
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical generation of acid-degradable poly(enol ether)s†
Jinghui
Yang
 and
Yan
Xia
*
and
Yan
Xia
*
Department of Chemistry, Stanford University, Stanford, California 94305, USA. E-mail: yanx@stanford.edu
First published on 12th February 2021
Abstract
In an effort to develop polymers that can undergo extensive backbone degradation in response to mechanical stress, we report a polymer system that is hydrolytically stable but unmasks easily hydrolysable enol ether backbone linkages when force is applied. These polymers were synthesized by ring-opening metathesis polymerization (ROMP) of a novel mechanophore monomer consisting of cyclic ether fused bicyclohexene. Hydrogenation of the resulting polymers led to significantly enhanced thermal stability (Td > 400 °C) and excellent resistance toward acidic or basic conditions. Solution ultrasonication of the polymers resulted in up to 65% activation of the mechanophore units and conversion to backbone enol ether linkages, which then allowed facile degradation of the polymers to generate small molecule or oligomeric species under mildly acidic conditions. We also achieved solid-state mechano-activation and polymer degradation via grinding the solid polymer. Force-induced hydrolytic polymer degradability can enable materials that are stable under force-free conditions but readily degrade under stress. Facile degradation of mechanically activated polymechanophores also facilitates the analysis of mechanochemical products.
Introduction
Polymer mechanochemistry explores the use of mechanical force to direct selective chemical transformations in force-responsive molecules called mechanophores.1,2 Embedding properly designed mechanophores in polymer backbones can induce nondestructive covalent bond transformations and generate materials with a wide range of force-responses.3–11 Mechanical activation of mechanophores is usually accompanied by nonselective chain scission, and chain scission can halt further activation of mechanophores.12 While it has long been known that high molecular weight (MW) polymers undergo undesired mechanical degradation under shear stress,13,14 mechanically triggered chemical degradation over an extended strand of polymer (rather than a single-point chain degradation) is an interesting and desired mechanical response. It can enhance the extent of polymer degradation to result in much smaller species. Such polymers may enable effective self-destruction and minimize their environmental impact. For fundamental study of mechanochemistry, incorporating force-triggered degradable units in polymechanophores may facilitate the analysis of the mechanochemical products and provide information on the continuity (or discontinuity) of polymechanophore activation.In 2020, the Craig and Wang groups simultaneously reported strategies for mechanically “unlocking” polymer degradability by fusing hydrolysable lactone and cyclic ketal linkages with cyclobutane mechanophores in the polymer backbone. Upon ring-opening of cyclobutane, these hydrolysable moieties become backbone linkages and enable hydrolytic degradation of the mechanochemically activated polymers (Scheme 1A and B).15,16 Subsequently, the Craig group reported a clever alternative design of a tetrasubstituted [4.2.0]bicyclooctene based mechanophore that, upon mechanochemical ring-opening into 1,3-cyclooctadiene, can spontaneously undergo intramolecular lactonization and result in backbone cleavage (Scheme 1C).17 These recent examples showcased mechanically enhanced polymer degradation as a new modality of response, but in these designs, the hydrolysable functionalities already exist in the polymers before mechanoactivation. As a result, deleterious hydrolysis prior to the desired mechanoactivation would inevitably hydrolyze these same functionalities in the precursor polymers and change their properties, leading to the generation of undesired scissile cyclobutane linkages or premature polymer cleavage that disrupts the transduction of force and thus limits the extent of mechanoactivation. We herein report a new design of polymechanophores that overcomes these limitations and only contains completely chemically inert linkages, but upon mechanoactivation, easily hydrolysable and degradable linkages are unmasked. Taking advantage of the olefins generated from mechanochemical ring-opening of cyclobutane, we sought to mechanically convert chemically inert alkyl ether linkages to acid-labile enol ether linkages. The resultant poly(enol ether)s can be rapidly degraded under mildly acidic conditions but remain stable under basic conditions.18–20
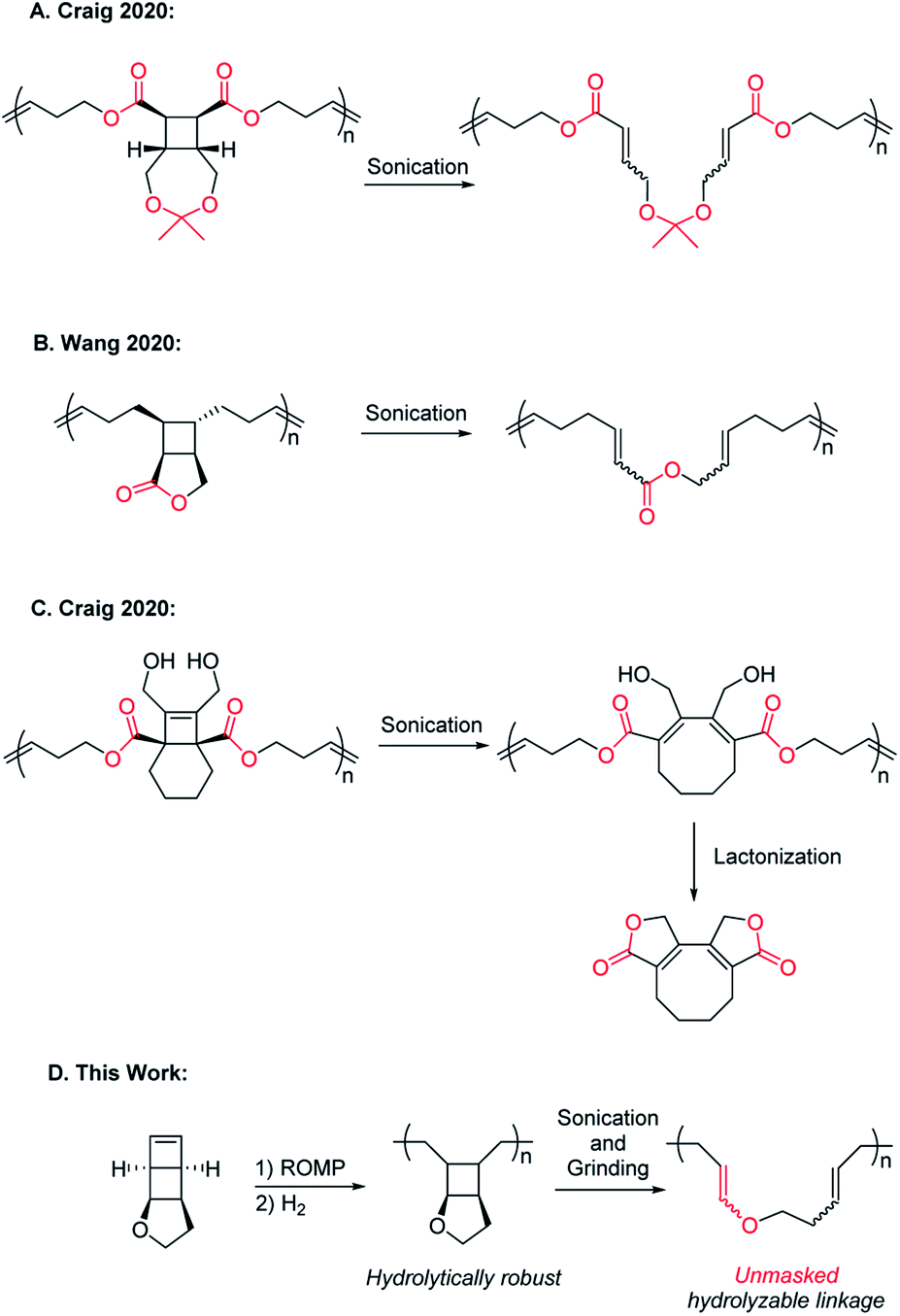 | ||
| Scheme 1 Designs of polymers that can mechanochemically generate degradable backbone linkages. | ||
Results and discussions
We have developed a general strategy to directly polymerize mechanophore monomers in a controlled fashion to generate polymechanophores, and designed such polymechanophores to undergo dramatic mechanochemical transformation from non-conjugated structures to conjugated structures.21–24 Leveraging this strategy, we designed a tricyclic mechanophore monomer 1 that can be synthesized from commercially available chemicals at gram scales as outlined in Scheme 2. Thermally induced [2 + 2] cycloaddition between dimethyl acetylenedicarboxylate and 2,3-dihydrofuran produced bicyclic compound 2,25 which was subsequently hydrogenated and reduced to afford diol 3. 3 was found to have an exclusive endo configuration (Fig. S1†) due to the high facial selectivity in the hydrogenation step. Treatment of 3 with MsCl afforded dimesylate 4, which was then treated with Na2S followed by oxidation with H2O2 to yield sulfoxide 5. Chlorination of 5 by one equivalent of SO2Cl2 generated α-chlorosulfoxide as a mixture of different regioisomers and stereoisomers, which underwent sulfoxide Ramberg–Bäckland olefination to generate 1 as a volatile liquid.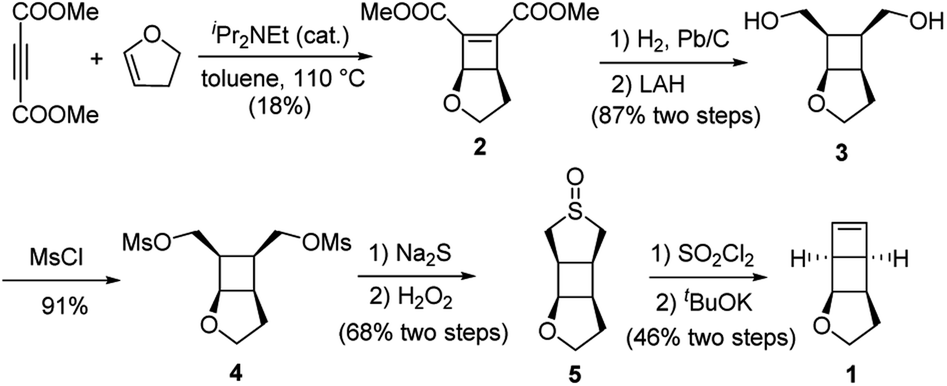 | ||
| Scheme 2 Synthesis of monomer 5. | ||
The ring opening metathesis polymerization (ROMP) of 1 was then explored using the fast-initiating Grubbs catalyst G3. G3 was added into a solution of 1 in CHCl3 ([1]0 = 1 M) at room temperature with a target degree of polymerization (DP) of 1000. 1H NMR analysis of the crude reaction mixture indicated complete monomer conversion within 15 min. Despite the fast polymerization, gel permeation chromatography (GPC) of the polymerization solution showed an asymmetric peak skewed to the low MW side and the measured MW was lower than the expected value (Fig. S2†). We have previously observed reduced stability of the propagating Ru in ROMP of ladderenes, but the stability is improved at lower temeratures.22 Delightfully, lowering the polymerization temperature to 0 °C yielded a symmetric peak with expected MW and low dispersity. The measured MWs agreed with the theoretical values and could be controlled by the ratio of [1]0/[G3]0 (P1 and P2 in Fig. 1b and Table 1). However, if the quenched polymer solution was left under ambient conditions, low MW polymers were progressively formed over a few hours (Fig. S3†), suggesting decomposition of the ROMP polymer. Differential scanning calorimetry (DSC) analysis of the isolated ROMP polymer showed two overlapping irreversible exothermic peaks starting from 46 °C (Fig. S4†). The limited thermal stability was also corroborated by thermogravimetric analysis (TGA), which showed gradual mass loss starting from ∼100 °C (Fig. 1c). To probe the thermal decomposition, poly(1) was dissolved in CDCl3 and heated at 65 °C. Additional olefinic peaks at 6.0–6.2 ppm emerged, which were tentatively assigned to conjugated olefins generated from ring-opening of cyclobutanes (Fig. S8†). Interestingly, the peak intensity of new olefins quickly saturated after 3 h of heating and remained unchanged after additional 37 h of heating at the same temperature. Further increasing the temperature to 90 °C for another 24 h did not result in much change in the peak intensity (Fig. S9†). This result implies that a fraction of cyclobutanes in the polymer are less thermally stable than the rest, which also agrees with the two overlapping exothermic peaks at different temperatures from the DSC analysis (Fig. S4†). To investigate whether the configuration of backbone olefins would influence the thermal stability of cyclobutanes, we synthesized a Z-olefin rich polymer using a Z-selective Grubbs catalyst26 and examined the thermal stability using the same 1H NMR analysis. Surprisingly, almost identical thermal decomposition profile was observed despite the distinct difference in backbone olefin configurations (Fig. S10†). At this point, the reason behind this unusual thermal behavior remains unknown.
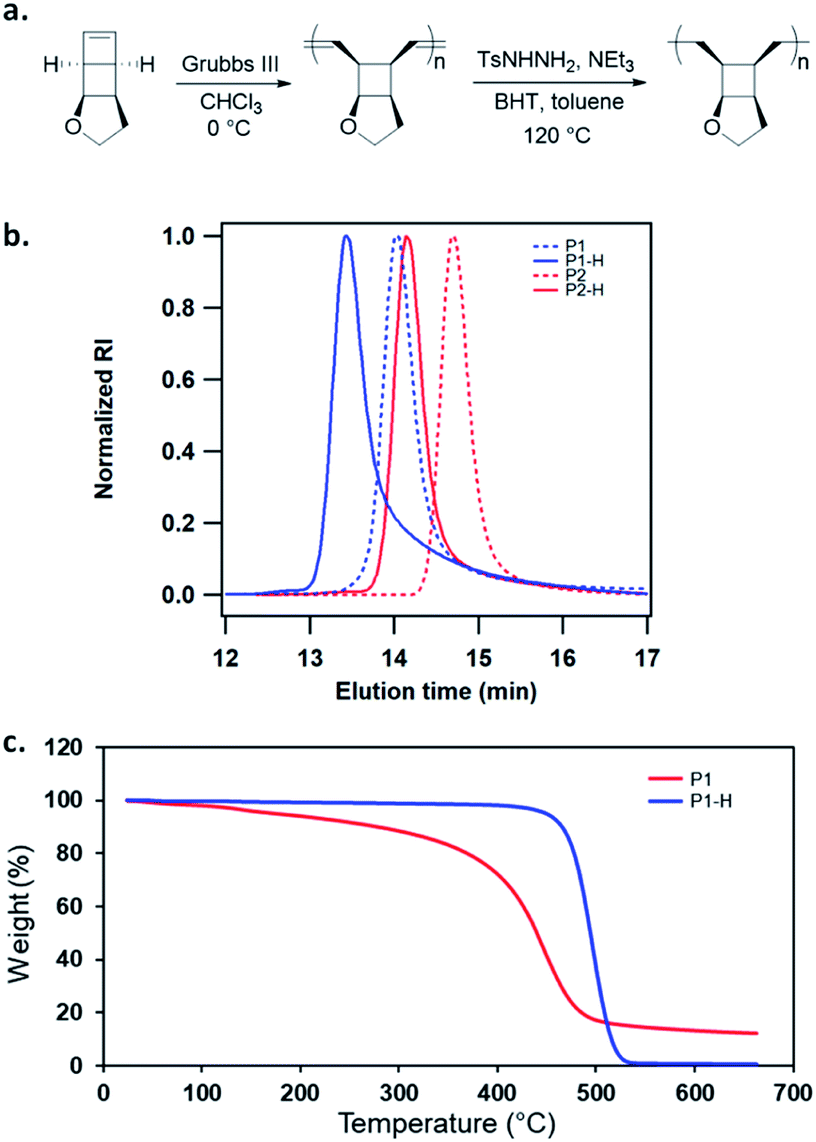 | ||
| Fig. 1 (a) ROMP of monomer 1 and subsequent hydrogenation (the specified stereochemistry refers to that of the repeat unit, not suggesting the polymer tacticity). (b) GPC traces of poly(1) at different DPs and their hydrogenated polymers. (c) TGA traces of poly(1) and hydrogenated poly(1). | ||
Recently, Bielawski and coworkers reported poly(dewar benzene), which has the same divinyl cyclobutane backbone structure.27 This polymer was also found to be unstable, but was stabilized after hydrogenation of the backbone olefins. Therefore, we hydrogenated the backbone olefins of poly(1) using tosylhydrazide to improve the thermal stability. Hydrogenation also simplifies the analysis of mechanically generated olefins by eliminating the formation of conjugated backbone olefins, which have complex configurations and broad 1H NMR signals. Freshly prepared poly(1) was hydrogenated using tosyl hydrazide (5 equiv. to olefin) with added NEt3. 1H NMR analysis of the hydrogenated polymers (P1-H and P2-H) showed complete disappearance of olefinic peaks and unnoticeable ring-opening of cyclobutane (Fig. S46†). Their GPC traces exhibited narrow-disperse peaks with the same distribution as the ROMP polymers and MWs close to the theoretical values (Fig. 1b, Table 1). The hydrogenated polymers showed significantly improved thermal stability with a Td over 400 °C (Fig. 1c). DSC analysis showed that P1-H is amorphous with a Tg of 94 °C (Fig. S5†).
Hydrogenated poly(1) also exhibited excellent stability towards acidic or basic conditions. Treating their solutions with either 100 mM HCl or KOH resulted in no change in GPC traces (Fig. 3c) or 1H NMR spectra (Fig. S13†) over multiple days. The absence of any labile linkages in both polymer backbone and side chain is attributed to the high chemical stability prior to the mechanoactivation of the polymers.
To study the mechanoactivation, we subjected a dilute solution of P1-H in THF (1 mg mL−1) to pulsed ultrasonication. Aliquots removed at regular time intervals were analyzed by both GPC and 1H NMR spectroscopy. The GPC traces showed a rapid decrease of the original polymer peak and appearance of lower MW peak (Fig. 2b). The Mn decreased steadily and reached an ultimate MW of 18.0 kDa after 480 min of ultrasonication. Sonicating a shorter polymer P2-H under the same conditions resulted in a similar ultimate Mn of 18.7 kDa. 1H NMR analysis showed a continuous decrease in cyclobutane signals along with a concomitant increase in two sets of alkenyl peaks at the same intensity, corresponding to the generated enol ether olefin and the other olefin as assigned in Fig. 2c. The activation ratio increased steadily overtime and reached up to 65% and 55% for P1-H and P2-H, respectively, after 480 min of ultrasonication based on the 1H NMR integrations (Fig. S14, S15, Tables S2 and S3†). These degrees of activation are notably higher than our other polymechanophore systems that were similarly synthesized by ROMP of mechanophore monomers but generated conjugated polymers,21–24 which typically show a final degree of activation around 40%. The lower degree of activation observed in our other systems may be attributed to the variable extent of microscale aggregation and potentially occasional chemical crosslinking of the generated conjugated segments, which may disrupt the force transduction to some regions of the polymer strand. The absence of extended conjugation in the activated P1-H also gives them well resolved 1H NMR signals, in contrast to the broad and partly suppressed signals for the mechanically generated conjugated polymers.
 | ||
| Fig. 2 (a) Mechanochemical activation of P1-H to generate poly(enol ether). (b) GPC traces of P1-H at different sonication times. (c) 1H NMR spectrum of activated P1-H after 240 min of sonication. | ||
The generated poly(enol ether)s consisted of nearly all Z olefins with an E/Z ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 regardless of the MW and remained unchanged over the course of ultrasonication (Fig. 2c). The predominant formation of Z olefins represents stereochemical retention in the mechanochemical ring-opening. Interestingly, other studies of cyclobutane mechanochemical cycloreversion by Craig28 and Wang15 groups reported higher fractions of stereochemical inversion. The stereochemical distribution of the mechanochemically generated diene can be ascribed to the instability of the diradical transition state upon the first bond scission and dynamic effect,29,30 and warrants comprehensive computational studies. 22% and 28% mechanophore activation per scission cycle was achieved for P1-H and P2-H, respectively (Fig. S17†), agreeing with the previous report that shorter polymers are more selective in mechanophore activation over nonselective chain cleavage.31
:100 regardless of the MW and remained unchanged over the course of ultrasonication (Fig. 2c). The predominant formation of Z olefins represents stereochemical retention in the mechanochemical ring-opening. Interestingly, other studies of cyclobutane mechanochemical cycloreversion by Craig28 and Wang15 groups reported higher fractions of stereochemical inversion. The stereochemical distribution of the mechanochemically generated diene can be ascribed to the instability of the diradical transition state upon the first bond scission and dynamic effect,29,30 and warrants comprehensive computational studies. 22% and 28% mechanophore activation per scission cycle was achieved for P1-H and P2-H, respectively (Fig. S17†), agreeing with the previous report that shorter polymers are more selective in mechanophore activation over nonselective chain cleavage.31
The activated polymers after 480 min of sonication were then degraded by the addition of 10 mM TFA. 1H NMR analysis showed the complete disappearance of enol ether signals as well as the appearance of multiple aldehyde peaks at 9.7–9.8 ppm (Fig. 3b), as expected from several aldehyde-containing degradation products at different MWs. Several small molecule hydrolysis products may be expected due to the regio-irregular placement of the pendent cyclic ether rings in the polymers (Fig. S21†). GPC analysis of the hydrolyzed polymer showed a bimodal distribution with a relatively narrow-disperse high MW peak representing the unactivated fraction of the polymer strand (Punactivated), which is presumably located toward the ends of pre-activated polymer, and a low MW population corresponding to the generated oligomers and small molecules from hydrolysis(Poligomer), which were formed due to the noncontinuous activation of mechanophore repeat units. Theoretically, the distribution of force on a polymer chain under elongational flow can be described using a bead-rod model, where force is maximal around the chain center and gradually decreases toward chain ends.32 Presumably, mechanophores are activated more continuously under high forces and less continuously as the force approaches activation threshold (Fig. 2a). Intriguingly, the Mn of both Punactivated and Poligomer decreased gradually over the course of sonication (Fig. 3d). Notably, the Punactivated fraction of P1-H and P2-H after 480 min sonication had similar Mn of 9.6 and 11.4 kDa, respectively, despite their different original Mn of 141 and 63.1 kDa for the full-length polymers. However, most interestingly, the MW distribution of the Poligomer fraction for these two samples were noticeably different (Fig. 3e). Longer P1-H resulted in shorter oligomers after degradation, indicating a shorter distance between the activated units or less discontinuous activation for the longer polymer.
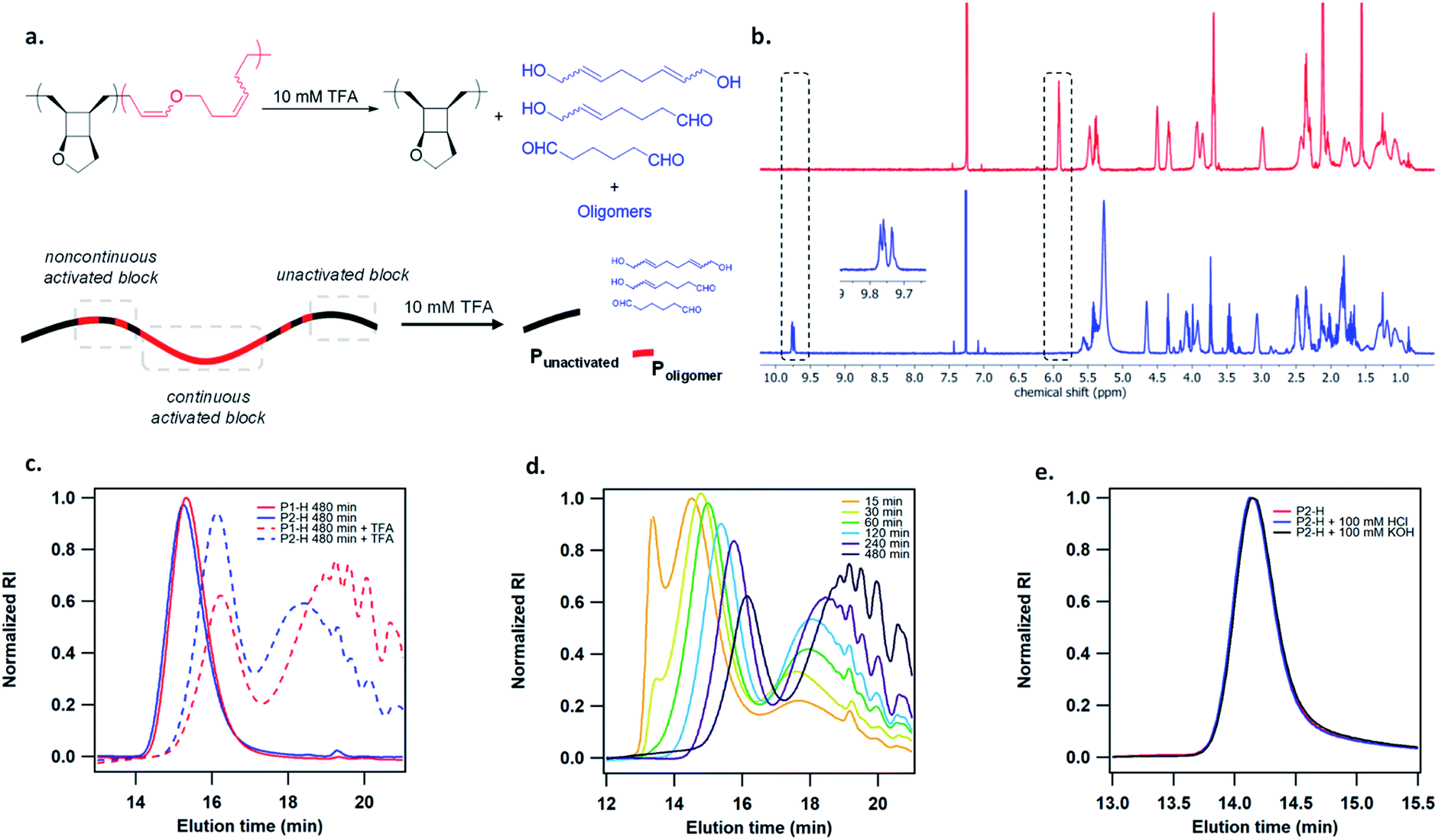 | ||
| Fig. 3 (a) Acid degradation of mechanically activated polymer. (b) 1H NMR spectra of mechanically activated poly(1) before (top, red trace) and after (bottom, blue trace) 10 mM TFA treatment. (c) GPC traces of hydrogenated poly(1) showing resistance to acidic and basic hydrolysis. (d) GPC traces of activated P1-H at different times after 10 mM TFA treatment. (e) Comparison of GPC traces of P1-H and P2-H after 480 min of sonication and 10 mM TFA treatment. | ||
We next attempted activation of poly(1) in the solid state. The facile copolymerization of 1 with substituted norbornenes provides a simple means to tune the solid state properties of the copolymers. Poly(1) is a glassy polymer with Tg of 94 °C. We copolymerized 1 with NBE-EtHex to lower Tg and generate more viscoelastic polymers (Fig. 4a). Copolymer P3 of 1 with NBE-EtHex at 1:1 ratio was easily synthesized in low dispersity and controlled MW (Table 1). P3 was completely hydrogenated using tosyl hydrazide without side reactions on the ester side chains as evidenced by 1H NMR analysis (Fig. S25†). The hydrogenated copolymer P3-H showed a Tg of 56 °C (Fig. S26†). To demonstrate the solid state activation, we ground P3-H in a mortar for about 10 min. The GPC trace of ground P3-H showed slight tailing resulting from mechanical degradation of the polymer. 10 mM TFA was then added to P3-H before and after grinding. While the GPC trace for P3-H without grinding essentially remained unchanged, a considerable fraction of lower MW polymers was observed for the ground P3-H (Fig. 4b), suggesting the formation of hydrolysable enol ether linkages.
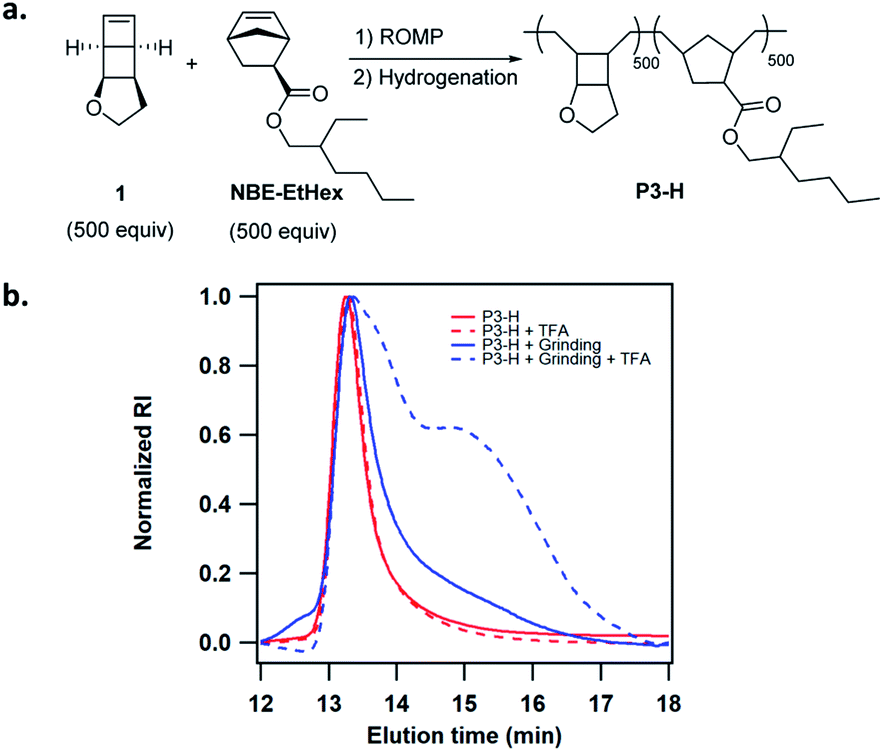 | ||
| Fig. 4 (a) Synthesis of copolymer P3-H. (b) Changes of GPC traces of P3-H before and after grinding for 10 min, followed by treatment with 10 mM TFA in THF. | ||
Conclusions
In summary, we have developed a new polymechanophore system that is hydrolytically robust but uncovers a large number of hydrolytically labile bonds in the polymer backbone under mechanical force. The mechanophore monomer can be rapidly polymerized by ROMP to yield homopolymers or copolymers. Hydrogenation of the backbone olefins of the ROMP polymers yielded saturated polymers with greatly enhanced thermal stability and excellent hydrolytic stability. The degradability of these polymers is turned on by mechanochemically transforming a previously non-degradable ether bond into a degradable enol-ether bond under stress applied by either sonication of the solution or grinding of the solid polymer. This design can impart additional chemical degradability to polymers beyond conventional mechanical chain scission and yield low MW oligomers and small molecules when under both stress and acidic conditions. We envision that the mechanophore monomer 1 can also be directly polymerized with other mechanophore monomers to allow facile degradation of the mechanically activated copolymers and facilitate detailed analysis of mechanochemical products in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Army Research Office under grant number W911NF-15-1-0525. J. Y. is supported by a Stanford Graduate Fellowship. We thank Matias Horst for help with this manuscript. Part of the work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-1542152. We thank Zhiao Yu for assistance with DSC and TGA data collection and Jana Maclaren and Conor M. Galvin for assistance with crystallography.Notes and references
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755 CrossRef CAS.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181 CrossRef CAS.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68 CrossRef CAS.
- A. Piermattei, S. Karthikeyan and R. P. Sijbesma, Nat. Chem., 2009, 1, 133 CrossRef CAS.
- Y. Chen, A. J. Spiering, S. Karthikeyan, G. W. Peters, E. W. Meijer and R. P. Sijbesma, Nat. Chem., 2012, 4, 559 CrossRef CAS.
- A. L. Ramirez, Z. S. Kean, J. A. Orlicki, M. Champhekar, S. M. Elsakr, W. E. Krause and S. L. Craig, Nat. Chem., 2013, 5, 757 CrossRef CAS.
- M. B. Larsen and A. J. Boydston, J. Am. Chem. Soc., 2013, 135, 8189 CrossRef CAS.
- K. Imato, A. Irie, T. Kosuge, T. Ohishi, M. Nishihara, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2015, 54, 6168 CrossRef CAS.
- Y. Sagara, M. Karman, A. Seki, M. Pannipara, N. Tamaoki and C. Weder, ACS Cent. Sci., 2019, 5, 874 CAS.
- T. Matsuda, R. Kawakami, R. Namba, T. Nakajima and J. P. Gong, Science, 2019, 363, 504 CrossRef CAS.
- X. R. Hu, T. Zeng, C. C. Husic and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 15018 CrossRef CAS.
- P. A. May and J. S. Moore, Chem. Soc. Rev., 2013, 42, 7497 RSC.
- H. Staudinger, Ber. Dtsch. Chem. Ges. A, 1930, 63, 921 CrossRef.
- J. M. Lenhardt, M. T. Ong, R. Choe, C. R. Evenhuis, T. J. Martinez and S. L. Craig, Science, 2010, 329, 1057 CrossRef CAS.
- T.-G. Hsu, J. Zhou, H.-W. Su, B. R. Schrage, C. J. Ziegler and J. Wang, J. Am. Chem. Soc., 2020, 142, 2100 CrossRef CAS.
- Y. Lin, T. B. Kouznetsova and S. L. Craig, J. Am. Chem. Soc., 2020, 142, 2105 CrossRef CAS.
- Y. Lin, T. B. Kouznetsova, C.-C. Chang and S. L. Craig, Nat. Commun., 2020, 11, 4987 CrossRef CAS.
- H. Shimomoto, T. Mori, T. Itoh and E. Ihara, Macromolecules, 2019, 52, 5761 CrossRef CAS.
- J. D. Feist and Y. Xia, J. Am. Chem. Soc., 2020, 142, 1186 CrossRef CAS.
- X. Sui, T. Zhang, A. B. Pabarue, L. Fu and W. R. Gutekunst, J. Am. Chem. Soc., 2020, 142, 12942 CrossRef CAS.
- Z. Chen, J. A. M. Mercer, X. Zhu, J. A. H. Romaniuk, R. Pfattner, L. Cegelski, T. J. Martinez, N. Z. Burns and Y. Xia, Science, 2017, 357, 475 CrossRef CAS.
- J. K. Su, J. D. Feist, J. Yang, J. A. M. Mercer, J. A. H. Romaniuk, Z. Chen, L. Cegelski, N. Z. Burns and Y. Xia, J. Am. Chem. Soc., 2018, 140, 12388 CrossRef CAS.
- J. Yang, M. Horst, J. A. H. Romaniuk, Z. Jin, L. Cegelski and Y. Xia, J. Am. Chem. Soc., 2019, 141, 6479 CrossRef CAS.
- J. Yang, M. Horst, S. H. Werby, L. Cegelski, N. Z. Burns and Y. Xia, J. Am. Chem. Soc., 2020, 142, 14619 CrossRef CAS.
- K. C. Nicolaou, C. K. Hwang, M. E. Duggan and K. B. Reddy, Tetrahedron Lett., 1987, 28, 1501 CrossRef CAS.
- B. K. Keitz, A. Fedorov and R. H. Grubbs, J. Am. Chem. Soc., 2012, 134, 2040 CrossRef CAS.
- J. Seo, S. Y. Lee and C. W. Bielawski, Macromolecules, 2020, 53, 3202 CrossRef CAS.
- Z. S. Kean, Z. Niu, G. B. Hewage, A. L. Rheingold and S. L. Craig, J. Am. Chem. Soc., 2013, 135, 13598 CrossRef CAS.
- M. Wollenhaupt, C. Schran, M. Krupička and D. Marx, ChemPhysChem, 2018, 19, 837 CrossRef CAS.
- Z. Chen, X. Zhu, J. Yang, J. A. M. Mercer, N. Z. Burns, T. J. Martinez and Y. Xia, Nat. Chem., 2020, 12, 302 CrossRef CAS.
- J. M. Lenhardt, A. L. Black Ramirez, B. Lee, T. B. Kouznetsova and S. L. Craig, Macromolecules, 2015, 48, 6396 CrossRef CAS.
- A. Keller and J. A. Odell, Colloid Polym. Sci., 1985, 263, 181 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed descriptions of synthetic procedures, 1H and 13C NMR spectra of new compounds, GPC traces and single crystal structure. Crystallographic data for compound S2. CCDC 2023034. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00001b |
| This journal is © The Royal Society of Chemistry 2021 |