
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deep eutectic solvents: alternative reaction media for organic oxidation reactions
Graziano
Di Carmine
 ab,
Andrew P.
Abbott
c and
Carmine
D'Agostino
*b
ab,
Andrew P.
Abbott
c and
Carmine
D'Agostino
*b
aDipartimento di Scienze Chimiche e Farmaceutiche, Università di Ferrara, Via Luigi Borsari, 46, I-44121 Ferrara, Italy
bDepartment of Chemical Engineering and Analytical Science, The University of Manchester, The Mill, Sackville Street, Manchester, M13 9PL, UK. E-mail: carmine.dagostino@manchester.ac.uk
cSchool of Chemistry, University of Leicester, Leicester, LE1 7RH, UK
First published on 20th January 2021
Abstract
Deep eutectic solvents (DESs) have emerged as an alternative to ionic liquids (ILs). DESs share with ILs some appealing features, such as low vapor pressure, capability to dissolve reagents insoluble in common organic solvents, and the possibility to tune the overall pH of the medium by replacing one of the constituents of the mixture. Furthermore, DESs can be prepared by combining molecules that come from natural sources (i.e., glycerol, glucose), making them biodegradable. DESs have already been used for a variety of reactions and protocols since they were reported for the first time by A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70, and among the reactions studied, organic oxidation has recently gained much attention. In particular, the recyclability of these ionic compounds makes it possible to achieve anchoring of organic oxidants, such as TEMPO and peroxydisulfate, directly onto one species of the DES mixture components. In addition, their solubility properties play a crucial role in organic oxidation since DESs have the ability to dissolve both organic lipophilic and hydrophilic species, making the oxidation of organic compounds mediated by hydrogen peroxide more efficient. Herein we report the state of the art of this developing field, focusing on the benefits of substituting common organic solvents with DESs, especially in terms of sustainability, enhancement of reactivity, and recyclability.
 Graziano Di Carmine | Graziano Di Carmine obtained his Bachelor's Degree from the University of Rome “Tor Vergata”, a Master's Degree at the University of Bologna and a PhD in chemistry at the University of Ferrara under the supervision of Prof. Olga Bortolini working on umpolung reactivity promoted by organocatalysts. He was a Research Fellow at the ISOF CNR of Bologna and then moved to the University of Manchester as a Research Associate in the group of Dr Carmine D'Agostino working on NMR methods for the investigation of reactions promoted by solid-supported organocatalysts. He is currently a Research Fellow at the University of Ferrara. |
 Andrew Abbott | Andrew Abbott is Professor of Physical Chemistry at the University of Leicester where he has been since 1993. He started research into ionic liquids and deep eutectic solvents in 1999 and launched the spin-out company Scionix in 2000. He has research interests in electrodeposition, mineral and natural product extraction and biomaterials. |
 Carmine D'Agostino | Carmine D'Agostino received his BEng and MEng in Chemical Engineering at the Universita' di Napoli “Federico II” and a PhD in Chemical Engineering at the University of Cambridge under the supervision of Prof. Lynn Gladden. He is currently a Lecturer in Chemical Engineering at the University of Manchester. His research focuses on diffusion, dynamics and adsorption of complex fluids and fluids within porous structures and catalysts. He received several awards, including the Young Scientist Award at the International Congress on Catalysis, the Reaction Chemistry & Engineering Emerging Investigator award and a prestigious Junior Research Fellowship at the University of Cambridge. |
1. Introduction
Ionic liquids (ILs) have been of interest for more than 40 years as alternative media for a wide variety of applications1 and they have attracted significant attention in the past two decades in many areas of chemistry such as materials,2 gas separation,3 electrochemistry,4 biomass valorization5 and catalysis.6 While they were initially championed as being ‘green’ due to their low vapor pressure, their use is seen primarily as enabling processes which are difficult in molecular solvents. Primarily their unusual solvent properties and phase behavior have been utilized. A prime example of this is the BASIL process which uses an imidazole to neutralize an acid by-product from a reaction. The process intensification enabled by the formation of a liquid imidazolium chloride by-product decreases solvent use, negates a filtration process and results in a significant increase in process yield.7Early-generation ILs were formed by metals such as aluminium, tin and zinc and these were used in part as liquid catalysts for Lewis acid-catalyzed reactions. When the reagents and products were liquids then these were effectively solvent-free processes. Further research was then carried out to find metal-free alternatives, which maintained the required solvent properties, and a new class of ILs was designed by combining metal-free cations, mainly imidazolium analogues, with several coordinating and non-coordinating anions such as Cl−, BF4−, PF6− and NTf2−.8 While these have been proposed as green solvents, the fact that they are toxic, expensive to produce, and do not in many cases easily biodegrade has negated many opinions of their green credentials.9
The term ionic liquid was initially rigorously applied to systems that only contained ionic species which melted below 100 °C, but this definition has slowly been relaxed to include a wider range of fluids where ionic character tends to dominate solvent–solute interactions.10 Deep eutectic solvents (DESs) are just one of these ion-dominated systems which have been found to be useful for a variety of applications. DESs are easily formed by mixing the components, usually an ammonium salt and a hydrogen bond donor (HBD). DESs have the toxicological properties of the two components and so the choice of benign starting materials can produce materials of low toxicity. The majority of these components are low-cost and biodegradable. Many can be extracted from biomass, increasing their sustainability credentials.11 Among the ammonium salts, choline chloride (ChCl) is one of the most commonly used components. It is used on a large scale in a variety of commodity products including as an animal feed additive as it is a pro-vitamin. It is therefore biodegradable, available at low cost, has low toxicity and can be accessible from biomass. Choline chloride forms a eutectic mixture when blended with a HBD such as urea, glucose and oxalic acid. The addition of the HBDs decreases the lattice energy of both homocouples, reducing the freezing point. In recent years, a plethora of HBDs have been reported in the literature, producing a large library of ChCl-based DESs that covers a wide spectrum of physical and chemical properties. In addition, several alternatives to ChCl have been presented and the tuning of the two components seems to be the key to create the suitable medium required for each particular process. Some examples of salts and HBDs are summarized in Table 1.12
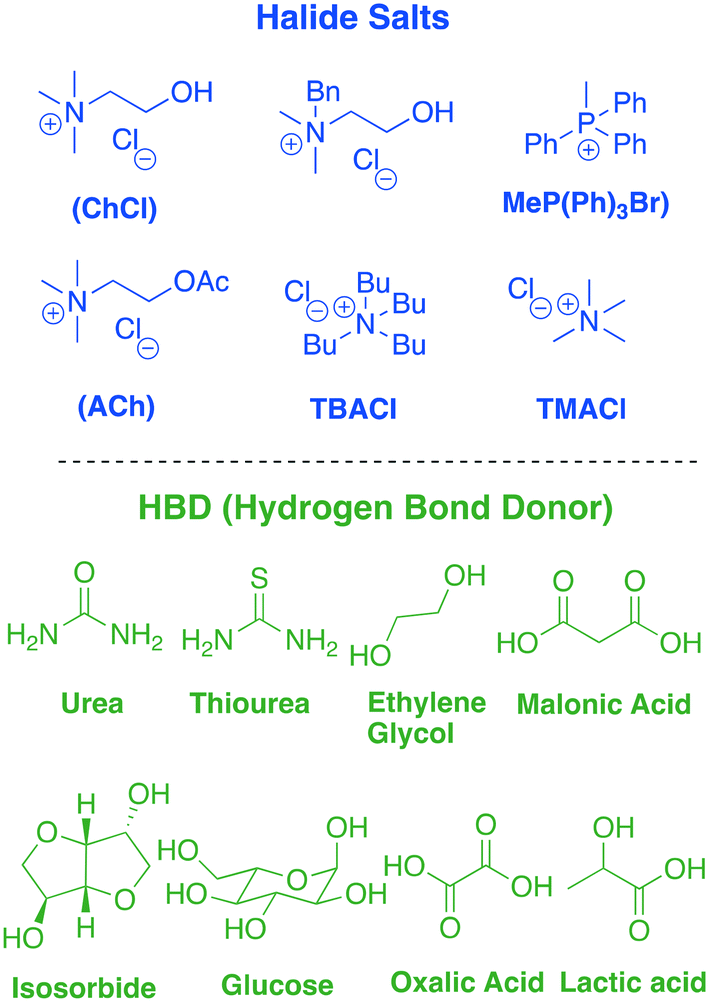
|
While the first definition of DESs involved quaternary ammonium salt and hydrogen bond donor mixtures, they can be more generally described as mixtures of Brønsted and Lewis acids and bases. In all these systems strong hydrogen bonds develop and these deviate from ideal mixtures, resulting in a significant depression of freezing point. Similar mixtures of Brønsted and Lewis acids and bases are commonly found in nature and the term natural DESs has been coined, although this should only be applied to naturally occurring mixtures rather than those synthesized from naturally occurring starting materials.13
Table 2 lists the different types of Brønsted and Lewis acid and base mixtures which have been studied together with examples of the components used.
| Eutectic | Acid | Base |
|---|---|---|
| 1 | Lewis | Lewis |
| MClx M = Zn, Sn, Fe,14 Al,15 Ga,16 In (ref. 17) | R4NCl | |
| 2 | Lewis | Lewis |
| MClx·yH2O, M = Cr, Co, Cu, Ni, Fe (ref. 18) | R4NCl | |
| 3 | Brønsted | Lewis |
| RZ, Z = CONH2, COOH, OH (ref. 19) | R4NCl | |
| 4 | Lewis | Lewis |
| MClx or MClx·yH2O M = Al,20 Zn,21 Cr (ref. 22) | RZ, Z = CONH2, COOH, OH | |
| 5 | Brønsted | Lewis |
| ROH RZ, Z = COOH (ref. 23) | RZ, Z = CONH2, COOH, OH |
Generally, an appreciable depression of the freezing point is observed and the magnitude of that depression is related to the strength of the bond formed between the two components, i.e. AlCl3 will exhibit a greater depression of freezing point than an alcoholic HBD such as glucose.
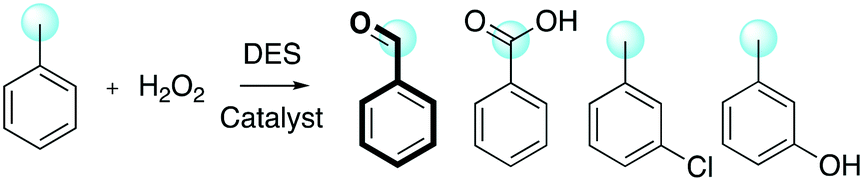
DESs have been used in numerous applications, such as for metal–organic framework (MOF) synthesis, nanoparticle preparation and gasoline desulfurization and as a medium for organic reactions.11b,24,25 Among organic reactions, oxidation reactions are well-known to be fundamental transformations in both industry and academia due to their capability to turn inert molecules into more reactive analogues prone to undergo further transformations. For example, hydrogen atom transfer (HAT) in free radical halogenation is a well-known protocol to prepare chlorinated solvents (chloroform, hexachlorobutadiene and dichloromethane) starting from fossil resources;26 ozonolysis is generally employed to fragment alkynes to prepare carboxylic acids,27 and the hydroxylation of the C–H bond is used to convert lipophilic compounds into hydrophobic analogues for environmental and pharmaceutical purposes (Scheme 1).28
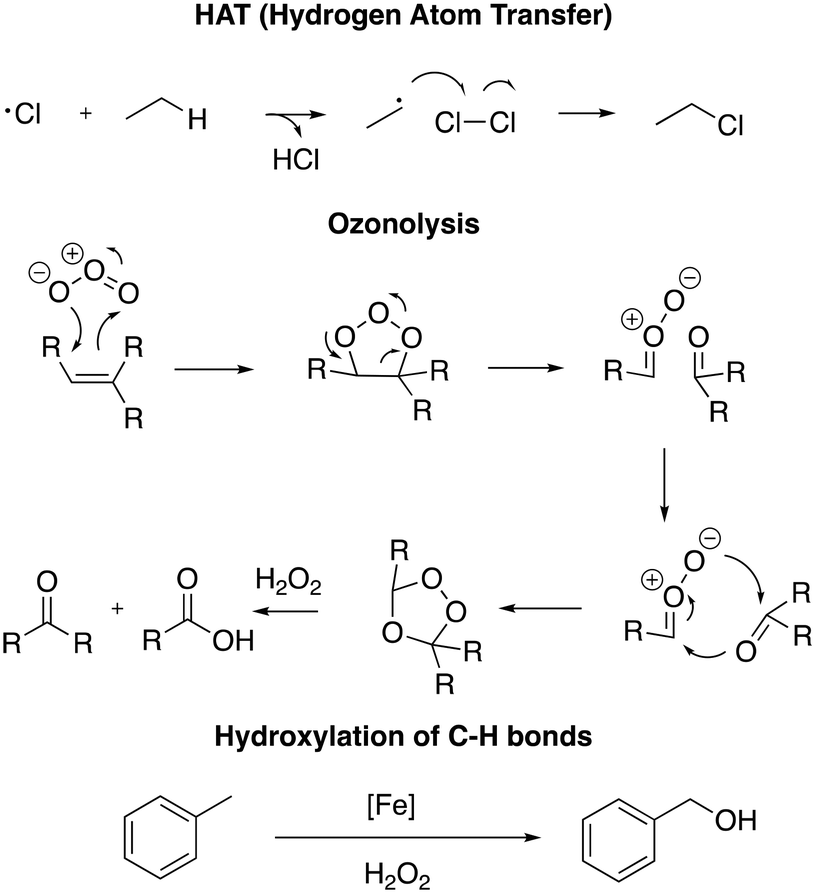 | ||
| Scheme 1 Selected examples of key oxidation processes in organic chemistry. | ||
2. Physicochemical properties of DESs
Ionic fluids have different solvent–solvent–solute interactions and this results in significantly different physicochemical properties. These properties are easily tuned by changing the type and composition of the constituents. Among these, freezing point, viscosity, conductivity and pH have to be well characterized since they can significantly affect the reactivity and mass transport.Freezing point and vapor pressure
Freezing point is among the most important properties as this can limit the temperature range during the reaction screening in the optimization stage. Freezing-point depression upon formation of DESs is related to the magnitude of the interaction between the components being mixed. For example, in the formation of reline, which is a mixture of ChCl and urea, the lattice energy of ChCl, which is a measure of the cohesive forces that bind the ions to each other, is perturbed by the urea molecules present in the mixture, which disrupt the original framework by mutual interaction between halide anions (ChCl) and the hydrogen atoms attached to the nitrogen atom (urea). When choline chloride and urea are mixed together (in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), the resulting freezing point of the mixture is 12 °C, whereas the melting points of ChCl and urea are 302 and 133 °C, respectively.11a Generally, room temperature DESs are the most desirable ones because they allow performing reactions at lower temperature for minimizing the energy requirements of the process. Unfortunately, the number of DESs that are liquid at room temperature is still very limited. In Table 3 the freezing point of several DESs reported in the literature is shown.
:2), the resulting freezing point of the mixture is 12 °C, whereas the melting points of ChCl and urea are 302 and 133 °C, respectively.11a Generally, room temperature DESs are the most desirable ones because they allow performing reactions at lower temperature for minimizing the energy requirements of the process. Unfortunately, the number of DESs that are liquid at room temperature is still very limited. In Table 3 the freezing point of several DESs reported in the literature is shown.
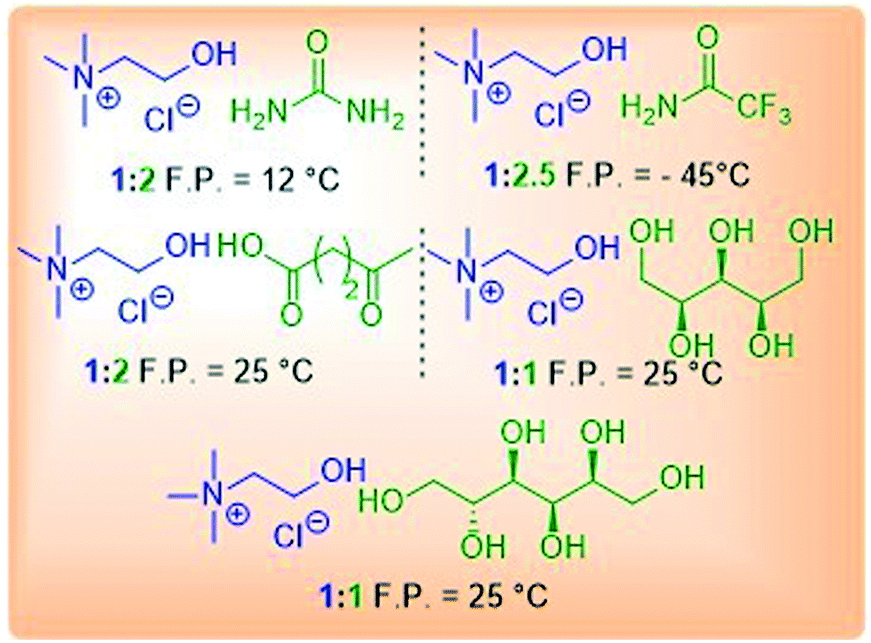
|
It is clear, by looking at the structure of the HBDs capable of forming room-temperature DESs that molecules able to sequestrate the halide anion by a strong hydrogen bond are good candidates (urea, 2,2,2-trifluoroacetamide, levulinic acid, xylitol, D-sorbitol, etc.). In the context of organic reactions, the capability of the solvent to remain liquid at low temperatures is sometimes crucial in order to minimize the side reaction pathways. Among the physical properties, vapor pressure is closely related to environmental concerns. Solvents exhibiting a low vapor pressure can be more easily separated from much more volatile reaction species, hence recovered. This makes DESs ideal from this perspective since they have a negligible vapor pressure.
Viscosity
Most DESs have viscosities in the range of 10 to 1000 cP at room temperature, which is significantly higher than that of molecular solvents, typically 1 cP. The high viscosity results from the large size of the solvent molecules (ions) and the extensive hydrogen bonding network which results in a low molar volume and a small free volume in the liquid. The viscosity of DESs has been modelled using hole theory which assumes that the movement of charges is limited by the availability of free volumes large enough to accept the ions.29 Most reactions are mass transport limited so solvent viscosity is important in controlling reaction rates.30,31 The viscosity of DESs can be significantly decreased by the addition of even a small amount of a molecular solvent, particularly water or even the reagent itself.It has been shown that molecular liquids are heterogeneous with DESs. Water is found to form a microemulsion when mixed with DESs using dynamic light scattering and NMR spectroscopy.30b,32 This has been shown to be important for the electropolymerization of aniline and could control the mechanism of other reactions in mixed media.33
Conductivity
Numerous studies have shown that the conductivity of ionic liquids and DESs does not follow the classical model for ionic strength but instead it has been shown by numerous groups that conductivity is proportional to fluidity (1/viscosity). Accordingly, since the liquids are relatively viscous they have conductivities which are significantly lower than that expected for a solution with an ionic strength in the range 4–6 mol dm−3. Typically the conductivities of DESs are in the range 0.1 to 5 mS cm−1. The conductivity of ionic liquids and DESs has been accurately predicted using hole theory.34Polarity
Polarity is another property that must be carefully evaluated before designing a reaction, especially organic reactions, since it can affect the solubility of the reactants and change the activation energy drastically. It is possible to understand the polarity effect on some reactions considering as examples the SN1 and SN2 reaction mechanisms. SN1 reactions occur considerably faster in polar solvents since the first step involves the formation of a carbocation, which can be stabilized by a polar solvent decreasing the activation energy Δ, whereas no significant effect can be observed in SN2 reactions, which do not involve charged intermediates.35 Owing to their composition, DESs have high polarity, which can be evaluated using the ET (30) scale (electronic transition energy of a probe dye in a solvent).36 Similar data have been obtained for Kamlet and Taft polarity parameters (Table 4) and these also show that all DESs have similarly high values for all parameters.37| DES | pH | π* | α | β |
|---|---|---|---|---|
| 1ChCl:1 oxalic acid |
1.32 | — | — | — |
| 1ChCl:1 malonic acid |
2.39 | — | — | — |
| 1ChCl:1 citric |
2.84 | — | — | — |
| 1ChCl:2 ethylene glycol |
6.89 | 0.96 ± 0.001 | 1.02 ± 0.02 | 0.33 ± 0.007 |
| 1ChCl:2 glycerol |
7.50 | 0.96 ± 0.04 | 1.04 ± 0.06 | 0.32 ± 0.01 |
| 1ChCl:2 urea |
8.91 | 0.98 ± 0.02 | 1.05 ± 0.02 | 0.30 ± 0.04 |
pH
While pH is a difficult concept to characterize in non-aqueous solutions, a recent study38 used a classic colorimetric indicator, bromophenol blue, to quantify the pKa of organic acids in ionic liquids and deep eutectic solvents. The pKa values for 9 organic acids were found to be about 0.5 units higher than that of the corresponding aqueous solution which was rationalized in terms of the different concentrations of bases when comparing DESs with water. As would be expected, the addition of more ChCl to the DES makes the mixture more basic. The same was found for more traditional ionic liquids where the basicity of the anion controlled the overall pH, although [Bmim] [BF4] was found to be more acidic than expected due to the hydrolysis of the anion forming HF. pH values for pure DESs were determined using a glass electrode and found to be in good agreement with colorimetric measurements. Some examples of DES pH are given in Table 4. Clearly, HBDs which are carboxylic acids have a much lower pH than HBDs with OH moieties. Those with amides tend to be slightly alkaline as might be expected.Recently the dissolution behavior of a variety of d- and p-block metal oxides was studied in DESs and it was found that for non-coordinating acids the solubility of the metal oxides was directly correlated with the acid strength as the proton acts as an oxygen acceptor. This understanding of DES pH should be directly transferrable to an understanding of organic reactivity.
The following sections provide an overview of the state of the art of organic oxidations performed in DESs. Through selected examples found in the most recent literature, we highlight the benefits of substituting volatile organic solvents with DESs, not only in terms of overall sustainability achieved but also in terms of improved efficiency and recyclability.
3. Organic oxidation in DESs
3.1. Metal-promoted oxidation in DESs
Due to the increasing demand of new transformations of renewable resources, many efforts have been dedicated to the design of new processes for a more sustainable valorization of raw materials. Within this realm, the processing of lignocellulosic biomass has gained attention as a renewable alternative to fossil resources.39 In this context, Mu and co-workers reported an efficient catalytic method to convert cellulose, the most abundant component in lignocellulosic biomass, into gluconic acid in DESs, employing FeCl3 as a catalyst for the aerobic oxidation step. The reaction is followed by a spontaneous self-precipitation of the products, which makes the process appealing for industrial purposes.40 Aware of the capability of highly concentrated FeCl3 aqueous solutions to catalyze the oxidation of cellulose into gluconic acid, the authors designed a protocol using DESs as reaction media in order to overcome the reuse of the FeCl3 and the separation of the gluconic acid, which is not trivial.41Scheme 2 shows the reaction proposed by Mu et al.40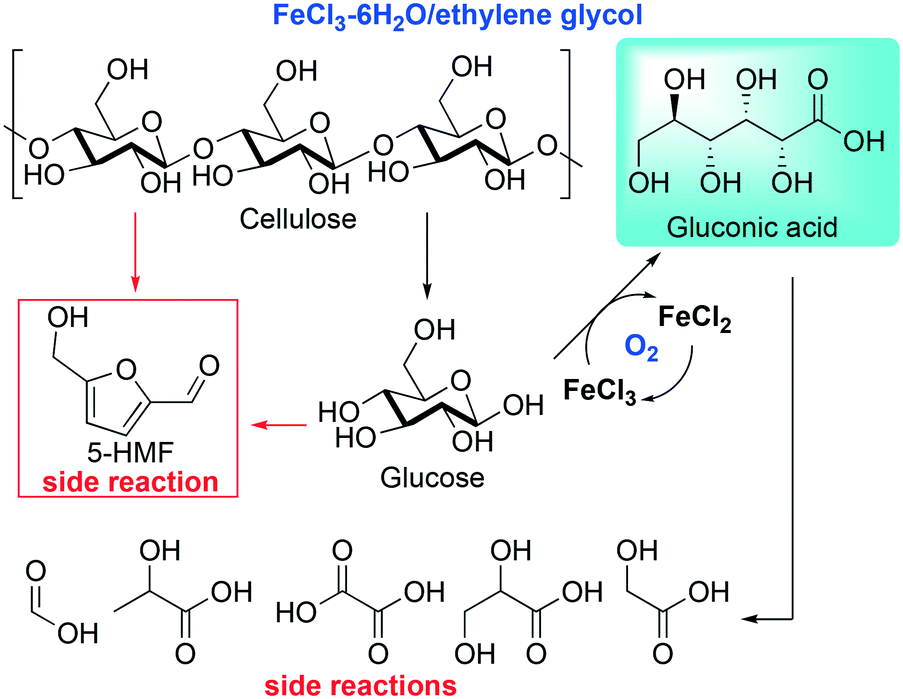 | ||
| Scheme 2 Cellulose conversion into gluconic acid catalyzed by FeCl3 proposed by Mu et al.40 | ||
The authors prepared several DESs by mixing FeCl3 with different HBDs (ethylene glycol (EG), malonic acid, glycine and xylitol, to mention some), obtaining DESs with low viscosity and good conductivity when compared to other common DESs. Furthermore, these DESs exhibited a relatively high acidity, a crucial feature to promote the first step of the reaction, that is, the hydrolysis of cellulose into monomeric glucose. The temperature effect was investigated as well, and it was found that the best conditions in terms of conversion and selectivity were achieved using FeCl3–EG as a DES at 120 °C. Full conversion was achieved in one hour with a 52.7% yield of gluconic acid that can be simply collected by filtration due to its insolubility in the medium after cooling.
Despite the DESs used exhibiting a low freezing point (−55 °C to −66 °C), high temperatures were required in order to increase the selectivity; the concentration of cellulose played an important role as well, as shown in Fig. 1.
 | ||
| Fig. 1 Temperature (A), reaction time (B) and concentration effect (C) on the yield of reaction and recycling test (D). Reproduced from ref. 40 with permission from The Royal Society of Chemistry. | ||
Among the oxidation reactions catalyzed by FeCl3, oxidation of toluene into benzaldehyde is another important key transformation in the chemical industry, toluene being the cheapest and primary source of aromatic compounds. Unfortunately, the selective partial oxidation still remains a great challenge, partly due to the more favorable tendency of the aldehyde to undergo overoxidation to benzoic acid. Operationally this requires performing the reaction while keeping the concentration of toluene very high.42 Guajardo and co-workers reported a protocol to convert toluene into benzaldehyde with hydrogen peroxide in DESs.43 Despite hydrogen peroxide being often recognized as a green reactant, it is poorly soluble in organic solvents and, in some cases, the reaction must be performed under a PTC (phase transfer catalysis) regime, with water having a detrimental effect on the selectivity by promoting the oxidation of benzaldehyde into the undesirable benzoic acid. It is well-known that ILs allow the capture of water formed, decreasing the side reaction.44 The work disclosed by Guajardo represents an alternative in which the more desirable DESs are used instead of ILs. Sodium tungstate (ST), iron(III) chloride (FeCl3), phosphotungstic acid (PT), and phosphomolybdic acid (PM) were tested as catalysts in different DESs. FeCl3 was selected as the most promising for the next screening step. The results of such solvent screening are summarized in Table 5.
|
|
||||||
|---|---|---|---|---|---|---|
| DES | Cat | S Bzh [%] | S BzAc [%] | S BzCl [%] | S BzOH [%] | Conv [%] |
| a ChCl/urea. b ChCl/ethylene glycol. c ChCl is a DES obtained by mixing choline chloride and FeCl3. All reactions were performed at 60 °C. | ||||||
| None | FeCl3 | 69 | 0 | 26.5 | 4.4 | 8.4 |
| URAa | FeCl3 | 87 | 0 | 9 | 4 | 5 |
| ETAb | FeCl3 | 73.5 | 13.9 | 10.1 | 2.5 | 18 |
| ChClc | FeCl3 | 66.3 | 5.3 | 5.3 | 2.8 | 10.5 |
The authors studied the intrinsic catalytic activity of DES but no reactivity was observed. Iron(III) chloride was the only catalyst able to promote the oxidation, presumably by a Fenton-like mechanism. The ETA–FeCl3 system showed higher conversion (SBzh = 73%, conv = 18%) when compared to the neat system (SBzh = 69%, conv = 8.4%), but the URA–FeCl3 couple was able to give the highest selectivity, despite lower conversion (SBzh = 87%, conv = 5%). The authors investigated the temperature effect as well, observing conversion decreasing when reaching temperatures above 60 °C. This is probably due to the faster decomposition of hydrogen peroxide.
Iron-based catalysis finds several applications in organic synthesis and C–C bond formation reactions are undoubtedly among key transformations due to their intrinsic capability to increase the framework complexity of molecules. In particular, C–C bond formation through C–H activation is one of the great challenges in organic synthesis.45 One example is the mechanism of C(sp3)–H bond activation at the α-position of nitrogen, which is well-known and proceeds through two consequent single electron transfer (SET) reactions generating an iminium intermediate, as shown in Scheme 3.46
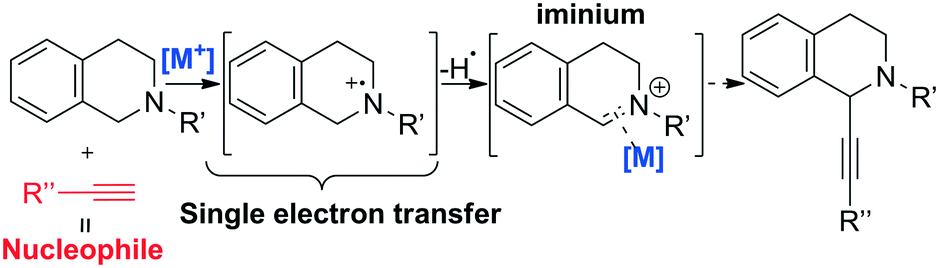 | ||
| Scheme 3 Mechanism of generic C–C coupling in vicinal nitrogen C–H activation by metal. | ||
In this context, Zhang reported the synthesis of imidazo[1,2-a]pyridines promoted by CuFeO2 nanoparticles in DES.47 In 2016, Ramón et al. presented for the first time the cross-dehydrogenative coupling between 1,2,3,4-tetrahydroisoquinoline and phenylacetylene through C–H bond activation promoted by copper impregnated on magnetite (CuFeO2) in DES using atmospheric oxygen as the oxidant.48 The optimized reaction is shown in Scheme 4.
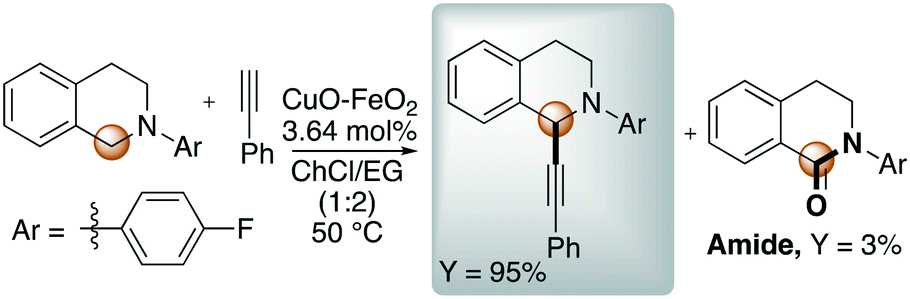 | ||
| Scheme 4 Cross-dehydrogenative coupling reported by Ramón et al. through C–H bond activation promoted by copper impregnated on magnetite (CuFeO2) in ChCl:EG.48 | ||
To prove the essential role of DESs in the reaction the authors reported the reactivity in other common organic solvents. They observed that in all the cases the yield is lower than that in the reaction performed in the DES. Furthermore, a better selectivity was observed for the reaction in the DES as shown in Fig. 2.
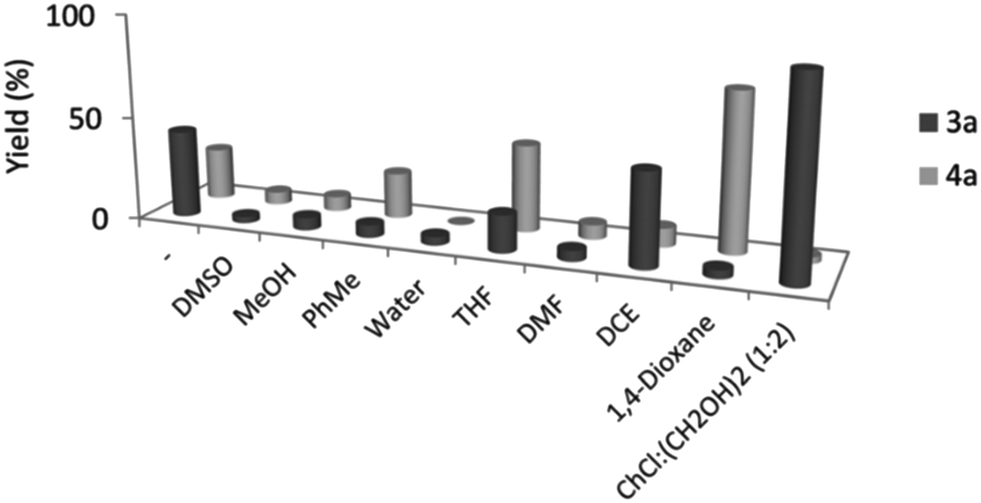 | ||
| Fig. 2 Comparison of yield of reaction and selectivity between DES and common organic solvents used for cross-dehydrogenative coupling performed by Ramón et al. Reproduced from ref. 48 with permission from The Royal Society of Chemistry. | ||
It is worth noting how the conductivity affects the reaction output following the trend observed by the authors. Fig. 3 shows the plot of the yield versus conductivity for different DESs tested. The trend can be reasonably explained by considering that the iminium intermediate is a charged ion and a better conductivity can enhance the movement of ions in the medium, explaining the increase in reactivity.
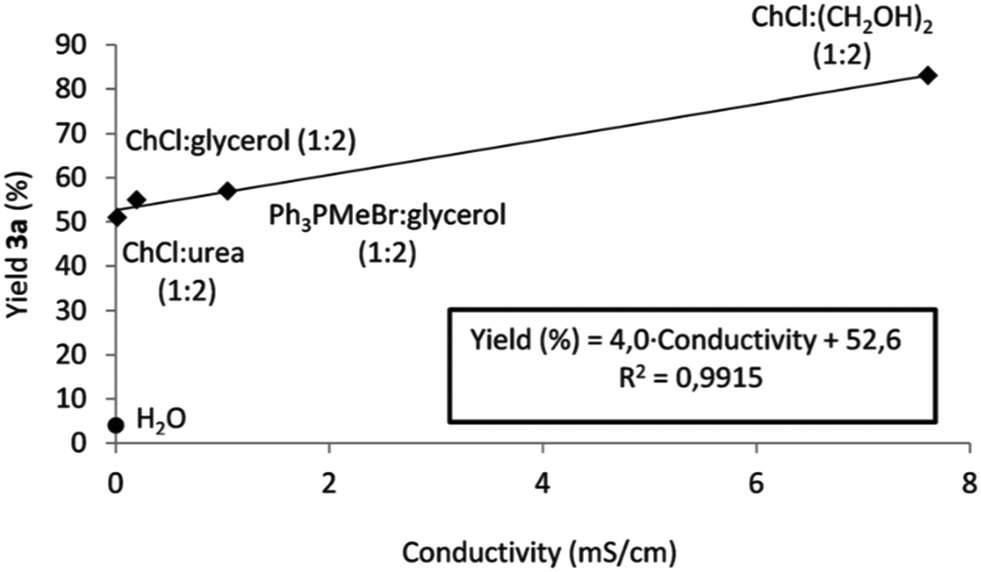 | ||
| Fig. 3 Relationship between conductivity of the solvent and yield. Reproduced from ref. 48 with permission from The Royal Society of Chemistry. | ||
Finally, the recyclability of the DES–catalyst mixture was tested as well; the results showed that the mixture could be reused up to ten times without appreciable decrease of reactivity.
Solubility issues are dominant in the design of an organic reaction and when among the reactants are present both lipophilic and hydrophilic compounds the reaction has to be often performed in the heterogeneous phase. Owing to the presence of organic chains, and the ionic nature, DESs are capable, after a suitable tailoring of their structures, of dissolving both lipophilic and hydrophilic substances.
FeCl3 also plays a crucial role in the synthesis of polymers in modern industrial chemistry. Some particular features of these materials are required nowadays, particularly in specific applications such as photoluminescence and electrochemistry.49 For example, poly(3-alkylthione)s are a class of important polymers that exhibit electro- and photo properties, which make them attractive for manufacturing of semiconductor polymers and dyes. FeCl3 oxidative polymerization is the protocol mainly adopted to prepare these polymers on a large-scale process, affording the desired product in high yield employing a low-cost catalyst.50
In 2016, Lee and co-workers reported an efficient protocol to polymerize alkylthiophenes using FeCl3 in DESs.51 In their previous work they reported a similar protocol employing an IL (1-butyl-3-methylimidazolium hexafluoroantimonate([BmIm][SbF6])) as a medium and they pointed out how replacing chloroform, the organic solvent commonly used, with the IL can have remarkable advantages in terms of polymer growth and yield.52 Aware of the general benefits gained by substituting ILs with DESs the authors decided to investigate the transformation in DESs. Table 6 reports the results, which show the remarkable advantages given by DESs (up to 100% yield and MW of 44179) compared to the outcome of the reaction performed in chloroform (yield = 87, MW = 34672), in addition to their well-known intrinsic economic and sustainable aspects.
|
|
||||
|---|---|---|---|---|
| Solvent | Time [h] | M w [g mol−1] | PDI [Mw/Mn] | Yield [%] |
| ChCl:urea |
1 | 13466 |
6.7 | 59 |
| ChCl:urea |
4 | 27787 |
13.7 | 81 |
| ChCl:urea |
10 | 44179 |
12.9 | 100 |
| [Bmim][SbF6] | 48 | 42584 |
15.9 | 99 |
| Chloroform | 48 | 34672 |
12.9 | 87 |
FeCl3 (4 equiv.) reaction performed at 25 °C.
The highest Mw for poly-3-octylthiophene (P3OT) was obtained with ChCl/urea (Mw = 44179) when all the monomer is consumed. The highest reactivity was achieved using the DES (Y = 100% in 10 hours), whereby the yields in IL and chloroform were 99% and 87% in 48 hours, respectively. The temperature effect was investigated as well. As shown in Fig. 4, the polymerization rates in DESs or ILs were enhanced by increasing the temperature, which is not always possible when volatile solvents are used.
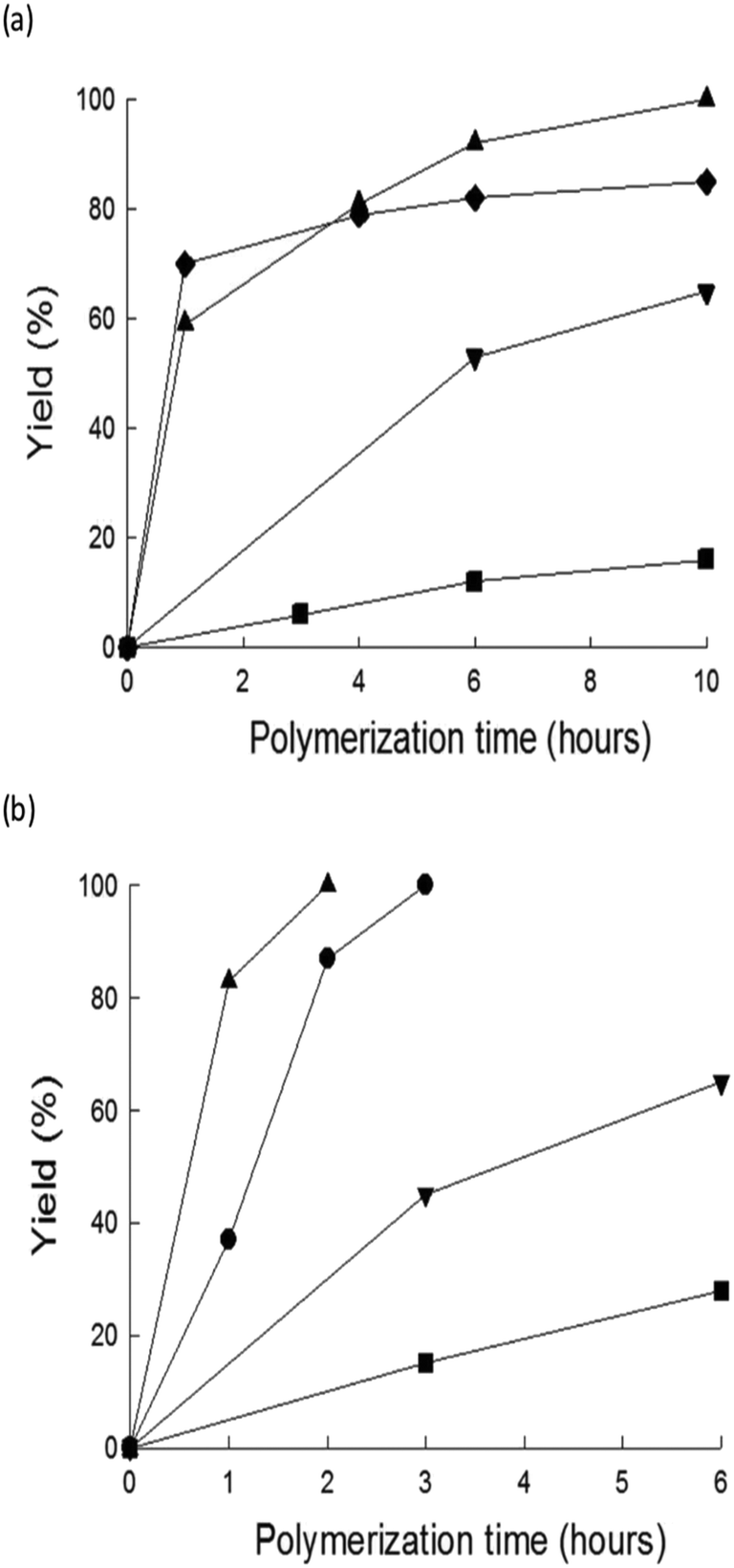 | ||
| Fig. 4 Time–yield curves for P3OT prepared by FeCl3-catalyzed oxidative polymerization (a) at 25 °C in DESs, IL and chloroform and (b) at 50 °C in DESs and IL (▲: [Ch]Cl:U, ●: [Ch]Cl:U:TU, ▼: [BmIm][SbF6], ■: [Ch]Cl:F:TU, ◆: CHCl3). Reproduced from ref. 51 with permission from The Royal Society of Chemistry. | ||
The results summarized in Fig. 4 and Table 6 show that ChCl/urea is the most efficient solvent for the FeCl3-promoted oxidative polymerization of 3-octylthiophene, performing better than ILs and chloroform.
As mentioned in the introduction section, the intrinsic alkalinity of DESs is an important property that can be exploited in the design of organic reactions in DESs. An interesting correlation between the basicity parameter and reactivity in different DESs was observed. The yield of P3OT was seen to increase with decreasing values of pH. A similar effect has already been observed in the polymerization of 3OT performed in ILs. This finding indicates that the basicity of the solvent plays an important role in the FeCl3-catalyzed oxidative polymerization of 3OT.
Despite iron being more preferable than palladium for abundancy reasons, the latter remains ubiquitous in organic synthesis due to its versatility. In 2017, Ramón et al. reported the palladium catalyzed C–S bond formation in the presence of sodium metabisulfite as a SO2 source.53 Exploiting the ability of DESs to dissolve organic substrates, catalysts, ligands and the inorganic salt, the authors obtained an improvement when the reaction was performed in DESs instead of common organic solvents. The most promising DES was a mixture of ChCl and acetamide (1:2), whereas, 2-(dicyclohexylphosphaneyl)-1-phenylpyridin-1-ium chloride was selected as the best ligand. The reaction is shown in Scheme 5.
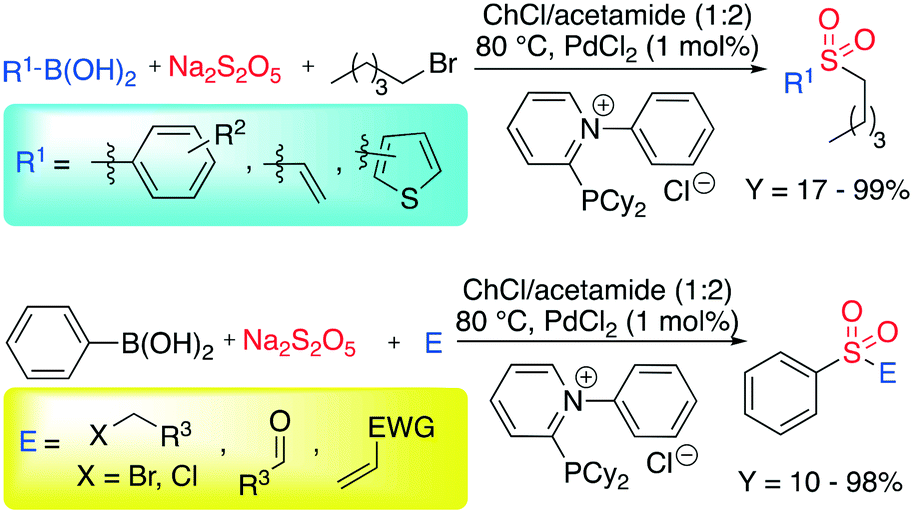 | ||
| Scheme 5 Palladium-catalyzed C–S bond formation in DESs.53 | ||
In their studies, the authors focused on the C–S coupling, keeping the pentyl bromide as the electrophile and investigating the scope of the reaction by varying the aryl boronic acid. Aryl boronic acid-based compounds bearing both electron-withdrawing groups (EWGs) and electron-donating groups (EDGs) were tested, showing that the reactivity varies from poor to excellent (17–99%). Other electrophiles were tested among the most commonly used in organic synthesis such as Michael acceptors, alkyl chloride and aldehydes, showing the great flexibility of the protocol.
The recycling of the catalyst was also studied. The products were extracted using 2-MeTHF, a sustainable organic solvent, whereas the catalyst remained in the DES. The authors observed that the DES–catalyst medium could be reused three times without adding the catalyst, ligand or solvent.
3.2. One-pot oxidative processes in DESs
One-pot processes involve two or more chemical reactions which take part in a single reactor and the product of the previous reaction acts as a substrate for the next one. These processes are highly desirable since they eliminate the need for time- and energy-consuming separation processes and purification of the intermediates.54 Recently, Capriati and co-workers reported the synthesis of 2,5-diarylpyrazines employing DESs as media with the purpose of exploring a well-known reaction using non-conventional reaction solvents.55 2,5-Diarylpyrazines are extensively studied scaffolds due to their properties in medicinal chemistry, such as anti-inflammatory, anti-diabetic, anti-bacterial and anti-fungal, and as dyes.56 The authors envisaged the possibility of performing a one-pot process starting from affordable substituted ketones for obtaining the desired product in two steps followed by two spontaneous transformations (dimerization and aerobic oxidation) as shown in Scheme 6.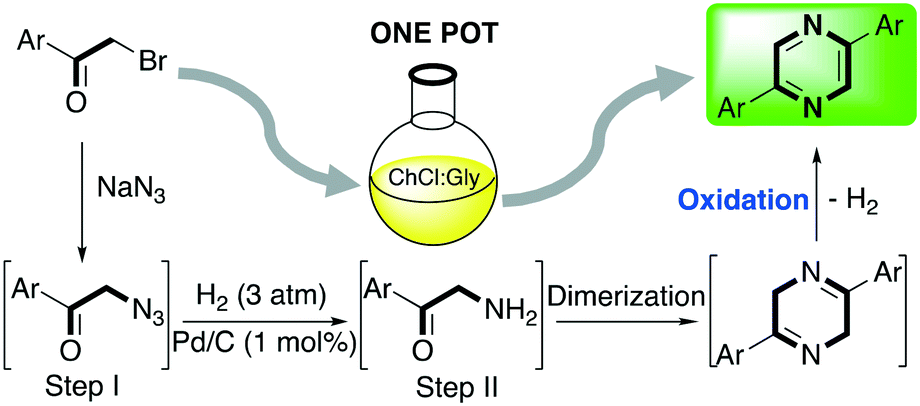 | ||
| Scheme 6 One-pot (2 steps) synthesis of 2,5-diarylpyrazynes in DES starting from 2-bromo-1-arylethan-1-ones.55 | ||
They started investigating the nucleophilic substitution of the halide in DES after considering that the reaction works very well in ILs and that it is classically performed in solvents with high polarity.57 The use of a ChCl/Gly eutectic mixture (1:2) at room temperature allowed obtaining the desired product in 4 h with 97% yield (Scheme 7).
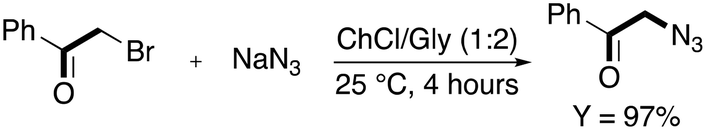 | ||
| Scheme 7 Synthesis of 2-azido-1-phenylethan-1-one from 2-bromo-1-phenylethan-1-one in ChCl/Gly.55 | ||
The authors suggested that a cooperative effect of the ChCl is involved in the mechanism, observing that in the presence of pure Gly the yield drops down to 23%. After examining the scope and limitation of this reaction, varying the substituents on the aromatic ring and finding that no detrimental effects were observed with both electron-poor and electron-rich rings, the authors carried out a subsequent investigation of the chemoselective reduction of the azido functional group. Classical reductive systems were tested such as Zn/ammonium formate, Zn/HCl, Zn/NH4Cl, Ph3P, and D-glucose/KOH without any significant results. The use of SnCl2·2H2O smoothly promoted the reduction of the azido group, whilst the subsequent use of Pd/C (1 mol%) in the presence of hydrogen (3 atm) promoted the dimerization/oxidation to 2,5 diphenylpyrazine, obtained in 89% yield (Scheme 8).
 | ||
| Scheme 8 Telescoped one-pot synthesis of 2,5-diphenylpyrazine in a ChCl/Gly deep eutectic mixture.55 | ||
Finally, the authors presented the scope and limitations of the telescoped one-pot synthesis tested using different bromide ketones as shown in Table 7.
|
|
||
|---|---|---|
 Y = 64 Y = 64 |
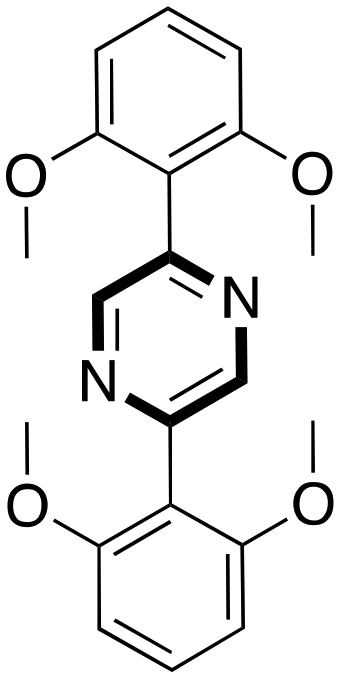 Y = 95 Y = 95 |
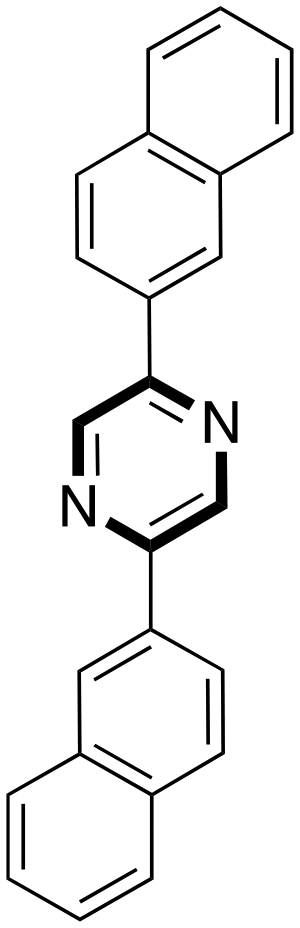 Y = 65 Y = 65 |

The authors did not investigate the reuse of the DES, probably because the mixture, which was contaminated with palladium, necessary to trigger the next step, was not as efficient as the pure one for the nucleophilic substitution.
Continuing with the preparation of heteroaromatic compounds, pyrimidine is broadly recognized as a privileged scaffold for important drugs, which exhibit biological activities in several diseases. In addition, pyrimidine analogues find broad applications in polymer chemistry and materials science.58 A variety of protocols has been reported during these years on the preparation of pyrimidines. However, the reported methods often suffer from drawbacks such as the need to use rare or toxic metals as catalysts, expensive or moisture-sensitive starting materials, longer reaction times, harsh reaction conditions and poor recycling of the catalyst. Thus, new economical and environmentally friendly methods for the synthesis of substituted pyrimidines are much sought-after. Recently, Chaskar and co-workers disclosed a new and affordable method to prepare this class of molecules in DESs.59 The authors selected the reaction of 1,3-diphenyl-2-en-1-one with benzamidine hydrochloride as a model system (Scheme 9).
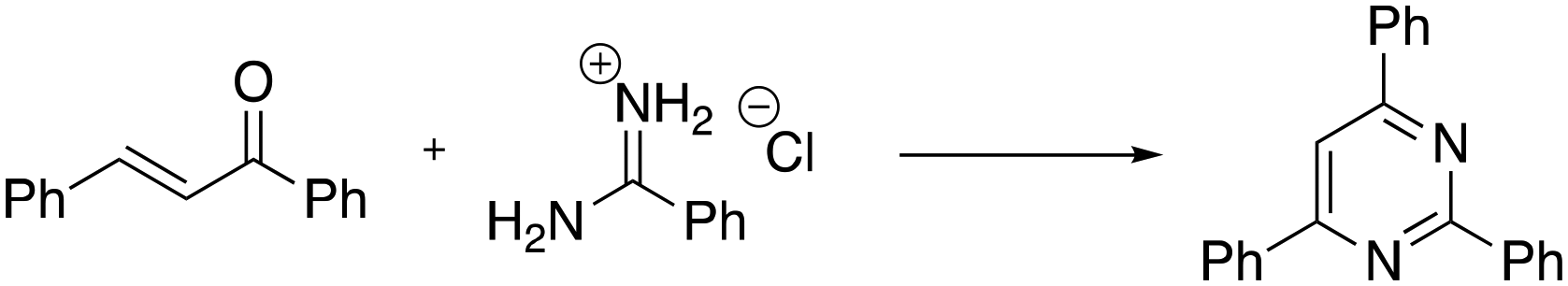 | ||
| Scheme 9 Model reaction chosen for the preliminary study of the pyrimidine one-pot protocol in DESs reported by Chaskar et al.59 | ||
The authors investigated the reactivity by screening several common organic solvents such as EtOH, toluene, DMF and acetonitrile. They reported that the reaction affords the desired product up to 83% yield using a strong base such as NaH and NaOH at very high temperature (80–110 °C) in a reasonably short reaction time (1–2 hours). The reaction was seen to be more efficient in choline hydroxide (ChOH) (Table 8). In particular, in ChOH the reaction gave a yield of 90% in a shorter reaction time (30 min) and under milder conditions (60 °C) without strong bases as additive.
|
|
|||
|---|---|---|---|
| Time [h] | Additive/solvent | Temp [°C] | Yield [%] |
| Reaction conditions: 1,3-diphenyl-2-en-1-one (1.0 mmol), benzamidine hydrochloride (1 mmol), base (2 mmol) and solvent 5 mL. | |||
| 24 | EtOH | 80 | NR |
| 1.5 | NaOH/EtOH | 80 | 83 |
| 5 | NaH/THF | 80 | 59 |
| 2.5 | Toluene | 110 | 43 |
| 0.5 | ChOH | 60 | 90 |
The recyclability of the DESs was also studied showing that it was possible to reuse the DES five times without any appreciable loss of activity (90% to 87%). The authors proposed a mechanism whereby ChOH seems to play a crucial role in promoting the reaction (Scheme 10).
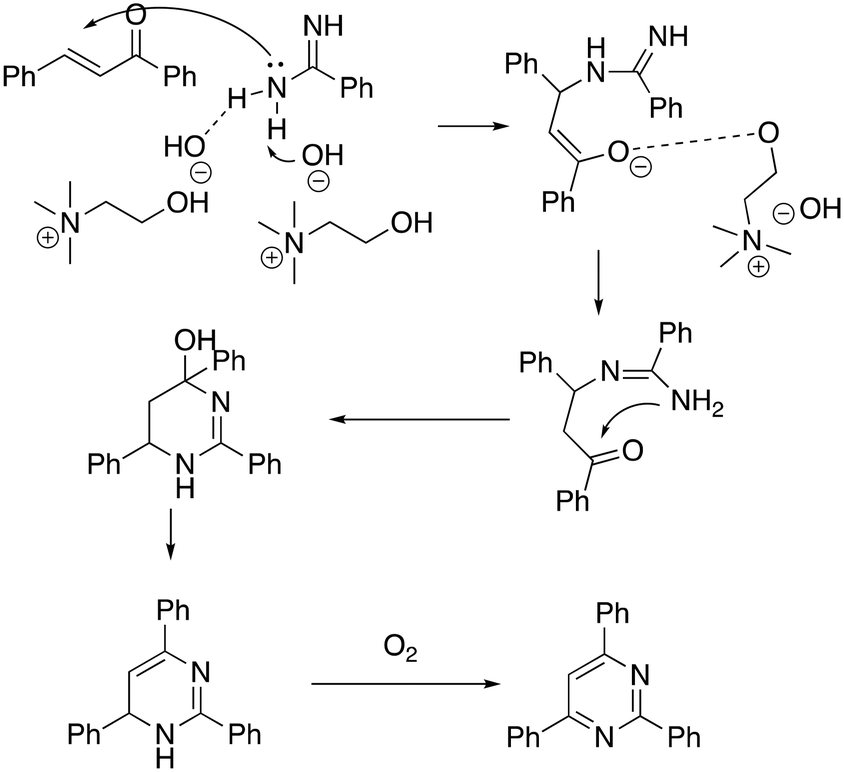 | ||
| Scheme 10 Proposed mechanism for the [3 + 3] tandem pyrimidine synthesis in ChOH.59 | ||
Regarding diazo-heterocyclic compounds, in 2016 Nagarajan and co-workers reported a metal-free DES-mediated cyclization strategy to prepare substituted and unsubstituted quinazolinones, well-known molecular scaffolds that can be further elaborated for the synthesis of various natural products and drugs.60 They screened several sugar-based DES mixtures such as citric acid/N,N′-dimethylurea, D-(−)-fructose/N,N′-dimethylurea, L-(+)-tartaric acid/N,N′-dimethylurea and mannose/N,N′-dimethylurea/NH4Cl employing anthranilamide and o-tolualdehyde as model substrates. L-(+)-tartaric acid/N,N′-dimethylurea (3:7) (F.P. 90 °C) was found to be the most promising DES for the reaction, affording the desired product in 78% yield. The reaction required to be performed in an open air atmosphere to allow the aromatization of the initially formed dihydroquinazolinone to quinazolinone through aerobic oxidation (Scheme 11).
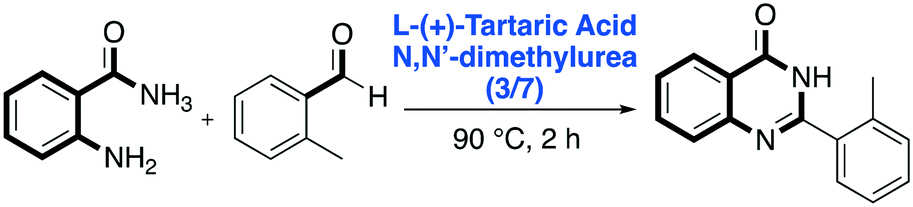 | ||
| Scheme 11 Dihydroquinazolinone one-pot synthesis in L-(+)-tartaric acid/N,N′-dimethylurea.60 | ||
The robustness and the limitations of the methods were tested by varying the substituents on the reagents. Interestingly, the authors proved the utility of the protocol in preparing APIs (active pharmaceutical ingredients) (Scheme 12).61
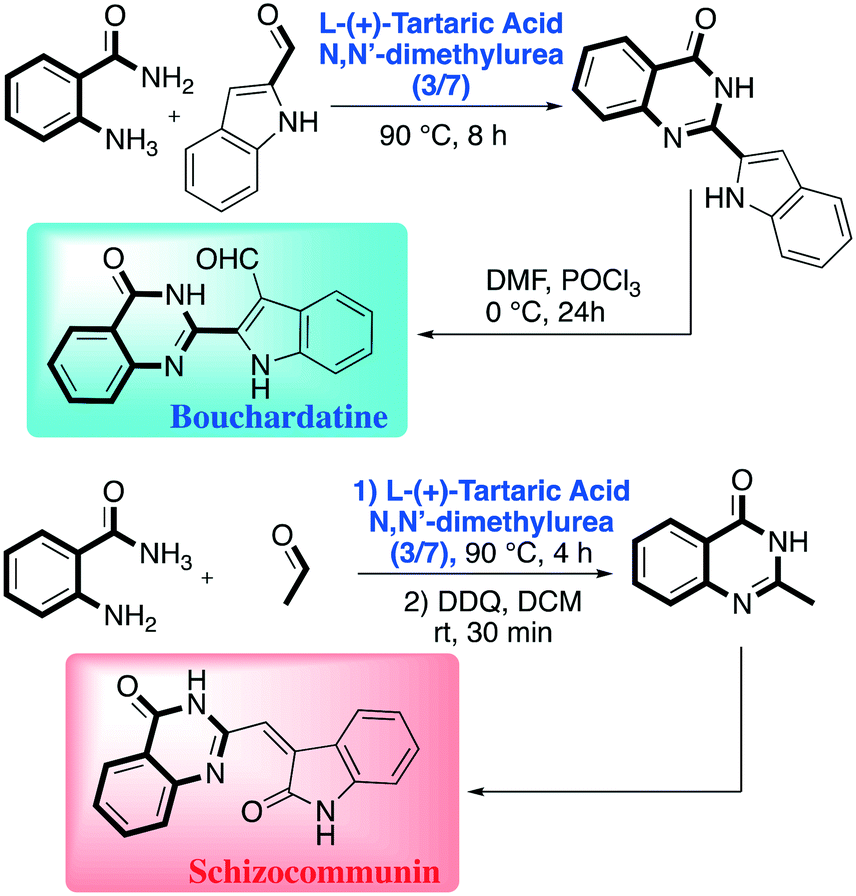 | ||
| Scheme 12 Dihydroquinazolinone derivate elaboration towards pharmaceutically relevant compounds reported by Nagarajan et al.60 | ||
Pyrroles are another class of molecules of interest in several areas of chemistry and materials science. They have found promising applications as organic semiconductors and in OLED (organic light-emitting diode) applications.62 Chaskar and co-workers reported a one-pot method employing DESs for the synthesis of these important scaffolds.63 The one-pot procedure consists of a [3 + 2] cycloaddition between a chalcone and a isocyanoacetate promoted by a copper(I) salt followed by aerobic oxidation to access the tri-substituted pyrrole (Scheme 13).
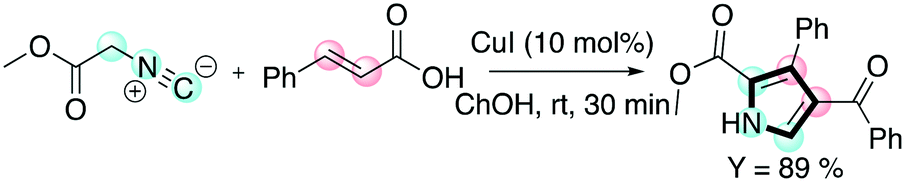 | ||
| Scheme 13 The one-pot protocol disclosed by Chaskar for the preparation of tri-substituted pyrroles.63 | ||
In the reported paper, the authors showed the benefit of moving from classical to DES-based solvents. The reaction in DESs proceeded faster and under milder conditions when compared with the use of classical organic solvents (THF, DMF, MeOH) that all require strong bases to trigger the reaction. The authors investigated the scope of the reaction as well, as shown in Table 9.
|
|
||
|---|---|---|
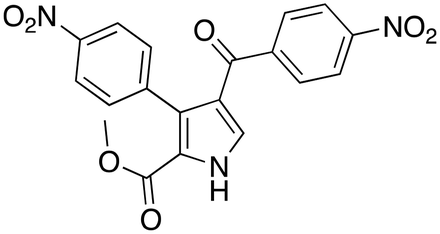 (25 min, 90%) (25 min, 90%) |
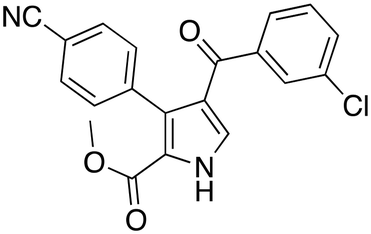 (30 min, 88%) (30 min, 88%) |
|
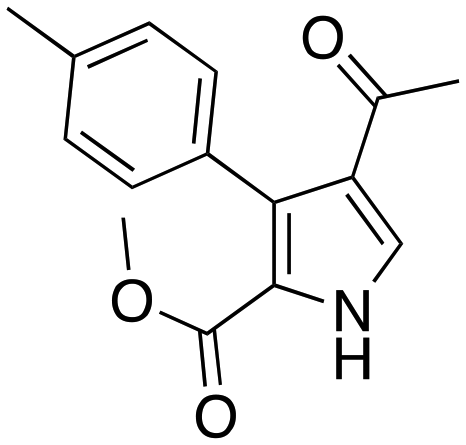 (40 min, 69%) (40 min, 69%) |
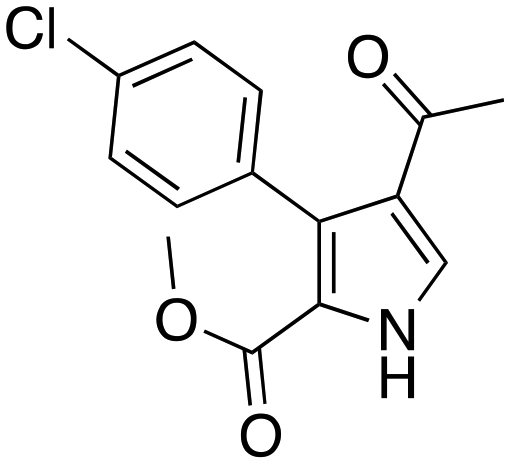 (45 min, 73%) (45 min, 73%) |
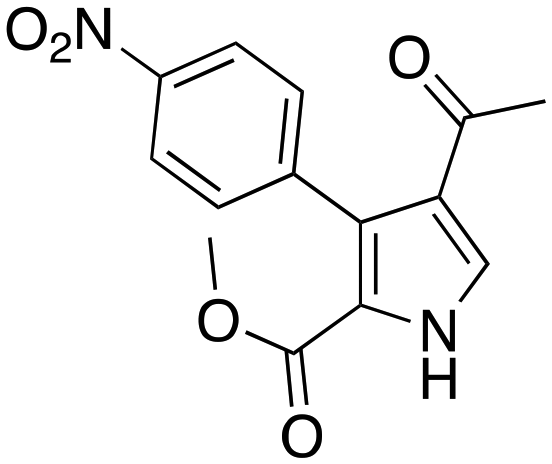 (40 min, 78%) (40 min, 78%) |
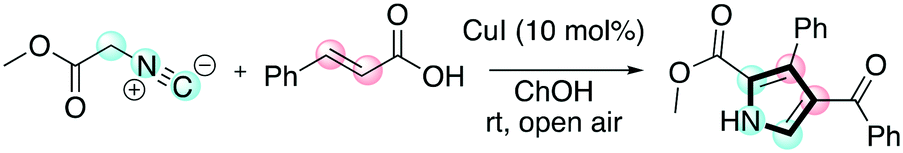
The protocol was shown to be robust and applicable for both electron-withdrawing and electron-donating groups bearing on the aryl moiety of the olefin. Replacing the aryl moiety with the alkyl onto the ketone did not change dramatically the reactivity. The recycling test showed that the DESs could be reused five times without loss of reactivity. The mechanism proposed by the authors was also reported (Scheme 14), whereby the role of the ChOH is thought to be the deprotonation of the isocyanoacetate in order to generate the dipole for the cyclization.
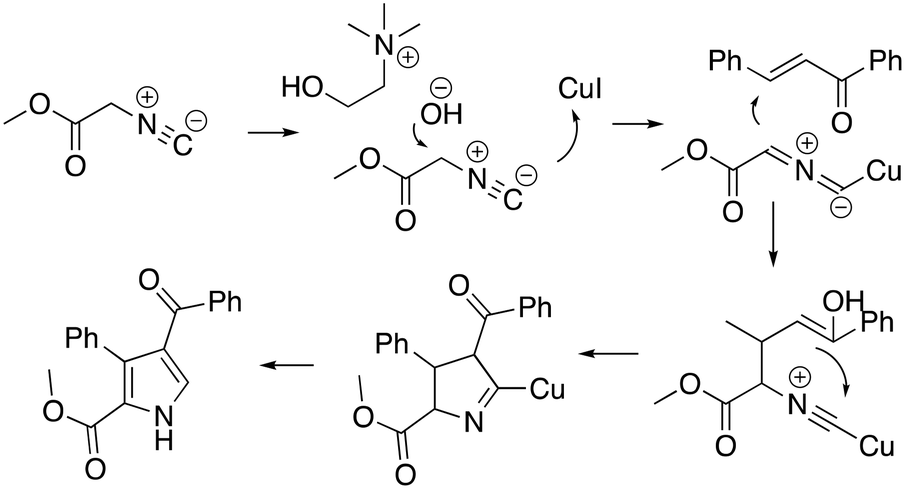 | ||
| Scheme 14 Proposed mechanism for the one-pot pyrrole synthesis in ChOH.63 | ||
C–N and C–C bond oxidation by atmospheric oxygen is easy to observe when a re-aromatization occurs. However, in other cases a stronger oxidant is required. Oxidation by hypervalent iodine compounds is one of the most used methods to oxidize selectively organic compounds. This class of reactions has been widely used in research laboratories since the first reactions, under mild conditions, were discovered by Dess and Martin.64 Within this class, o-iodoxybenzoic acid is one of the most prominent and interesting reactants. It is one of the cheapest hypervalent iodine reagents and it can be reused by treating the derived reduced by-product with aqueous oxone, after recovery and separation by the crude reaction. In 2017, Sharma and co-workers, intrigued by previously reported studies on oxidation in DESs, decided to explore the oxidation of primary amines into imines promoted by IBX (2-iodoxybenzoic acid),65 extending the concept to the Ugi reaction, a one-pot process featuring reorganization of imine, organic acid and isocyanide to obtain bis-amide analogues in situ. Selected results about the first step of the Ugi collected by Sharma are summarized in Table 10.
|
|
|||
|---|---|---|---|
| Entry | Solvent | Additive | Yield [%] |
| Reaction conditions: benzylamine, IBX (0.1 equiv.), additive (0.1 equiv.). Solvent (0.4 M). BA (benzoic acid), DCM (dichloromethane). | |||
| 1 | DCM | None | 17 |
| 2 | DCM | BA | 54 |
| 3 | CH3CN | BA | 39 |
| 4 | ChCl/urea | BA | 84 |
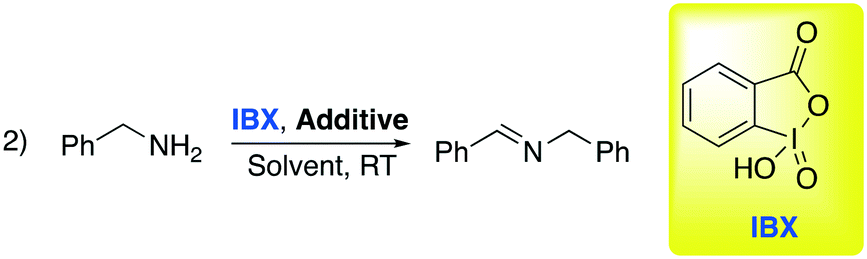
The authors found that the addition of an additive (benzoic acid) is crucial to obtain the desired product. Otherwise, the absence of the acid afforded a mixture of the corresponding aldehyde and nitrile. It is worth noting that substitution of classical organic solvents (entries 1, 2 and 3) with DESs significantly boosted the reactivity (entry 4 in Table 10). The authors rationalized this behavior, observing a better solubility of IBX in the DES compared to some organic solvents (DCM, CH3CN, toluene). After finding the optimized condition for the oxidation, the authors investigated its implementation in the Ugi reaction. Ugi reaction involves the condensation of a carbonyl and an amine in order to generate an imine as an intermediate followed by the addition of isocyanide and a carboxylic acid. Some examples are shown in Scheme 15.
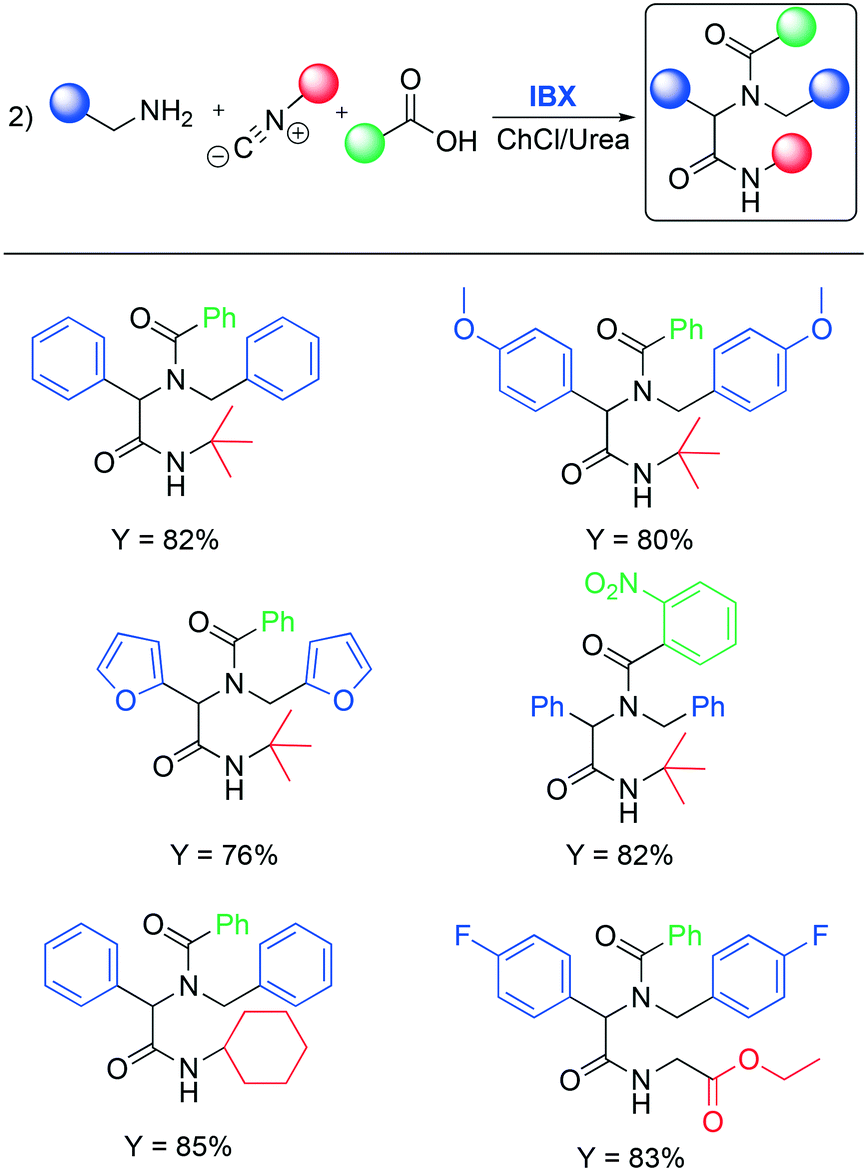 | ||
| Scheme 15 Selected examples of the Ugi reaction performed in deep eutectic solvents.65 | ||
Phenylamine and benzylamine bearing electron-donating substituents (p-methoxy) reacted under standard conditions and afforded the target product in very good yield (82% and 80%, respectively). The authors reported that the reaction remains effective when varying the substituents on both the acid and the isocyanide species. A complete study was performed to investigate the reusability of the DESs. Water was added to the reaction mixture to separate the corresponding reduced IBX and the reaction products. The water was then removed by distillation and the DES was reused five times without any significant loss in reactivity. The desired products and the iodosylbenzoic acid (IBA, reduced by IBX) were separated by dissolving the crude product in ethyl acetate followed by filtration.
Fast and selective oxidation occurs smoothly not only for heterocyclic compounds. Recently, Capriati and co-workers disclosed a one-pot methodology to prepare α- and β-hydroxy-phosphine oxides in a very short reaction time (Scheme 16).66
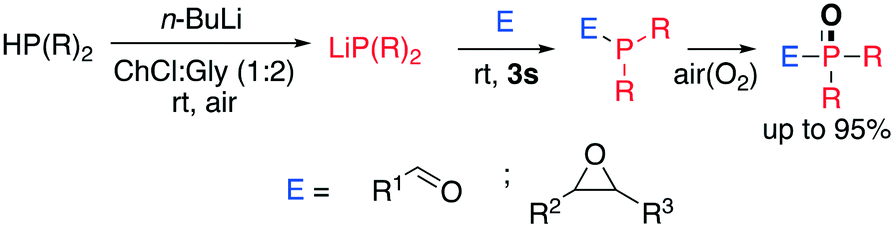 | ||
| Scheme 16 α- and β-hydroxy-phosphine oxide synthesis through a one-pot two-step protocol disclosed by Capriati et al.66 | ||
The protocol consists of three steps. Firstly, the reactive lithium phosphide is generated by adding 1 equivalent of n-BuLi to a solution of phosphine in DES (ChCl:Gly, 1:2); the electrophile (epoxy or aldehyde) is then added, affording either the α- or β-hydroxy-phosphine analogue, which spontaneously undergoes oxidation at the phosphorus centre in excellent yield (up to 95%). Surprisingly, the reaction works in DESs containing a HBD despite the lithium phosphide being well-known to be sensitive to moisture and incompatible with protic solvents. The authors investigated the fate of the lithium phosphide in the DES and observed a drastic decrease of reactivity by increasing the time lag between the n-BuLi addition and the electrophile addition. This suggests a detrimental effect of the solvent which is however not significant if the addition occurs fast.
3.3. Modified DESs bearing oxidative units
When the recycling of the DESs is possible, the strategy of anchoring a catalyst or a reactivable reagent onto DESs can be an elegant way to carry out chemical reactions. In recent years, some protocols have been reported in this area. Shankarling et al. disclosed a method to selectively oxidize benzyl alcohol derivates to aldehydes in choline peroxydisulfate using ultrasound as the energy source.67 The aim of the authors was to develop an environmentally benign oxidation reaction with high yield, facile to reproduce, and with minimization of energy requirements. They explored the oxidizing activity of the biodegradable choline peroxydisulfate (ChPS) (Scheme 17) comparing the energy consumption in the cases of thermal and ultrasound energy transfer. The preparation of the DES and a comparison with some common oxidative reactants has also been reported in a previous paper published by the same authors.68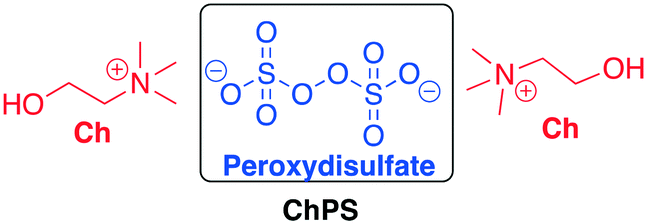 | ||
| Scheme 17 Choline peroxydisulfate prepared by Shankarling et al. The oxidative species is part of the DES (peroxydisulfate).67 | ||
The authors chose benzyl alcohol as a model reactant and they found the optimal conditions using 2 equiv. of DES in water (2 M). The reaction occurred more easily under ultrasound conditions (5 min) compared with thermal conditions (30 min). They also compared their protocol with standard protocols reported in the literature (H2O2 and Fe(DS)3, NaOCl, and FeCl3/HNO3) and showed that their protocol was more efficient and sustainable.68 The scope of the reaction was also investigated, proving the robustness of the procedure (Scheme 18).
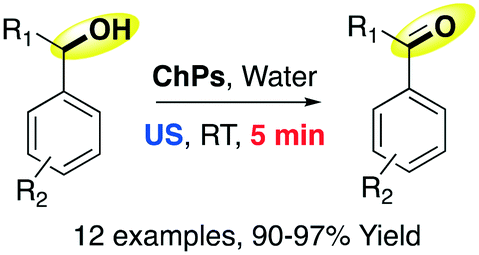 | ||
| Scheme 18 Choline peroxydisulfate-promoted oxidation of benzyl alcohol derivates to aldehydes in ultrasound.67 | ||
Fig. 5 shows the energy required (kJ) for the oxidation of benzyl alcohol per unit weight of the material consumed/obtained (g) under thermal (conventional) and ultrasound conditions. The total energy required per unit weight benzaldehyde synthesis was 0.958 kJ g−1 for the thermal method and 0.125 kJ g−1 for the ultrasound method, proving that the latter is more sustainable for this reaction.
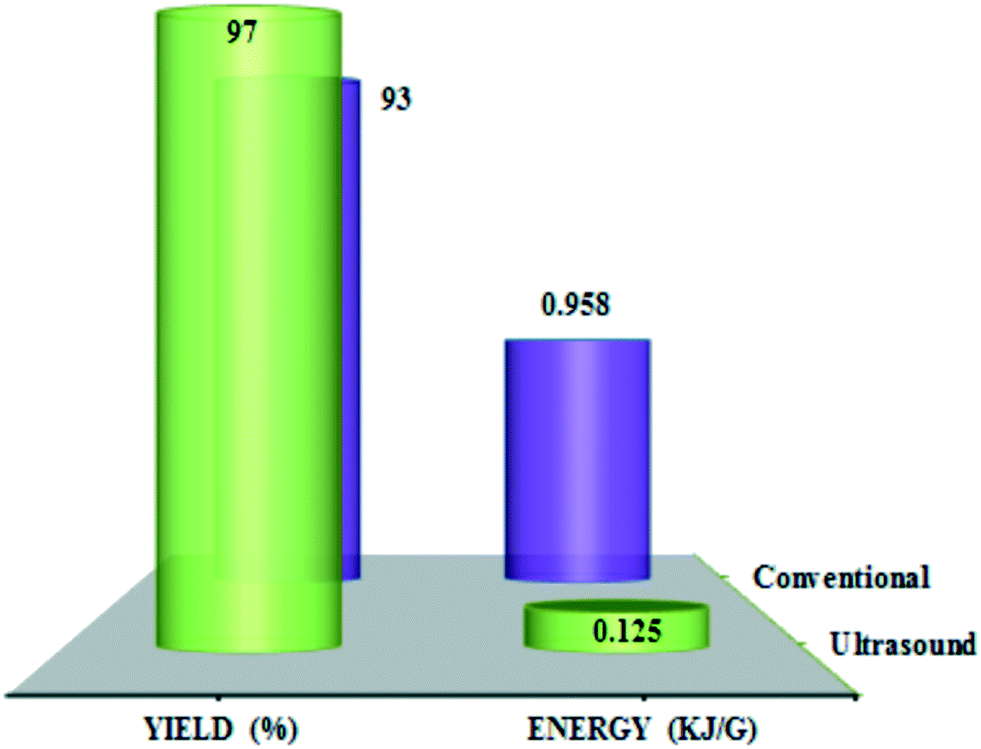 | ||
| Fig. 5 Yield (%) and energy (kJ g−1) comparison using thermal and ultrasound methods in benzyl alcohol oxidation promoted by ChPS·H2O. Reproduced from ref. 67 with permission from American Chemical Society. | ||
In general, the selective oxidation of alcohols to aldehydes in DESs is a topic that is currently attracting much interest.69 The group of Zhao reported a selective oxidation of both primary and secondary alcohols to aldehydes and ketones promoted by TEMPO supported onto a DES employing oxygen as a sacrificial reactant.70 The authors reported the synthesis of the modified DES as well (Scheme 19).
 | ||
| Scheme 19 Preparation of DES-supported TEMPO reported by Zhao et al.70 | ||
The protocol is rather simple. The TEMPO is treated with a di-bromide alkyl chain in the presence of a strong base (NaH), then a long-chain tertiary amine is added to the resulting intermediate to give the quaternary salt that mixed with the urea affords the desired DESs with the anchored TEMPO.
This DES was tested in the aerobic oxidation of benzyl alcohol in the presence of several metallic co-catalysts with air at ambient pressure (Table 11).
|
|
|||
|---|---|---|---|
| Time [h] | Co-catalyst | Conv. [%] | Selec [%] |
| 1 | None | 0.4 | >99 |
| 1.5 | FeCl3·6H2O | 3.1 | >99 |
| 1.5 | Fe(NO2)3·9H2O | >99 | >99 |
| 17.5 | Cu(NO2)3·3H2O | 24.0 | >99 |
| 1.5 | Co(NO2)3·6H2O | 3.1 | >99 |

Reaction conditions: benzyl alcohol 10 mmol, 1.25% DES–TEMPO, 3% salt(s), atmospheric oxygen pressure.
The co-catalyst is crucial to trigger the reaction and in its absence the conversion after 1 hour was below 1%. Among the most used metals in the TEMPO oxidation Fe(NO2)3·6H2O was the most active, boosting the reactivity to 99% conversion, whereas the selectivity remained the same as that of the other entries.
The applicability of the protocol was studied by testing more than 20 types of alcohols, both primary and secondary, showing a broad range of application with conversion over 90% and almost full selectivity towards the aldehyde or ketone products. A recyclability study was reported as shown in Table 12. The system solvent/catalyst was reused up to five times without significant loss of reactivity, although fresh co-catalyst (Fe(NO2)3·6H2O) had to be added after each run. After the seventh cycle, the conversion decreased more drastically and the DES-supported TEMPO could not be used again.
| Run | Conversion [%] | TOF [h−1] | Selec [%] |
|---|---|---|---|
| 1 | >99 | 26.4 | >99 |
| 2 | >99 | 26.4 | >99 |
| 3 | >99 | 26.4 | >99 |
| 4 | >99 | 26.4 | >99 |
| 5 | >99 | 26.4 | >99 |
| 6 | 85 | 22.7 | >99 |
| 7 | 66 | 17.6 | >99 |
| 8 | 27 | 7.2 | >99 |
3.4. Oxidation of alcohols and aldehydes
Carboxylic acids are recognized as pivotal intermediates in organic and industrial chemistry and they allow access to more complex frameworks. Carboxylic acids are prepared mainly using sacrificial reagents and transition metal catalysts starting from alcohols or aldehydes.71 Hydrogen peroxide is one of the most commonly used oxidative agents along with atmospheric oxygen. It is low-cost and the by-product generated is water, which does not pose particular issues in terms of disposal.In 2018, the Nagarkar's group reported the oxidation of aldehydes to carboxylic acids using H2O2 in DESs.72 The authors used benzaldehyde as a model substrate and found that by maintaining the reaction temperature at 50 °C in ChCl/urea the number of equivalents of hydrogen peroxide dramatically affected the reactivity. The conversion was seen to increase from 38% to 100% when moving from 1 equiv. to 1.5, respectively. Keeping the reaction running for more than 2 hours resulted in a drop of yield due to degradation of the benzoic acid previously formed. A comparative study showing the effect of replacing common solvents with the DESs was performed (Table 13). The recycling experiments showed that the activity within the DESs reaction media remained the same for five cycles.
|
|
|||
|---|---|---|---|
| Entry | Solvent | Conv. [%] | Select. [%] |
| Reaction conditions: benzaldehyde (1 mmol), solvent (2.0 mL), 30% H2O2 (1.5 equiv.), reaction time 2 h. | |||
| 1 | ChCl/urea | 100 | 100 |
| 2 | MeCN | 66 | 66 |
| 3 | MeOH | 55 | 55 |
| 4 | Toluene | 49 | 49 |
| 5 | H2O | 50 | 50 |
The preparation of organic acids, especially those used as bulk chemicals, from renewable resources is a very attractive topic nowadays. In this context, maleic acid and fumaric acid along with their anhydrides are well-known as important C4 chemical intermediates within the chemical industry, such as resins, lubricant additives, surface coatings and pharmaceuticals. In 2013 Yan and co-workers disclosed a protocol to convert furfural, one of the most important intermediates which comes from lignocellulose, into maleic and fumaric acid employing ChCl–oxalic acid DESs as both solvent and catalyst (Scheme 20).73
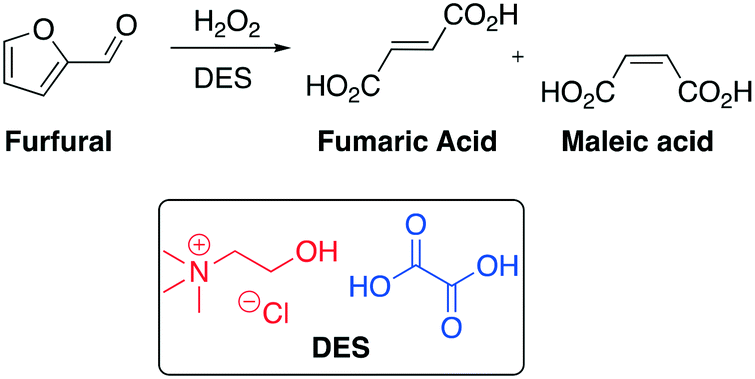 | ||
| Scheme 20 Furfural conversion into fumaric acid and maleic acid promoted by hydrogen peroxide and ChCl–oxalic acid DES.73 | ||
Carboxylic acids play a crucial role in the oxidation of furfural due to their capability to be oxidized to organic peracids by H2O2 and then efficiently oxidize furfural to maleic acid and fumaric acid. Based on this observation, the authors designed a DES with one organic acid that can catalyze the reaction. They selected oxalic acid as the key component of the DES due to its strong acidity (pKa = 1.13). A conversion of 100% of furfural into maleic and fumaric acid was achieved in six hours under mild conditions (50–60 °C).
In 2014 the Azizi group reported a very fast and selective oxidation of primary and secondary alcohols promoted by N-bromosuccinimide (NBS) in DESs.74 The reaction was observed to be faster in DESs than in common organic solvents when 1-phenylethanol was used as a model starting material (Table 14).
|
|
|||
|---|---|---|---|
| Entry | Solvent | Time [min] | Yield [%] |
| Reaction conditions: 1-phenylethanol (1 mmol) and NBS (1 mmol), solvent (0.5 mL), temperature 60 °C. | |||
| 1 | ChCl/urea | 5 | 100 |
| 2 | DCM | 80 | 45 |
| 3 | MeOH | 80 | 65 |
| 4 | MeCN | 80 | 55 |
| 5 | H2O | 80 | 80 |

The authors found that the ChCl/urea DES was a more efficient medium compared to other solvent media, remarking the absence of overoxidative side-products. Interestingly, the high chemoselectivity of the protocol towards primary alcohols was proved in an experiment in which a mixture of benzyl alcohol and 1-phenylethanol was used (Scheme 21).
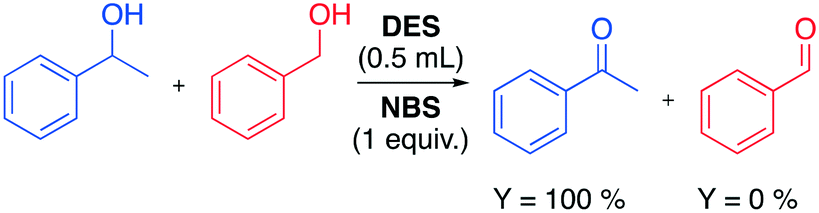 | ||
| Scheme 21 Chemoselective test for the oxidation of alcohols promoted by NBS in ChCl/urea DES.74 | ||
3.5. Phenol preparation in DESs
The preparation of phenols has always held a role of great importance in industrial chemistry and continues to attract the attention of the chemical and engineering communities. Recently, several reports have been disclosed about aromatic boronic acids as synthons for the synthesis of phenols through catalytic oxidative hydroxylation. In 2013, He and co-workers reported the oxidation of boronic acid into phenol in DESs employing hydrogen peroxide in the absence of a catalyst.75 Phenylboronic acid was chosen as a model reagent, with the reaction proceeding at room temperature in water in the presence of catalytic amounts of DES ChCl/urea (yield = 92%). The selected screening of reaction conditions is shown in Table 15, whereby the reaction in DESs appears faster and requires less amount of hydrogen peroxide in water, whereas the absence of DESs results in a drop of reactivity in any solvent.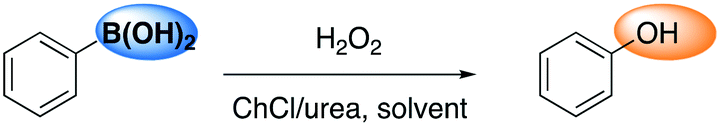
The authors explored the scope of the reaction using different aryl boronic acid compounds by varying the substituent on the aromatic ring. Electron-rich aryl boronic acids exhibited high yield (90 to 96%), showing complete consumption of the reagents in five minutes, whereas electron-poor boronic acids required a longer reaction time (15 minutes) with lower yield (82 to 90%). The authors proposed a mechanism to explain the role of the DES as the catalyst (Scheme 22) in which the DES activates the hydrogen peroxide by polarization of the peroxide bond through hydrogen bonding.
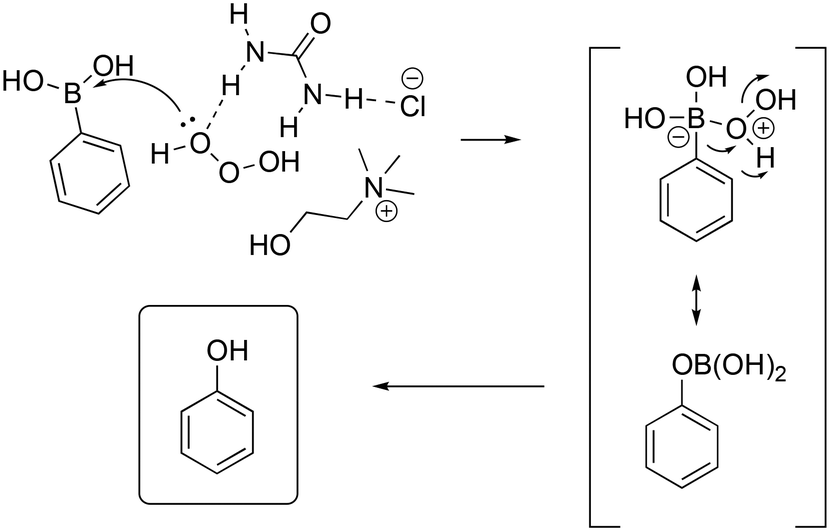 | ||
| Scheme 22 Proposed mechanism for the oxidation of aryl boronic acids promoted by hydrogen peroxide and ChCl/urea.75 | ||
A similar protocol using ChCl/HFIP (Scheme 23) was reported, studying in this case the reuse of the catalyst, which showed a reusability for five cycles.76
 | ||
| Scheme 23 Preparation of the ChCl/HFIP.76 | ||
4. Summary and outlook
Deep eutectic solvents (DESs) have emerged as more affordable alternatives to ionic liquid (IL) solvents, whilst sharing with them some desirable physicochemical properties, such as similar viscosities, pH, and capability to dissolve both organic and hydrophilic compounds. Their use as solvent media for organic reactions has been growing rapidly, with organic oxidations carried out in DESs emerging in recent years. The work reported in this area has shown promising results, highlighting several benefits in using DESs over conventional ILs and fossil fuel-based organic solvents, which can be summarized as:■ Enhancement in reactivity.
■ Easier recyclability.
■ Possibility to bind organic reactants, such as oxidative reagents, and catalyst functionalities to the DES framework.
■ Easier separation and purification of reaction products.
■ Possibility to operate under a wide temperature range.
Whilst some issues remain for a much wider use at a larger scale, such as the high viscosity of some DESs, commercial applications seem to be on the doorstep and we believe that their use for organic oxidations will lead to new processes for the manufacture of chemical commodities.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Carmine D'Agostino and Graziano Di Carmine would like to thank the EPSRC New Investigator Award (EP/S019138/1) for supporting their research activities.References
- T. Welton, Coord. Chem. Rev., 2004, 248, 2459 CrossRef CAS.
- (a) R. Fortunato, C. A. M. Afonso, M. A. M. Reis and J. G. Crespo, J. Membr. Sci., 2004, 242, 197 CrossRef CAS; (b) L. C. Branco, J. G. Crespo and C. A. M. Afonso, Angew. Chem., Int. Ed., 2002, 41, 2771 CrossRef CAS.
- P. Scovazzo, J. Kieft, D. A. Finan, C. Koval, D. DuBois and R. Noble, J. Membr. Sci., 2004, 238, 57 CrossRef CAS.
- D. A. Walsh, K. R. J. Lovelock and P. Licence, Chem. Soc. Rev., 2010, 39, 4185 RSC.
- (a) A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550 RSC; (b) P. Halder, S. Kundu, S. Patel, A. Setiawan, R. Atkin, R. Parthasarthy, J. Paz-Ferreiro, A. Surapaneni and K. Shah, Renewable Sustainable Energy Rev., 2019, 105, 268 CrossRef CAS.
- (a) Q. Zhang, S. Zhang and Y. Deng, Green Chem., 2011, 13, 2619 RSC; (b) B. Karimi, M. Tavakolian, M. Akbari and F. Mansouri, ChemCatChem, 2018, 10, 3173 CrossRef CAS.
- M. Freemantle, Chem. Eng. News, 2003, 18, 9 Search PubMed.
- (a) F. Javed, F. Ullah, M. R. Zakaria and H. M. Akil, J. Mol. Liq., 2018, 271, 403 CrossRef CAS; (b) V. A. Pereira, P. V. Mendonça, J. F. J. Coelho and A. C. Serra, Polym. Chem., 2019, 10, 4904 RSC; (c) D. Zheng, L. Dong, W. Huang, X. Wu and N. Nie, Renewable Sustainable Energy Rev., 2014, 37, 47 CrossRef CAS.
- (a) M. Bystrzanowska, F. Pena-Pereira, Ł. Marcinkowski and M. Tobiszewski, Ecotoxicol. Environ. Saf., 2019, 174, 455 CrossRef CAS; (b) M. Deetlefs and K. R. Seddon, Green Chem., 2010, 12, 17 RSC.
- D. R. MacFarlane, A. L. Chong, M. Forsyth, M. Kar, R. Vijayaraghavan, A. Somers and J. M. Pringle, Faraday Discuss., 2018, 206, 9–28 RSC.
- (a) A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70 RSC; (b) E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060 CrossRef CAS.
- Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108 RSC.
- Y. Dai, J. Van Spronsen, J. Witkamp, R. Verpoorte and Y. H. Choi, J. Nat. Prod., 2013, 76, 2162 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, H. Munro, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2001, 2010 RSC.
- F. H. Hurley and T. P. Wier, J. Electrochem. Soc., 1951, 98, 207 CrossRef CAS.
- W.-G. Xu, X.-M. Lü, Q.-G. Zhang, J.-S. Gui and J.-Z. Yang, Chin. J. Chem., 2006, 24, 331 CrossRef CAS.
- J.-Z. Yang, P. Tian, L.-L. He and W.-G. Xu, Fluid Phase Equilib., 2003, 204, 295 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies and R. K. Rasheed, Eur. J. Chem., 2004, 10, 3769 CrossRef CAS.
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142 CrossRef CAS.
- H. M. A. Abood, A. P. Abbott, A. D. Ballantyne and K. S. Ryder, Chem. Commun., 2011, 47, 3523 RSC.
- A. P. Abbott, J. C. Barron, K. S. Ryder and D. Wilson, Eur. J. Chem., 2007, 13, 6495 CrossRef CAS.
- A. P. Abbott, A. A. Al-Barzinjy, P. D. Abbott, G. Frisch, R. C. Harris, J. Hartley and K. S. Ryder, Phys. Chem. Chem. Phys., 2014, 16, 9047 RSC.
- (a) M. Francisco, A. van den Bruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074 CrossRef CAS; (b) D. O. Abranches, M. A. R. Martins, L. P. Silva, N. Schaeffer, S. P. Pinho and J. A. P. Coutinho, Chem. Commun., 2019, 55, 10253 RSC.
- (a) S. Honga, Y. Yuana, Q. Yanga, L. Chena, J. Denga, W. Chena, H. Liana, J. D. Mota-Moralesc and H. Liimatainen, Carbohydr. Polym., 2019, 220, 211 CrossRef; (b) S. Hong, H. Lian, X. Sun, D. Pan, A. Carranza, J. A. Pojman and J. D. Mota-Morales, RSC Adv., 2016, 6, 89599 RSC; (c) F. S. G. Bagh, K. Shahbaz, F. S. Mjalli, M. A. Hashim and I. M. AlNashef, J. Mol. Liq., 2015, 204, 76 CrossRef.
- (a) F.-F. Cheng, J.-N. Zhu, M.-Y. Zhao, Z.-J. Ma and W.-W. Xiong, J. Solid State Chem., 2019, 278, 120904 CrossRef CAS; (b) A. Abo-Hamad, M. Hayyan, M. A. AlSaadi and M. A. Hashim, Chem. Eng. J., 2015, 273, 551 CrossRef CAS; (c) M. F. Majid, H. F. M. Zaid, C. F. Kait, K. Jumbri, L. C. Yuan and S. Rajasuriyan, J. Mol. Liq., 2020, 306, 112870 CrossRef CAS.
- F. A. Carey, Organic Chemistry, McGraw Hill, New York, NY, 6th edn, 2006 Search PubMed.
- P. S. Bailey, Ozonation in Organic Chemistry. 2, Academic Press, New York, NY, 1982, ch. 2 Search PubMed.
- D. Astruc, C-H Activation and Functionalization of Alkanes and Arenes, in Organometallic Chemistry and Catalysis, Springer, Berlin, Heidelberg, 2007 Search PubMed.
- A. P. Abbott, ChemPhysChem, 2004, 5, 1242 CrossRef CAS.
- (a) A. F. Olea and J. K. Thomas, J. Am. Chem. Soc., 1988, 110, 4494 CrossRef CAS; (b) C. D'Agostino, R. C. Harris, A. P. Abbott, L. F. Gladden and M. D. Mantle, Phys. Chem. Chem. Phys., 2015, 17, 15297–15304 RSC; (c) A. P. Abbott, R. C. Harris, K. S. Ryder, C. D'Agostino, L. F. Gladden and M. D. Mantle, Green Chem., 2011, 13, 82 RSC.
- A. P. Abbott, R. C. Harris and K. S. Ryder, J. Phys. Chem. B, 2007, 111, 4910 CrossRef CAS.
- R. Häkkinen and A. Abbott, Green Chem., 2019, 21, 4673 RSC.
- S. M. Alabdullah, H. K. Ismail, K. S. Ryder and A. P. Abbott, Electrochim. Acta, 2020, 354, 136737 CrossRef CAS.
- A. P. Abbott, Phys. Chem., 2005, 6, 2502 CAS.
- Y. Kim, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 9109 CrossRef CAS.
- H. Ghaedi, M. Ayoub, S. Sufian, S. M. Hailegiorgis, G. Murshid and S. N. Khan, J. Chem. Thermodyn., 2018, 116, 50 CrossRef CAS.
- (a) A. P. Abbott, A. Y. M. Al-Murshedi, O. A. O. Alshammari, R. C. Harris, J. H. Kareem, I. B. Qader and K. Ryder, Fluid Phase Equilib., 2017, 448, 99 CrossRef CAS; (b) X. Li, M. Hou, B. Han, X. Wang and L. Zou, J. Chem. Eng. Data, 2008, 53, 548 CrossRef CAS.
- A. P. Abbott, S. S. M. Alabdullah, A. Y. M. Al-Murshedia and K. S. Ryder, Faraday Discuss., 2018, 206, 365 RSC.
- S. Gillet, M. Aguedo, L. Petitjean, A. R. C. Morais, A. M. da Costa Lopes, R. M. Łukasik and P. T. Anastas, Green Chem., 2017, 19, 4200 RSC.
- F. Liu, Z. Xue, X. Zhao, H. Mou, J. He and T. Mu, Chem. Commun., 2018, 54, 6140 RSC.
- (a) H. Zhang, N. Li, X. Pan, S. Wu and J. Xie, ACS Sustainable Chem. Eng., 2017, 5, 4066 CrossRef CAS; (b) N. Ríos-Lombardía, L. Cicco, K. Yamamoto, J. A. Hernández-Fernández, F. Morís, V. Capriati, J. García-Álvarez and J. González-Sabín, Chem. Commun., 2020, 56, 15165 RSC.
- F. Konietzni, U. Kolb, U. Dingerdissen and W. F. Maier, J. Catal., 1998, 176, 527 CrossRef CAS.
- N. Guajardo, C. Carlesi and A. Aracena, ChemCatChem, 2015, 7, 2451 CrossRef CAS.
- K. R. Seddon and A. Stark, Green Chem., 2002, 4, 119 RSC.
- B. G. Hashiguchi, S. M. Bishof, M. M. Konnick and R. A. Periana, Acc. Chem. Res., 2012, 45, 885 CrossRef CAS.
- C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS.
- J. Lu, X.-T. Li, E.-Q. Ma, L.-P. Mo and Z.-H. Zhang, ChemCatChem, 2014, 6, 2854 CrossRef CAS.
- X. Marset, J. M. Pérez and D. J. Ramón, Green Chem., 2016, 18, 826 RSC.
- P.-O. Morin, T. Bura and M. Leclerc, Mater. Horiz., 2016, 3, 11 RSC.
- V. M. Niemi, P. Knuuttila and J.-E. Österholm, Polymer, 1992, 33, 1559 CrossRef CAS.
- T.-J. Park and S. H. Lee, Green Chem., 2017, 19, 910 RSC.
- T.-J. Park, Y. S. Kim, E. Kanc and S. H. Lee, RSC Adv., 2015, 5, 25590 RSC.
- X. Marset, G. Guillena and D. J. Ramón, Chem. – Eur. J., 2017, 23, 10522 CrossRef CAS.
- Y. Hayashi, Chem. Sci., 2016, 7, 866 RSC.
- P. Vitale, L. Cicco, F. Messa, F. M. Perna, A. Salomone and V. Capriati, Eur. J. Org. Chem., 2019, 5557 CrossRef CAS.
- G. B. Barlin, The Chemistry of Heterocyclic Compounds, Wiley, New York, 1982, vol. 41 Search PubMed.
- N. M. T. Lourenço and C. A. M. Afonso, Tetrahedron, 2003, 59, 789 CrossRef.
- (a) D. J. Brown, R. F. Evans and W. B. Cowden, The Pyrimidines, ed. E. C. Taylor and A. Weissberger, John Wiley, New York, 1994, vol. 52 Search PubMed; (b) D. M. Bassani, J.-M. Lehn, G. Baum and D. Fenske, Angew. Chem., Int. Ed. Engl., 1997, 36, 1845 CrossRef CAS.
- K. S. Vadagaonkar, H. P. Kalmode, S. Prakash and A. C. Chaskar, New J. Chem., 2015, 39, 3639 RSC.
- S. K. Ghosh and R. Nagarajan, RSC Adv., 2016, 6, 27378 RSC.
- (a) C. Wattanapiromsakul, P. I. Forster and P. G. Waterman, Phytochemistry, 2003, 64, 609 CrossRef CAS; (b) M. Viji and R. Nagarajan, J. Chem. Sci., 2014, 126, 1075 CrossRef CAS.
- S. J. Evenson, M. J. Mumm, K. I. Pokhodnya and S. C. Rasmussen, Macromolecules, 2011, 44, 835 CrossRef CAS.
- H. P. Kalmode, K. S. Vadagaonkar, K. Murugan, S. Prakash and A. C. Chaskar, RSC Adv., 2015, 5, 35166 RSC.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155 CrossRef CAS.
- K. Singh, A. Kaur, V. S. Mithu and S. Sharma, J. Org. Chem., 2017, 82, 5285 CrossRef CAS.
- L. Cicco, A. Fombona-Pascual, A. Sánchez-Condado, G. A. Carriedo, F. M. Perna, V. Capriati, A. P. Soto and J. García-Álvarez, ChemSusChem, 2020, 13, 4967 CrossRef CAS.
- B. L. Gadilohar, D. V. Pinjari and G. S. Shankarling, Ind. Eng. Chem. Res., 2016, 55, 4797 CrossRef CAS.
- B. L. Gadilohar, H. S. Kumbhar and G. S. Shankarling, Ind. Eng. Chem. Res., 2014, 53, 19010 CrossRef CAS.
- (a) Y. Yu, B. Lu, X. Wang, J. Zhao, X. Wang and Q. Cai, Chem. Eng. J., 2010, 162, 738 CrossRef CAS; (b) J. M. Khurana, P. K. Sahoo, S. S. Titus and G. C. Maikap, Synth. Commun., 1990, 20, 1357 CrossRef CAS; (c) R. Naik, A. Nizam, A. Siddekha and M. A. Pasha, Ultrason. Sonochem., 2011, 18, 1124 CrossRef CAS.
- Y. Zhang, F. Lü, X. Cao and J. Zhao, RSC Adv., 2014, 4, 40161 RSC.
- (a) J. Sedelmeier, S. V. Ley, I. R. Baxendale and M. Baumann, Org. Lett., 2010, 12, 3618 CrossRef CAS; (b) Z. Yang, R. Luo, Z. Zhu, X. Yang and W. Tang, Organometallics, 2017, 36, 4095 CrossRef CAS; (c) Q. Tian, D. Shi and Y. Sha, Molecules, 2008, 13, 948 CrossRef CAS.
- R. Wagh, G. Nerurkar and J. Nagarkar, ChemistrySelect, 2018, 3, 9654 CrossRef.
- Y. Ni, Z. Bi, H. Su and L. Yan, Green Chem., 2019, 21, 1075 RSC.
- N. Azizi, M. Khajeh and M. Alipour, Ind. Eng. Chem. Res., 2014, 53, 15561 CrossRef CAS.
- L. Wang, D.-Y. Dai, Q. Chen and M.-Y. He, Asian J. Org. Chem., 2013, 2, 1040 CrossRef CAS.
- L. Wang, D.-Y. Dai, Q. Chen and M.-Y. He, J. Fluorine Chem., 2014, 158, 44 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |