C. Y. Mou, S. D. Lin, H. K. Cheng, J. M. Chen, J. F. Lee and T. S. Lin, J. Am. Chem. Soc., 2012, 134, 10251–10258 CrossRef CAS.
- J. Zhang, H. Wang, L. Wang, S. Ali, C. T. Wang, L. X. Wang, X. J. Meng, B. Li, D. S. Su and F. S. Xiao, J. Am. Chem. Soc., 2019, 141, 2975–2983 CrossRef CAS.
- Y. Zhang, X. Yang, X. Yang, H. Duan, H. Qi, Y. Su, B. Liang, H. Tao, B. Liu, D. Chen, X. Su, Y. Huang and T. Zhang, Nat. Commun., 2020, 11, 3185 CrossRef CAS PubMed.
- A. Beck, X. Huang, L. Artiglia, M. Zabilskiy, X. Wang, P. Rzepka, D. Palagin, M.-G. Willinger and J. A. van Bokhoven, Nat. Commun., 2020, 11, 3220 CrossRef CAS.
- H. L. Tang, Y. Su, B. S. Zhang, A. F. Lee, M. A. Isaacs, K. Wilson, L. Li, Y. G. Ren, J. H. Huang, M. Haruta, B. T. Qiao, X. Liu, C. Z. Jin, D. S. Su, J. H. Wang and T. Zhang, Sci. Adv., 2017, 3, e1700231 CrossRef.
- X. Du, Y. Huang, X. Pan, B. Han, Y. Su, Q. Jiang, M. Li, H. Tang, G. Li and B. Qiao, Nat. Commun., 2020, 11, 5811 CrossRef CAS.
- S. Bernal, J. J. Calvino, J. M. Gatica, C. Larese, C. LopezCartes and J. A. PerezOmil, J. Catal., 1997, 169, 510–515 CrossRef CAS.
- S. Penner and M. Armbrüster, ChemCatChem, 2015, 7, 374–392 CrossRef.
- S. W. Li, Y. Xu, Y. F. Chen, W. Z. Li, L. L. Lin, M. Z. Li, Y. C. Deng, X. P. Wang, B. H. Ge, C. Yang, S. Y. Yao, J. L. Xie, Y. W. Li, X. Liu and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 10761–10765 CrossRef CAS PubMed.
- A. Bruix, J. A. Rodriguez, P. J. Ramirez, S. D. Senanayake, J. Evans, J. B. Park, D. Stacchiola, P. Liu, J. Hrbek and F. Illas, J. Am. Chem. Soc., 2012, 134, 8968–8974 CrossRef CAS.
- M. G. Willinger, W. Zhang, O. Bondarchuk, S. Shaikhutdinov, H. J. Freund and R. Schlogl, Angew. Chem., Int. Ed., 2014, 53, 5998–6001 CrossRef CAS PubMed.
- J. Liu, L. Wang, F. Okejiri, J. Luo, J. Zhao, P. Zhang, M. Liu, S. Yang, Z. Zhang, W. Song, W. Zhu, J. Liu, Z. Zhao, G. Feng, C. Xu and S. Dai, ACS Catal., 2020, 10, 8950–8959 CrossRef CAS.
- X. Mao, A. C. Foucher, T. Montini, E. A. Stach, P. Fornasiero and R. J. Gorte, J. Am. Chem. Soc., 2020, 142, 10373–10382 CrossRef CAS PubMed.
- M. E. Strayer, J. M. Binz, M. Tanase, S. M. K. Shahri, R. Sharma, R. M. Rioux and T. E. Mallouk, J. Am. Chem. Soc., 2014, 136, 5687–5696 CrossRef CAS PubMed.
- S. Labich, A. Kohl, E. Taglauer and H. Knözinger, J. Chem. Phys., 1998, 109, 2052–2055 CrossRef CAS.
- H. L. Tang, F. Liu, J. K. Wei, B. T. Qiao, K. F. Zhao, Y. Su, C. Z. Jin, L. Li, J. Y. Liu, J. H. Wang and T. Zhang, Angew. Chem., Int. Ed., 2016, 55, 10606–10611 CrossRef CAS.
- J. Lee, S. P. Burt, C. A. Carrero, A. C. Alba-Rubio, I. Ro, B. J. O'Neill, H. J. Kim, D. H. K. Jackson, T. F. Kuech, I. Hermans, J. A. Dumesic and G. W. Huber, J. Catal., 2015, 330, 19–27 CrossRef CAS.
- M. Li, F. Yang, L. Ding, X. Liu, Z. Zhang, D. Zhang, X. Zhao, J. Yang and Y. Li, Carbon, 2017, 118, 485–492 CrossRef CAS.
- P. Sonstrom, D. Arndt, X. D. Wang, V. Zielasek and M. Baumer, Angew. Chem., Int. Ed., 2011, 50, 3888–3891 CrossRef.
- H. Wang, L. Wang, D. Lin, X. Feng, Y. Niu, B. Zhang and F.-S. Xiao, Nat. Catal., 2021, 4, 418–424 CrossRef CAS.
- T. Lunkenbein, J. Schumann, M. Behrens, R. Schlçgl and M. G. Willinger, Angew. Chem., Int. Ed., 2015, 54, 4544–4548 CrossRef CAS.
- V. Schott, H. Oberhofer, A. Birkner, M. Xu, Y. Wang, M. Muhler, K. Reuter and C. Woll, Angew. Chem., Int. Ed., 2013, 52, 11925–11929 CrossRef CAS PubMed.
- X. G. Guo, G. Z. Fang, G. Li, H. Ma, H. J. Fan, L. Yu, C. Ma, X. Wu, D. H. Deng, M. M. Wei, D. L. Tan, R. Si, S. Zhang, J. Q. Li, L. T. Sun, Z. C. Tang, X. L. Pan and X. H. Bao, Science, 2014, 344, 616–619 CrossRef CAS PubMed.
- Y. Zhang, J. Zhang, B. Zhang, R. Si, B. Han, F. Hong, Y. Niu, L. Sun, L. Li, B. Qiao, K. Sun, J. Huang and M. Haruta, Nat. Commun., 2020, 11, 558 CrossRef CAS.
- H. G. Zhu, C. D. Liang, W. F. Yan, S. H. Overbury and S. Dai, J. Phys. Chem. B, 2006, 110, 10842–10848 CrossRef CAS PubMed.
- V. Sudheeshkumar, A. Shivhare and R. W. J. Scott, Catal. Sci. Technol., 2017, 7, 272–280 RSC.
- L. Wang, E. Guan, Y. Wang, L. Wang, Z. Gong, Y. Cui, X. Meng, B. C. Gates and F. S. Xiao, Nat. Commun., 2020, 11, 1033 CrossRef CAS.
- F. Peng, Y. Liu, R. L. Cui, D. L. Gao, F. Yang and Y. Li, Chin. Sci. Bull., 2012, 57, 225–233 CrossRef CAS.
- A. E. Islam, P. Nikolaev, P. B. Amama, S. Saber, D. Zakharov, D. Huffman, M. Erford, G. Sargent, S. L. Semiatin, E. A. Stach and B. Maruyama, Nano Lett., 2014, 14, 4997–5003 CrossRef CAS.
- M. Li, X. Liu, X. Zhao, F. Yang, X. Wang and Y. Li, Top. Curr. Chem., 2017, 375, 29 CrossRef.
- F. Yang, M. Wang, D. Zhang, J. Yang, M. Zheng and Y. Li, Chem. Rev., 2020, 120, 2693–2758 CrossRef CAS.
- S. Penner, D. Wang, D. S. Su, G. Rupprechter, R. Podloucky, R. Schlögl and K. Hayek, Surf. Sci., 2003, 532–535, 276–280 CrossRef CAS.
- J.-S. Park, D. K. Sohn, Y. Kim, J.-U. Bae, B. H. Lee, J. S. Byun and J. J. Kim, Appl. Phys. Lett., 1998, 73, 2284–2286 CrossRef CAS.
- L. D. Deng, H. Miura, T. Shishido, S. Hosokawa, K. Teramura and T. Tanaka, Chem. Commun., 2017, 53, 6937–6940 RSC.
- D. S. Su, B. Zhang and R. Schlögl, Chem. Rev., 2015, 115, 2818–2882 CrossRef CAS.
- F. Yang, X. Wang, D. Zhang, J. Yang, D. Luo, Z. Xu, J. Wei, J.-Q. Wang, Z. Xu, F. Peng, X. Li, R. Li, Y. Li, M. Li, X. Bai, F. Ding and Y. Li, Nature, 2014, 510, 522–524 CrossRef CAS.
- F. Yang, X. Wang, M. Li, X. Liu, X. Zhao, D. Zhang, Y. Zhang, J. Yang and Y. Li, Acc. Chem. Res., 2016, 49, 606–615 CrossRef CAS PubMed.
- W. Yuan, B. Zhu, X.-Y. Li, T. W. Hansen, Y. Ou, K. Fang, H. Yang, Z. Zhang, J. B. Wagner, Y. Gao and Y. Wang, Science, 2020, 367, 428–430 CrossRef CAS.
- X. Zhang, S. Han, B. Zhu, G. Zhang, X. Li, Y. Gao, Z. Wu, B. Yang, Y. Liu, W. Baaziz, O. Ersen, M. Gu, J. T. Miller and W. Liu, Nat. Catal., 2020, 3, 411–417 CrossRef CAS.
- L. Luo, Y. Nian, S. Wang, Z. Dong, Y. He, Y. Han and C. Wang, Angew. Chem., Int. Ed., 2020, 59, 2505–2509 CrossRef CAS.
- X. Zhang, F. Yang, D. Tian, H. Zhao, R. Wang and W.-M. Lau, Adv. Mater. Interfaces, 2020, 7, 2001112 CrossRef CAS.
- W. Yuan, B. Zhu, K. Fang, X. Y. Li, T. W. Hansen, Y. Ou, H. Yang, J. B. Wagner, Y. Gao, Y. Wang and Z. Zhang, Science, 2021, 371, 517–521 CrossRef CAS PubMed.
- F. Yang, H. Zhao, X. Wang, X. Liu, Q. Liu, X. Liu, C. Jin, R. Wang and Y. Li, J. Am. Chem. Soc., 2019, 141, 5871–5879 CrossRef CAS.
- F. Yang, H. Zhao, W. Wang, Q. Liu, X. Liu, Y. Hu, X. Zhang, S. Zhu, D. He, Y. Xu, J. He, R. Wang and Y. Li, CCS Chem., 2021, 3, 154–167 CrossRef.
- Q. Fu and T. Wagner, Surf. Sci. Rep., 2007, 62, 431–498 CrossRef CAS.
- Y. T. Pan, Y. Yan, Y. T. Shao, J. M. Zuo and H. Yang, Nano Lett., 2016, 16, 6599–6603 CrossRef CAS PubMed.
- A. R. Harutyunyan, G. Chen, T. M. Paronyan, E. M. Pigos, O. A. Kuznetsov, K. Hewaparakrama, S. M. Kim, D. Zakharov, E. A. Stach and G. U. Sumanasekera, Science, 2009, 326, 116–120 CrossRef CAS.
- S. J. Tauster, S. C. Fung, R. T. K. Baker and J. A. Horsley, Science, 1981, 211, 1121–1125 CrossRef CAS.
- F. Sadi, D. Duprez, F. Gerard and A. Miloudi, J. Catal., 2003, 213, 226–234 CrossRef CAS.
- L. C. A. van den Oetelaar, A. Partridge, S. L. G. Toussaint, C. F. J. Flipse and H. H. Brongersma, J. Phys. Chem. B, 1998, 102, 9541–9549 CrossRef CAS.
- M. Crespo-Quesada, F. Cardenas-Lizana, A. L. Dessimoz and L. Kiwi-Minsker, ACS Catal., 2012, 2, 1773–1786 CrossRef CAS.
- B. Das, B. Balasubramanian, P. Manchanda, P. Mukherjee, R. Skomski, G. C. Hadjipanayis and D. Sellmyer, Nano Lett., 2016, 16, 1132–1137 CrossRef CAS PubMed.
- C. T. Koch, Ph.D. thesis, Arizona State University, May, 2002.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03480d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |