
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of naringenin and its combination with cisplatin in cell death, proliferation and invasion of cervical cancer spheroids†
Oswaldo Pablo Martínez-Rodríguez a,
Alejandro González-Torresa,
Luis Marat Álvarez-Salasb,
Humberto Hernández-Sáncheza,
Blanca Estela García-Pérezc,
María del Rocío Thompson-Bonillad and
María Eugenia Jaramillo-Flores*a
a,
Alejandro González-Torresa,
Luis Marat Álvarez-Salasb,
Humberto Hernández-Sáncheza,
Blanca Estela García-Pérezc,
María del Rocío Thompson-Bonillad and
María Eugenia Jaramillo-Flores*a
aLaboratorio de Biopolímeros, Departamento de Ingeniería Bioquímica, Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional, Av Wilfrido Masseiu Esq. Manuel Stampa S/N, Unidad Profesional Adolfo López Mateos, CP 07738, Ciudad de México, México. E-mail: jaramillo_flores@hotmail.com; mjarfl@ipn.mx
bDepartamento de Genética y Biología Molecular, Centro de Investigación y de Estudios Avanzados del I.P.N., Av. I.P.N. 2508, CP 07360, Ciudad de México, México
cDepartamento de Microbiología, Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional, Carpio y Plan de Ayala. Casco de Santo Tomás, CP 11340, Ciudad de México, México
dLaboratorio de Medicina Genómica, Hospital 1ro de Octubre, Instituto de Seguridad y Servicios Sociales de los Trabajadores del Estado, Av. I.P.N. 1669, CP 07300 Ciudad de México, México
First published on 22nd December 2020
Abstract
The main treatment alternative for cervical cancer is cisplatin chemotherapy. However, the resistance of tumor cells to cisplatin, in addition to side effects, limits its use. The flavonoid naringenin has shown cytotoxic effects on tumor cells and may be considered as a coadjuvant in the treatment of cervical cancer. In the present study, the effect of naringenin on cell viability, cytotoxicity, proliferation, apoptosis and invasion was evaluated in HeLa spheroid cultures. Naringenin impaired the cell viability as indicated by low ATP levels and caused concentration- and time-dependent cytotoxicity via the loss of cell membrane integrity. Furthermore, it did not activate caspases 3, 7, 8, and 9, suggesting that the cytotoxic effect was by necrotic cell death instead of apoptosis. Additionally, proliferation in the G0/G1 phase of the cell cycle was inhibited. Cell invasion also decreased as time progressed. Later, we determined if naringenin could improve the anti-tumor effect of cisplatin. The combination of naringenin with low concentrations of cisplatin improved the effect of the drug by significantly decreasing cell viability, potentiating the induction of cytotoxicity and decreasing the invasive capacity of the spheroids. Since these effects are regulated by some key proteins, molecular docking results indicated the interaction of naringenin with RIP3 and MLKL, cyclin B and with matrix metalloproteases 2 and 9. The results showed the anti-tumor effect of naringenin on the HeLa spheroids and improved effect of the cisplatin at low concentrations in combination with naringenin, placing flavonoids as a potential adjuvant in the therapy against cervical cancer.
Introduction
Cervical cancer was ranked fourth in incidence in women worldwide in 2018.1 Infection by the human papillomavirus (HPV) is the most important risk factor for cervical cancer development,2 in addition to other risk factors that contribute to the development of this cancer.3 In the early stages of cancer, surgery is the recommended option,4 whilst in advanced stages, treatment involves brachytherapy in combination with platinum complex chemotherapy,5,6 such as cis-diamminedichloroplatinum(II), or cisplatin (Fig. 1A), is a well-known alkylating agent with anti-cancer properties. Depending on the concentration, cisplatin induces cytotoxicity, by interfering with DNA transcription and/or replication mechanisms by binding to guanine bases, causing damage and cell cycle arrest.7,8 In cervical cancer, it is more effective in advanced stages,6 however, the side effects that it also causes, such as damage to non-cancerous cells, nausea, vomit, decreased blood cells, platelets in the bone marrow, immunosuppression, nephrotoxicity, infertility, ototoxicity, and neurotoxicity, it limits its long-term use.7,8 On the arduous path of discovering new drugs, natural products (extracts and isolated molecules) serve as key resources for the development of candidates for new drugs.9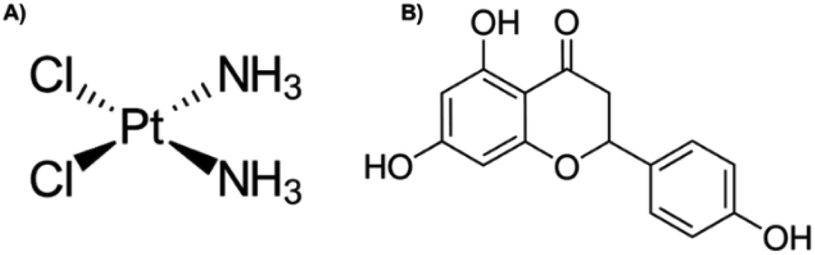 | ||
| Fig. 1 Chemical structure of (A) cisplatin and (B) naringenin. | ||
Flavonoids are found abundantly in fruits, vegetables, tea, seeds, grains, nuts, and some traditional medicinal herbs.10 In addition to their physiological functions in plants, flavonoids are important components of the human diet, and although they are not considered nutrients, possess various biological activities,11 including antioxidant, hepatoprotective, anti-nephrotoxicity, anti-antibacterial, anti-inflammatory, anti-obesity, antiviral, and anti-tumour activity.10 Naringenin (4′,5,7-trihydroxyflavanone) (Fig. 1B) contains three hydroxyl groups on the 4′,5,7 carbons and is the aglycone of naringin, and it is found mainly in citrus fruits, such as grapefruit and orange.12,13 Naringenin cytotoxic effect has been demonstrated at different concentrations in monolayer cultures of different types of cancer, such as leukaemia, pancreas, breast, stomach, liver, and colon.14 In cervical cancer, naringenin and naringin induce apoptosis and arrest the cell cycle in HeLa and SiHa cells, respectively.13,14 The induction of apoptosis with naringenin nanoparticles is carried out by altering the potential of the mitochondrial membrane.15
However, it is now recognized that the 2D model of cultured cells, cannot simulate the microenvironment of tumours, which grow in three dimensions.15 3D culture models are especially beneficial for investigating drug resistance and mechanism processes in tumour cells. Furthermore, a variety of molecular mechanisms in the study of cancer cells in 3D models suggest that tumour cells grown in 2D monolayer do not respond to anticancer compounds in a similar way.16 Therefore, the objective of the present study was to evaluate the anti-tumour capacity of naringenin in a 3D in vitro model of cervical cancer and to demonstrate that it may be an adjunctive agent in the therapy with cisplatin against cervical cancer.
Materials and methods
Cell culture
The cervical cancer cell line HeLa (ATCC® CCL-2™) was grown in a DMEM high glucose GIBCO™ medium (Thermo Fisher Scientific Inc., Waltham, MA) supplemented with 10% GIBCO™ fetal bovine serum (FBS) (Thermo Fisher Scientific) and 1% GIBCO™ antibiotic-antimycotic (Thermo Fisher Scientific), with 5% CO2 at 37 °C.3D-spheroids culture
The spheroids were performed according to the methodology proposed by González-Torres,17 with slight modifications. Briefly, in Corning® U-bottom 96-well ultra-low attachment plates (Corning Inc., Corning, NY), 1.2 × 103 were seeded for cell staining assays and 300 cells for ATP viability assays and caspase activity, and incubated in DMEM high glucose GIBCO™ medium (Thermo Fisher Scientific) supplemented with 10% FBS GIBCO™ (Thermo Fisher Scientific) free from antibiotic-antimycotic for 24 h. The spheroids were then treated with 100, 300, and 500 μM naringenin (≥95% purity; mp 247–250 °C; Sigma-Aldrich, Inc., St. Louis, MO USA) dissolved in 0.1% DMSO alone or in combination with cisplatin (mp 270 °C; PiSA SA de CV, Mexico).Viability/cytotoxicity tests in 3D culture
The effect of naringenin alone or in combination with cisplatin on cell viability was determined by the CellTiter-Glo® 3D luminescence assay (Promega, Madison, WI) following the manufacturer's specifications. Briefly, after 24 h incubation with naringenin alone or in combination with cisplatin, CellTiter-Glo® reagent was added to the spheroids and they were incubated for 25 minutes to immediately quantify the luminescence in a plate reader (SYNERGY H1, BioTek). The obtained values were reported as relative light units that are proportional to ATP levels.Additionally, cytotoxicity was determined by the addition of SYTOX Green® 250 nM (Thermo Fisher Scientific) and Hoechst 33342 at 1 μg mL−1 (Thermo Fisher Scientific) with an additional 4 h incubation. Fluorescence was analyzed in a plate reader (SYNERGY H1, Biotek) with an excitation/emission of 350/461 and 504/523 nm respectively. Viability was calculated by subtracting SYTOX Green® dye (impermeable) fluorescence from Hoechst 33342 dye (permeable) fluorescence, while the cytotoxicity (represented as the ratio of dead cells in relation to total cells), was determined by dividing the fluorescence values of SYTOX Green® by the fluorescence values of Hoechst 33342. Changes in the spheroid morphology and SYTOX Green® staining were monitored by using a Zeiss Axiovert 25 inverted epifluorescence microscope (Carl Zeiss, GA, Oberkochen, Germany) with 10× magnification and the ImageJ software.
Cell division assay in 2D culture
To determine the effect of naringenin on cell division, the Click-iT™ EdU Pacific Blue™ Flow Cytometry Assay Kit (Thermo Fisher Scientific) was used, following the manufacturer's specifications and paring it to the methodology proposed by Yuan et al.18 For the epifluorescence microscopy analysis, 180 × 103 cells per well were seeded in chamber slides. Subsequently, the cells were treated with 500 μM naringenin, DMEM without FBS (serum starvation) as a negative proliferation control and the vehicle (0.1% DMSO) as a positive proliferation control, keeping in incubation for 48 h. Then, 10 μM of 5-ethynyl-2′-deoxyuridine (EdU) were added for 2 h. The Click-iT reaction with azide-Pacific Blue was performed to label the synthesized DNA, additional to the Propidium Iodide (PI) staining. After, cells were mounted in the chamber slides with Vecta-Shield® (Vector Laboratories Inc., Burlingame, CA) as the mounting reagent and they were analysed with a Nikon epifluorescence microscope (Nikon, Tokyo, Japan) and the ImageJ software (National Institutes of Health, Bethesda, USA).For the flow cytometry analysis, 180 × 103 cells per well were seeded in six-well plates and the same protocol used for epifluorescence microscopy with slight variations was performed. Briefly, cells were incubated for 24 h in DMEM without FBS, to synchronize the cell cycle. After this time, complete medium (FBS) and vehicle (0.1% DMSO) were added to the positive proliferation controls; to the negative proliferation control, DMEM medium without FBS, and the treatments with 500 μM naringenin and the 500 μM naringenin-EC25 (16 μM) cisplatin combination were incubated for 24 and 48 h. Subsequently, the cells were harvested, fixed with 4% PFA and permeabilized to immediately add 100 μM RNAse and stained with PI. 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 events were analyzed by flow cytometry (BD FACSAria™ III from the UDIBI, ENCB – IPN) and the results were analyzed in the ModFit LT v5.0 software.
000 events were analyzed by flow cytometry (BD FACSAria™ III from the UDIBI, ENCB – IPN) and the results were analyzed in the ModFit LT v5.0 software.
Caspase activity assays in 3D culture
Caspase activity was determined by using the Caspase-Glo® 3/7 kit (Promega), following the manufacturer's specifications. For this, the Caspase-Glo® 3/7 reagent was added to the spheroids previously treated with 500 μM naringenin for 8, 12, 24, and 72 h, whose reaction was incubated 1.5 h to immediately measure the luminescence in a plate reader (SYNERGY H1, BioTek). Additionally, in order to determine the possible apoptosis pathway, caspase inhibitors (Merck-Millipore, Billerica, MA) Z-IETD-FMK (caspase-8), Z-LEHD-FMK (caspase-9), and Z-VAD-FMK (pancaspase) were used at 40 μM, along with the CellTiter-Glo® reagent (Promega). In both assays, cisplatin was used as an apoptosis inducer.Cell invasion assay in 3D culture
The invasion assays were performed according to the methodology proposed by González-Torres,17 with slight modifications. Briefly, after seeding the spheroids, naringenin was added to 100, 300, 500 μM naringenin alone or in combination with cisplatin and 100 μL of Matrigel® Matrix (Corning) were added, allowing its solidification for 1 h at 37 °C to immediately overlaid with100 μL of DMEM. The spheroids were incubated for 24 h and 48 h and the invasion rate was determined by using an inverted microscope (Zeiss) and the ImageJ software. Invasion was calculated by measuring the area between the spheroid edge and the leading edge of the invasive cells.In silico assay
The interaction of naringenin with proteins involved in the, death cell, cell cycle and extracellular matrix (ECM) remodelling proteins was studied by molecular docking in the Docking Server (http://www.dockingserver.com), through the methodology proposed by Anguiano-Sevilla.19 Briefly, proteins were selected from the Protein Data Bank (PDB) TNF-R1 (Code PDB 2ZJC), RIP3 (Code PDB 4M66), MLKL (Code PDB 4M67), IGF-R1 (Code PDB 1K3A), MMP2 (Code PDB 1CK7), MMP9 (Code PDB 1L6J), E-cadherin (Code PDB 1Q1P), N-Cadherin (Code PDB 1NCG), Cyclin B1 (Code PDB 2B9R), CDK2 (Code PDB 1HCL). The naringenin structure was downloaded from PubChem, to which Gaesteiger partial charges were added. Nonpolar hydrogen atoms were fused and rotary bonds were defined. The MMFF94 force field was used for energy minimization of the naringenin molecules. Docking was performed by using the Lamarckian Genetic Algorithm, and each experiment was derived from 100 different runs that were set to end after a maximum of 2500000 energy evaluations. The population size was set at 150. During the search, a translational step of 0.2 Å and quaternion and torsion steps of five were applied.
Results
Naringenin decreases ATP levels and reduces cell viability in HeLa spheroids
The analysis of the effect of naringenin on cell viability of spheroids at three concentrations (100, 300, and 500 μM) and 3 times (1, 4, and 7 days), showed on the first day a significant decrease in ATP levels from 300 and 500 μM. Over time, ATP levels decreased ∼18000 RLUs from 0 and 100 μM naringenin at 4 days. At day 7, the negative control continued to grow and in the naringenin-treated spheroids (100, 300, and 500 μM), ATP levels evidently decreased to ∼250 RLUs with 500 μM (Fig. 2), indicating the decrease in cell viability.
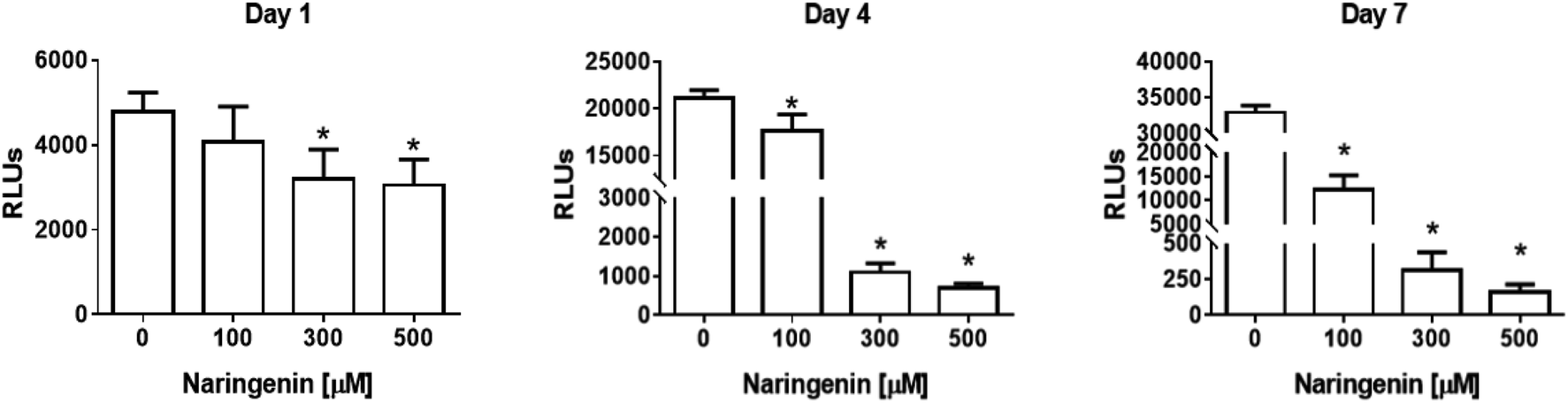 | ||
| Fig. 2 Reduction of cell viability in HeLa cell spheroids by naringenin. ATP levels of spheroids exposed to 1, 4, and 7 days with 100, 300, and 500 μM of naringenin. p ≤ 0.05 according to Tukey's mean test, n = 12. | ||
Naringenin cytotoxicity was determined through the integrity of the membrane, for which the spheroids were stained with Sytox Green® (impermeable) and Hoechst 33342 (permeable) (Fig. 3A). After 1 day, an increase in the fluorescence intensity produced by Sytox Green® with 500 μM of naringenin was observed; on the contrary, with Hoechst 33342 the fluorescence decreased from 35000 (negative control) to 1700 RFs, indicating induction of cell death in the spheroid and possible inhibition cell growth. On day 4, the fluorescence produced by Sytox Green® with 300 and 400 μM, showed the increase in cell death of the spheroid, while the fluorescence with Hoechst 33342 followed the tendency to decrease. On day 7, this effect was clearly observed, both Hoechst 33342 and Sytox Green® fluorescence decrease with different concentrations of naringenin, causing the decrease in viability, and these results also suggest that the flavonoid can modulate growth of concentration- and time-dependent manner. It is also necessary to emphasize that the decrease viability and fluorescence of Hoechst 33342 in the spheroids, in addition to being caused by cell death, may also be due to the possible effect of flavonoid modulation on proliferation.
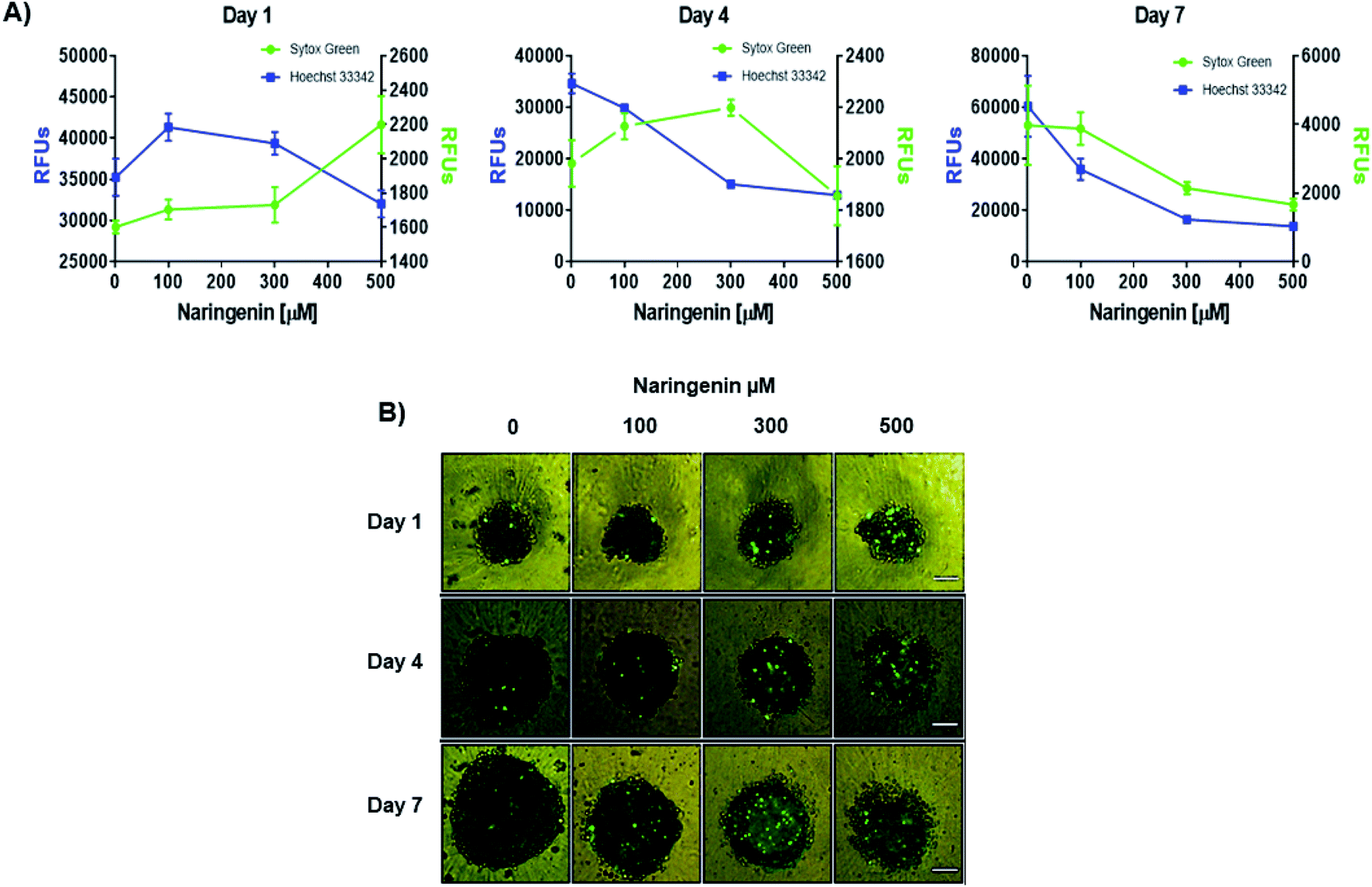 | ||
| Fig. 3 Cytotoxic and cytostatic effect of naringenin in HeLa cell spheroids. (A) Relative fluorescence units (RFU) of staining with Sytox Green® (dead cells) and Hoechst 33342 (total cell population) in HeLa cell spheroids exposed to 1, 4, and 7 days with 100, 300, and 500 μM of naringenin. (B) Epifluorescence microscopy images of spheroids treated with naringenin (100, 300, and 500 μM) during 1, 4, and 7 days stained with Sytox Green® (dead cells). p ≤ 0.05 according to Tukey's mean test, n = 12. Scale bar: 100 μm; amplification: 10×. | ||
By microscopy, the morphological damage and induction of death of the spheroid caused by naringenin was observed by increasing the fluorescence due to staining with Sytox Green® (Fig. 3B). These results, both decreased viability, cytotoxicity through membrane integrity, and decreased spheroid size, demonstrate the effect of naringenin concentration- and time-dependent. The effect of naringenin was also measured in fibroblasts and an increase in cell viability was detected (data not showed). These results indicate that naringenin is selective in cervical cancer cells.
Naringenin arrest the cell cycle in the G0/G1 (24 h) and S (48 h) phase of cervical cancer cells
The effect of naringenin on the cell cycle of HeLa cells was analyzed by quantifying total DNA by flow cytometry using PI. The cells were treated with 500 μM naringenin and the combination naringenin-EC25 cisplatin (16 μM) for 24 and 48 hours.Cell division was observed by epifluorescence microscopy, through Pacific Blue staining and total DNA with PI staining. In the cells of the positive control (vehicle DMSO 0.1%), fluorescence due to Pacific Blue staining was observed, signaling de la division cellular, while in the cells treated with naringenin, the incorporation of the EdU was even less than the cells in negative control (serum starvation), which were in quiescent stage since the cells stopped dividing (Fig. 4).
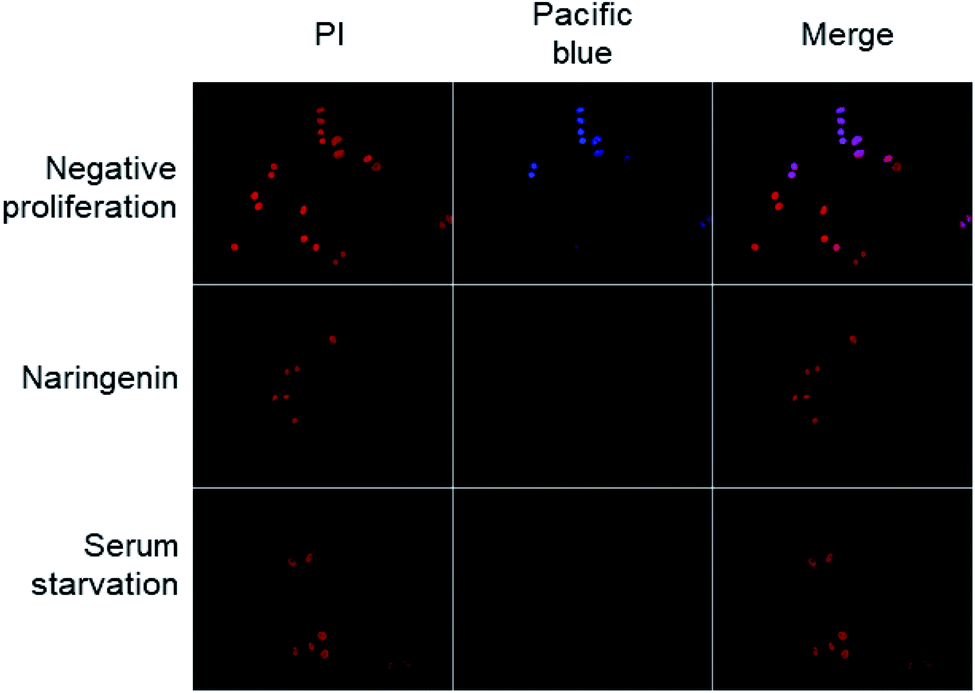 | ||
| Fig. 4 Epifluorescence microscopy images of decreased cell division of HeLa cells; the total DNA content was detected with PI, while proliferation was observed by staining with Pacific Blue; scale bar 20 μm amplification: 40×. | ||
30000 events per sample were analyzed by flow cytometry (Table 1). At 24 h, the vehicle concentrated the majority of the population in the G0/G1 phase (57.50 ± 2.62%), while in the S and G2/M phases 11.55 ± 2.27% and 30.95 ± 0.70% was found respectively. Most of the starvation cells remained in the G0/G1 phase with 60.81 ± 1.28%, followed by the G2/M (22.20 ± 0.56%) and S phases (16.99 ± 1.50%).
| Treatments | Cells (%) | ||
|---|---|---|---|
| G0/G1 | S | G2/M | |
| a Statistically significant values (negative control), p ≤ 0.05 according to Tukey's mean test, n = 6. | |||
| 24 h | |||
| Vehicle | 57.50 ± 2.62 | 11.55 ± 2.27 | 30.95 ± 0.70 |
| Starvation | 60.81 ± 1.28 | 16.99 ± 1.50a | 22.20 ± 0.56a |
| Naringenin 500 μM | 50.63 ± 3.69 | 24.78 ± 3.94a | 24.59 ± 0.41a |
| Naringenin + EC25 CP | 63.13 ± 1.37a | 8.44 ± 2.10 | 28.43 ± 1.41a |
|
|||
| 48 h | |||
| Vehicle | 59.28 ± 0.97 | 13.94 ± 1.30 | 26.82 ± 0.95 |
| Starvation | 62.74 ± 3.36 | 37.16 ± 3.43a | 0.09 ± 0.15a |
| Naringenin 500 μM | 11.62 ± 4.02a | 55.66 ± 7.89a | 32.72 ± 4.58 |
| Naringenin + EC25 CP | 18.06 ± 1.67a | 37.93 ± 4.56a | 44.0 ± 3.90a |
The cells treated with naringenin compared to the vehicle, in the G0/G1 (50.63 ± 3.69%) and G2/M (24.59 ± 0.41%) phases, decreased by 7% and 5% respectively, on the contrary, they doubled in the S phase (24.78%), indicating that the flavonoid arrest the cell cycle in the G0/G1 phase, with a significant increase in S phase. The combination naringenin-EC25 CP (16 μM) concentrated most of the cell population in G0/G1 phase with 63.13 ± 1.37%, showing a very important reduction in S phase, being the lowest value of the groups (8.44 ± 2.10%) with 28.43 ± 1.41% of cells in G2/M phase (Table 1 and Fig. 1S†). At 48 h, the vehicle maintained the same behavior as at 24 h, observing that the majority of the population was found in the G0/G1 phase (59.28 ± 0.97%), in S 13.94 ± 1.30% and in G2/M 26.82 ± 0.95%. Starvation cells also maintained the majority in G0/G1 with 62.74 ± 3.36%, with an increase in S (37.16 ± 3.43%) and a decrease in G2/M to 0.09 ± 0.15%. On the other hand, naringenin showed a decrease in the G0/G1 phase to 11.62 ± 4.02%, while they increased in S (55.66 ± 7.89%) and in G2/M (32.72 ± 4.58%), showing cell cycle arrest in S phase with increase in G2/M. With naringenin-EC25 CP combination the cells decreased to 18.06 ± 1.67% in G0/G1 and increased both in S and G2/M to 37.93 ± 4.56% and 44.0 ± 3.90% respectively, being in this last phase where the majority of cells was concentrated, showing that naringenin-EC25 CP combination modulates the cell cycle in differently way, arresting at G2/M, unlike naringenin alone which the arrest is at G0/G1 with an increase in S phase (Table 1 and Fig. 1S†).
In both cases, at 24 and 48 h, the flavonoid, in addition to having a cytotoxic effect on HeLa cells, can also modulate the cell cycle, arresting in G0/G1 phase with an increase in S at 24 h, while at 48 h it arrests in phase S with an increase in G2/M. The combination naringenin-EC25 CP at 24 h arrest the cycle in the G0/G1 phase and at 48 h in G2/M phase (Table 1 and Fig. 1S†). These results demonstrate that the effect of naringenin on the cell cycle is time dependent and, in both cases, proliferation is arrested.
Naringenin inhibits the activity of caspases in HeLa cells spheroids
Since naringenin apparently induced cell death in the spheroids by loss membrane integrity, the activity of caspases 3, 7, 8 and 9 were measured. The EC25 (16 μM) and EC50 (33 μM) of cisplatin were previously determined. The effect of naringenin on the activity of apoptosis effector caspases, caspases 3 and 7 of the spheroids was measured by luminescence. The higher the RLU, the higher the caspase activity, as shown in Fig. 5A. Even though naringenin caused cell death in the spheroids, no activity of these caspases was observed at 12, 24, and 72 h of exposure, showing the same behavior as the spheroids of the negative control (0 h). In contrast, in the spheroids treated with cisplatin (33 μM) an evident increase in the activity of these caspases is observed up to 1500 RLUs, indicating induction of apoptosis. To confirm the lack of apoptosis activation, the activity of initiator caspases 8 and 9 was measured through the effect of specific inhibitors on cell viability of the spheroid. Relative to naringenin-treated spheroids, viability decreased compared to the negative control, as expected. The same phenomenon occurred in the spheroids treated with naringenin in the presence of the inhibitors of both caspases, obtaining the same viability as the spheroids treated without inhibitors (Fig. 5B).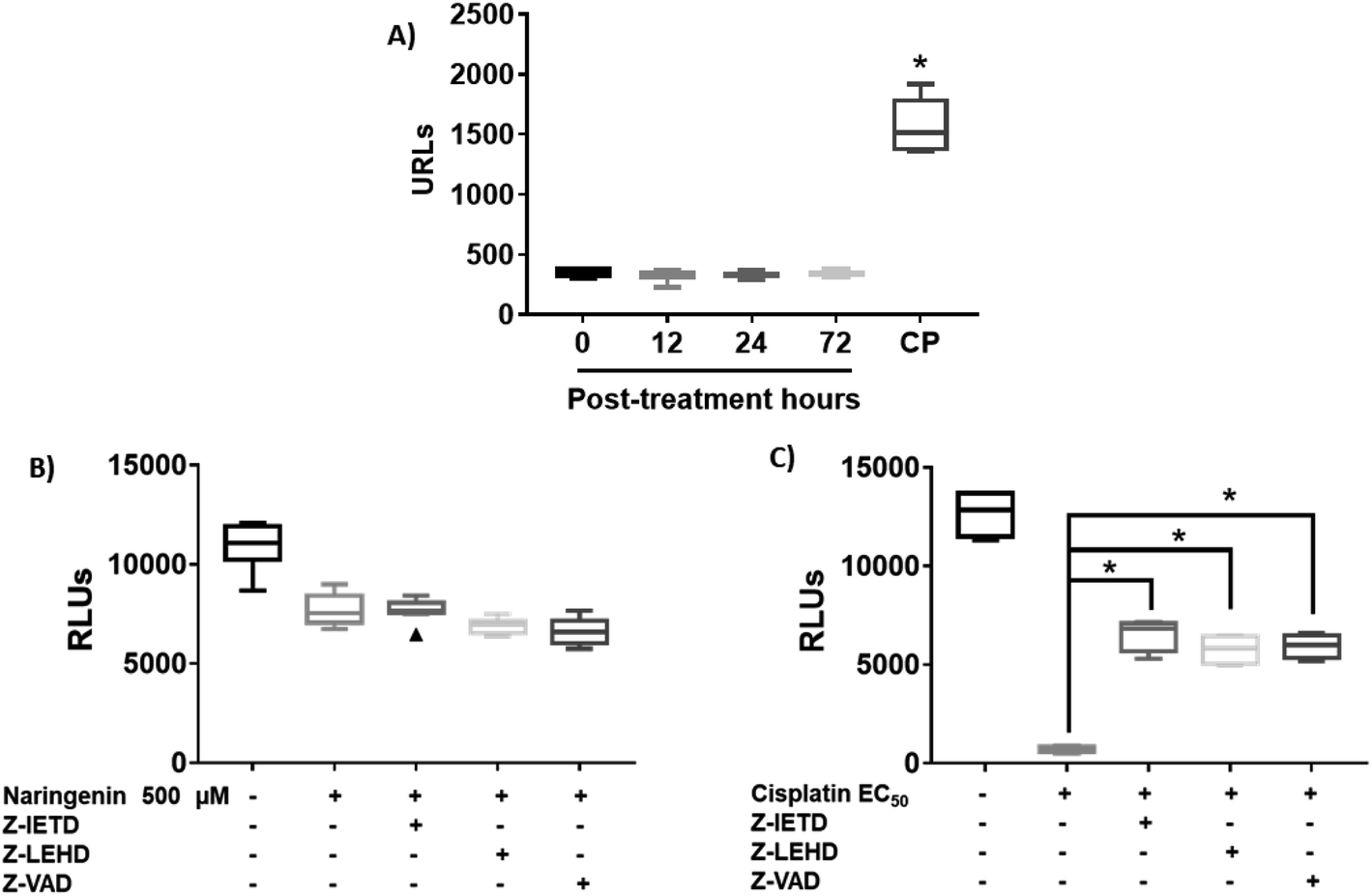 | ||
| Fig. 5 Naringenin does not alter caspase activity in HeLa cell spheroids. Activity of caspases 3, 7, 8, and 9 in HeLa cell spheroids exposed to 500 μM of naringenin. CP, apoptosis-inducing cisplatin as a positive control. (A) Activity of caspases 3 and 7 at 12, 24, and 72 h exposure with naringenin; CP at EC50 (33 μM). (B) Alteration of cell viability due to the inhibition of caspases 8 and 9 in the presence of 500 μM of naringenin; (C) alteration of cell viability due to the inhibition of caspases 8 and 9 in the presence of EC50 (33 μM) of cisplatin. p ≤ 0.05 according to Tukey's mean test, n = 12. | ||
Fig. 5B shows the assay with caspase inhibitors in the presence only of naringenin, while Fig. 5C shows the same assay but only with cis platinum. Z-IETD reagent is a caspase 8 inhibitor, Z_LEHD is a caspase 9 inhibitor, and Z-VAD is an all caspase inhibitor. In Fig. 5B, the first column is the cell culture without inhibitors, the second column is naringenin, the third column is naringenin + caspase 8 inhibitor, the fourth column is naringenin + caspase 9 inhibitor, and the fifth column is naringenin + inhibitor general of caspases. Fig. 5C in the first column is the culture without inhibitors, the second is cisplatin + caspase 8 inhibitor, the third column is cisplatin + caspase 8 inhibitor, the fourth column is cisplatin + caspase 9 inhibitor and finally, the fifth column is cisplatin + general inhibitor of caspases. What can be appreciated is that naringenin reduces of cell viability, which is the same when the caspases responsible for apoptosis are inhibited. On the other hand, with cisplatin it is observed that it kills almost all the cells (column 2, Fig. 5C), but in the presence of caspase inhibitors, the reduction in cell viability is reduced by almost 50% (column 5, Fig. 5C). In the presence of inhibitors of all caspases, a decrease in the viability of spheroids is observed, and since one of the mechanisms of cisplatin to induce death is to form covalent adducts between nucleotide bases during DNA synthesis, thus managing to stop the cell cycle in the checkpoints, to immediately induce apoptosis, the percentage of living cells observed when inhibiting all caspases (both initiators and effectors), is probably due to the fact that the cells are arrested in some phase of the cell cycle, compared with positive control that continues to proliferate.
In spheroids treated with cisplatin (33 μM) and without caspase inhibitors, the viability decreased as expected, since the drug is an inducer of death through apoptosis, however, in the presence of caspase inhibitors 8 and 9, this decrease was not as marked, being statistically different from the EC50 cisplatin, which clearly indicates that by inhibiting caspases, there is no induction of apoptosis. Importantly, the results suggest that death caused by naringenin does not involve caspase activity (Fig. 5C).
Naringenin inhibits spheroids cell invasion
Cellular invasion was determined by the invaded area of Matrigel®. At 24 h, the invasion was significantly decreased with 100 μM of naringenin (Fig. 6A), this being more evident at 300 and 500 μM. At 48 h, while the negative control increased the invasion, the naringenin decreased it significantly from 100 μM, to 300 and 500 μM (Fig. 6B). Significant morphological damage was also observed in the spheroid due to flavonoid cytotoxicity (Fig. 6C).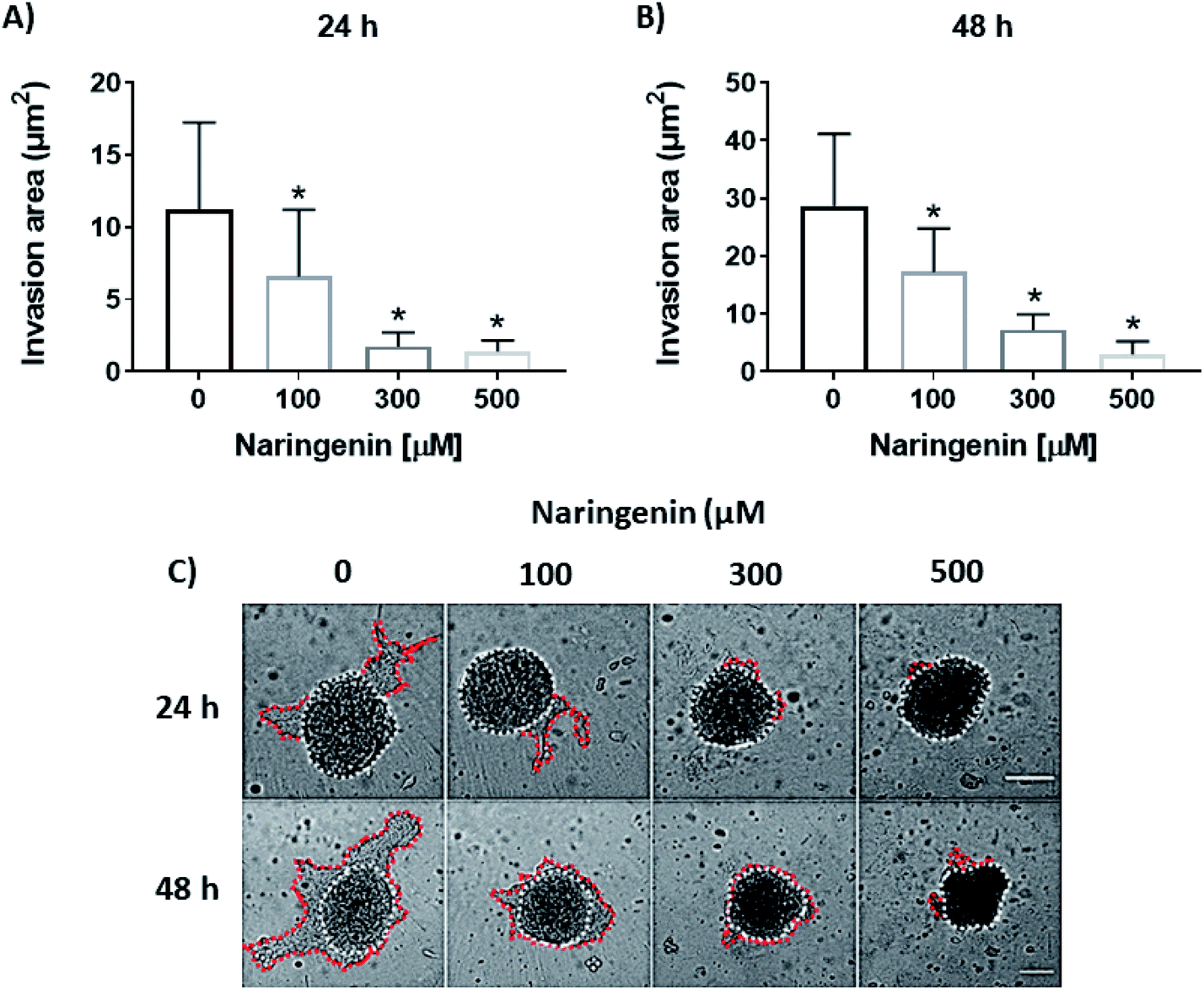 | ||
| Fig. 6 Reduction of cell invasion by naringenin. Area invaded by HeLa cell spheroids, exposed to 1 and 2 days with 100, 300, and 500 μM of naringenin. (A) Relative area of invasion of spheroids at 24 h. p ≤ 0.05 according to the Tukey's mean test, n = 12; (B) relative area of invasion of spheroids at 48 h. p ≤ 0.05 according to the Tukey's mean test, n = 12; (C) microscopy images of cell invasion of spheroids. Scale bar: 100 μm; amplification: 10×. | ||
Naringenin enhances the effect of cisplatin on spheroids
To determine the combined effect of naringenin with cisplatin, combinations of the flavonoid at 500 μM with cisplatin were performed at the previously determined EC25 (16 μM) and EC50 (33 μM). The viability decreased significantly by combining naringenin and cisplatin with EC25 (2856 RLUs), compared to viability of spheroids treated with EC25 alone (4268 RLUs) (Fig. 7A). This combination reduced ATP viability, indicating that the activity of the drug is enhanced at low concentrations. When measuring cytotoxicity through membrane integrity, there was a significant increase in dead cells respect with to total population, confirming that the reduction of ATP by the combination of naringenin and cisplatin EC25 is due to the synergy between the drug and the flavonoid. Similarly, the naringenin-EC50 cisplatin combination followed the same trend in decreasing the cell viability of the spheroid, this being less than the naringenin-EC25 cisplatin combination, as well as the viability observed only with the EC50 of cisplatin (Fig. 7B and C).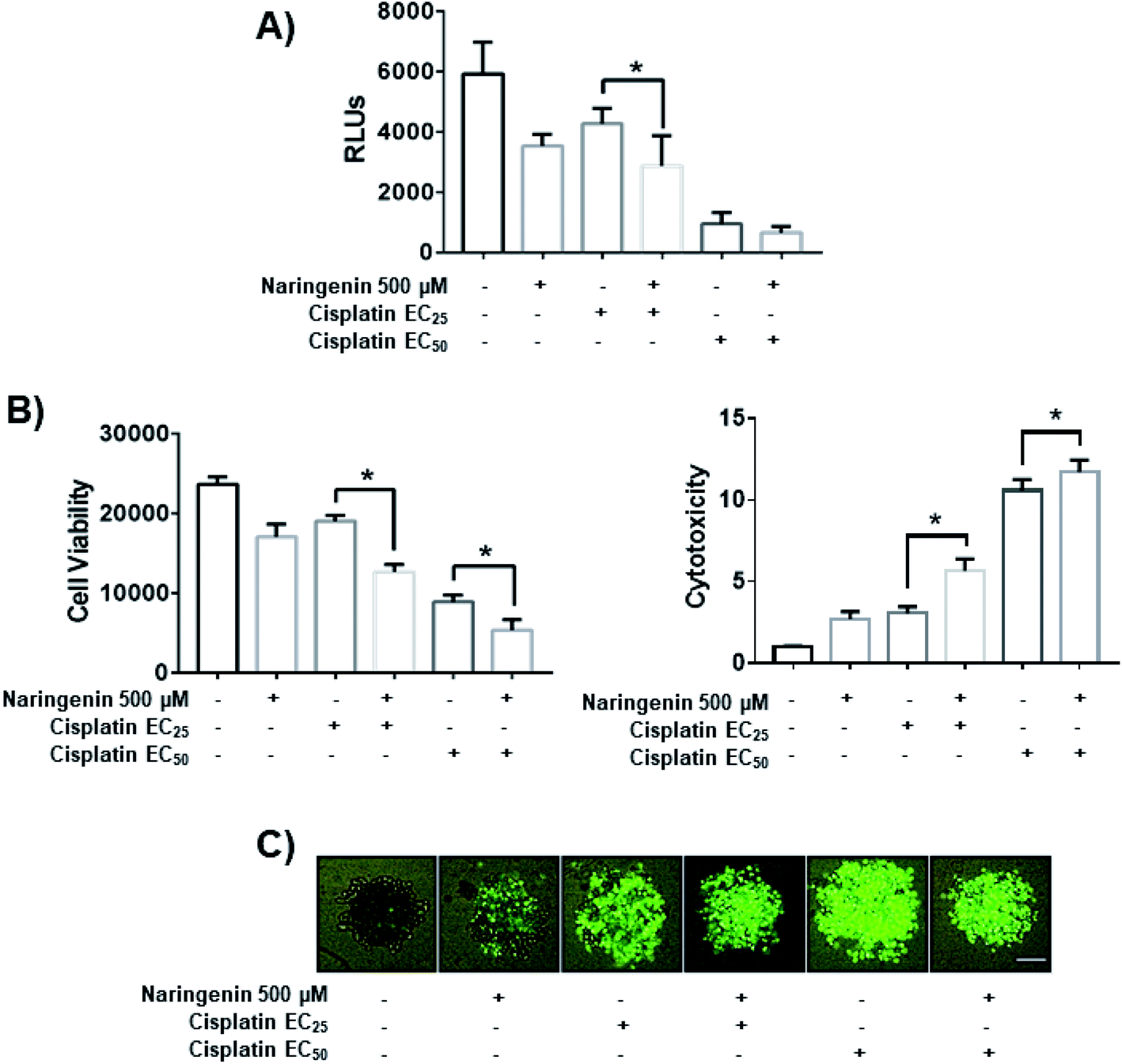 | ||
| Fig. 7 Effect of the combination of naringenin with cisplatin on the viability of HeLa cell spheroids at 24 h. (A) Decreased ATP levels of spheroids; (B) decreased viability and increased cytotoxicity in spheroids, through loss of membrane integrity, by staining with Sytox Green and Hoechst 33342, p ≤ 0.05 according to the Tukey's mean test, n = 12; (C) morphological changes in spheroids and fluorescence of Sytox Green indicating cell death. Scale bar: 100 μm; amplification: 10×. | ||
Activity of caspases 3 and 7 by combination of naringenin and cisplatin in spheroids
In cisplatin-treated spheroids, the activity of effector caspases 3 and 7 increased with both concentrations (16 and 33 μM), being statistically different from naringenin-treated spheroids, which did not present cell death by apoptosis. In contrast, with the combination of naringenin with both EC25 and EC50 of cisplatin, the activity of caspases −3 and −7 was similar to the spheroids of the negative control and with naringenin (Fig. 8). These data corroborate the previous results, where it was observed that naringenin does not cause apoptosis.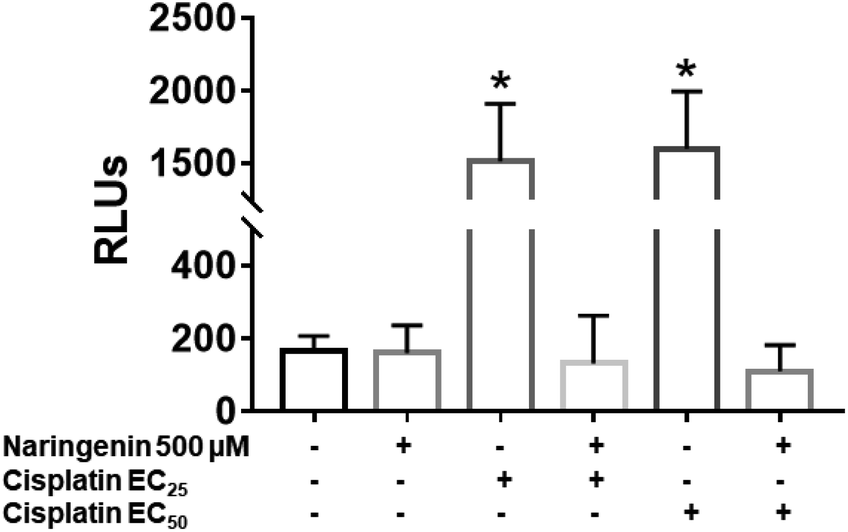 | ||
| Fig. 8 Activity of caspases 3 and 7 in HeLa cell spheroids, due to the combination of 500 μM of naringenin with EC25 and EC50 of cisplatin. RLU, relative fluorescence units. The higher the number of RLUs, the higher caspase activity. p ≤ 0.05 according to Tukey's mean test, n = 12. | ||
Cell invasion of spheroids by the effect of the combination of naringenin and cisplatin
Treatment with EC25 of cisplatin led to cell invasion of the spheroids at 24 h. However, when combining this concentration of cisplatin with 500 μM naringenin, the invasion decreased significantly, being statistically equal to the invasion of the spheroids treated with the EC50 of cisplatin (Fig. 9A). Conversely, when the EC50 was applied alone and in combination with naringenin, the invasion did not show significant changes, being the same as the invasion observed with the combination of EC25 cisplatin–naringenin (Fig. 9B).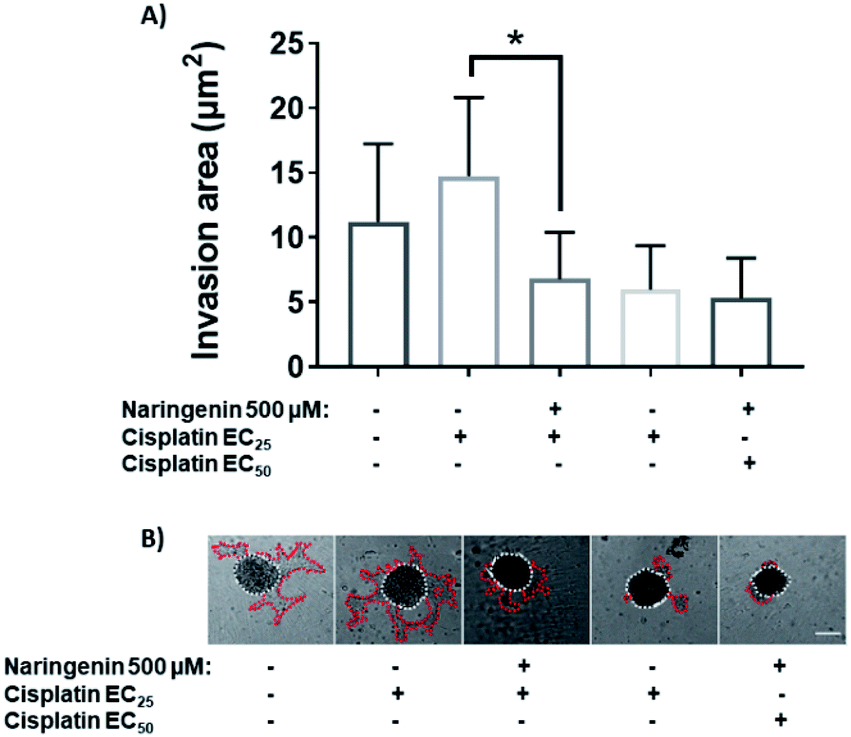 | ||
| Fig. 9 Area invaded by HeLa cell spheroids, exposed to 1 and 2 days with 100, 300, and 500 μM of naringenin. (A) Relative area of invasion of spheroids; (B) microscopy images of cell invasion of spheroids. p ≤ 0.05 according to Tukey's mean test, n = 12. Scale bar: 100 μm; amplification: 10×. | ||
Molecular docking of the effect of naringenin on proteins involved in cell death, cell invasion, and cell cycle
Since the effects mentioned above are controlled by different proteins, through molecular docking, the possible interaction of naringenin with proteins involved in cell death other than apoptosis, such as necroptosis (TNF-R1, RIP3, MLKL) and paraptosis (IGF-R1) was sought. Likewise, with ECM remodeling proteins (MMP2, MMP9, E-cadherin, N-cadherin) and also with proteins of the cell cycle (Cyclin B1 and CDK2) was carried out. Table 2 shows the results of molecular docking.| Protein | Free energy (kcal)/Ki (μM) | Hydrogen bonds | Polar interactions | Hydrophobic interactions | |||
|---|---|---|---|---|---|---|---|
| Atom/Aa | Distance [Å] | Atom/Aa | Distance [Å] | Atom/Aa | Distance [Å] | ||
| TNF-R1 | +9.53/— | — | — | — | — | — | — |
| RIP3 | −6.16/30.73 | O2–Lys158 | 3.42 | O1–Glu71 | 3.13 | C3–Leu75 | 3.79 |
| — | — | O5–Arg96 | 3.72 | C9–Leu75 | 3.54 | ||
| — | — | H3–Arg96 | 3.17 | C13–Leu75 | 3.34 | ||
| — | — | H2–His46 | 3.55 | C15–Leu75 | 3.47 | ||
| — | — | O3–Arg96 | 3.61 | C14–Leu75 | 3.19 | ||
| — | — | H2–Arg96 | 3.17 | C10–Leu75 | 3.55 | ||
| — | — | O4–Arg96 | 3.88 | C4–Leu75 | 3.48 | ||
| — | — | — | — | C5–Leu75 | 3.49 | ||
| — | — | — | — | C7–Leu75 | 3.68 | ||
| — | — | — | — | C1–Leu75 | 3.73 | ||
| — | — | — | — | C6–Leu75 | 3.45 | ||
| — | — | — | — | C8–Leu75 | 3.74 | ||
| — | — | — | — | C11–Leu77 | 3.49 | ||
| — | — | — | — | C12–Leu77 | 3.40 | ||
| MLKL | −4.81/299.59 | — | — | O3–Arg210 | 3.39 | C11–Leu338 | 3.69 |
| — | — | H1–Arg210 | 3.63 | C11–Ala348 | 3.70 | ||
| — | — | O1–Lys230 | 3.43 | C12–Ala348 | 3.54 | ||
| — | — | O4–Glu250 | 3.10 | — | — | ||
| — | — | H2–Glu250 | 2.14 | — | — | ||
| — | — | O1–Asn336 | 3.45 | — | — | ||
| — | — | O2–ASn336 | 3.71 | — | — | ||
| — | — | H3–Glu351 | 2.15 | — | — | ||
| — | — | O5–Glu351 | 2.93 | — | |||
| IGF-R1 | −5.29/132.75 | O4–Ser979 | 2.93 | O1–Lys1003 | 3.52 | C14–Val1033 | 3.39 |
| H2–Ser979 | 2.97 | O2–Asn1110 | 3.42 | C13–Met1049 | 3.89 | ||
| — | — | O3–Asn1110 | 2.72 | C15–Met1049 | 3.33 | ||
| — | — | H1–Asn1110 | 2.21 | C14–Met1049 | 3.31 | ||
| — | — | H1–Asp1123 | 3.85 | C10–Met1049 | 3.88 | ||
| — | — | — | — | C2–Met1112 | 3.27 | ||
| — | — | — | — | C10–Met1112 | 3.67 | ||
| MMP-2 | −8.68/432.76 | — | — | H1–Thr428 | 3.55 | C13–Cys102 | 3.84 |
| — | — | — | — | C15–Cys102 | 3.82 | ||
| — | — | — | C14–Val400 | 3.73 | |||
| — | — | — | C2–Leu420 | 3.61 | |||
| — | — | — | — | C5–Leu420 | 3.47 | ||
| — | — | — | C8–Ala422 | 3.69 | |||
| — | — | — | — | C12–Ala422 | 3.89 | ||
| MMP-9 | −4.56/456.54 | — | — | O4–Asp131 | 3.06 | C11–Met338 | 3.57 |
| — | — | H2–Asp131 | 2.15 | — | — | ||
| — | — | O1–Asp206 | 3.43 | — | — | ||
| — | — | O2–Asp207 | 3.16 | — | — | ||
| — | — | O3–Asp207 | 3.05 | — | — | ||
| — | — | H1–Asp207 | 2.22 | — | — | ||
| E-cadherin | −4.59/432.12 | — | — | O5–Arg68 | 3.15 | C14–Val139 | 3.25 |
| — | — | H3–Arg68 | 3.67 | C8–Ala144 | 3.42 | ||
| — | — | O5–Asp137 | 2.83 | — | — | ||
| — | — | H3–Asp137 | 1.95 | — | — | ||
| N-cadherin | −6.16/30.70 | — | — | O3–Glu89 | 3.53 | C14–Ile4 | 3.69 |
| — | — | H1–Glu89 | 3.56 | C10–Ile4 | 3.43 | ||
| — | — | — | — | C14–Ile4 | 3.89 | ||
| — | — | — | — | C10–Ile24 | 3.69 | ||
| — | — | — | — | C4–Ile92 | 3.51 | ||
| — | — | — | — | C6–Ile92 | 3.46 | ||
| — | — | — | — | C7–Ile92 | 3.61 | ||
| — | — | — | — | C8–Ile92 | 3.33 | ||
| — | — | — | — | C11–Ile92 | 3.79 | ||
| — | — | — | — | C12–Ile92 | 3.72 | ||
| — | — | — | — | C5–Ile92 | 3.78 | ||
| Cyclin B1 | −5.83/53.45 | — | — | O1–Arg68 | 3.42 | C7–Leu17 | 3.81 |
| — | — | O3–Arg135 | 2.51 | C11–Leu17 | 3.57 | ||
| — | — | H1–Arg135 | 3.34 | — | — | ||
| — | — | O4–Asn130 | 3.56 | — | — | ||
| — | — | H2–Asn130 | 3.40 | — | — | ||
| CDK2 | −4.12/955.86 | O1–Thr14 | 2.71 | O5–Tyr15 | 3.67 | — | — |
| O1–Lys129 | 3.07 | H3–His125 | 3.78 | — | — | ||
| — | — | O4–Gln131 | 3.84 | — | — | ||
From the signaling pathway of necroptosis, the interaction of the flavonoid with the TNF-R1, RIP3, and MLKL proteins, which are characteristic of this type of death, was simulated. Although TNF-R1 did not interact with naringenin, RIP3 was activated with a free energy of −6.16 kcal through hydrogen bonds with Lys158, polar interactions with Glu71 and Arg96, and hydrophobic interactions with Leu75. Another key protein is MLKL, the simulation showed the possible polar interactions between naringenin-MLKL with Arg10, Lys230, Glu250, Asn336, and Glu351, while the hydrophobic interactions occurred with Leu338, ala348, and Ala348. By being active, the RIP3 and MLKL proteins form the protein complex that leads to death by necroptosis. Another type of cell death, where caspases are also not involved, is paraptosis. A protein of this type of death is IGF-R1, necessary for the activation of the signaling pathway that leads the cell to paraptosis. The simulation was carried out with this protein, resulting a free energy of −5.29 kcal and with interaction through hydrogen bonds with ser979, polar interactions with Lys1003, Asn1110, and Asp1123, hydrophobic interactions mainly with Val1033, Met1049, and Met1112 (Table 2 and Fig. 10A).
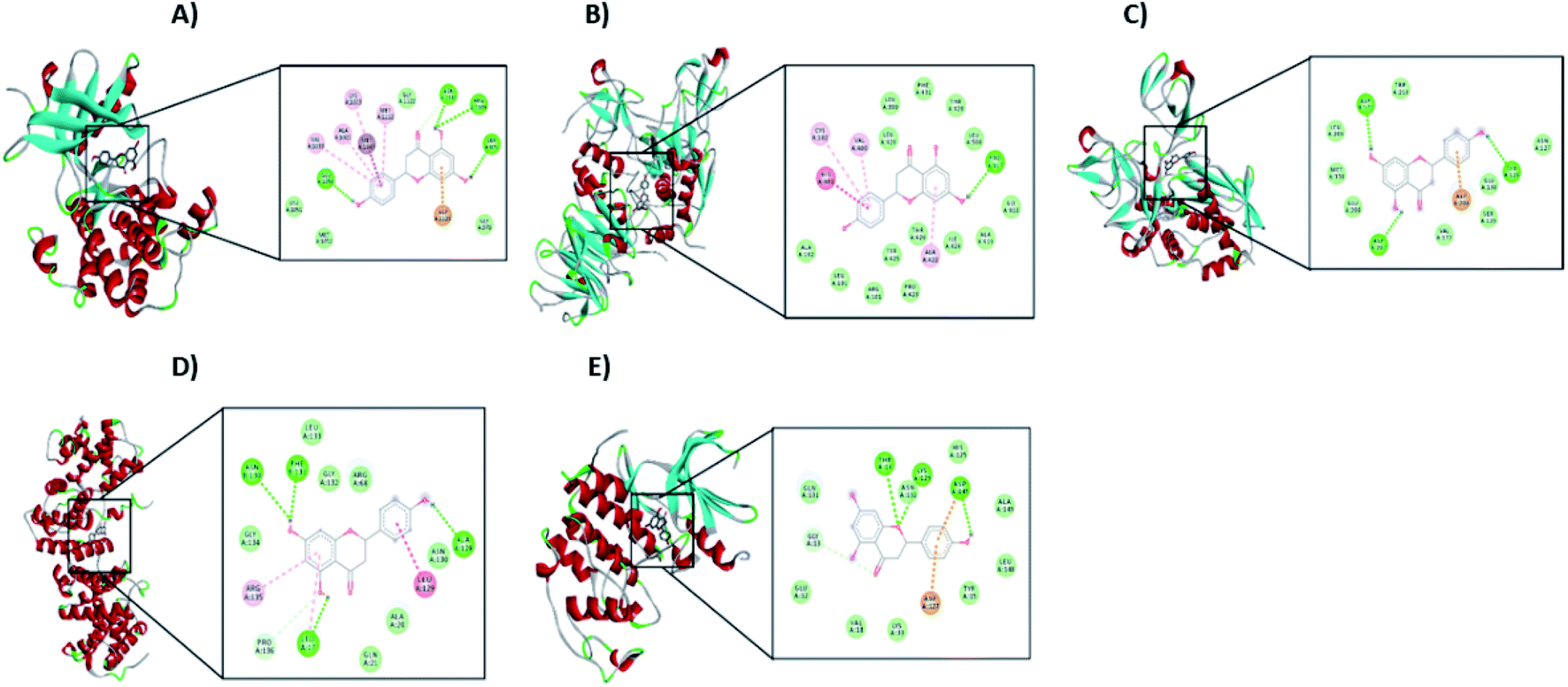 | ||
| Fig. 10 Interaction of naringenin with (A) IGF-R1, (B) MMP2, (C) MMP9, (D) Cyclin B1, and (E) CDK2. In each case, the amino acid residues involved in the flavonoid–protein interaction are shown. | ||
Matrix metalloproteinases (MMP) are proteases responsible for degrading the ECM during the invasion of tumour cells. Simulation with these matrix metalloproteinases was performed between naringenin and MMPs2/9, resulting in a free energy of −8.68 and −4.56, respectively. Naringenin–MMP2 interactions occurred through polar interactions with Thr428 and hydrophobic interactions with Cys102, Val400, Leu420, and Ala422 (Fig. 10B), while in the naringenin–MMP9 simulation, the polar interactions were with Glu89 and the hydrophobic interactions were with residues Ile4, Ile24, and Ile92 (Table 2 and Fig. 10C).
Cell cycle progression and cell division are regulated by cyclins and cyclin dependent kinases (CDKs), which function at specific phases of the cell cycle. Due to the results obtained where the arrest of the cell cycle by naringenin in the G2/M phase was observed, the interaction of naringenin with cyclin B1, which is necessary for DNA replication and mitosis to take place, was simulated. The results showed a free energy of −5.83 kcal (Table 2), with polar interactions in Arg68, Arg136, and Asn130, and hydrophobic interactions with Leu17 (Fig. 10D). Regarding the results where an increase in the G0/G1 phase was observed, the simulation was performed with CDK2, having a free energy of −4.12 kcal (Fig. 10E).
Discussion
In the present study, the results showed the effect of naringenin on the viability of HeLa cells spheroids, and it is shown that cytotoxicity in the spheroid is time- (1, 4, and 7 days) and concentration-dependent (100, 300, and 500 μM), noting that at high concentrations, naringenin decreases the viability of the spheroids significantly and as time progresses, low concentrations of naringenin are required to achieve the desired effect. Naringenin at 10 μM showed an cytotoxic effect in HeLa cells that expressed ERα and Erβ, by activating p38/MAPK leading in turn to activation of caspase-3 and cleavage of the enzyme poly(ADP-ribose) polymerase.20 Finding that the IC50 for naringenin was 100 μM.21 Naringenin and its derivatives with E-oxime (synthesized with hydroxylamine hydrochloride in the presence of potassium hydroxide) have been tested in HeLa and SiHa cell lines, inhibiting 20% growth at 25 μM and 23.9% with 50 μM respectively.22 In HeLa cells the effect of gold nanoparticles naringenin (50 μg mL−1 (183 μM)) is enhanced.23 To date there are a few studies reported on the effect of flavonoids on spheroids of different types of cancer cells. Regarding cervical cancer, apigenin has also been tested on spheroids. Liu et al.24 tested the inhibitory effect of this flavonoid at 10, 20, and 40 μM in the formation of HeLa cell spheroids and by using the MTT assay, they observed the inhibition of viability, leading to the reduction of the formation of spheroids in a time and concentration dependent manner. Similarly, apigenin negatively regulated the expression of the CK2α protein (casein kinase 2α), resulting in the inhibition of the self-renewal of the spheroids of HeLa cervical cancer cells.Loss of cell cycle control leads to proliferation of cancer cells.25 One of the reported mechanisms of naringenin is the regulation of the cell cycle, inhibiting proliferation,24 and therefore, this effect has been reported at different concentrations in cells of prostate cancer,26 breast cancer,27 and cervical cancer.21 In PC3 and LNCaP cells of prostate cancer, Lim et al.26 reported the effect of naringenin alone and in combination with paclitaxel (50 μM and 10 μM, respectively) on cell proliferation, finding that the flavonoid and the drug had a similar effect in PC3 cells, but without synergy, in addition that the effect was independent of the AKT/MAPK transduction pathways. In contrast, LNCaP cells did observe a synergistic effect of naringenin with paclitaxel. In breast cancer, Zhao et al.28 reported the effect of naringenin on cell proliferation in MDA-MB-231 cells in a concentration- and time-dependent manner, arresting the cell cycle in the G1 phase at 6.25 and 12.5 μg mL−1 (22.95 and 91.82 μM, respectively) at 12 h and 24 h, respectively. Likewise, after 48 h, cell deposition began in the sub-G1 phase with all concentrations of naringenin. The effect of naringenin has also been evaluated in different cervical cancer cell lines. Larasati et al.21 observed that naringenin in combination with doxorubicin, 100 and 0.5 μM, respectively, arrested the cell cycle of HeLa cells in the S phase with an increase in the sub-G1 phase. One of the mechanisms of doxorubicin, is the intercalation with the DNA and unlike cisplatin, it causes the inhibition of the enzyme topoisomerase II causing death or arrest of the cell cycle.29 In this study, naringenin inhibits cell division by arresting the cell cycle in the G0/G1 results related to the decrease in cell viability from the first 24 h of treatment, and S phase at 48 h. This clearly indicates that naringenin has different mechanisms of action than the drugs used to fight cervical cancer, such as cisplatin and doxorubicin, since these drugs arrest the cell cycle in G1 and S phases, while naringenin at the concentration used modulates the cell cycle in G0/G1 and S phases.
When treating the spheroids with 500 μM of naringenin, no changes in the activity of caspases 3, 7, 8, and 9 were observed at 12, 24, and 72 h. There are some reports of apoptosis death in HeLa cells, such as the study by Larasati.21 However, our results demonstrated the opposite in a HeLa cell spheroid culture. This may be because the cytotoxic effect of naringenin did not allow the activation of the apoptosis signalling pathway. The morphological changes observed in the spheroids and the permeabilization of the membrane suggested a type of instantaneous death, such as necrosis or necroptosis, which is described as regulated necrosis with morphological characteristics, such as cell swelling, cytoplasmic granulation with rupture of the cell membrane that leads to the release of cell components to the extracellular environment.30 At molecular level, necroptosis occurs with the activation of death receptors, such as TNF-R1, the inhibition of the activity of caspase 8 and the formation of the necrosome, a complex between RIP1/RIP3/MLKL.30,31 Some studies on necroptosis have revealed that MLKL mediates the rupture of the plasma membrane.32 Therefore, the results of docking molecular indicated the interaction of naringenin with RIP3 and MLKL key proteins, suggesting that the type of cell death could be necroptosis rather than apoptosis. Although the results suggest this type of death, it is necessary to carry out experiments that confirm the results obtained by docking molecular.
In metastasis, tumour cells secrete MMP2/9 that degrade the ECM of the basement membrane33 to migrate through the surrounding vasculature and to invade distant tissues and/or organs to form new tumour masses.34 In this study, naringenin reduced HeLa cell spheroid invasion, dependent on concentration and time. The inhibitory effect of naringenin on MMP2/9 has been reported by Chang et al.35 in a monolayer culture of A549 lung cancer cells in a concentration-dependent manner. This same effect has also been observed in glioblastoma cells 8901 and 8401, by negatively regulating cell invasion, and this was reported by Chen et al.36 These data may corroborate the results of molecular docking of the interactions between naringenin and MMP2/9, suggesting the inhibition of the activity of these enzymes and therefore, the decrease in the cellular invasion of the HeLa cell spheroids.
The EMT allows degradation of the ECM and increases cell invasiveness. This occurs mainly due to the loss of adhesion molecules of epithelial cells, such as E-cadherin and the positive regulation of the expression of mesenchymal proteins, such as N-cadherin and vimentin.34,37 The molecular docking results showed the interaction of naringenin with E-cadherin and N-cadherin respectively. Lou et al.37 showed that naringenin decreased the expression of vimentin, N-cadherin, and MMP2/9 in Aspc-1 and panc-1 cells of prostate cancer, while the expression of E-cadherin improved with naringenin treatment, decreasing cell invasion. Although these results were reported in prostate cancer cell lines, they support the results of molecular docking, which suggest the interaction of naringenin with adhesion molecules involved in the EMT process, leading the inhibition of cell invasion.
One of the main disadvantages within the strategy against cervical cancer and other types of cancer is cellular resistance to drugs, in addition to the side effects caused by these. Decreasing the doses of the drug in combination with natural compounds such as flavonoids, can overcome this barrier to eliminate cervical cancer.38 The combination of different anti-cancer agents can generate a synergistic effect, increasing cytotoxicity in tumour cells by decreasing the concentrations of the drugs used.39 The results of this study demonstrate that the combination of naringenin with low concentrations of cisplatin improves the activity of the drug by increasing the cytotoxic effect, close to that of EC50, also modifying the death mechanism, since there is no activity of caspases 3 and 7, which shows that cell death is not due to apoptosis, since the naringenin–cisplatin combination does not activate these caspases, the same as naringenin alone. Once cisplatin interacts with DNA and forms covalent adducts with purine, it induces the cell to apoptosis.40 However, when combined with naringenin, there is no activation of apoptosis and another type of necrotic cell death occurs. This means that, naringenin does not allow activation of apoptosis signalling pathways, although the cytotoxicity results with the naringenin–cisplatin combinations indicate a positive effect, causing a cytostatic state and increasing cell death in the spheroids.
One of the phenomena that was observed with the EC25 of cisplatin, was an increase in cell invasion of the spheroid, even greater than the negative control, this may be because these consequences of cisplatin, cancer cells can activate molecular pathways, such as PI3K/AKT, or inhibit the expression of the tumour suppressor gene Schlafen 11 (SLFN11), increasing markers such as Snail to ensure cell invasion.41 At low concentrations of cisplatin (2 μg mL−1), prostate cancer DU145 cells have been shown to positively regulate EMT, improving invasiveness capacity, by reducing the expression of E-cadherin, while the expression of vimentin, Snail, Slug, and MMP9 increased.42 In this case, the combination of naringenin with cisplatin greatly reduced invasion of the spheroids. The results of molecular coupling suggest that naringenin may inhibit MMP2/9 activity and N-cadherin expression. In this way, the EMT caused by the low concentrations of cisplatin could be reversed, and thus, the invasion of the HeLa cell spheroids could be reduced.
Conclusions
The results demonstrated the anti-tumor effect of naringenin in 3D spheroid cultures of cervical cancer cells, causing cell death, arrest of the cell cycle in the G0/G1 (24 h) and S phase (48 h) and decreased cell invasion. The combination of naringenin with low concentrations of cisplatin, locate this flavonoid as a coadjuvant in the treatment of cervical cancer, by significantly improving the effect of cisplatin at low concentrations.Conflicts of interest
The authors declare that they have no conflicts of interests.Acknowledgements
This article was funded by SIP-2018048; SIP-20195333; SIP-20200734, Lic. Luis Miguel Victoria Ranfla. Presidente del Sindicato Nacional de Trabajadores del ISSSTE. SIP-COFAA-EDI-IPN and SNI-CONACYT. O. P. M. R. is grateful for the PhD scholarship 621723 from CONACYT and BEIFI-IPN.References
- M. Gultekin, P. T. Ramirez, N. Broutet and R. Hutubessy, World Health Organization call for action to eliminate cervical cancer globally, Int. J. Gynecol. Cancer, 2020, 1–2 Search PubMed
.
- P. Olusola, H. N. Banerjee, J. V. Philley and S. Dasgupta, Human Papilloma Virus-Associated Cervical Cancer and Health Disparities, Cells, 2019, 8(6), 622 CrossRef CAS
- D. L. Richardson, Cervical Cancer, in Williams GYNECOLOGY, ed. Hoffman B. L., Schorge J. O., Bradshaw K. D., Halvorson L. M., Schaffer J. I. and Corton M. M., McGraw-Hill Education, 3rd edn, 2016, pp. 657–679 Search PubMed
- M. de la Torre, Neoadjuvant chemotherapy in woman with early or locally advanced cervical cancer, Rep. Pract. Oncol. Radiother., 2018, 23(6), 528–532 CrossRef
- W. Small, M. A. Bacon, A. Bajaj, L. T. Chuang, B. J. Fisher and M. M. Harkenrider, et al., Cervical cancer: a global health crisis, Cancer, 2017, 123(13), 2404–2412 CrossRef
- H. Zhu, H. Luo, W. Zhang, Z. Shen, X. Hu and X. Zhu, Molecular mechanisms of cisplatin resistance in cervical cancer, Drug Des., Dev. Ther., 2016, 10, 1885–1895 CrossRef CAS
- N. Baek, O. W. Seo, J. Lee, J. Hulme and S. S. A. An, Real-time monitoring of cisplatin cytotoxicity on three-dimensional spheroid tumor cells, Drug Des., Dev. Ther., 2016, 10, 2155–2165 CrossRef CAS
- A. M. Florea and D. Büsselberg, Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects, Cancers, 2011, 3, 1351–1371 CrossRef CAS
- Q. Cui, D. H. Yang and Z. S. Chen, Special issue: natural products: anticancer and beyond, Molecules, 2018, 23(6), 11–14 CrossRef
- V. M. Patil and N. Masand, Anticancer Potential of Flavonoids: Chemistry, Biological Activities, and Future Perspectives, Studies in Natural Products Chemistry, Elsevier B.V., 1st edn, 2019, vol. 59. pp. 401–430, DOI:10.1016/B978-0-444-64179-3.00012-8
- P. Venkateswara Rao, S. Kiran, P. Rohini and P. Bhagyasree, Flavonoid: a review on Naringenin, J. Pharmacogn. Phytochem., 2017, 6(5), 2778–2783 Search PubMed
- H. J. Park, Y. J. Choi, J. H. Lee and M. J. Nam, Naringenin causes ASK1-induced apoptosis via reactive oxygen species in human pancreatic cancer cells, Food Chem. Toxicol., 2017, 99, 1–8 CrossRef CAS
- I. Koyuncu, A. Kocyigit, A. Gonel, E. Arslan and M. Durgun, The Protective Effect of Naringenin-Oxime on Cisplatin-Induced Toxicity in Rats, Biochem. Res. Int., 2017, 2017, 9478958 Search PubMed
- N. Rani, S. Bharti, B. Krishnamurthy, J. Bhatia, C. Sharma and M. Amjad Kamal, et al., Pharmacological Properties and Therapeutic Potential of Naringenin: A Citrus Flavonoid of Pharmaceutical Promise, Curr. Pharm. Des., 2016, 22(28), 4341–4359 CrossRef CAS
- Y. Imamura, T. Mukohara, Y. Shimono, Y. Funakoshi, N. Chayahara and M. Toyoda, et al., Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer, Oncol. Rep., 2015, 33, 1838–1843 CrossRef
- C. J. Lovitt, T. B. Shelper and V. M. Avery, Advanced cell culture techniques for cancer drug discovery, Biology, 2014, 3, 345–367 CrossRef
- A. González-Torres, E. G. Bañuelos-Villegas, N. Martínez-Acuña, E. Sulpice, X. Gidrol and L. M. Alvarez-Salas, MYPT1 is targeted by miR-145 inhibiting viability, migration and invasion in 2D and 3D HeLa cultures, Biochem. Biophys. Res. Commun., 2018, 507(1–4), 348–354 CrossRef
- Z. Yuan, D. Chen, X. Chen, H. Yang and Y. Wei, Overexpression of trefoil factor 3 (TFF3) contributes to the malignant progression in cervical cancer cells, Cancer Cell Int., 2017, 17(1), 1–13 CrossRef
- L. A. Anguiano-Sevilla, E. Lugo-Cervantes, C. Ordaz-Pichardo, J. L. Rosas-Trigueros and M. E. Jaramillo-Flores, Apoptosis induction of agave lechuguilla torrey extract on human lung adenocarcinoma cells (SK-LU-1), Int. J. Mol. Sci., 2018, 19, 3765 CrossRef
- P. Totta, F. Acconcia, S. Leone, I. Cardillo and M. Marino, Mechanisms of naringenin-induced apoptotic cascade in cancer cells: Involvement of estrogen receptor α and β signalling, IUBMB Life, 2004, 56(8), 491–499 CrossRef CAS
- L. Larasati, I. Kusharyanti, A. Hermawan, R. A. Susidarti and E. Meiyanto, Naringenin Enhances the Anti-Tumor Effect of Doxorubicin on HeLa Cervical Cancer Cells Through Cytotoxic Activity and Apoptosis Induction, Indonesian Journal of Cancer Chemoprevention, 2011, 325 CrossRef
- A. D. Latif, T. Gonda, M. Vágvölgyi, N. Kúsz, Á. Kulmány and I. Ocsovszki, et al., Synthesis and in vitro anti-tumor activity of naringenin oxime and oxime ether derivatives, Int. J. Mol. Sci., 2019, 20(9), 2184 CrossRef CAS
- N. Krishnakumar, N. Sulfikkarali, N. RajendraPrasad and S. Karthikeyan, Enhanced anticancer activity of naringenin-loaded nanoparticles in human cervical (HeLa) cancer cells, Biomed. Prev. Nutr., 2011, 1(4), 223–231 CrossRef
- J. Liu, X. C. Cao, Q. Xiao and M. F. Quan, Apigenin inhibits HeLa sphere-forming cells through inactivation of casein kinase 2α, Mol. Med. Rep., 2015, 11, 665–669 CrossRef CAS
- Z. Tavsan and H. A. Kayali, Flavonoids showed anticancer effects on the ovarian cancer cells: Involvement of reactive oxygen species, apoptosis, cell cycle and invasion, Biomed. Pharmacother., 2019, 116, 109004 CrossRef CAS
- W. Lim, S. Park, F. W. Bazer and G. Song, Naringenin-Induced Apoptotic Cell Death in Prostate Cancer Cells Is Mediated via the PI3K/AKT and MAPK Signaling Pathways, J. Cell. Biochem., 2017, 118(5), 1118–1131 CrossRef CAS
- J. Ramos, T. Hatkevich, L. Eanes, I. Santos-Sanchez and Y. M. Patel. Naringenin Inhibits Proliferation and Survival of Tamoxifen-Resistant Breast Cancer Cells, in Breast Cancer, ed. Pham P Van, IntechOpen, Rijeka; 2017, DOI:10.5772/66698
- Z. Zhao, G. Jin, Y. Ge and Z. Guo, Naringenin inhibits migration of breast cancer cells via inflammatory and apoptosis cell signaling pathways, Inflammopharmacology, 2019, 0123456789 Search PubMed
- H. M. Alkuraishy, A. I. Al-gareeb and H. A. Al-hussaniy, Doxorubicin-Induced Cardiotoxicity: Molecular Mechanism and Protection by Conventional Drugs and Natural Products, Int J Clin Oncol Cancer Res, 2017, 2, 31–44 Search PubMed
- L. Galluzzi and I. Vitale, Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on, Cell Death, 2018, 486–541 CrossRef
- M. N. Messmer, A. G. Snyder and A. Oberst, Comparing the effects of different cell death programs in tumor progression and immunotherapy, Cell Death Differ., 2019, 26, 115–129 CrossRef
- Y. Zhang, X. Chen, C. Gueydan and J. Han, Plasma membrane changes during programmed cell deaths, Cell Res., 2018, 28(1), 9–21 CrossRef CAS
- A. Albini, Extracellular matrix invasion in metastases and angiogenesis: commentary on the Matrigel “chemoinvasion assay”, Cancer Res., 2016, 76(16), 4595–4597 CrossRef CAS
- H. Amawi, C. R. Ashby, T. Samuel, R. Peraman and A. K. Tiwari, Polyphenolic nutrients in cancer chemoprevention and metastasis: role of the epithelial-to-mesenchymal (EMT) pathway, Nutrients, 2017, 9, 911 CrossRef
- H. L. Chang, Y. M. Chang, S. C. Lai, K. M. Chen, K. C. Wang and T. T. Chiu, et al., Naringenin inhibits migration of lung cancer cells via the inhibition of matrix metalloproteinases-2 and-9, Exp. Ther. Med., 2017, 13(2), 739–744 CrossRef CAS
- Y. Y. Chen, Y. M. Chang, K. Y. Wang, P. N. Chen, Y. C. Hseu and K. M. Chen, et al., Naringenin inhibited migration and invasion of glioblastoma cells through multiple mechanisms, Environ. Toxicol., 2019, 34(3), 233–239 CrossRef CAS
- C. Lou, F. Zhang, M. Yang, J. Zhao, W. Zeng and X. Fang, et al., Naringenin Decreases Invasiveness and Metastasis by Inhibiting TGF-β-Induced Epithelial to Mesenchymal Transition in Pancreatic Cancer Cells, PLoS One, 2012, 7(12), e50956 CrossRef CAS
- M. Salinas-Santander, P. Alvarez-Ortiz, J. Alberto-Ascacio Valdes, R. Rodriguez-Herrera, A. Zugasti-Cruz and R. Rangel-Zertuche, et al., Pharmacology Evaluation of Bioactive Compounds that Regulate Cervical Cancer Cells, in Pharmacognosy – Medicinal Plants, 2019 Search PubMed
- T. N. Aung, Z. Qu, R. D. Kortschak and D. L. Adelson, Understanding the effectiveness of natural compound mixtures in cancer through their molecular mode of action, Int. J. Mol. Sci., 2017, 18, 656 CrossRef
- S. Dasari and P. Bernard Tchounwou, Cisplatin in cancer therapy: molecular mechanisms of action, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS
- M. Ashrafizadeh, A. Zarrabi, K. Hushmandi, M. Kalantari, R. Mohammadinejad and T. Javaheri, et al., Association of the Epithelial–Mesenchymal Transition (EMT) with Cisplatin Resistance, Int. J. Mol. Sci., 2020, 21(11), 4002 CrossRef CAS
- Y. Q. Liu, G. A. Zhang, B. C. Zhang, Y. Wang, Z. Liu and Y. L. Jiao, et al., Short low concentration cisplatin treatment leads to an epithelial mesenchymal transition-like response in du145 prostate cancer cells, Asian Pac. J. Cancer Prev., 2015, 16(3), 1025–1028 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07309a |
| This journal is © The Royal Society of Chemistry 2021 |