
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTraceless parallel peptide purification by a first-in-class reductively cleavable linker system featuring a safety-release†
Robert
Zitterbart‡
 *a,
Nadja
Berger‡
a,
Oliver
Reimann
a,
Gavin T.
Noble
b,
Stephan
Lüdtke
a,
Dominik
Sarma‡
a and
Oliver
Seitz
c
*a,
Nadja
Berger‡
a,
Oliver
Reimann
a,
Gavin T.
Noble
b,
Stephan
Lüdtke
a,
Dominik
Sarma‡
a and
Oliver
Seitz
c
aBelyntic GmbH, Richard-Willstätter-Str. 11, 12489 Berlin, Germany. E-mail: Robert.Zitterbart@belyntic.com
bBachem (UK) Ltd., Delph Court, Sullivans Way, St. Helens, Merseyside WA9 5GL, UK
cHumboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany
First published on 3rd February 2021
Abstract
Hundreds of peptides can be synthesized by automated parallel synthesizers in a single run. In contrast, the most widely used peptide purification method – high-pressure liquid chromatography (HPLC) – only allows one-by-one processing of each sample. The chromatographic purification of many peptides, therefore, remains a time-consuming and costly effort. Catch-and-release methods can be processed in parallel and potentially provide a remedy. However, no such system has yet provided a true alternative to HPLC. Herein we present the development of a side-reaction free, reductively cleavable linker. The linker is added to the target peptide as the last building block during peptide synthesis. After acidic cleavage from synthetic resin, the linker-tagged full-length peptide is caught onto an aldehyde-modified solid support by rapid oxime ligation, allowing removal of all impurities lacking the linker by washing. Reducing the aryl azide to an aniline sensitizes the linker for cleavage. However, scission does not occur at non-acidic pH enabling wash out of reducing agent. Final acidic treatment safely liberates the peptide by an acid-catalysed 1,6-elimination. We showcase this first-in-class reductively cleavable linker system in the parallel purification of a personalized neoantigen cocktail, containing 20 peptides for cancer immunotherapy within six hours.
Introduction
Peptides have high therapeutic potential due to their pivotal role in nature. Currently, 60 approved peptide drugs are on the market and 157 active clinical trials are ongoing.1,2 Yet, peptide-based medicines have limitations, such as short plasma half-life and low oral bioavailability.2 To enhance pharmacokinetic and pharmacodynamic properties many peptides that enter the clinic today are chemically altered.2 The trend to longer and chemically modified peptides imposes increased challenges for peptide synthesis and purification. Solid-phase peptide synthesis (SPPS)3 is the preferred method of peptide production, and reversed-phase high-performance liquid chromatography (RP-HPLC) is the most widely used method for peptide purification.4 Commercially available automated peptide synthesizers allow the parallel synthesis of several hundreds of peptides in a single run. HPLC, in contrast, is a linear process and even with the most modern set-up, requires over 15 min per peptide rendering purification the bottleneck in peptide manufacturing.5 As a consequence, researchers mostly employ crude peptide libraries and, as such, accept higher risk for false-positive responses, which hampers the speed of pharmaceutical development.6,7Catch and release (c&r) methods could facilitate the purification process in parallel, since precise pumps, massive columns, collector units, and high solvent volumes become obsolete. The great potential of c&r was first recognized by Krieger, Erikson and Merrifield in 1976.8 A key component of c&r (Scheme 1a) is a purification tag that is usually installed as the last residue at the N-terminus during SPPS. Employing capping steps for acetylation of truncations, arising from incomplete amino acid coupling, allows the selective installation of the purification tag onto the full-length product. Subsequently, the target peptide is immobilised onto a solid support (catch step) via interactions with the purification handle. Washing removes all non-tagged substances like capped truncations, salt burden, or protecting group debris. Finally, a chemical stimulus releases the purified peptide from the support (release step). For example, treatment with imidazole or acid has been used to liberate hexahistidine-tagged peptide from metal affinity solid supports.9,10 Alternatively, O-methoxyamine was used for releasing peptides captured by means of thiazolidine/oxazolidine-11 or oxime-forming reactions.12
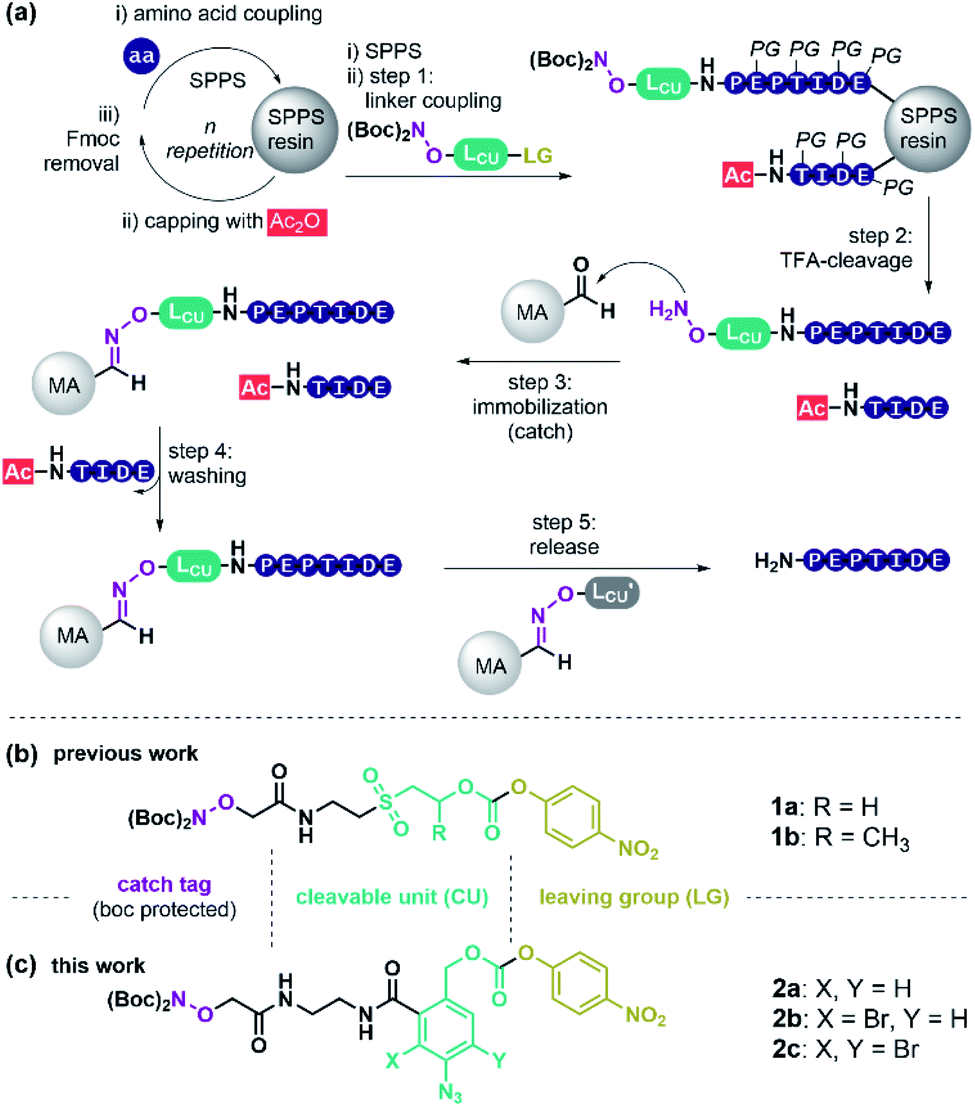 | ||
| Scheme 1 (a) General scheme of oxime-based c&r purification of peptides. (b) Molecular structure of previously reported base-labile linker molecules 1a, 1b and (c) of the novel reductively cleavable linker units 2a, 2b and 2c described in this paper. aa: amino acid; PG: protecting group; MA: modified agarose beads; LCU: cleavable linker unit; LG: leaving group. | ||
Instead of utilising peptide residues for immobilisation that remain in the product, purification handles which represent tracelessly cleavable linkages offer more versatility. We identified 30 independent linker-based c&r systems, that may be categorised by their cleavage stimulus. Acid-labile c&r linkers based on 4-alkyl- or 4-alkoxybenzyloxycarbonyl ethers13,14 were used to capture fully protected crude peptides, however, dissolution and handling of fully protected peptides can be challenging. Base-labile linkers allow processing of unprotected crude peptides obtained after standard TFA cleavage, which typically have higher solubility than their protected counterparts. Base-labile scaffolds such as β-sulfonylethyloxycarbonyl15–22 and fluorenylmethoxy-carbonyl (Fmoc)-linkers23–26 provided TFA-stability while allowing cleavage upon basic conditions. Both required pH > 11 for cleavage, which can be harmful to specific peptide sequences. We previously introduced a method termed Peptide Easy Clean (PEC, Scheme 1), that used a β-sulfonylethyloxycarbonyl linker to purify pharmaceutically relevant peptides in high yield and purity.27 However, base-induced side reactions, such as oxazolidinone-formation were observed using linker 1a or 1b (Scheme 1b) with certain peptides, questioning the general applicability of this c&r system.
Some c&r linkers rely on nucleophile-induced cleavage. Typical linkers included phenyl sulfonate,28 dimedone-enamines (Dde)-type29–31 and keto-acyl linkers,32,33 which are susceptible to cleavage upon treatment with internal Cys residue, hydrazine/NH2OMe32 or o-phenylenediamine,33 respectively. However, with exception of Cys residue-mediated cleavage of the phenyl sulfonate linker,28 the cleavage agents remain in the product, and a subsequent purification step such as HPLC is required to obtain a “cleavage agent-free” product. Contamination with release agents also accompanies linker cleavage reactions induced by electrophiles such as CNBr,8,34 or oxidizing agents such as sodium periodate.35,36 Light is a reagent-free trigger for the release of ortho-nitrobenzyl-carbamate linkers and has been reported.37,38 However, upscaling is difficult due to the limited penetration of light through solid- or gel-based capture materials.
Surprisingly, no reductively cleavable linker has yet been described for peptide purification. Reductively cleavable linker molecules could enable widespread application of c&r within peptide manufacturing since reductive conditions are generally mild and compatible with all canonical residues.
Katzenellenbogen et al. introduced a 4-amidobenzyl carbamate prodrug in 1981 that liberated the model drug aniline after tryptic cleavage of the 4-amido-group.39 The resulting amino-group triggered a 1,6-elimination towards azaquinonemethide, CO2, and aniline.40 Similar concepts with reductive stimuli followed.41,42 Machida et al. brought this concept to the realm of oligomer synthesis, by developing a C-terminal linker to screen and release a library of peptides conjugated to peptide nucleic acids (PNAs).43 The PNA-peptide-PNA sequences were synthesized on a novel C-terminal para-azido benzyl carbamate linker on water-swellable amino-methyl resin, whereas over 1000 peptide sequences where generated by split-and-mix.44 After TFA-cleavage the linkage stayed intact enabling on-bead screening, where the PNA hybridization formed a hairpin structure presenting the peptides. Positive hits were analysed by azide reduction with PMe3 of the linker leading to 1,6-elimination and release from support.
Herein we report the development of a first-in-class reductively cleavable linker molecule for peptide purification that provides unprotected peptides without contamination of cleavage agents. The linker contains a para-azidobenzyl carbamate core 2 (Scheme 1c). This molecular arrangement enables a safety-release reaction sequence, in which azide reduction is used to activate the linker for mild acid-induced fragmentation. Of note, reducing agents can be removed by filtration prior to release enabling a side-reaction free purification of SPPS peptides via a truly traceless c&r process.
Results and discussion
The first reductively cleavable linker for peptide purification
To retain the working principles of linker coupling and immobilisation from our previously published base-labile linker (Scheme 1b), we constructed the new linker with an oxyamine catch tag, a para-nitrophenol leaving group, and a reductively cleavable para-azido-benzyl unit (Scheme 1c). The reductively cleavable linker 2a was accessible in gram-scale via a four-step synthesis from 6-amino-phthalide (Scheme S1†).Scheme 2 shows the adapted c&r pathway with reductively cleavable linkers. The linker molecule 2 was connected to the N-terminus of resin-bound peptides within 120 minutes, facilitated by Oxyma. After that, TFA cleavage and subsequent ether precipitation provided linker-tagged peptides 3 (Scheme 2). Subsequent dissolution for immobilization of hydrophobic, aggregating, and hydrogel-forming peptides can be tedious or impossible. A screen with different solvent systems (S4.2†) revealed that a set of test peptides dissolved well in pure dimethyl sulfoxide (DMSO). After adding 10 vol% of a 0.1 M Na citrate buffer containing chaotropic agent (7 M guanidium chloride (GdmCl), pH 3.5), no hydrogel formation or aggregation occurred. This dissolution mixture enables quantitative immobilization on aldehyde-modified agarose beads (MA) within 90 min by forming oxime 4 (Scheme 2). Dissolution and immobilisation in pure hexafluoroisopropanol is a useful option for very hydrophobic sequences (Fig. S7†).
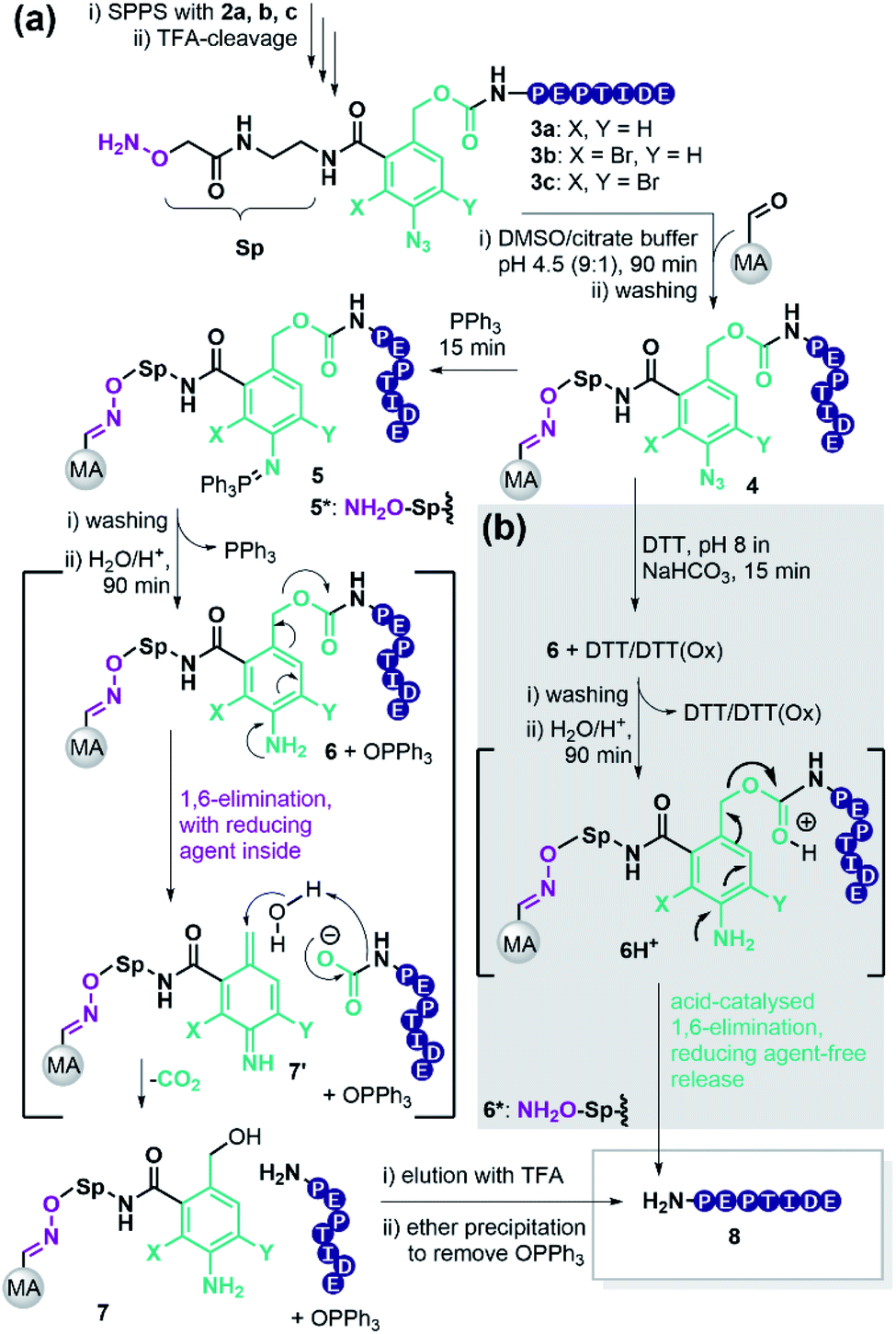 | ||
| Scheme 2 (a) Application of reductively cleavable linker 2a, b, c in c&r purification of peptides with PPh3 as reductive stimulus; MA: modified agarose beads, GdmCl: Guanidinium chloride. (b) Usage of dithiothreitol (DTT) as reductive stimulus, *: linker-modified peptide in solution. | ||
Next, we sought a reagent that effectively reduces the para-azido benzyl moiety to its aniline derivative. Triphenylphosphine (PPh3) has frequently been used in Staudinger reactions.45 Removal of excess PPh3 is facile with linker 2a, because the iminophosphorane 5 afforded by reduction of the azide 4, is stable enough to allow the washing out of excess PPh3.
Aqueous treatment then induces the acid-catalysed hydrolysis of the iminophosphorane and provides the aniline 6 releasing Ph3PO. The nitrogen lone pair triggers a 1,6-elimination freeing the target peptide as a carbamate that spontaneously decomposes to CO2 and the desired peptide 8. According to literature, the linker forms the azaquinonemethide 7′ upon scission, whereas water attacks at the benzylic position to form the stable benzyl alcohol 7.46
This strategy yielded the purified peptides as a white solid after elution with TFA and precipitation in ether. Importantly, Ph3PO partitioned to the ether phase, though traces remained. The described procedure enables fast work up since no lyophilization is required.
We assessed the performance of the method by analysing a panel of four peptides (Table 1) in c&r purification with reductively cleavable linker 2a and the previously used base-labile capture tag 1a.27
| Name | Peptide sequencea | Expected base-induced side product | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Bold underlined motif underwent base-induced side reactions earlier. b The mass deviation to the native peptide when side product is formed. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P1 | H-![[T with combining low line]](https://www.rsc.org/images/entities/b_char_0054_0332.gif) RYQAKPVNRSTPISTGKEG-OH RYQAKPVNRSTPISTGKEG-OH |
Oxazolidinone (+26 Da)b | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P2 |
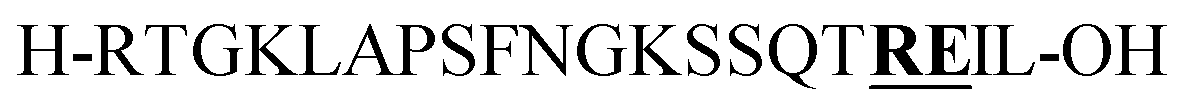
|
Citrulline (+1 Da)b | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P3 |
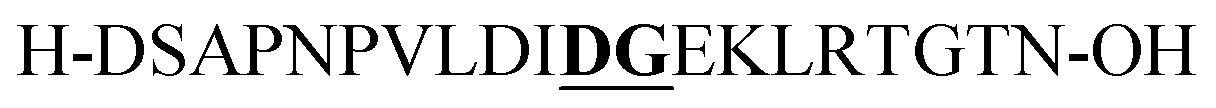
|
Aspartimide (−18 Da)b | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P4 | H-ARTKQTARKSTGGKA-OH | None | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
From the tested sequences, three (P1–3) are prone to base-induced side-reactions, as reported previously.27P1 can form an oxazolidinone upon deprotonation of alcohol side-chains of N-terminal Ser and Thr residues. P2 is susceptible to citrulline formation at the Arg–Glu motif and the sequence from Miraculine (1–20) P3 yields an aspartimide at the critical Asp–Gly junction. To prevent aspartimide formation during SPPS of P3 the peptide was synthesized using Fmoc-Asp(OMpe)-OH and Fmoc-Asp(OtBu)-(Dmb)Gly-OH building blocks as precautionary measures. The histone H3 (1–15) sequence P4 is an example of a peptide that is difficult to purify by HPLC; at least two HPLC runs with different acidic modifiers (TFA and heptafluorobutyric acid) were required (data not shown).
A TFA cleavage with a fraction of each peptidyl resin was performed after solid phase synthesis. Ultra-performance liquid chromatography (UPLC) analyses revealed impurities, mostly from capped truncations that were present prior to the introduction of the cleavable capture tags (Fig. 1(i)). In case of P4, these impurities were closely or co-eluting with P4. After coupling of the capture tags 1a and 2a and TFA-cleavage, the main products were in all cases the linker-modified target peptides (Fig. 1(ii)). However, a substantial amount (∼30%) of unmodified peptide remained when employing linker 2a. Next, the target peptides were bound to the solid support (modified agarose, MA), while most impurities remained in the supernatant (Fig. S2–S5†).
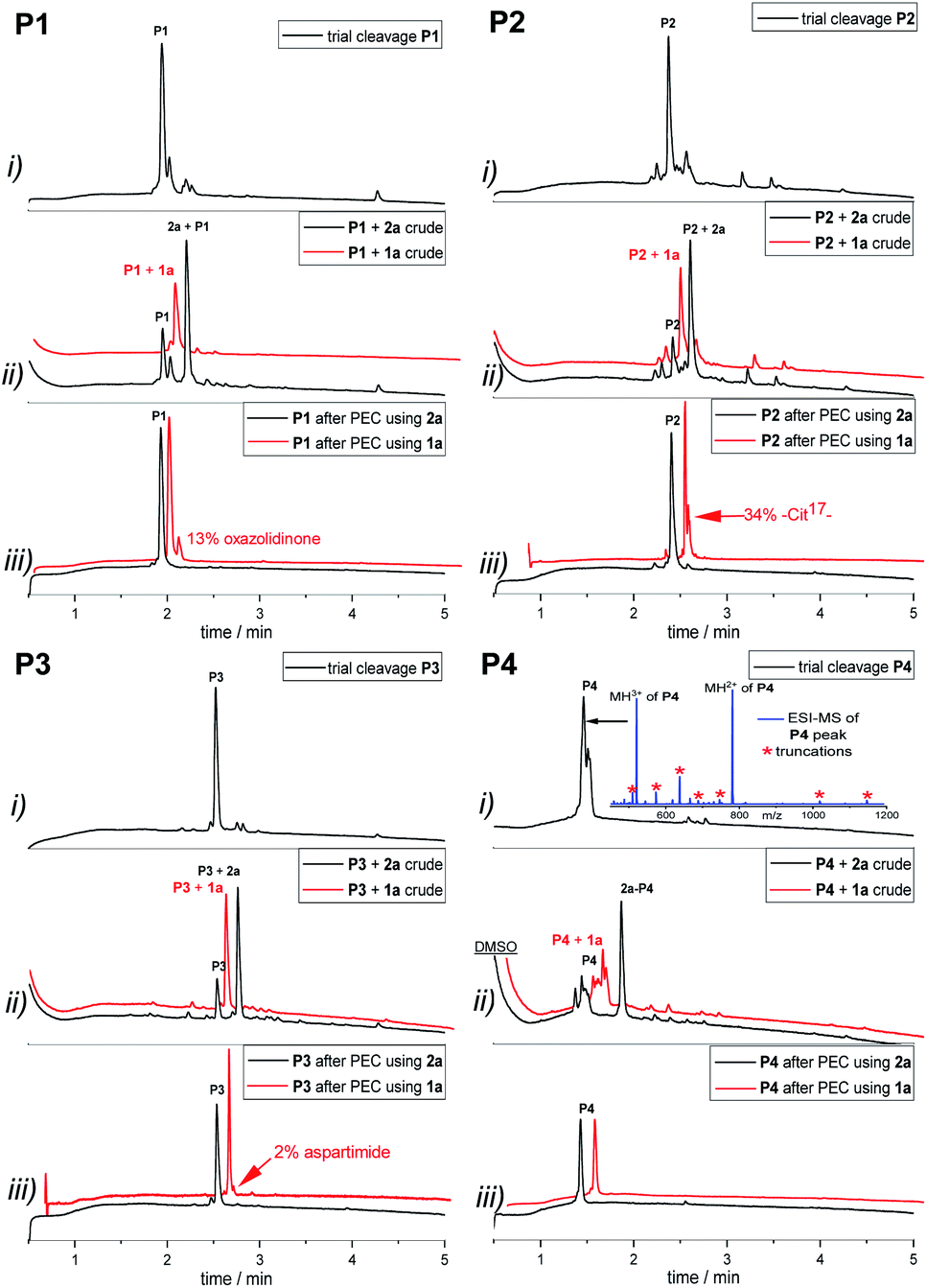 | ||
| Fig. 1 PEC-purification of base-sensitive peptides P1–3 and Histone H3 (1–15) peptide P4. (i) Trial cleavage of peptide before linker coupling; (ii) full cleavage after linker coupling of 1a or 2a, (iii) after PEC-process using 1a or 2a. *Assigned masses of co-eluting truncated peptides in P4 trial cleavage; red traces using base-labile 1a and black traces using reductively cleavable linker 2a. | ||
After washing the purified peptides were released by basic treatment with MeNH2 (pH 11) when 1a or by reduction with PPh3 when linker 2a was used. Base induced side reactions, such as oxazolidinone formation (13%) at the N-terminal threonine of P1, citrulline formation (34%) at arginine in P2, and aspartimide formation (2%) in P3, were confirmed. Gratifyingly, the formation of side products did not occur when the peptides were detached via PPh3 reduction in combination with linker 2a (black traces of products in P1–3Fig. 1(iii)). Independent of the linker system used, for P4, no side-products or co-eluting impurities remained after purification.
Moreover, the comparative study showed that the new reductively cleavable linker 2a yields higher peptide purity than the base-labile linker 1a (Table 2). However, in contrast to the purities, recoveries of peptides purified with 2a were lower (22–40% compared with 32–74% with 1a). As mentioned above, UPLC traces of crudes obtained after introduction of linker 2a, showed around 30% unmodified peptide (Fig. 1(ii)) even though coupling of the linker proceeded quantitatively according to a negative chloranil test.47 This pointed to a loss of linker during TFA cleavage.
| No. | Crude puritya | Puritya after PEC using linker | Recoveryb using linker | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 2a | 2b | 2c | 1a | 2a | 2b | 2c | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Purity determined by integration of UPLC-MS traces at 210 nm. b (Final weight × purity after PEC)/(molecular weight × synthetic scale × crude purity) see S3.6. c Purity and recovery with dithiothreitol as reducing agent. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P1 | 67% | 77% | 91% | 91% | 91% | 74% | 40% | 74% | 44% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P2 | 42% | 67% | 83% | 89% | 92% | 60% | 40% | 70% | 42% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P3 | 75% | 84% | 88% | 87% | 88% | 46% | 23% | 56% | 32% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P4 | 36% | 92% | 89% | 96% | 91%c | 32% | 22% | 58% | 29%c | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
To test the assumption, we analysed the linker stability in solution. The linker was coupled to n-propylamine and the resulting conjugate 9a (Fig. 2a) was subjected to TFA cleavage conditions. UPLC analysis (Fig. 2b, black lines) exposed a vulnerability of the benzyl carbamate structure.
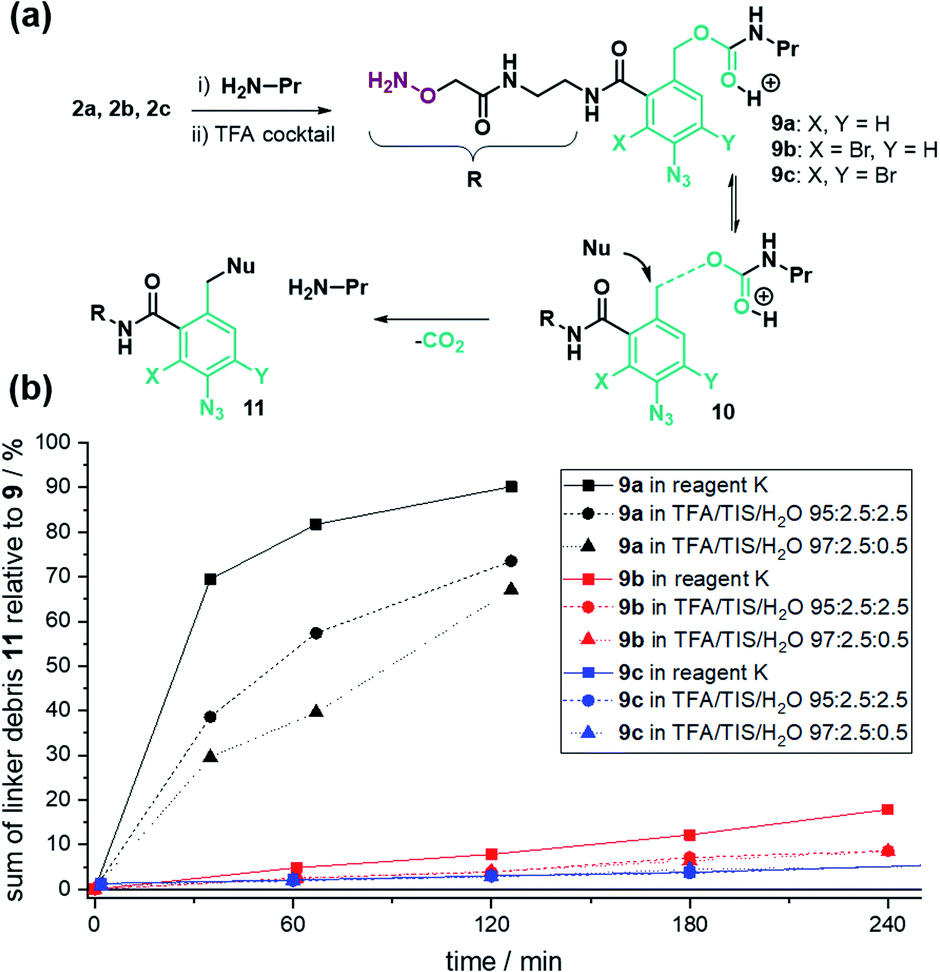 | ||
| Fig. 2 (a) Generation of propylamine carbamates 9a–c from 2a–c. (b) TFA-stability analysis of carbamates 9a–c relative to the sum of UV-UPLC integrals of TFA-cleavage products 11. reagent K: TFA/H2O/PhOH/PhSMe/ethanedithiol 82.5 : 5 : 5 : 5 : 2.4. | ||
We assume that a cleavage mechanism48 similar to the acidic deprotection of the carboxybenzyl (Cbz) protecting group (Fig. 2a) takes place.49 In that case, protonation of the urethan moiety 9 facilitates a nucleophilic attack at the benzylic position, inducing the detachment of N-propyl-carbamic acid, subsequently decomposing to CO2 and propylamine. The ortho-amide may provide anchimeric assistance and promotes the scission of the benzylic carbamate.
2nd generation reductively cleavable linker molecules
We expected the acid stability of the linker to correlate with the electron density of the benzylic carbon atom. Using Hammett-values, we reasoned that the electron-withdrawing properties of the ortho-amide (σo/p: 0.35)§50 and para-azide (σp: 0.08)50 did not sufficiently destabilize the transition state 10a (Fig. 2a) to hinder decomposition in TFA.An additional electron-withdrawing substituent was therefore required to increase the TFA stability. From the rich portfolio of listed Hammett values, we chose halogenation due to the simplicity of introduction and its relatively high electron-withdrawing effect (e.g. Br(σm): 0.39).50 The synthesis of 2-bromo (2b) and 2,4-di-bromo (2c) linker followed the same scheme as linker 2a, except starting with an additional bromination step of 6-amino phthalide using either one or two equivalents of N-bromosuccinimide (NBS, Schemes S2 and S3†). The stability test with linker-coupled n-propylamine confirmed an increased TFA-stability for the mono- 9b and di-bromo carbamate 9c (Fig. 2b). After 2 hours of treatment with TFA cocktail reagent K,51 92% of 9b and 97% of 9c remained intact (Fig. S10†). The stability for all linker conjugates was higher in TFA/TIS/H2O cleavage cocktails (Fig. 2b).
Encouraged by these results, we repeated the purification of peptide P1–4 with the stability-enhanced linkers 2b and 2c. The absence of unmodified peptides after global TFA deprotection provided testimony for the increased TFA-stability using the brominated linkers 2b and 2c (Fig. S2–S5†). Accordingly, after PEC purification, the amounts of obtained peptides were twice as high on average when using mono-bromo linker 2b compared with linker 2a (Table 2). Surprisingly, with di-bromo linker 2c the recoveries of the PEC-process were as low as with linker 2a. We assumed that di-bromination might kinetically hamper the reductive release (Scheme 2).
Consequently, we investigated the azide reduction, iminophosphorane hydrolysis, and 1,6-elimination of linker-coupled model sequences in solution (from 3a–c to 8, Schemes 2 and S7†). First, we found that 20 eq. PPh3 in MeCN/AcOH/H2O is sufficient for a quantitative reduction of 3a, 3b and 3c after 15 min (Fig. S11, Tables S8 and S9†). For all cases, the iminophosphorane intermediate 5*a–c was absent in UPLC-MS analysis indicating that hydrolysis is as fast as described,52 and the elimination is the rate-limiting step. Interestingly, we found significant differences between linker-derivates, judged by the amount of aniline-linker-modified peptides 6*a–c. The linker peptide conjugate 6a* without bromination was only present in traces, while the brominated aniline derivates 6b* and 6c* were the main products after reduction. Quantitative release of free peptide 8 could be triggered for 6b* by addition of 10 vol% TFA (Table S8†). The di-bromo linker conjugate 6c* gave only 88% free peptide 8 after treatment with TFA amounts as high as 75% (Table S9†).
In parallel, we investigated dithiothreitol (DTT) as a non-hazardous reducing reagent alternative to PPh3. The model sequence (P5: AKADEVSLHKWYG) was modified with all linker-types 2a–c. Analysis of the reduction of linker modified peptides 3a–c in solution showed that a 15 min treatment with 0.32 M DTT at pH 8 induced the quantitative reduction for all linker types (Fig. S12†).
In contrast to the reduction with PPh3 at acidic pH, aniline linker peptide conjugates 6*a–c showed higher stability at pH 8 (Fig. S12D†). Thus, a safety-release became conceivable, where the stability of brominated linker conjugates at neutral to basic pH is allowing reductive agent wash-out with subsequent acidic release of peptide.
Brominated linker 2b enables safety-release with DTT
To test the safety-release hypothesis, we quantified the stability of linker-conjugates 6a–c on solid support. Peptide P5 was linker modified to 3a–c and immobilized. Reduction with DTT at pH 8 (Scheme 2b) was carried out as in solution yielding 6a–c. Monitoring the UV-integrals of the free peptide 8 (P5) in reduction supernatant (Fig. 3, violet lines) and during acidic treatment (green lines) was used to quantify the lability of 6a–c during these steps.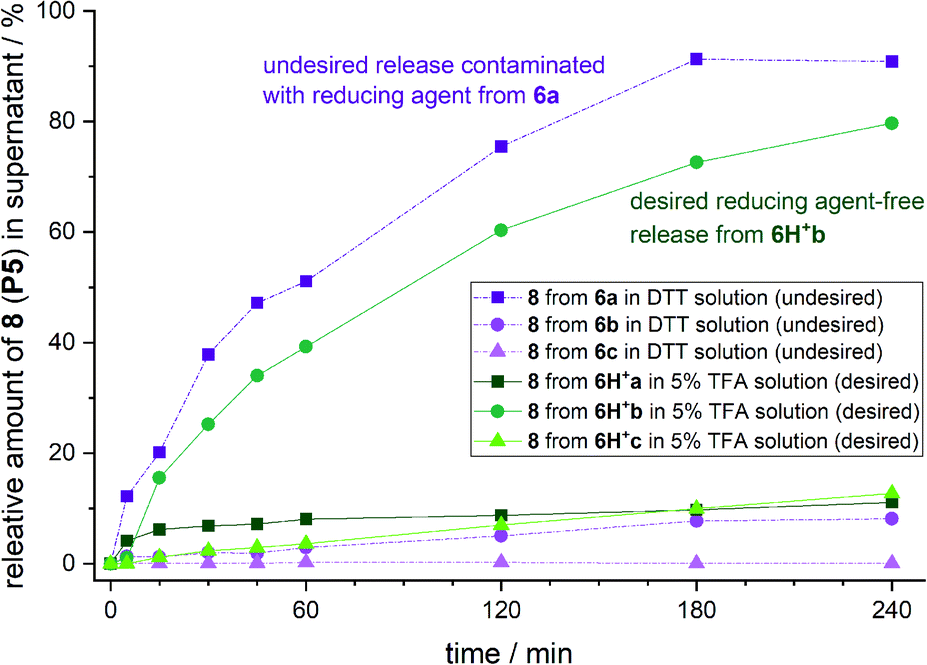 | ||
| Fig. 3 Amount of released peptide P5 in DTT supernatant (undesired) and after DTT wash-out in 5% aq. TFA supernatant (desired) calculated by the UV(210 nm) integral of product peak relative to the sum of DTT and TFA released peptide from 6a. | ||
The brominated linker conjugates 6b and 6c remained stable during the reduction of the azide at pH 8, while 6a decomposed gradually, releasing 90% 8 (P5) after 4 h. Removal of excess reducing agent and oxidized by-products is, therefore, only feasible for 6b and 6c.
After washing away DTT/DTT(Ox.) with water and acetonitrile, the resin was treated with 5% TFA in water (pH 0.41). UPLC analysis of the supernatant indicated that 79% of free peptide 8 was released from 6b, and only 10% from 6c, after 4 h. Hence, the mono-bromo linker 2b provides sufficient stability of the aniline species for wash-out of DTT/DTT(Ox.) while allowing the peptide release by a final acid treatment.
Density functional theory (DFT) calculations helped to understand the effect of bromination on linker stability in the various states of the PEC process. We calculated proton affinities and partial charges of the linker-propylamine conjugate 9a–c and its reduced aniline derivates 12a–c (Fig. S18, S19 and S22†). Partial charge analysis suggests, that the brominated aniline species 12b,c are stable towards a spontaneous 1,6-elimination due to reduced electron density of the benzylic carbon. Protonation of the carbamate oxygen pulls electron density from the aromatic core towards the benzylic carbon enabling 1,6-elimination of the brominated linkers and thus release of the peptide under acidic conditions.
Purification of a personalized peptide vaccine set of 20 peptides within a single day
As for conventional peptide synthesis, the manufacturing of peptidic neoantigen vaccines for cancer immunotherapy consists of two main steps: (1) parallel SPPS and (2) subsequent one-by-one purification by RP-HPLC. The purification step is usually the most time-consuming production step of such peptide libraries. Timely delivery of neoantigen sets in an affordable manner is critical for immunotherapy success. Currently, three to four months pass from the moment of tumour biopsy until the delivery of the vaccine formulation.53 The ability to purify peptides in parallel would massively increase the manufacturing speed of the 20–30 neoantigen peptides typically used per cocktail. Therefore, we aimed to show that PEC could accelerate peptide purification compared with linear HPLC purification. We chose 20 peptides from a previously reported vaccination cocktail, which has been tested on 16 patients in phase I clinical trials for glioblastoma immunotherapy (Table 3).54| No. | Sequencea | Crude purityb | Final purity | Recoveryc | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Fmoc-Lys(Trt)-OH has been used as building block for Lys residues. b UV-purity (A210 nm) on UPLC-MS of crude linker-modified peptides after SPPS and TFA-cleavage. c (final weight × purity after PEC)/(molecular weight × synthetic scale × crude purity) see S3.6. d Fmoc-Cys(StBu)-OH has been used as building block for Cys residues. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P6 | H-GWVKPIIIGHHAYGDQYRAT-NH2 | 56% | 93% | 45% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P7 | H-TLYEQEIEV-NH2 | 79% | 94% | 56% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P8 | H-HGSRKNITDMVEGAKKANG-NH2 | 42% | 91% | 52% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P9 | H-SLLNQPKAV-NH2 | 77% | 99% | 100% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P10 | H-EDPYLFELPVLKYLDMGTT-NH2 | 52% | 66% | 73% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P11 | H-ALAVLSNYDA-NH2 | 76% | 92% | 93% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P12 | H-TMEDKIYDQQVTKQCdLCdF-NH2 | 71% | 98% | 69% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P13 | H-YSYPETPLYMQTASTSYYE-NH2 | 38% | 69% | 69% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P14 | H-KVGYTERQRWDFLSEASIM-NH2 | 6% | 99% | 81% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P15 | H-RLRMREHMMKNVDTNQD-NH2 | 35% | 99% | 76% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P16 | H-VYEKNGYIYF-NH2 | 48% | 98% | 90% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P17 | H-ALAVLCdNYDA-NH2 | 73% | 85% | 81% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P18 | H-ALVPPSKRKMWVVSPAEKA-NH2 | 56% | 99% | 72% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P19 | H-ISTPTPTIVHPGSLPLHLG-NH2 | 57% | 90% | 60% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P20 | H-IVQENNTPGTYLLSVSARD-NH2 | 49% | 81% | 69% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P21 | H-RFHMKVSVYLLAPLREALS-NH2 | 45% | 99% | 57% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P22 | H-ENLKQNDISAEFTYQTKDA-NH2 | 45% | 90% | 82% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P23 | H-YMMPVNSEV-NH2 | 71% | 90% | 100% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P24 | H-TNDVKTLADLNGVIEEEFT-NH2 | 43% | 75% | 63% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P25 | H-SAWLFRMWYIFDHNYLKPL-NH2 | 35% | 66% | 49% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
We used optimized TFA cleavage cocktails (Fig. S14†) together with Fmoc building blocks Fmoc-Lys(Trt)-OH (Fig. S15†) and Fmoc-Cys(StBu)-OH (Fig. S16†) to minimize side products in the full-length chain. The peptides were synthesized in parallel on an automated peptide synthesiser followed by automated coupling of linker 2b. TFA-cleavage and PEC-purification were performed in parallel within 6 hours.
The purities (determined by analysis of UPLC and detection at 210 nm) of the 20 crude peptides with an average length of 17 residues after SPPS and coupling of linker 2b varied significantly, ranging from 6% for P14 to 79% to for P7, with a mean purity of 53% (Fig. 4a). After the PEC purification, the mean purity was 89%, ranging from 66% to 99%, with an average recovery of 72% (45–100%). Impressively, the purity of peptide P14 improved from 6% to 99%, which would have been difficult to achieve by HPLC due to closely eluting impurities (Fig. 4b). The majority of the purified peptides (14 of 20) had a final purity of over 90%.
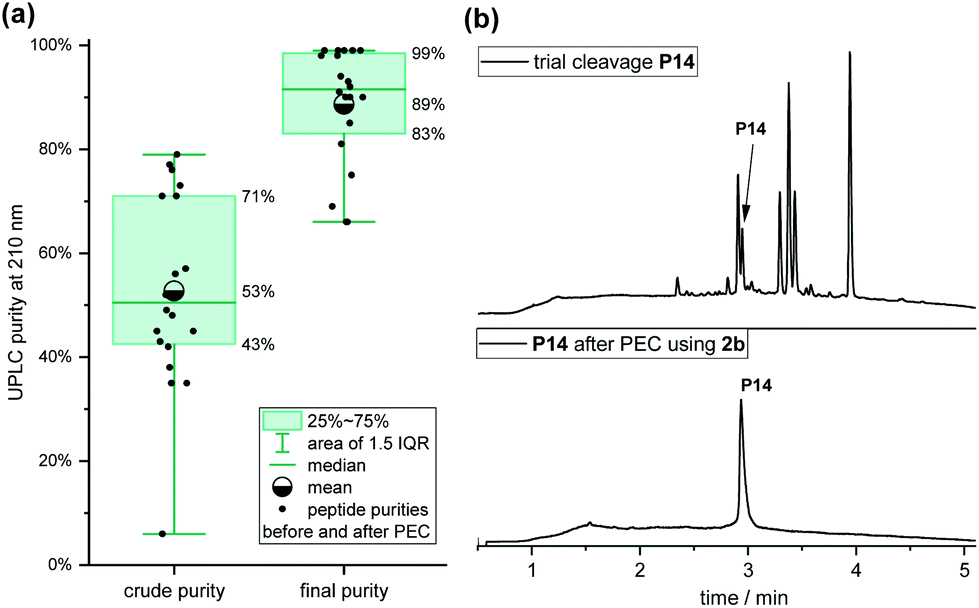 | ||
| Fig. 4 (a) Box-diagram of crude vs. final purities of the 20 neoantigen peptides. IQR: interquartile range. (b) UPLC chromatograms before (i) and after the PEC process (ii) of peptide P14 from a personalized peptide set of 20 peptides purified in parallel. | ||
Conclusions
In this report we have described the synthesis and development of a first-in-class reductively cleavable linker system for peptide purification and applied it successfully to parallel peptide purification. Bromination on the para-azido-benzyl core led to increased TFA-stability and enabled a safety-release mechanism. The linker 2b enabled a contamination-free traceless release, where the reductive agent could be washed out prior to acid-catalysed liberation of desired peptides by 1,6-elimination (safety-release). The utility of the method was demonstrated by the rapid purification (within 6 hours) of a neoantigen peptide cocktail of 20 peptides for personalized cancer immunotherapy. The results presented demonstrate the great potential of PEC as part of the rapid production of peptide sets. Furthermore, we anticipate that the presented PEC technology will be useful for purification of peptide libraries or for accelerating the peptide manufacturing process in general.Conflicts of interest
The authors declare competing financial interests: the technology described in the manuscript is part of pending patent applications. Kit products containing linker molecule and aldehyde-modified agarose beads as presented here can be purchased at http://belyntic.com.Acknowledgements
We thank the company CreativeQuantum GmbH for performing the computational investigations, and their assistance throughout all aspects of the DFT study. This work was financially supported by the EXIST Forschungstransfer program from the German Bundesministerium für Wirtschaft und Energie (BMWi) and the European Social Fund (ESF) and by the ProFIT program of the Investitionsbank Berlin financially supported by the European Regional Development Fund (ERDF).Notes and references
- A. C. Lee, J. L. Harris, K. K. Khanna and J. H. Hong, Int. J. Mol. Sci., 2019, 20, 2383–2404 CrossRef.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2017, 2700–2707 Search PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- M.-I. Aguilar, HPLC of Peptides and Proteins, Springer, Totowa, NJ, 2004 Search PubMed.
- R. Pipkorn, C. Boenke, M. Gehrke and R. Hoffmann, J. Pept. Res., 2002, 59, 105–114 CrossRef CAS.
- J. R. Currier, L. M. Galley, H. Wenschuh, V. Morafo, S. Ratto-Kim, C. M. Gray, L. Maboko, M. Hoelscher, M. A. Marovich and J. H. Cox, Clin. Vaccine Immunol., 2008, 15, 267–276 CrossRef CAS.
- J. W. de Beukelaar, J. W. Gratama, P. A. Smitt, G. M. Verjans, J. Kraan, T. M. Luider and P. C. Burgers, Rapid Commun. Mass Spectrom., 2007, 21, 1282–1288 CrossRef CAS.
- D. E. Krieger, B. W. Erickson and R. B. Merrifield, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 3160–3164 CrossRef CAS.
- D. Bang and S. B. H. Kent, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5014 CrossRef CAS.
- S. F. Loibl, R. Zitterbart, Z. Harpaz and O. Seitz, Chem. Sci., 2016, 7, 6753–6759 RSC.
- M. Villain, J. Vizzavona and K. Rose, Chem. Biol., 2001, 8, 673–679 CrossRef CAS.
- C. E. Murar, F. Thuaud and J. W. Bode, J. Am. Chem. Soc., 2014, 136, 18140–18148 CrossRef CAS.
- D. V. Filippov, D. J. van Zoelen, S. P. Oldfield, G. A. van der Marel, H. S. Overkleeft, J. W. Drijfhout and J. H. van Boom, Tetrahedron Lett., 2002, 43, 7809–7812 CrossRef CAS.
- M. Zhang, D. Pokharel and S. Fang, Org. Lett., 2014, 16, 1290–1293 CrossRef CAS.
- V. Aucagne, I. E. Valverde, P. Marceau, M. Galibert, N. Dendane and A. F. Delmas, Angew. Chem., Int. Ed., 2012, 51, 11320–11324 CrossRef CAS.
- S. Funakoshi, H. Fukuda and N. Fujii, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 6981–6985 CrossRef CAS.
- G. Delsolar, F. Albericio, R. Eritja and M. Espinosa, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5178–5182 CrossRef CAS.
- S. Funakoshi, H. Fukuda and N. Fujii, J. Chromatogr., A, 1993, 638, 21–27 CrossRef CAS.
- P. C. de Visser, M. van Helden, D. V. Filippov, G. A. van der Marel, J. W. Drijfhout, J. H. van Boom, D. Noort and H. S. Overkleeft, Tetrahedron Lett., 2003, 44, 9013–9016 CrossRef CAS.
- D. W. Anderson, G. J. Cotton, M. H. Alastair, W. A. Paul and W. Ian, Europe Pat., PCT/GB2011/000363, 16.03.2011, Almac Sciences (Scotland) Ltd., 2011 Search PubMed.
- L. E. Canne, R. L. Winston and S. B. H. Ken, Tetrahedron Lett., 1997, 38, 3361–3364 CrossRef CAS.
- L. E. Canne, P. Botti, R. J. Simon, Y. J. Chen, E. A. Dennis and S. B. H. Kent, J. Am. Chem. Soc., 1999, 121, 8720–8727 CrossRef CAS.
- P. Mascagni, H. L. Ball and G. Bertolini, Anal. Chim. Acta, 1997, 352, 375–385 CrossRef CAS.
- R. B. Merrifield and A. E. Bach, J. Org. Chem., 1978, 43, 4808–4816 CrossRef CAS.
- H. L. Ball, G. Bertolini, S. Levi and P. Mascagni, J. Chromatogr., A, 1994, 686, 73–83 CrossRef CAS.
- A. R. Brown, S. L. Irving and R. Ramage, Tetrahedron Lett., 1993, 34, 7129–7132 CrossRef CAS.
- O. Reimann, O. Seitz, D. Sarma and R. Zitterbart, J. Pept. Sci., 2019, 25, e3136 CrossRef.
- M. Kheirabadi, G. S. Creech, J. X. Qiao, D. S. Nirschl, D. K. Leahy, K. M. Boy, P. H. Carter and M. D. Eastgate, J. Org. Chem., 2018, 83, 4323–4335 CrossRef CAS.
- M. Galibert, V. Piller, F. Piller, V. Aucagne and A. F. Delmas, Chem. Sci., 2015, 6, 3617–3623 RSC.
- A. Jacob, J. Hoheisel, M. Dauber, M. Wiessler, P. Lorenz, H. Fleischhacker and H.-C. Kliem, International Pat., PCT/EP2011/005137, 13.10.2010, Deutsches Krebsforschungszentrum, 2012 Search PubMed.
- B. Kellam, W. C. Chan, S. R. Chhabra and B. W. Bycroft, Tetrahedron Lett., 1997, 38, 5391–5394 CrossRef CAS.
- N. Ollivier, R. Desmet, H. Drobecq, A. Blanpain, E. Boll, B. Leclercq, A. Mougel, J. Vicogne and O. Melnyk, Chem. Sci., 2017, 8, 5362–5370 RSC.
- T. Hara, A. Tainosho, K. i. Nakamura, T. Sato, T. Kawakami and S. Aimoto, J. Pept. Sci., 2009, 15, 369–376 CrossRef CAS.
- M. A. Roggero, C. Servis and G. Corradin, FEBS Lett., 1997, 408, 285–288 CrossRef CAS.
- J. Vizzavona, M. Villain and K. Rose, Tetrahedron Lett., 2002, 43, 8693–8696 CrossRef CAS.
- S. Pomplun, C. R. Shugrue, A. M. Schmitt, C. K. Schissel, C. E. Farquhar and B. L. Pentelute, Angew. Chem., Int. Ed., 2020, 59, 11566–11572 CrossRef CAS.
- O. Reimann, C. Smet-Nocca and C. P. Hackenberger, Angew. Chem., Int. Ed., 2015, 54, 306–310 CrossRef CAS.
- J. Olejnik, S. Sonar, E. Krzymañska-Olejnik and K. J. Rothschild, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7590 CrossRef CAS.
- P. L. Carl, P. K. Chakravarty and J. A. Katzenellenbogen, J. Med. Chem., 1981, 24, 479–480 CrossRef CAS.
- R. Erez and D. Shabat, Org. Biomol. Chem., 2008, 6, 2669–2672 RSC.
- P. D. Senter, W. E. Pearce and R. S. Greenfield, J. Org. Chem., 1990, 55, 2975–2978 CrossRef CAS.
- E. W. P. Damen, T. J. Nevalainen, T. J. M. van den Bergh, F. M. H. de Groot and H. W. Scheeren, Bioorg. Med. Chem., 2002, 10, 71–77 CrossRef CAS.
- T. Machida, S. Dutt and N. Winssinger, Angew. Chem., Int. Ed., 2016, 55, 8595–8598 CrossRef CAS.
- A. Furka, F. Sebestyén, M. Asgedom and G. Dibó, Int. J. Pept. Protein Res., 1991, 37, 487–493 CrossRef CAS.
- H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 635–646 CrossRef CAS.
- A. Alouane, R. Labruère, K. J. Silvestre, T. Le Saux, F. Schmidt and L. Jullien, Chem.–Asian J., 2014, 9, 1334–1340 CrossRef CAS.
- T. Groth, M. Grøtli and M. Meldal, J. Comb. Chem., 2001, 3, 461–468 CrossRef CAS.
- D. Ben-Ishai and A. Berger, J. Org. Chem., 1952, 17, 1564–1570 CrossRef CAS.
- M. Bergmann and L. Zervas, Ber. Dtsch. Chem. Ges., 1932, 65, 1192–1201 CrossRef.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- D. S. King, C. G. Fields and G. B. Fields, Int. J. Pept. Protein Res., 1990, 36, 255–266 CrossRef CAS.
- S.-Y. Pyun, Y.-H. Lee and T.-R. Kim, Kinet. Catal., 2005, 46, 21–28 CrossRef CAS.
- U. Sahin and Ö. Türeci, Science, 2018, 359, 1355–1360 CrossRef CAS.
- N. Hilf, S. Kuttruff-Coqui, K. Frenzel, V. Bukur, S. Stevanović, C. Gouttefangeas, M. Platten, G. Tabatabai, V. Dutoit, S. H. van der Burg, P. thor Straten, F. Martínez-Ricarte, B. Ponsati, H. Okada, U. Lassen, A. Admon, C. H. Ottensmeier, A. Ulges, S. Kreiter, A. von Deimling, M. Skardelly, D. Migliorini, J. R. Kroep, M. Idorn, J. Rodon, J. Piró, H. S. Poulsen, B. Shraibman, K. McCann, R. Mendrzyk, M. Löwer, M. Stieglbauer, C. M. Britten, D. Capper, M. J. P. Welters, J. Sahuquillo, K. Kiesel, E. Derhovanessian, E. Rusch, L. Bunse, C. Song, S. Heesch, C. Wagner, A. Kemmer-Brück, J. Ludwig, J. C. Castle, O. Schoor, A. D. Tadmor, E. Green, J. Fritsche, M. Meyer, N. Pawlowski, S. Dorner, F. Hoffgaard, B. Rössler, D. Maurer, T. Weinschenk, C. Reinhardt, C. Huber, H.-G. Rammensee, H. Singh-Jasuja, U. Sahin, P.-Y. Dietrich and W. Wick, Nature, 2019, 565, 240–245 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06285e |
| ‡ Contributed equally. |
| § σp are considered to be a good approximation for the effect of ortho-substituents. The electron-withdrawing effect of the o-amide may be mitigated by the asymmetric assistance in the acidic cleavage of the linker. |
| This journal is © The Royal Society of Chemistry 2021 |