
Metal-hydride hydrogen atom transfer (MHAT) reactions in natural product synthesis
Jinghua
Wu
and
Zhiqiang
Ma
 *
*
Key Lab of Functional Molecular Engineering of Guangdong Province, School of Chemistry & Chemical Engineering, South China University of Technology, Wushan Road-381, Guangzhou 510641, People's Republic of China. E-mail: cezqma@scut.edu.cn
First published on 9th October 2021
Abstract
The metal-hydride hydrogen atom transfer (MHAT) reaction plays an important role in the field of natural product synthesis. MHAT, as a powerful method, has been employed by chemists to construct C–C, C–H, and C–heteroatom bonds. In this review, we summarize the recent total synthesis of natural products using MHAT to functionalize olefins with first-row transition metal catalysts such as cobalt, manganese and iron.
 Jinghua Wu | Jinghua Wu was born in Fujian, China. He received his B.Sc. from Beijing University of Chemical Technology in 2016. Currently, Jinghua Wu is a graduate student in the group of Professor Zhiqiang Ma at South China University of Technology, where he focuses on natural product synthesis. |
 Zhiqiang Ma | Zhiqiang Ma obtained his BS in chemistry from Lanzhou University in 2001, and PhD in organic chemistry from Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences with Professor Hongbin Zhai in 2007. In the same year, he joined Prof. Chuo Chen's group at UT Southwestern Medical Center as a postdoctoral fellow. Since then, he has been working on the total synthesis of pyrrole-imidazole natural products. Currently, he is a faculty member in South China University of Technology. His research interests focus on natural product synthesis, target-oriented methodology development and medicinal chemistry. |
1. Introduction
Functionalization of olefins has been an important transformation in synthetic chemistry. In particular, functionalization of unactivated olefins is a difficult task. The metal-hydride hydrogen atom transfer (MHAT) reaction plays an important role in functionalization of olefins.In 1979, Tabushi achieved the hydration of cyclohexene under Mn-catalyzed conditions employing NaBH4 and air.1 In 1989, Mukaiyama reported Co-catalyzed hydration of olefins utilizing alcohol as the solvent and reductant,2 and he subsequently used silicon hydride as the hydrogen source instead of alcohols.3 In the following decades, the hydride atom transfer reaction using silicon hydride as the hydrogen source has been greatly developed, and diverse types of MHAT reactions have been reported. Researchers including Carreira,4 Shenvi,5 Herzon,6 Baran,7 and Boger8 and many others expanded the MHAT reaction. They reported the construction of carbon–carbon, carbon–heteroatom and carbon–hydrogen bonds from olefins, based on the generation of carbon-centered radicals. In light of the development, the application of MHAT reactions in total synthesis of natural products is booming, especially in recent years. In 2016 and 2018, Shenvi reviewed the development of metal-hydride hydrogen atom transfer reactions.9 There were also some related reviews disclosed afterward.10 This review focuses on the natural product synthesis employing the MHAT reaction as the key strategy, and basically covers the examples reported after 2016. We have basically divided the review into four sections based on the type of bond formation, including C–C, C–O, and C–H bonds, and isomerization as shown in Fig. 1. We will summarize the construction of C–C bonds from olefins through MHAT first, including conjugate addition, hydroarylation, and addition to carbonyl or cyano groups. Next, hydration and hydroperoxidation of olefins are discussed. Hydrogenation of olefins and isomerization are discussed last.
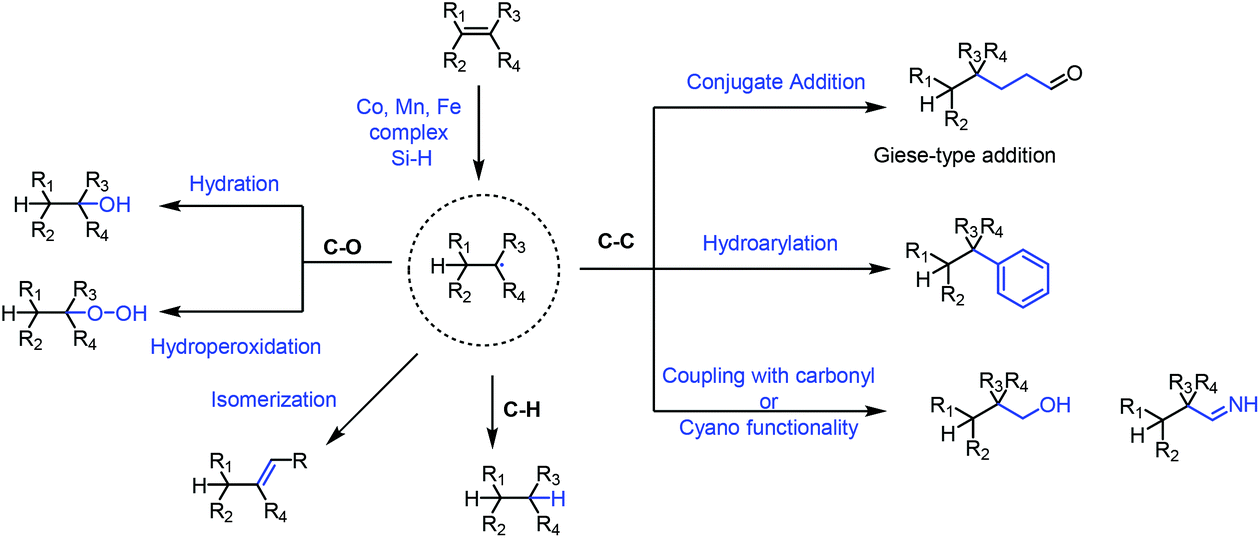 | ||
| Fig. 1 MHAT-based reactions of olefins. | ||
2. C–C bond formation
Based on the proposed mechanism of the MHAT reaction, carbon-centered radicals would be formed from olefins via MHAT, and diverse types of reactions, such as conjugate addition, hydroarylation, and addition to carbonyl or cyano groups, occur when different radical acceptors are present.2.1 Conjugate addition
Addition of nucleophilic radicals to electron-deficient olefins is called the Giese reaction,11 which is a useful approach for C–C bond construction. In 2010, Ishibashi reported iron-catalyzed radical cyclization of 1,6-dienes.12 Subsequently in 2014, Baran developed cross-coupling of unactivated olefins and electron-deficient olefins under mild conditions with Fe(acac)3 or Fe(dibm)3 as catalysts and PhSiH3 as the hydrogen source. Baran's work has greatly expanded the application of MHAT in the construction of C–C bonds. They further realized the cross-coupling reaction of heteroatom-substituted olefins with electron-deficient olefins.7This powerful method was uitilized by Pronin to accomplish the synthesis of emindole SB (13).13 In 2015, Pronin and co-workers employed iron-catalyzed intramolecular conjugate addition and aldol reaction to achieve polycyclization (Scheme 1). Their synthesis commenced with the cyclopentanone derivative 1, which was transformed to 2via a two-step sequence. Continuous oxidation gave 4. The authors then tried to achieve polycyclization of 4 to establish the tricyclic core of emindole SB (13) via Fe-catalyzed HAT reaction. Under the conditions (Fe(acac)3, Ph(i-PrO)SiH2, (CH2Cl)2, and EtOH), the polycyclization occurred through conjugate addition and aldol reaction, giving tricyclic core products 5 and 6 with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1 dr. The authors found that the C7 aldehyde of 4 existed predominantly in its hemiacetal form in alcoholic solvents, and then they proposed that reversible cyclic hemiacetal intermediates would be formed when a hydroxyl group instead of a ketone group is present in the precursor. Indeed, substrate 7 existed as a 1:2 mixture with the corresponding cyclic hemiacetal 8. The authors envisioned that the cyclic hemiacetal of 8 could allow for the desired stereocontrol over the initial radical cyclization. As expected, exposure of 7 to similar polycyclization conditions with lower temperature delivered a mixture of 9 and 10 with a 7.1:1 dr, favoring the desired scaffold 9. Accordingly, the authors speculated that the hemiaminal intermediate resulting from an indole N–H could induce similar stereoselective cyclization. A Fischer indole synthesis from 3 followed by oxidation furnished a dialdehyde, which was transformed to hemiaminal 11 in the presence of NaOH. Upon exposure of hemiaminal 11 to the conditions of Fe(acac)3 and Ph(i-PrO)SiH2, conjugate addition occurred, providing the pentacyclic intermediate 12 in 60% yield over two steps. In this conversion, three stereocenters were established with excellent selectivity. Lastly, a four-step sequence was performed to give emindole SB (13).
:1.1 dr. The authors found that the C7 aldehyde of 4 existed predominantly in its hemiacetal form in alcoholic solvents, and then they proposed that reversible cyclic hemiacetal intermediates would be formed when a hydroxyl group instead of a ketone group is present in the precursor. Indeed, substrate 7 existed as a 1:2 mixture with the corresponding cyclic hemiacetal 8. The authors envisioned that the cyclic hemiacetal of 8 could allow for the desired stereocontrol over the initial radical cyclization. As expected, exposure of 7 to similar polycyclization conditions with lower temperature delivered a mixture of 9 and 10 with a 7.1:1 dr, favoring the desired scaffold 9. Accordingly, the authors speculated that the hemiaminal intermediate resulting from an indole N–H could induce similar stereoselective cyclization. A Fischer indole synthesis from 3 followed by oxidation furnished a dialdehyde, which was transformed to hemiaminal 11 in the presence of NaOH. Upon exposure of hemiaminal 11 to the conditions of Fe(acac)3 and Ph(i-PrO)SiH2, conjugate addition occurred, providing the pentacyclic intermediate 12 in 60% yield over two steps. In this conversion, three stereocenters were established with excellent selectivity. Lastly, a four-step sequence was performed to give emindole SB (13).
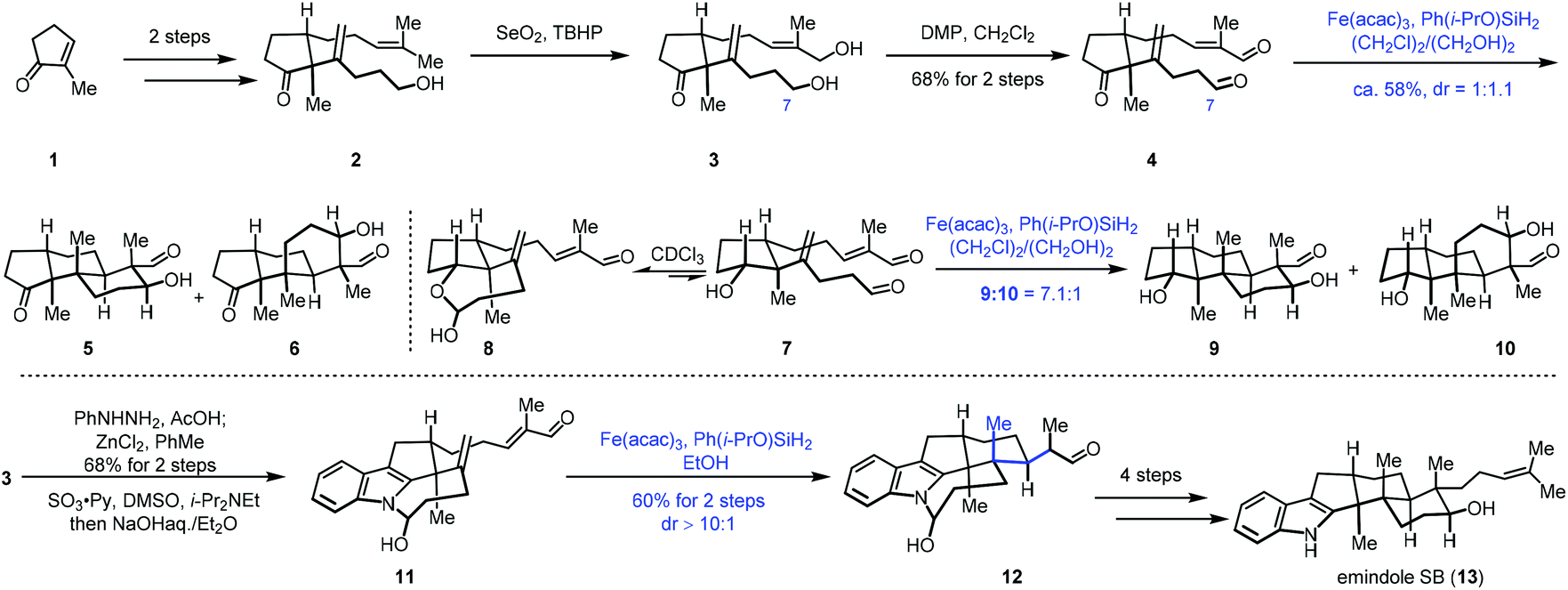 | ||
| Scheme 1 Pronin's synthesis of emindole SB (13). | ||
In 2017, Maimone and co-workers achieved the syntheses of andrastin D (17), preterrenoid (19), terrenoid (20), and terretonin L (21) (Scheme 2).14 Their syntheses started with 14, an intermediate in the synthesis of berkeleyone A.1514 could be oxidized to 15 with PCC, which was further converted into 16via a rearrangement reaction with the intermediacy of carbocations that were generated by the modified Shigehisa conditions (cat. Co-1, PhSiH3, and F1).16 The authors observed that increasing the equivalents of F1 and PhSiH3 from 1.0 to 2.5 could furnish 16 in 90% yield. The authors also tested a variety of Brønsted acids, and it turned out that these acidic conditions failed to induce the expected rearrangement. With 16 in hand, a Krapcho-type demethylation provided andrastin D (17). In 2008, Carreira reported the hydrochlorination of unactivated olefins under Co-catalyzed HAT in the presence of para-toluenesulfonyl chloride (TsCl).4f Compared to conventional hydrochlorination reactions, Carreira's method has good functional group compatibility. Under Carreira's conditions,4f Maimone and co-workers observed that a terretonin core skeleton (18) could be formed preferentially from 15, which was then advanced to preterrenoid (19), terrenoid (20), and terretonin L (21).
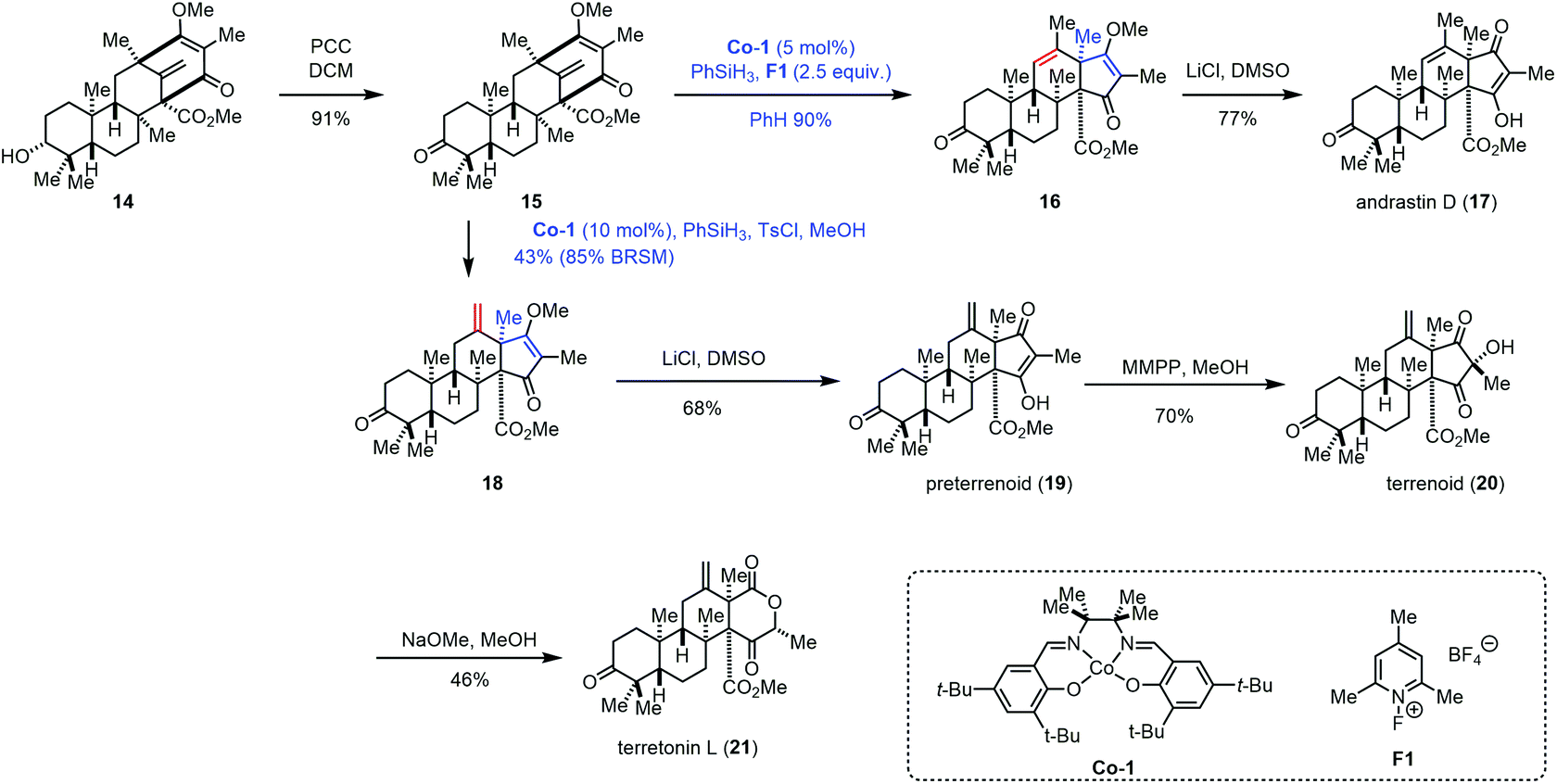 | ||
| Scheme 2 Maimone's synthesis of andrastin and terretonin meroterpenes. | ||
In 2017, Jahn employed Baran's reductive radical cyclizations to achieve the synthesis of 8-oxoasperparaline C (26) (Scheme 3).17 In the event, γ-methoxybutenolide 23 was prepared from 22via a four-step sequence. Treatment of 23 with Fe(acac)3 and PhSiH3 generated the corresponding tertiary radical, which underwent Giese-type radical conjugate addition with an unsaturated γ-butyrolactone moiety to give 25 and the undesired diastereomer 24. The authors proposed that the methoxy group controls the stereoselectivity of cyclization, in which the tertiary radical approached the butenolide moiety from the opposite face. A subsequent two-step sequence led to 8-oxoasperparaline C (26) from 25.
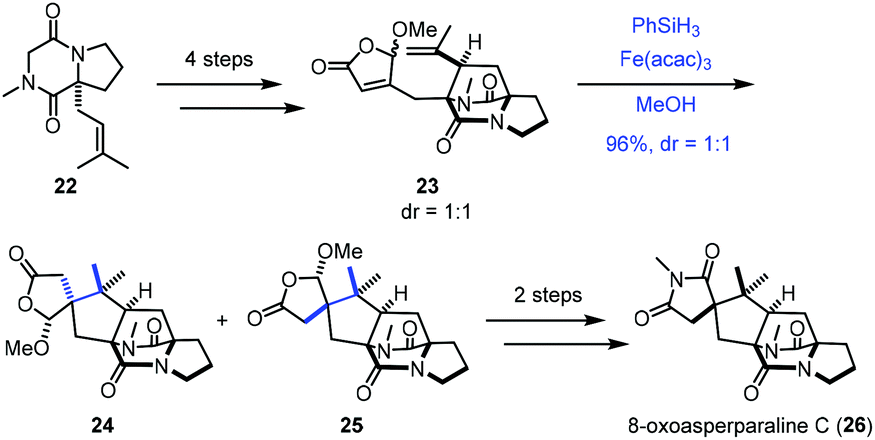 | ||
| Scheme 3 Jahn's synthesis of 8-oxoasperparaline C (26). | ||
In 2018, Li and co-workers employed MHAT-based radical cyclization to complete the synthesis of aplysiasecosterol A (34) (Scheme 4).18 α,β-Unsaturated enone 28 could be obtained from 27via a ten-step sequence. An intramolecular radical cyclization took place under the conditions of V-40 and (TMS)3SiH furnishing the tricyclic scaffold 29. Subsequently, a three-step sequence provided 30. Coupling of 30 and another segment 31 under the Oshima conditions let to an anti-aldol product, which was dehydrated to give 32. Initially, the authors tried the conditions (Fe(acac)3, PhSiH3, EtOH/(CH2OH)2) reported by Baran, and a mixture of four cyclization products (33a–33d) was obtained in 60% yield, in which the desired product 33a was obtained in just 25% yield due to the poor diastereoselectivity (33a:33b:33c:33d = 1:0.33:0.47:0.58). Switching the silicon reagent to the more reactive Ph(i-PrO)SiH2 used by Shenvi improved the diastereoselectivity (33a:(33b + 33c + 33d) = ca. 1:1). The authors then screened other iron complexes. They observed that the more bulky 1,3-diketone ligands like diisobutyrylmethanate (dibm) and dipivaloylmethanate (dpm) gave better stereoselectivity (33a:(33b + 33c + 33d) = ca. 1.7–2.5:1). Finally, under the optimal conditions (Fe(dpm)3, Ph(i-PrO)SiH2, and EtOH/DCE/(CH2OH)2), 33a was prepared in 56% yield. Aplysiasecosterol A (34) was obtained from 33a in two steps including acetonide hydrolysis and debenzylation.
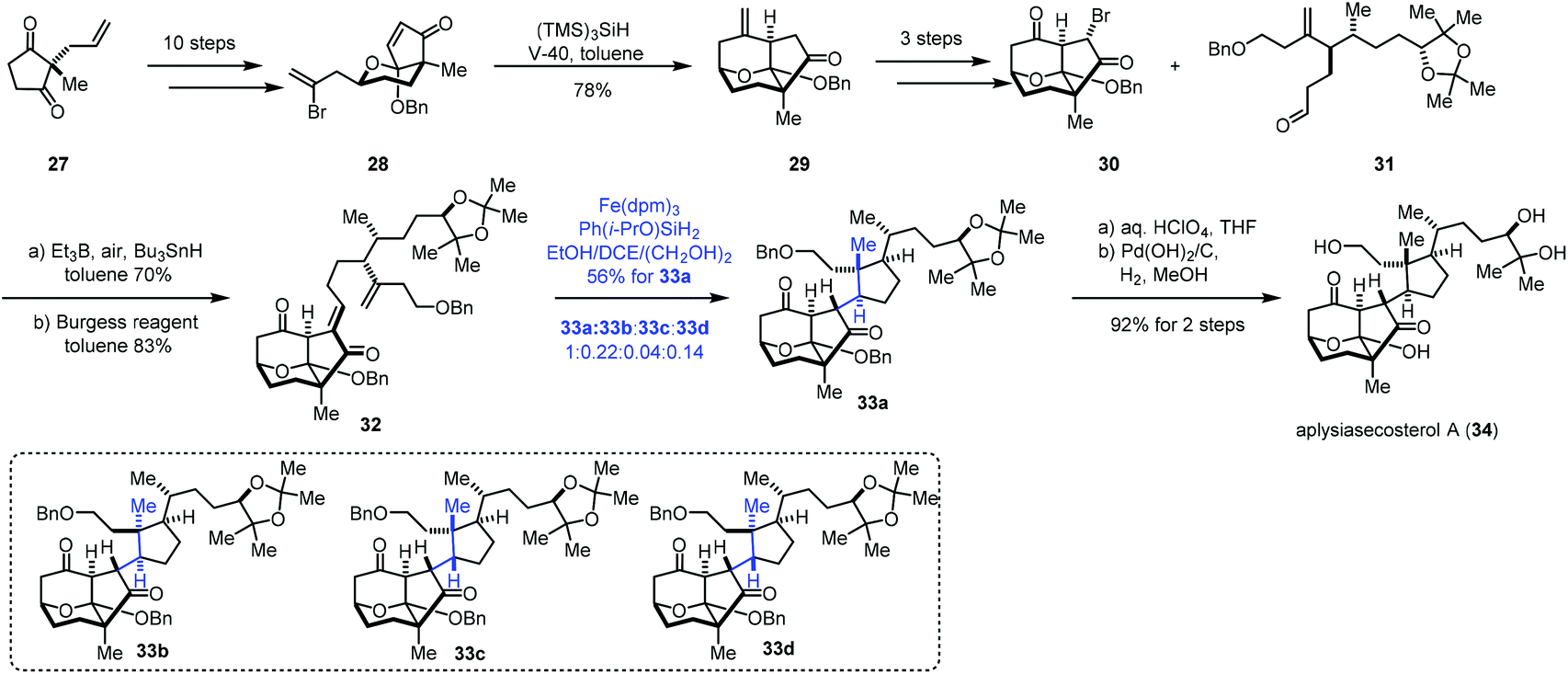 | ||
| Scheme 4 Li's synthesis of aplysiasecosterol A (34). | ||
In 2019, Snyder reported the synthesis of conidiogenol (37) by a quaternary-centre-guided synthetic strategy (Scheme 5).19 They employed radical conjugate addition initiated by iron-catalyzed HAT to furnish the bicyclic ketone 36 as a single diastereomer.
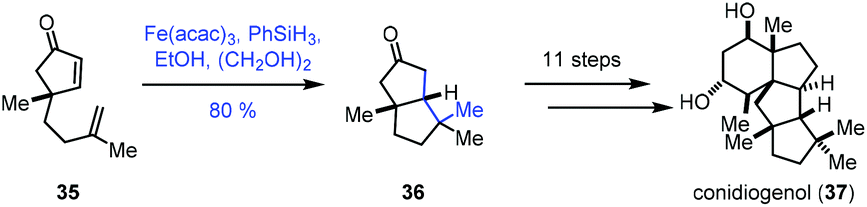 | ||
| Scheme 5 Snyder's synthesis of conidiogenol (37). | ||
In 2020, Li and co-workers employed radical cyclization via iron-catalyzed HAT to accomplish the synthesis of (−)-daphnezomines A (44) and B (45) (Scheme 6).20 In practice, carvone was transformed to 39via a five-step sequence. Upon exposure of 39 to 9-BBN, followed by Suzuki–Miyaura coupling with 40, desired 41 was obtained. Treatment of 41 with a six-step sequence gave 42. To build the core skeleton structure, they tried ene cyclization and anionic cyclization, which turned out to be unfruitful. They finally resorted to MHAT-based radical cyclization. Under the conditions developed by Baran (Fe(acac)3, PhSiH3, and EtOH), the trans-fused adduct 43 was afforded in 16% yield as a single product. The authors proposed that the C10–H of 43 was delivered from the ammonium proton of 42via a 1,5-proton transfer, which accounts for the trans-fused ring junction. Further screening of the conditions revealed that the desired cyclization product 43 could be prepared in 32% yield under the conditions of Fe(acac)3 and Ph(i-PrO)SiH2 in THF/EtOH. The fact that the radical pathway worked in this case is perhaps a testament to its insensitivity to steric crowding.7b Lastly, in the presence of TFA, 43 was transformed to daphnezomines A (44) and B (45).
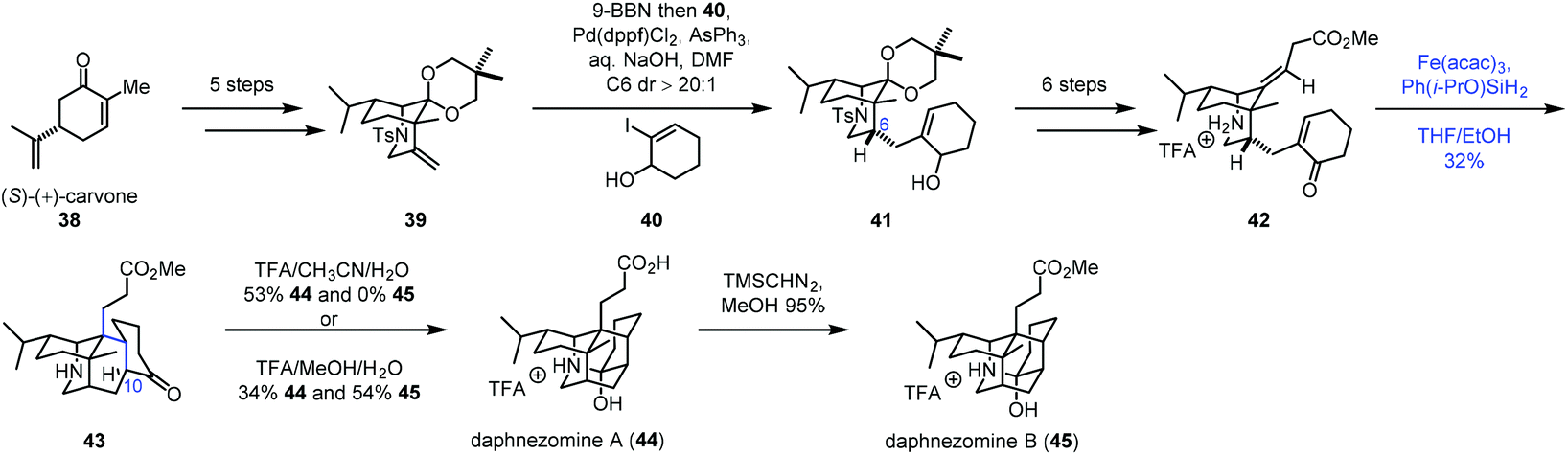 | ||
| Scheme 6 Li's synthesis of (−)-daphnezomines A (44) and B (45). | ||
In 2020, Boger and co-workers disclosed the synthesis of (−)-pseudocopsinine (50) and (−)-minovincinine (53) (Scheme 7).21 They employed the diene 48 as the common intermediate, from which different MHAT conditions were adopted to approach the target molecules. A sequential intramolecular [4 + 2] cycloaddition and [3 + 2] cycloaddition of 46 led to 47, which was followed by functional group transformations to give the key intermediate 48. Treatment of 48 with Fe(acac)3 and phenylsilane provided 49 and its undesired epimer in 80% yield with good diastereoselectivity (3:1), favoring 49. The authors observed that the yield of the cyclization increased with increasing Fe(acac)3 catalyst (1.5 vs. 0.5 equiv.) and time (2 vs. 16 h) and using a less reactive silicon reductant (PhSiH3vs. Ph(i-PrO)SiH2). (−)-Pseudocopsinine (50) was then obtained from 49 under conditions of Pt/C and H2. Meanwhile, treating 48 with the Co catalyst Co-2 furnished a pair of hydration products, the desired diastereomer 52 and its epimer in a nearly 1:1 ratio. Other attempted metal complexes such as Mn(acac)3, Fe2(ox)3, FePc, Fe(NO3)3, Fe2(SO4)3, Fe(acac)3, Fe(dpm)3 and Co(acac)2 all gave inferior results. (−)-Minovincinine (53) was formed after deprotection of 52.
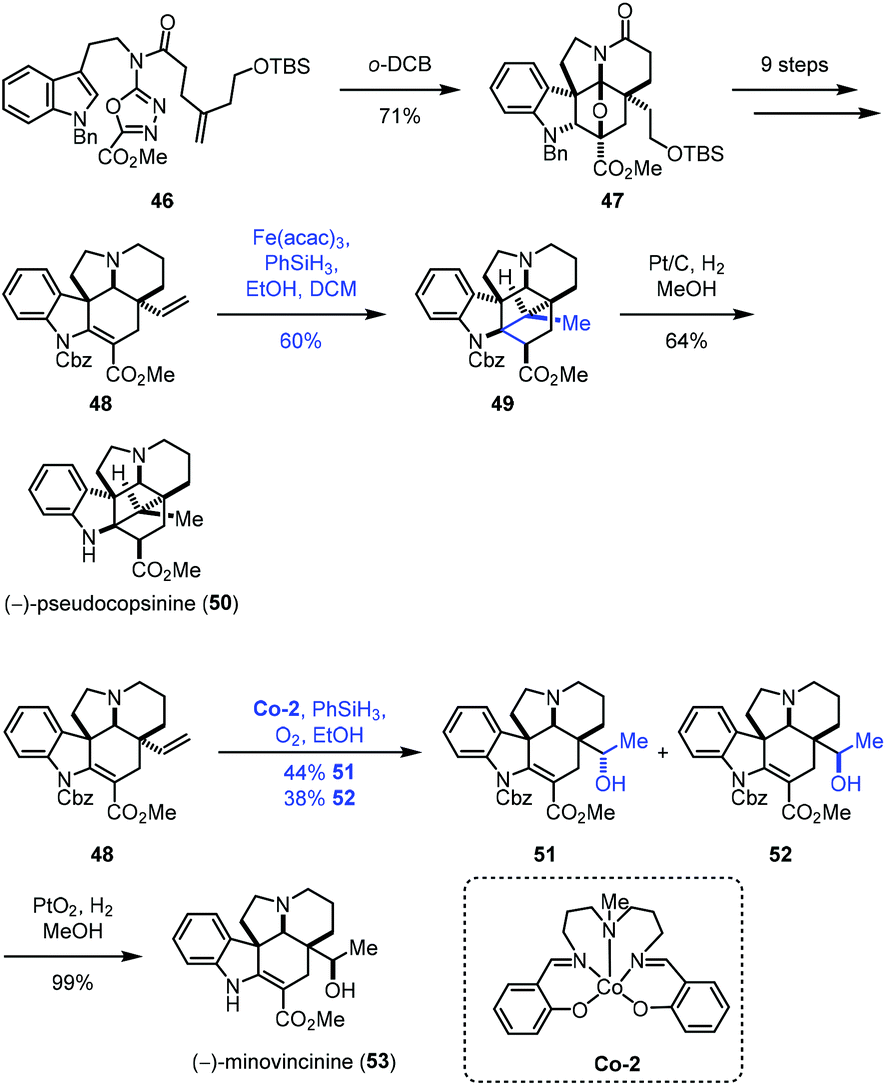 | ||
| Scheme 7 Boger's synthesis of (−)-pseudocopsinine (50) and (−)-minovincinine (53). | ||
In 2021, Ma and co-workers achieved the synthesis of dankasterones A (65) and B (66) and periconiastone A (67) (Scheme 8).22 They employed Co-catalyzed HAT-based radical cyclization to furnish a [6,6,6,5] tetracyclic core. Fragment 55 could be prepared from 54via a three-step sequence. In parallel, fragment 57 was afforded from 56 in six steps. 55 and 57 were coupled through halogen lithium exchange and nucleophilic addition, which was followed by oxidation to give enone 58. Exposure of the cis-fused C/D ring substrate 58 to Fe-catalyzed HAT conditions (Fe(acac)3, PhSiH3, EtOH) gave a pair of diastereomers 59 and 60 (dr = 1:2.9) via Giese-type radical conjugate addition, favoring undesired 60. Screening of different Fe(III) catalysts, silane reagents, additives, solvents and temperatures showed that the optimal conditions (Fe(dpm)3, Ph(i-PrO)SiH2, Na2HPO4, EtOH, and CF3Ph) resulted in better diastereoselectivity (dr = 1:1). Since the yield of the desired cyclization product 59 from a Giese type reaction mediated by Fe was still low, the authors extended the catalyst to Mn and Co. While the Mn-catalyzed systems used were messy, subjecting 58 to Co-3-catalyzed HAT conditions with an N-fluoropyridinium oxidant afforded the cycloisomerization product 61 in 23–27% yield. Screening of oxidants revealed that PhI(OAc)2 was optimal, delivering 61 in 46% yield. In this conversion, PhI(OAc)2 might serve as the oxidant to oxidize Co(II) to Co(III) and facilitate the dehydrogenation. It is noteworthy that, in contrast to the cis-fused C/D ring substrate 58, they found that the trans-fused C/D ring substrate 68 under Fe-catalyzed HAT conditions just gave the bis-reduced product 69 without cyclization. Enone 62 was then furnished from 61 in eight steps. Through C–H oxidation and radical rearrangement, 63 was afforded. Lastly, deacetylation, oxidation and Julia–Kocienski olefination gave dankasterone B (66), which could be transformed into dankasterone A (65) and periconiastone A (67).
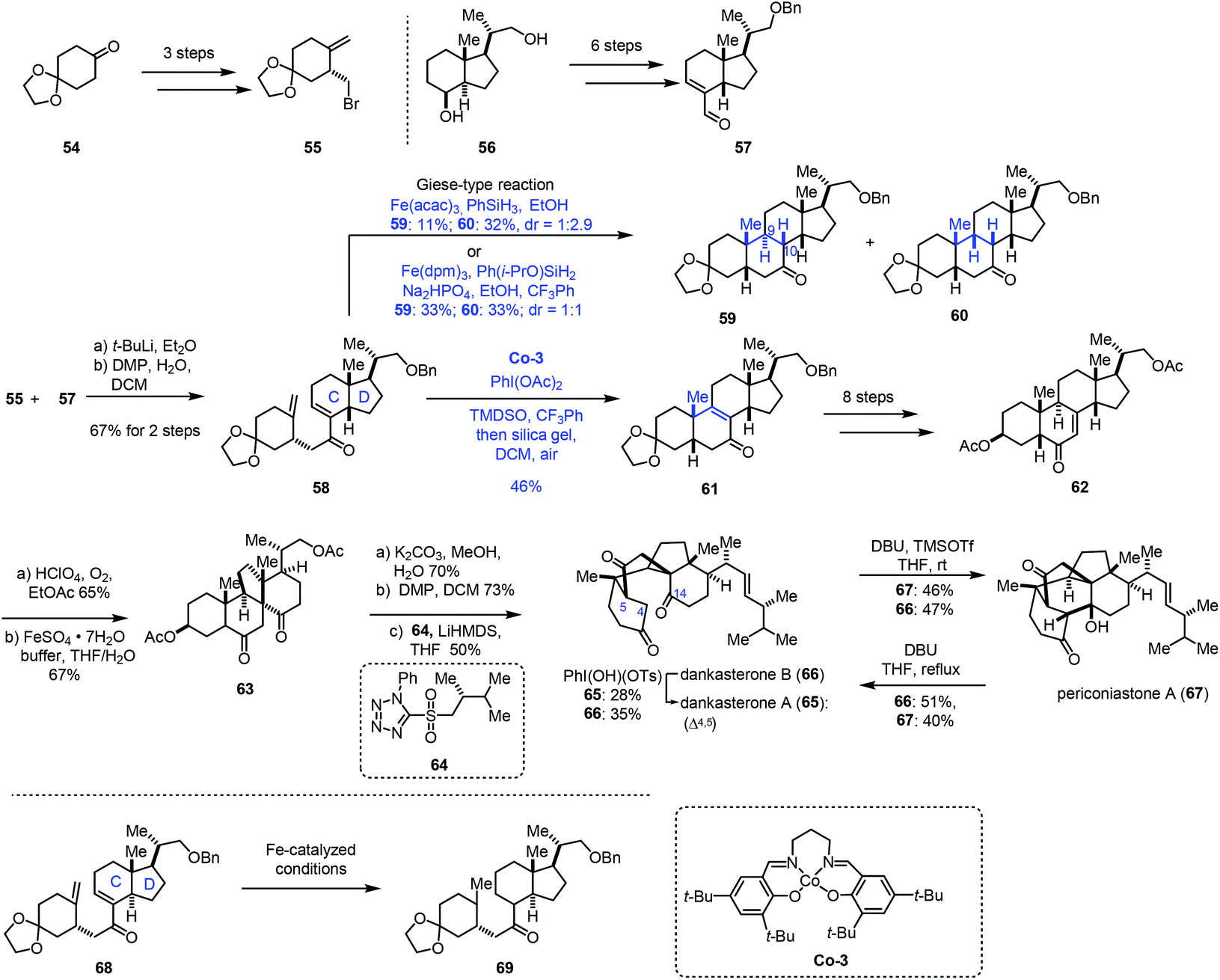 | ||
| Scheme 8 Ma's synthesis of dankasterones A (65) and B (66) and periconiastone A (67). | ||
In their synthetic studies towards wortmannin, Zhou and co-workers used HAT-based radical cyclization to install the tetracyclic core of wortmannin with an Fe catalyst (Scheme 9).23 Their findings also demonstrated that a cis-fused C/D ring was the key to the desired cyclization since they observed that the substrate containing a trans-fused C/D ring failed to produce the desired product under the same conditions.
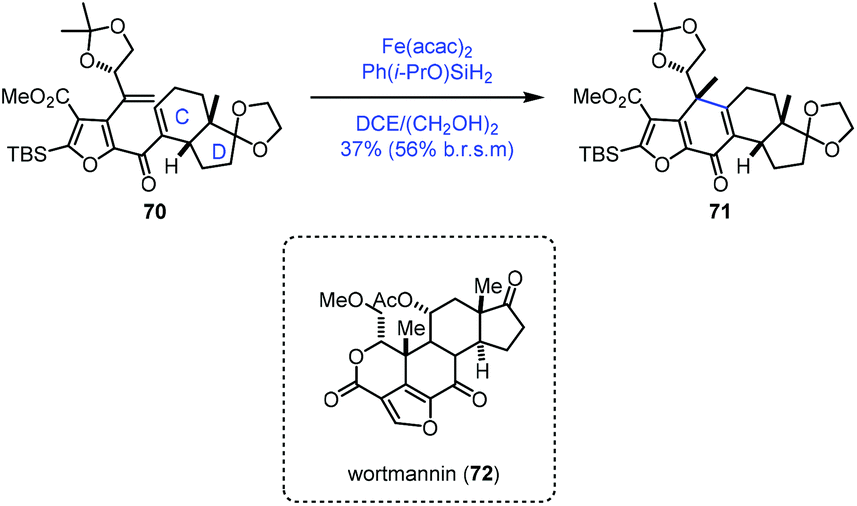 | ||
| Scheme 9 Zhou's synthesis of the tetracyclic core of wortmannin. | ||
In 2017, Liu and co-workers accomplished the synthesis of hispidanin A (82),24,25 in which they creatively employed iron-catalyzed radical polyene cyclization to furnish trans-decalin (Scheme 10). Compound 74 could be provided from 73 in four steps. Coupling 74 with Grignard reagent 75 in the presence of FeCl3 afforded 76. Oxidative operation of furan of 76 and a subsequent rearrangement provided the cyclization precursor 77. Under the conditions of Co(acac)2 or Mn(dpm)3 as the catalyst, no desired product was detected probably because of the failure of radical initiation at the terminal alkene. Switching the catalyst to Fe(acac)3 resulted in the desired radical polyene cyclization, giving the tricyclic compound 78 with 45% yield over 2 steps, along with an inseparable mixture of another three diastereomers in 19% combined yield. In this conversion, the radical initiated at the gem-disubstituted alkene went through a stereoselective cascade radical polycyclization process. As a result, the desired trans-decalin architecture bearing four contiguous stereogenic centers was obtained as the major product. In comparison, Pattenden's work26 showed that the cis-isomer was isolated as the major product when an acyl radical initiated the polyene process with a similar substrate. After nine steps of transformations from 78, 79 was obtained. At the same time, the dienophile fragment 80 was constructed using Yamamoto's cationic polyene cyclization. Next, a Diels–Alder cycloaddition between diene 79 and dienophile 80 occurred, furnishing 81. Reduction with NaBH4 followed by acetylation gave hispidanin A (82).
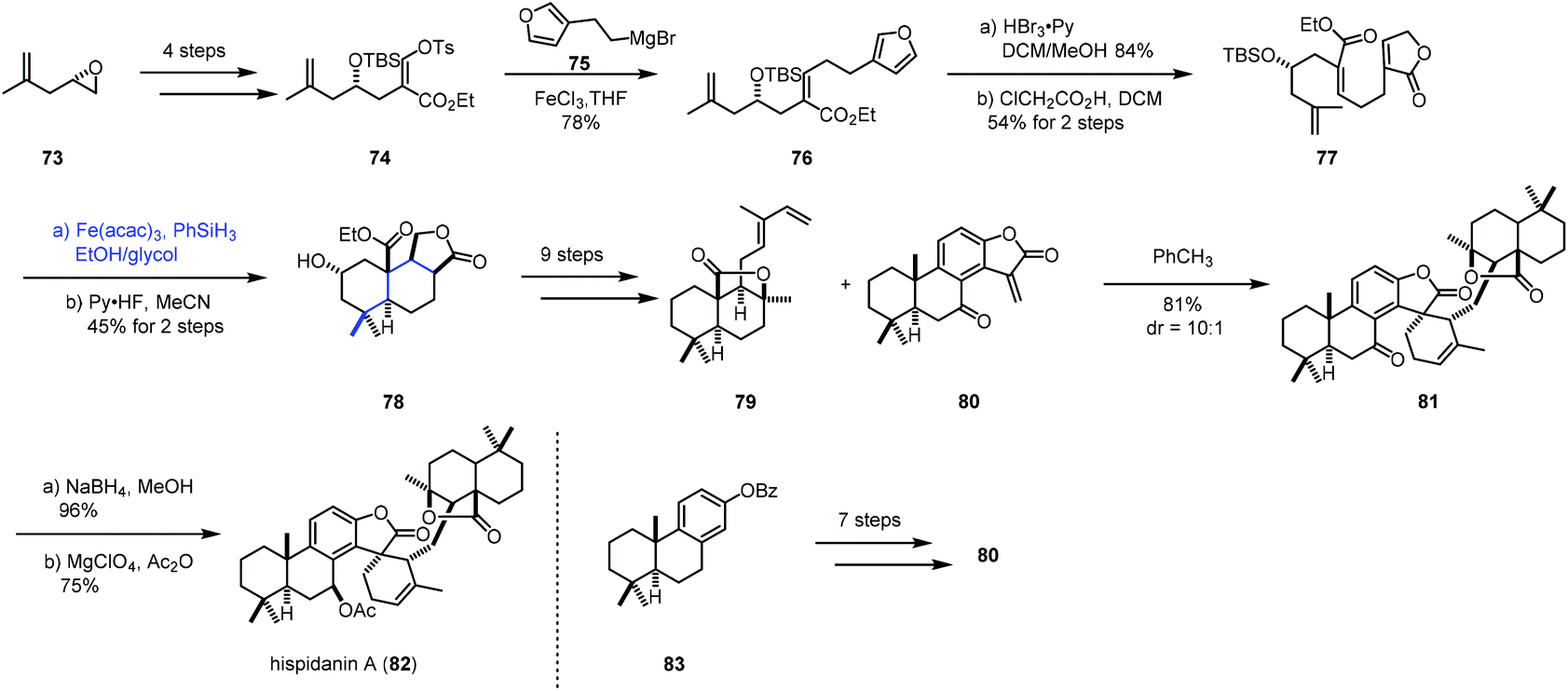 | ||
| Scheme 10 Liu's synthesis of hispidanin A (82). | ||
Very recently, Ding and co-workers communicated the synthesis of (+)-18-benzoyldavisinol (92) featuring a HAT-initiated transannular redox radical cyclization (Scheme 11).27 Commercially available cyclohexenone 84 could be advanced to 85 in six steps. An oxidative dearomatization-induced-Diels–Alder cycloaddition furnished 86. Additional transformations gave 87. Initially, the authors expected to achieve reductive radical cyclization with an epoxide precursor (epoxidation of C9–C11 double bonds); however, the epoxidation of 87 was difficult. As such, the authors resorted to MHAT-based radical cyclization to create the core skeleton. After using the Fe catalysts, the expected cyclization products were not obtained.
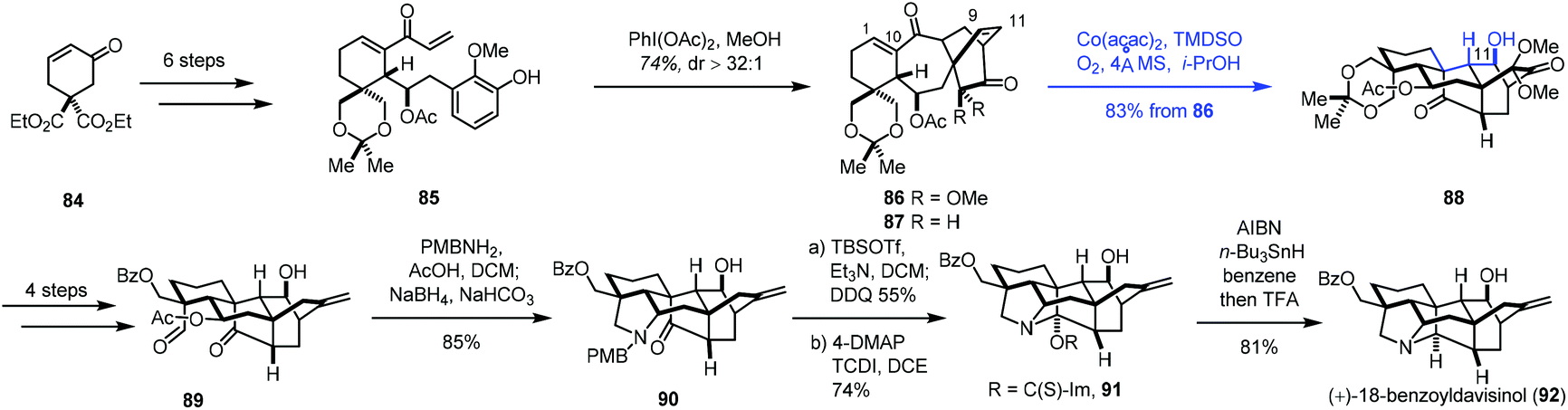 | ||
| Scheme 11 Ding's synthesis of (+)-18-benzoyldavisinol (92). | ||
They observed a hydration reaction occurring at the C1–C10 olefin of 86 under Mn(acac)2 conditions, giving the corresponding alcohol at the C10 position. Encouraged by the results, the authors began to investigate Co catalysts. They found that the expected radical cyclization reaction took place from 86 under the conditions (Co(acac)2, TMDSO, i-PrOH, O2, 4 Å MS), giving 88 in 83% yield. The reaction is remarkable as two highly strained rings and three contiguous stereogenic centers were created in the cascade process. With 88 in hand, a four-step sequence and a reductive amination reaction furnished 90. (+)-18-Benzoyldavisinol (92) was then prepared via hemiaminal formation and ensuing deoxygenation.
In 2019, Pronin and co-workers developed a new intermolecular annulation method between α,β- and γ,δ-unsaturated carbonyl compounds to furnish six-membered carbocycles through MHAT-initiated radical-polar crossover reaction. By taking advantage of the new method, they completed the synthesis of forskolin (102) (Scheme 12).28 A radical tandem reaction between γ,δ-unsaturated aldehyde 93 and enone 94 gave cyclization products 95 and desired 96 as a pair of diastereomers (dr = 1:1). Although the diastereoselectivity was low, the reaction was scalable and allowed for the production of multigram quantities of 96. In addition, the undesired epimer 95 could be converted to 96 after an oxidation and reduction process in 87% yield. Treatment of 96 with a two-step sequence including protection and retro-Diels–Alder reaction, followed by nucleophilic addition and epimerization, gave 99. After five steps, 99 could be transformed to 100. Forskolin (102) was then afforded through conjugate addition, deprotection and acetylation.
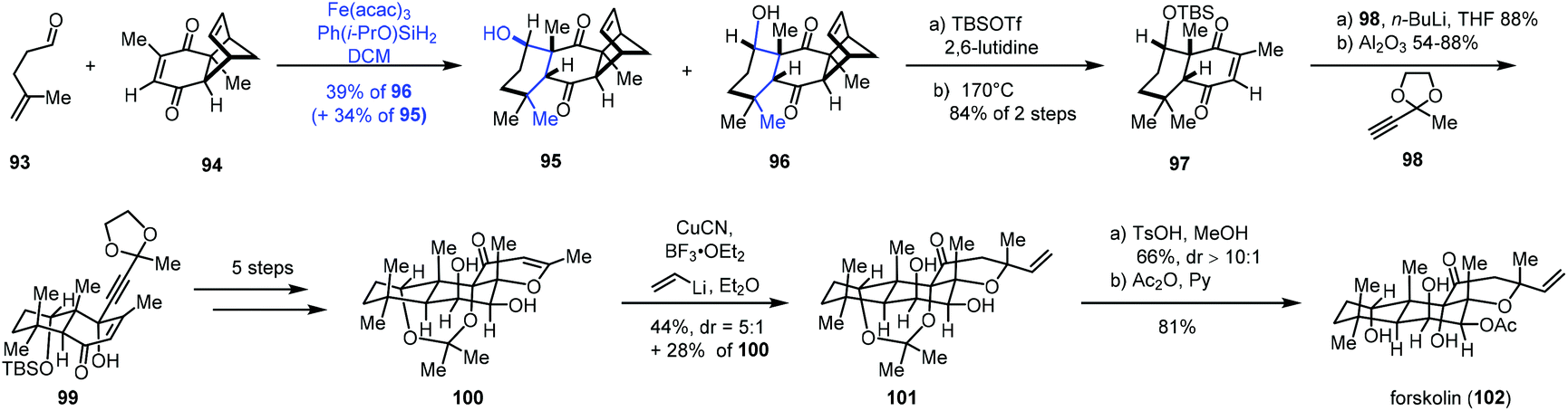 | ||
| Scheme 12 Pronin's synthesis of forskolin (102). | ||
2.2 Hydroarylation
In addition to electron-deficient olefins corresponding to conjugate addition, aryl groups could be used as the terminator in MHAT reactions. We will systematically introduce applications of hydroarylations of unactivated olefins triggered through MHAT-based reactions in this section. Traditional methods such as Friedel–Crafts alkylation usually require vigorous reaction conditions to activate olefins.29 There are also many reports showing transition metal-mediated addition of aromatic C–H bonds across olefins with high regio- and chemoselectivity.30 While metal catalysts usually involve expensive and toxic metals, compared to the above methods, MHAT-based hydroarylations would serve as an alternative to hydroarylation of unactivated olefins.In 2019, Gao and co-workers completed the total synthesis of viridin and viridiol,31 in which they employed Co-catalyzed hydroarylation reaction to close the C-ring (Scheme 13). Their synthesis commenced with 104, which was prepared from 103via a four-step sequence. Treatment of 104 with DMHH and i-PrMgBr afforded Weinreb amide, which was attacked by aryl lithium to furnish 105. Under conditions of Co-4 and PhSiH3, the intramolecular radical hydroarylation occurred to form the tetracyclic core 106 with excellent diastereoselectivity. The authors rationalized the stereochemistry of MHAT cyclization through analysis of the radical intermediates T1 and T2. There is a steric effect between the OTBS and the methyl in T1 and no such effect in T2. Therefore, the intermediate T2 is a favorable transition state to generate the tetracyclic core 106. Sequential deprotection and oxidation delivered 107. Five additional steps were carried out to give viridin (108). Viridin (108) could be converted to viridiol (109) by selective reduction.
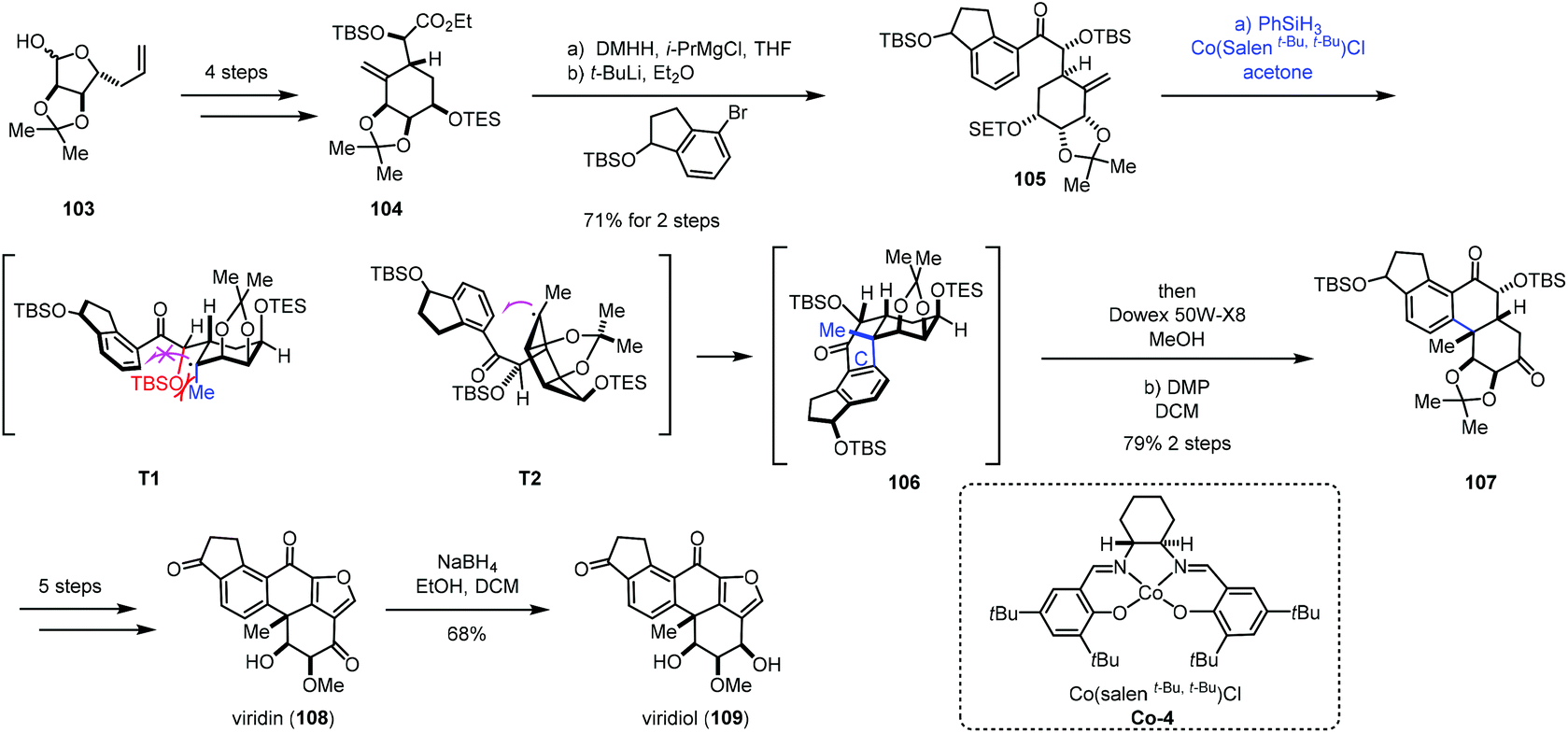 | ||
| Scheme 13 Gao's synthesis of viridin (108) and viridiol (109). | ||
Recently, Gao and co-workers accomplished the synthesis of norzoanthamine using the same hydroarylation strategy to create the tetracyclic core (Scheme 14).32 A carvone derivative could be converted into 111 in nine steps. Ueno–Stork radical cyclization furnished 112 through radical conjugate addition. 112 went through a three-step sequence to deliver aldehyde 113, which was reacted with Grignard reagent 114, followed by acetylation to afford compound 115. Treatment of 115 with Co-catalyzed hydroarylation conditions reported by Shenvi gave the tetracyclic core 116 after deprotection of TBS. Subsequent Birch reduction and acetylation led to 117. The following task was hydrogenation of the tetra-substituted olefin. The authors proposed that the desired product with the trans–anti–trans-fused perhydrophenanthrene A–B–C ring was more thermodynamically stable than other relative configurations, so the anti-addition of hydrogen was expected. Subjecting 117 to Shenvi's hydrogenation conditions (Mn(dpm)3 and PhSiH3) afforded 118 with a trans-fused A–B ring with good diastereoselectivity but low yield.
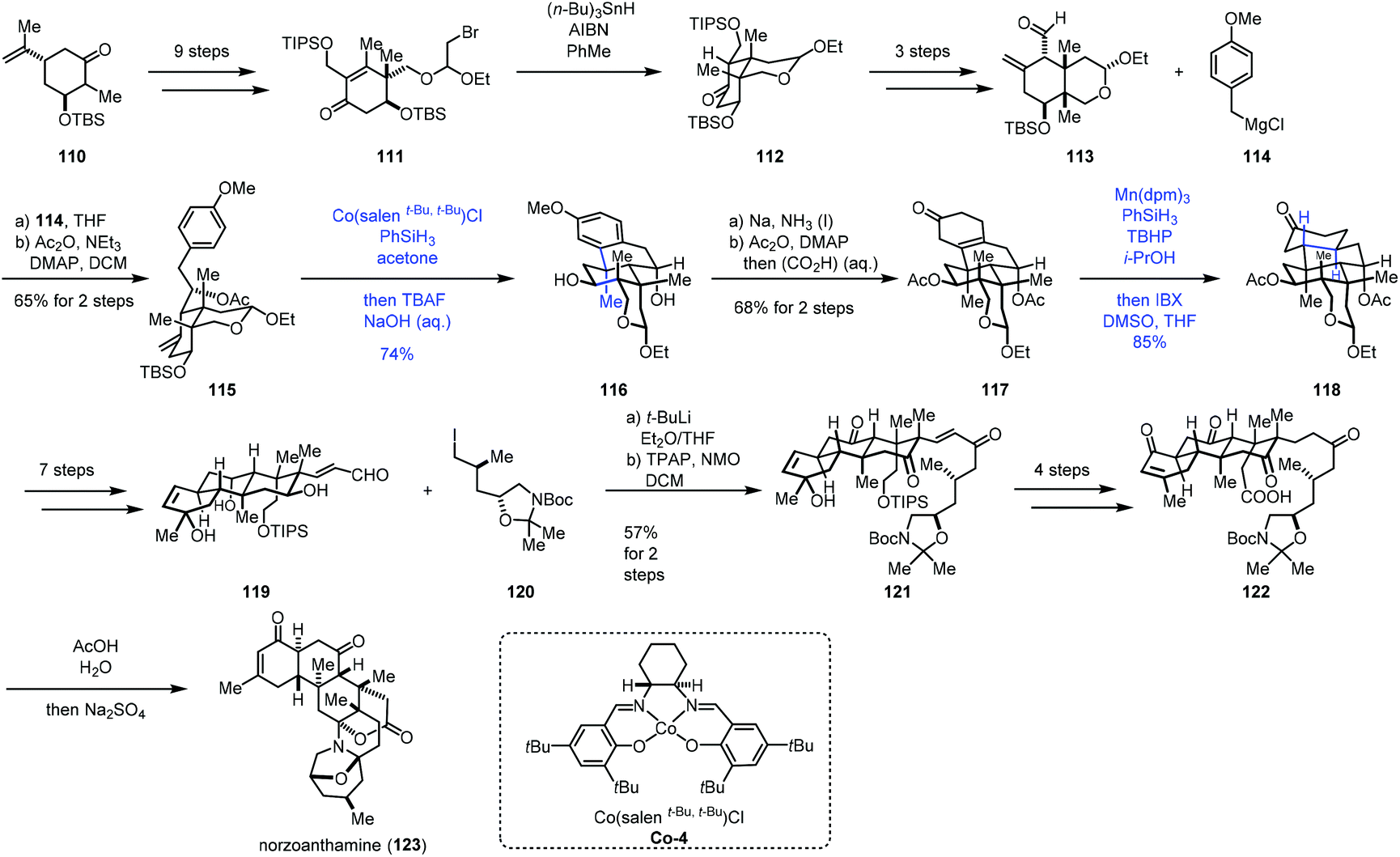 | ||
| Scheme 14 Gao's synthesis of norzoanthamine (123). | ||
After optimization, the authors discovered that excess of PhSiH3 (>4.0 equiv.) and t-butyl hydroperoxide (>4.0 equiv.) could ensure the complete conversion of 117, accompanied by the reduction of the carbonyl group. Subsequent oxidation gave 118 with 85% yield. 118 could be transformed to the unsaturated aldehyde 119 in seven steps. Reaction of 120 and aldehyde 119 followed by oxidation gave rise to ketone 121. 122 was then prepared from 121 in four steps. Deprotection of 122 followed by bis-aminoacetalization gave norzoanthamine (123).
Besides phenyl groups, indoles have also been used as radical acceptors in MHAT-based reactions. In 2016, Li and co-workers employed cobalt-catalyzed HAT reaction to achieve desired cyclization in the total syntheses of (+)-notoamides F (134), I (131), and R (132) and (−)-sclerotiamide (133) (Scheme 15).33 The starting material 124 was subjected to the reaction to give 125 in four steps. Under the optimal conditions, bicyclo[2,2,2]diazaoctane 126 was formed through oxidative aza-Prins cyclization with FeCl3. The reaction of 126 with 127 delivered ketone 128. With Shenvi's method, the desired product could not be obtained. While switching the cobalt catalyst to Co-5, the authors were able to obtain the desired products 130 along with 2,3-dihydroindole 129, both in poor yields. After optimization, they discovered that using a slight excess of PhSiH3 would increase the yields of 129 and 130, and they also envisioned that 129 could be elaborated to 130 through oxidation. Thus, when using the optimized conditions and quenching the reaction mixture with Dess–Martin periodinane (DMP), 130 was synthesized in 82% yield. In this reaction, the indole moiety is the radical acceptor. Compound 130 could be converted to (+)-notoamides F (134), I (131), and R (132) and (−)-sclerotiamide (133) featuring an allenyl Claisen rearrangement.
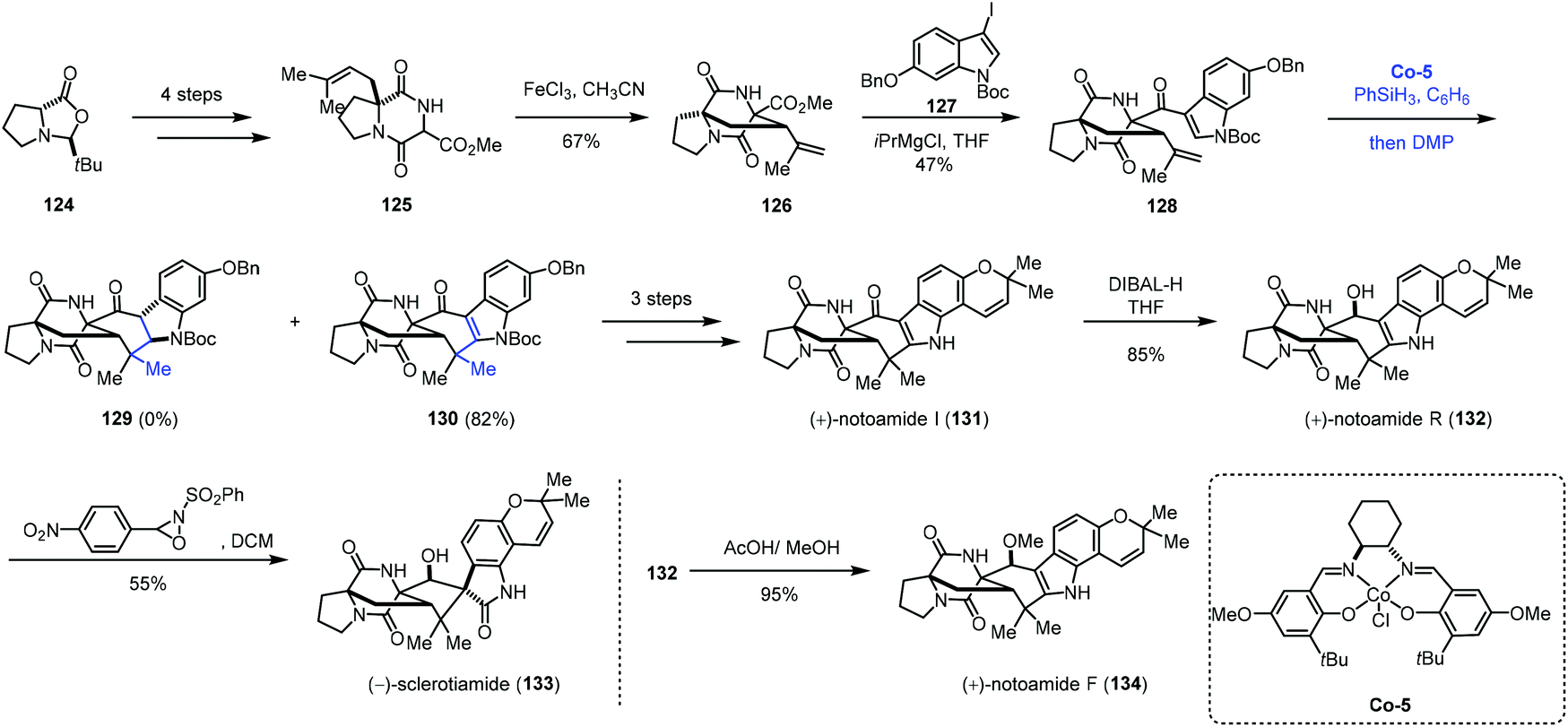 | ||
| Scheme 15 Li's synthesis of (+)-notoamides F (134), I (131), and R (132) and (−)-sclerotiamide (133). | ||
MHAT-initiated polyene cyclization was extensively investigated by Vanderwal and co-workers. They employed this strategy to complete the synthesis of a few oxidized abietane diterpenoids (Scheme 16).34 In their synthesis, ent-73 as the starting material was transformed to 135via epoxide ring opening and hydroxyl protection. Another fragment 136 was furnished from 144. Next, 135 was reacted with 136 to deliver the bicyclization precursor 137. With Co-1 as the catalyst, TMDSO as the hydrogen source and 2,6-di-tert-butylpyridine (DTBP) as the additive, a bicyclization took place via a radical process giving 138. The reaction process was similar to that in Liu's work,24,25 and the chiral center C2 would control the stereochemistry of the radical bicyclization. In particular, in their studies with model substrates, the authors found that oxygen substitution at C2 was associated with efficient polyene cyclization (>81% yield, dr > 20:1) for the equatorial group with both the free hydroxyl group and its corresponding silyl ether. The common intermediate 139 was then afforded through continuous reduction and deprotection. (+)-2-O-Deacetyl plebedipene A (140) was eventually obtained by oxidation of 139 with PIFA, and (+)-2-O-deacetyl plebedipene C (143) was obtained by oxidation with Ag2O. Plebedipene B (145) could be obtained from 136 through similar reactions.
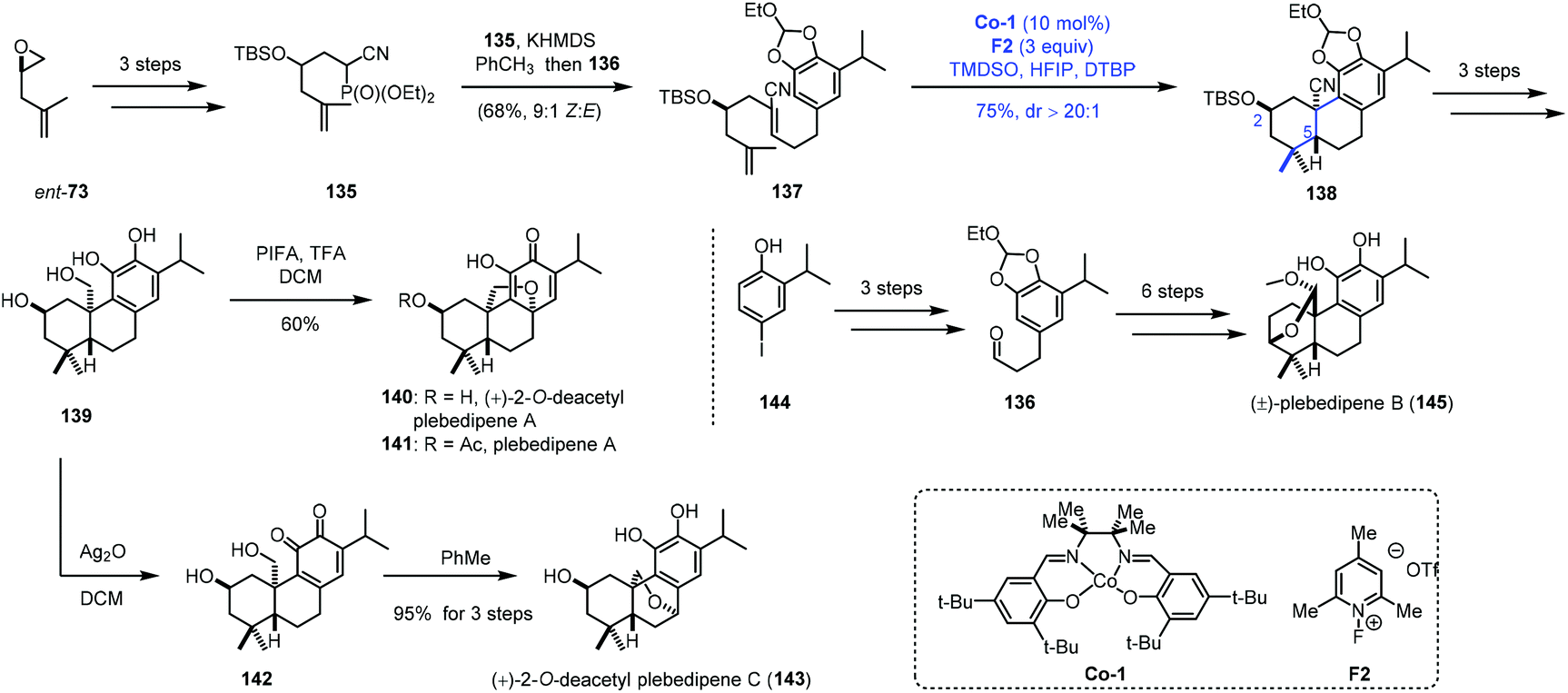 | ||
| Scheme 16 Vanderwal's synthesis of (+)-2-O-deacetyl plebedipenes A (140) and C (143), and plebedipene B (145). | ||
2.3 Reductive coupling with carbonyl or cyano functionalities
The reductive coupling of an olefin to a carbonyl functionality has been reported.35a Most of these reductive coupling reactions have been carried out under a hydrogen atmosphere in the presence of precious metal catalysts such as Rh. In 1989, Mukaiyama reported Co-catalyzed reductive coupling of α,β-unsaturated nitrile with aldehydes in the presence of phenylsilane.35bIn 2018, Bonjoch36a and Talbot36b independently disclosed their accomplishment in MHAT reactions. They extended the radical acceptor to ketone and cyano groups when unactivated olefins were employed. Bonjoch discovered that the radical-centered carbon center from MHAT to the olefin was able to attack the proximal ketone group giving the corresponding tertiary alcohol (Scheme 17a), while Talbot observed that the resulting carbon radical would attack the cyano group leading to a ketone via an imine intermediate (Scheme 17b).
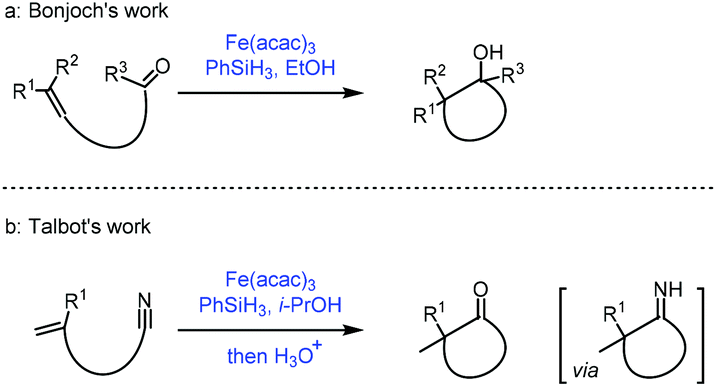 | ||
| Scheme 17 (a) Bonjoch's work and (b) Talbot's work. | ||
In 2021, Liu and co-workers applied iron-catalyzed reductive aldol reaction in the synthesis of rumphellclovane E (152) (Scheme 18).37 Cyclopropanation between ent-38 and 146 catalyzed by Rh2(esp)2 afforded 147, which could be transformed to 149via acylation and Michael addition. To assemble the tetracyclic core, they tried different reductive aldol conditions including Rh, Pd or Cu as catalysts, and all attempts failed. The authors thus resorted to MHAT-based conditions. The conditions of Mn and Co catalysts did not afford the desired cyclization product. Fortunately, a tetracyclic compound was obtained by the Fe-catalyzed radical process. After optimization, the authors observed that the treatment of 149 with Fe(acac)3 and PhSiH3 in i-PrOH led to the desired products 150/151. With respect to the mechanism, the authors proposed that MHAT to enone 149 provides the radical intermediate, which is further reduced by Fe(II) to provide the enolate, which reacts with the aldehyde through an aldol process to form tetracyclic 150/151. Next, a five-step transformation furnished rumphellclovane E (152).
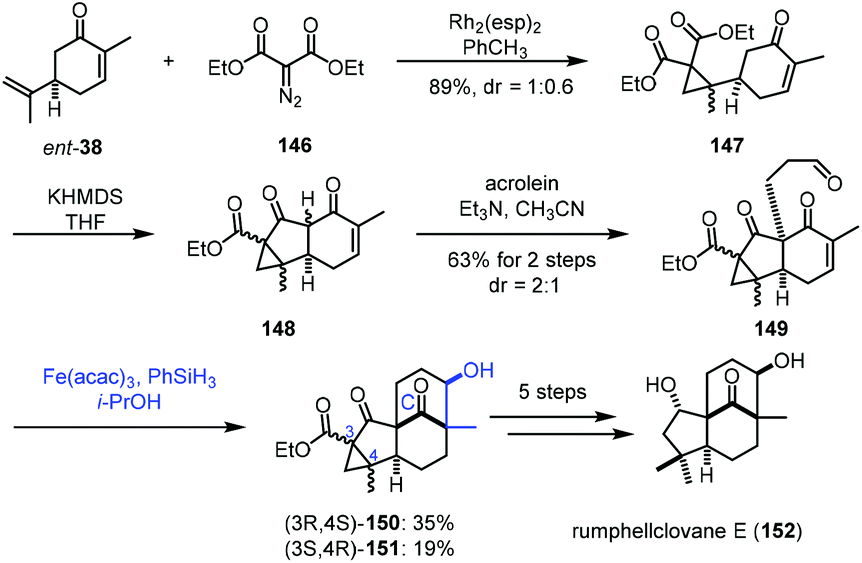 | ||
| Scheme 18 Liu's synthesis of rumphellclovane E (152). | ||
In 2018, Ma and co-workers reported the synthesis of navirine C (159) (Scheme 19).38 Their synthesis started with 153, which could be transformed to 154via a three-step sequence. Oxidative dearomatization of 154 and Diels–Alder cycloaddition gave 155. Then dinitrile 156 was obtained in six steps. Under MHAT reaction conditions (Mn(dpm)3, PhSiH3, and TBHP) reported by Shenvi and co-workers, the desired reductive cyclization occurred, delivering 158 in 68% yield, along with the reduction product 157 in 30% yield. The authors proposed that HMn(dpm)2 reacted with an alkene to produce the corresponding C14 radical intermediate, which might attack the cyano group to produce the cyclized intermediate. Further reduction generated 158. An additional six-step sequence gave avirine C (159).
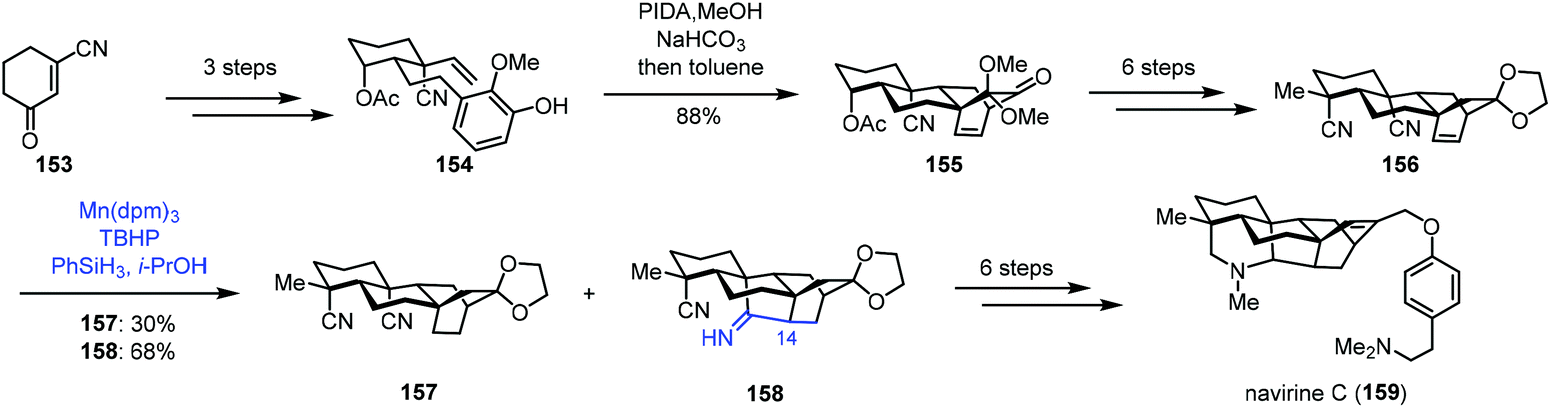 | ||
| Scheme 19 Ma's synthesis of navirine C (159). | ||
Talbot's method was subsequently used by Zhai and co-workers in their synthesis of (−)-conidiogenone B (166), (−)-conidiogenone (167), and (−)-conidiogenol (168) (Scheme 20).39 The starting material 160 could be transformed to 161via a three-step sequence involving Corey–Bakshi–Shibata reduction, tandem vinyl etherification/Claisen rearrangement, and van Leusen reaction. Under Talbot's conditions, alkene–nitrile cyclization was achieved, providing the bicyclic ketone 163, which was elaborated to deliver 164 by an additional nine steps. Subsequently, ozonolysis, followed by aldol reaction, provided (−)-conidiogenone B (166). After epoxidation and reduction, (−)-conidiogenone (167) was prepared. Reduction of (−)-conidiogenone (167) gave rise to (−)-conidiogenol (168).
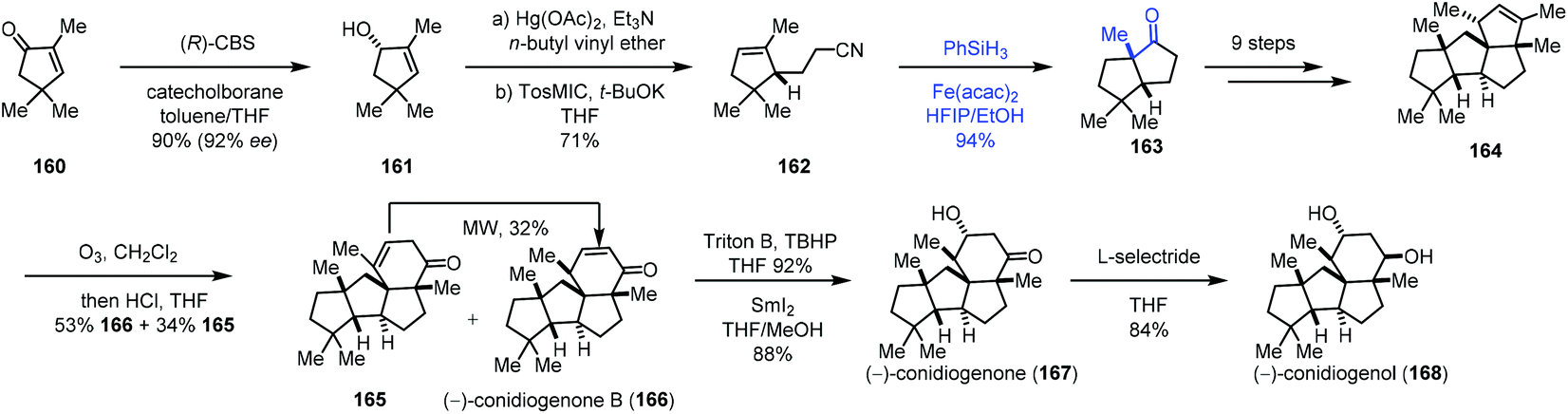 | ||
| Scheme 20 Zhai's synthesis of (−)-conidiogenone B (166), (−)-conidiogenone (167), and (−)-conidiogenol (168). | ||
3. C–O bond formation
Mukaiyama developed the Co-catalyzed hydration of olefins in the presence of silane and oxygen in 1989.3 Since then, the reaction has seen applications in natural product synthesis.40,41 In Shenvi's excellent review published in 2016, they carefully summarized examples of olefin hydration under MHAT conditions in natural product synthesis. So next we will mainly focus on the examples that appeared after 2016. Hydration and hydroperoxidation will be discussed separately.3.1 Hydration
Mukaiayama hydration reaction is a mild way to hydrate exocyclic double bonds to obtain tertiary hydroxyl groups. In 2017, Jia and co-workers applied Mukaiyama hydration in their synthesis of dihydrosetoclavine (Scheme 21).42 The starting material 169 was converted into 170 in six steps. Larock indole annulation of 170 afforded 171. Under Mn-catalyzed Mukaiyama hydration conditions, a diastereomeric mixture of tertiary hydroxyl 172 and 173 was furnished. The authors noticed that using a similar substrate (changing the tert-butanesulfinyl group of 171 to CO2Me) with hydration conditions failed to give the desired hydration product, indicating that the carbamate moiety has a negative effect on the hydration. Dihydrosetoclavine (174) and iso-dihydrosetoclavine (175) were then synthesized in three steps.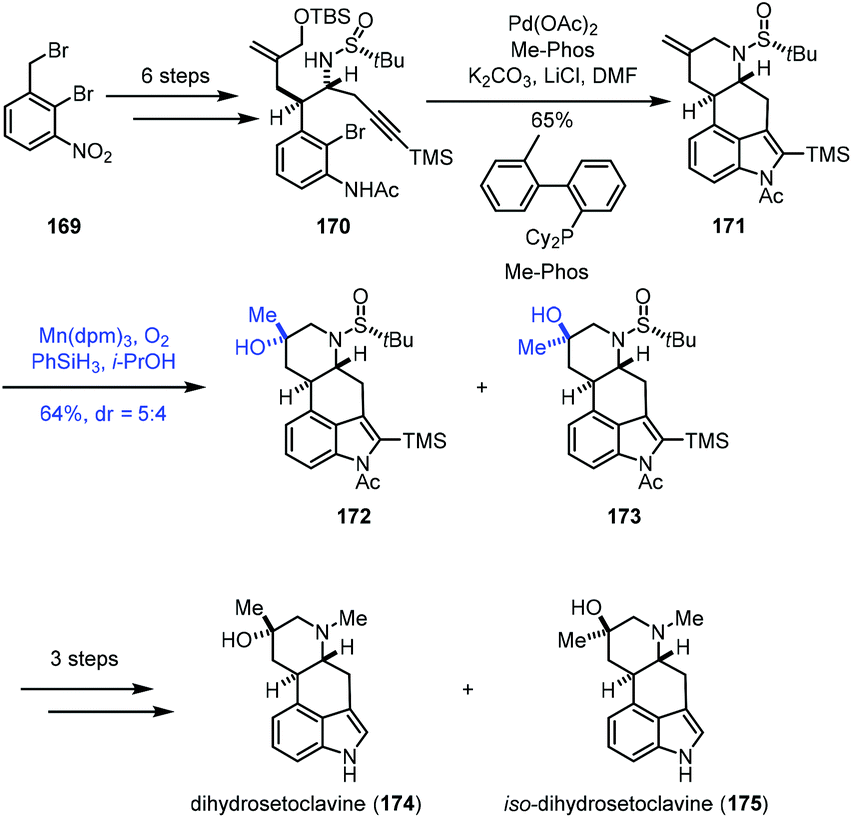 | ||
| Scheme 21 Jia's synthesis of dihydrosetoclavine (174). | ||
In 2017, Trauner and co-workers employed Mukaiyama hydration at the last step to furnish wickerol A (183) (Scheme 22).43 A Diels–Alder cycloaddition between the known enone 176 and diene 177 was performed to afford 178. The key intermediate 179 could be accessed from 178 in necessary functional group transformations. Treatment of 179 with KHMDS induced intramolecular alkylation to provide 180. Ketone 181 was afforded after the introduction of the missing methyl group. Initially, they carried out the methyl nucleophilic addition to the ketone carbonyl with methyl Grignard or methyllithium, but unfortunately, undesired 3-epi-wickerol A (184) was produced as the sole product. Addition of the Lewis acid MAD to the system still favored undesired 184 (183:184 = 1:4). The authors ultimately found that a two-step transformation including Wittig reaction and Mukaiyama hydration gave a better ratio. Olefination of ketone led to 182, which underwent Mukaiyama hydration reaction with Co(acac)2 as the catalyst to afford wickerol A (183) and 3-epi-wickerol A (184) in an almost 1:1 ratio.
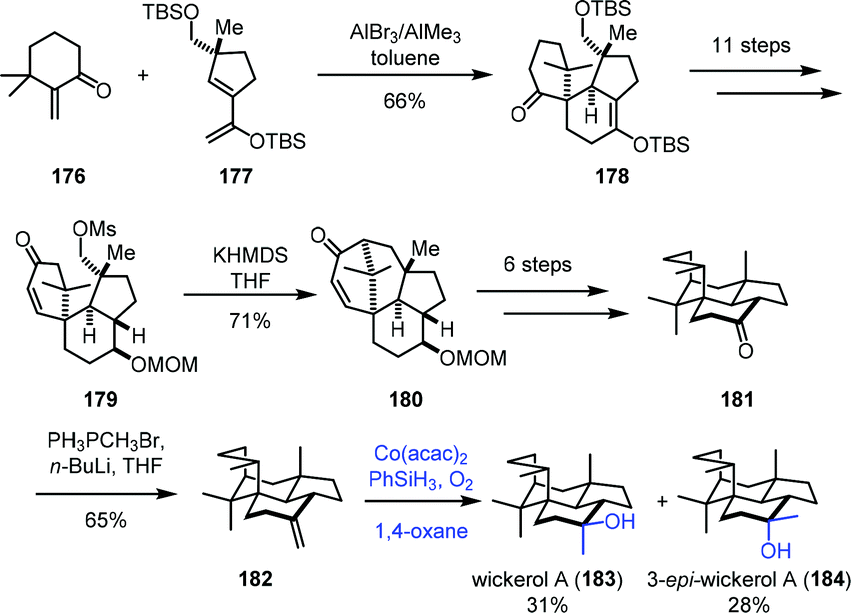 | ||
| Scheme 22 Trauner's synthesis of wickerol A (183). | ||
In 2018, a divergent total synthesis of enmein-type natural products was reported by Dong and co-workers (Scheme 23).44 The common intermediate 187 containing the A/B/C rings of (−)-enmein (192), (−)-isodocarpin (193), and (−)-sculponin R (191) was synthesized from diene 185 and anhydride 186 involving Diels–Alder cycloaddition and Birch reduction. γ-Hydroxylation was achieved with vinylogous enol ether formation and DMDO oxidation to afford ester 188, which was reduced by Luche reduction, followed by radical annulation to give 189. A three-step conversion furnished 190. Finally, functionalization of the exocyclic double bond was carried out under the Mn-catalyzed Mukaiyama hydration conditions (Mn(dpm)3, PhSiH3, O2, and EtOH) to furnish (−)-sculponin R (191) after deacetylation in 45% yield over two steps. The common intermediate 187 could be also converted to (−)-enmein (192) and (−)-isodocarpin (193) in a few steps.
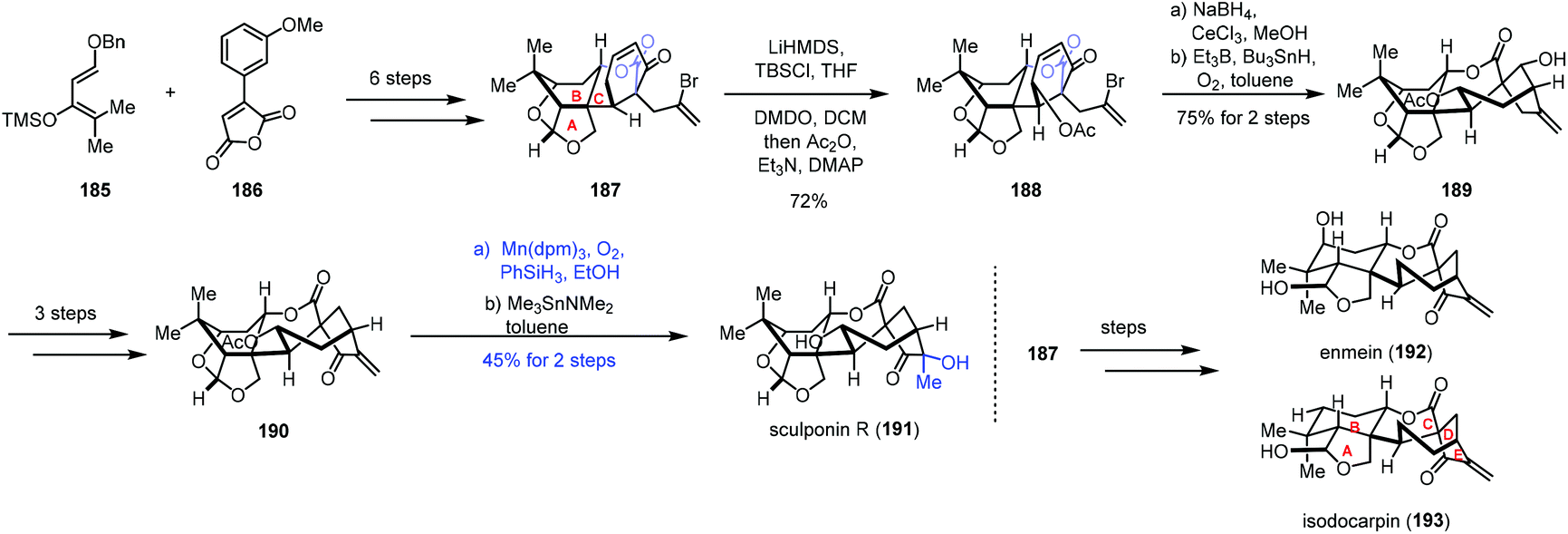 | ||
| Scheme 23 Dong's synthesis of (−)-enmein (192), (−)-isodocarpin (193), and (−)-sculponin R (191). | ||
In 2020, Li and co-workers reported the synthesis of bufospirostenin A (199), in which Mukaiyama hydration was adopted two times (Scheme 24).45 This synthesis began with 194, which was transformed to tetracyclic 195 in eight steps. Treatment of 195 with MOMCl, followed by a standard Mukaiyama hydration (Mn(acac)2, PhSiH3, PPh3, O2, and EtOH), gave the tertiary alcohol 196 in 80% yield. Afterwards, a two-step conversion furnished 197, poised for another Mukaiyama hydration. Then the tertiary alcohol 198 with the desired configuration was furnished via the same hydration process from the exocyclic double bond of 197 in 56% yield. A further eight steps of transformation afforded the natural product bufospirostenin A (199).
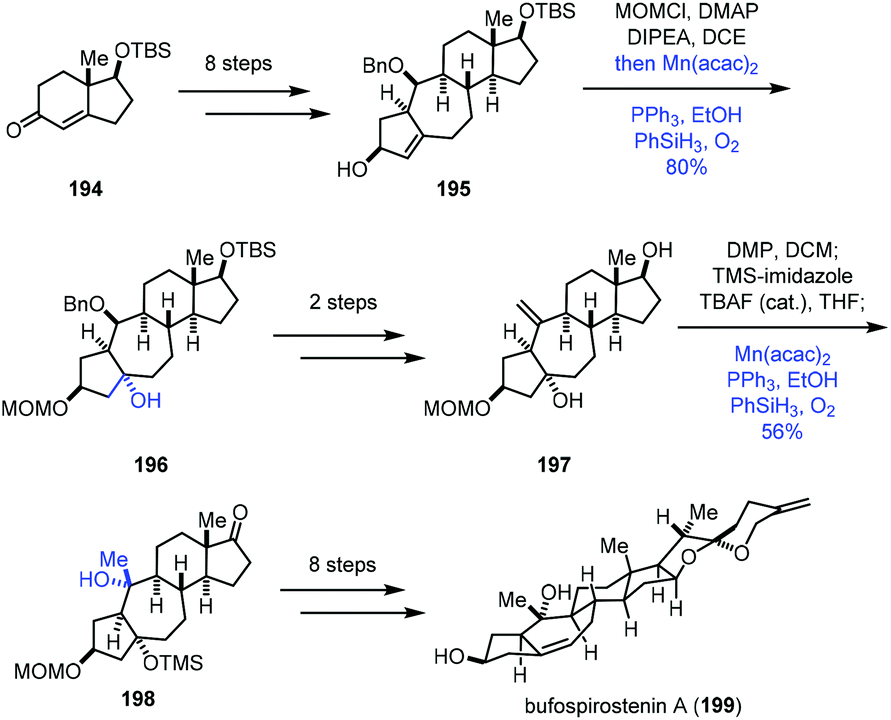 | ||
| Scheme 24 Li's synthesis of bufospirostenin A (199). | ||
In 2020, Shenvi reported the synthesis of picrotoxinin (204), which could be transformed to picrotin (205) by Mukaiyama hydration (Scheme 25).46 Their synthesis commenced with 200, which could be converted to 201via a four-step sequence. 201 went through a five-step continuous oxidation process to give 202. Treatment of 202 with AIBN and Bu3SnH removed the tertiary iodide, followed by the cleavage of the formyl group to furnish 203. Picrotoxinin (204) was obtained from 203 through functionalization of the methyl group and cleavage of bromoether. Finally, a Mukaiyama hydration under Co(acac)2, PhSiH3, O2, and i-PrOH conditions installed the tertiary hydroxyl from the exocyclic double bond of 204 to deliver the desired picrotin (205) in 84% yield.
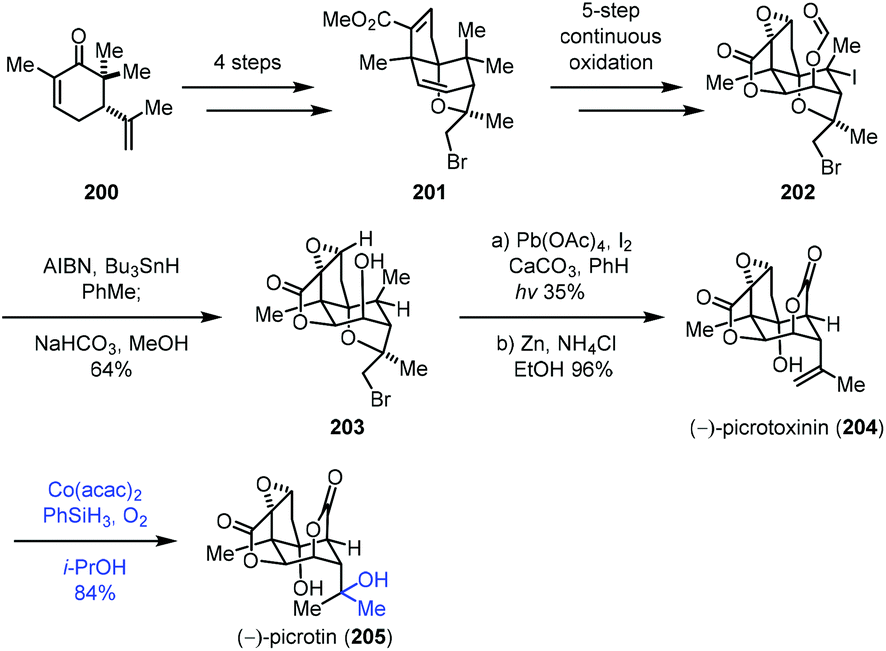 | ||
| Scheme 25 Shenvi's synthesis of picrotoxinin (204) and picrotin (205). | ||
In 2020, Overman and co-workers reported the synthesis of macfarlandin C (213), in which Mukaiyama hydration was used (Scheme 26).47 The oxalate radical precursor 207 was obtained from 206 after eleven steps of transformations. Irradiation of 207 and D-menthol-derived chlorobutenolide 208 in the presence of an Ir-catalyst installed the vicinal quaternary and tertiary stereocenters generating 209. Two more steps gave 211. Mukaiyama hydration with Mn(dpm)3 and Shenvi's more reactive Ph(i-PrO)SiH248 and O2 provided 212 in a regio- and stereoselective manner in 67% yield. In this case, the electron-rich olefin remained untouched. Macfarlandin C (213) was then obtained in five additional steps.
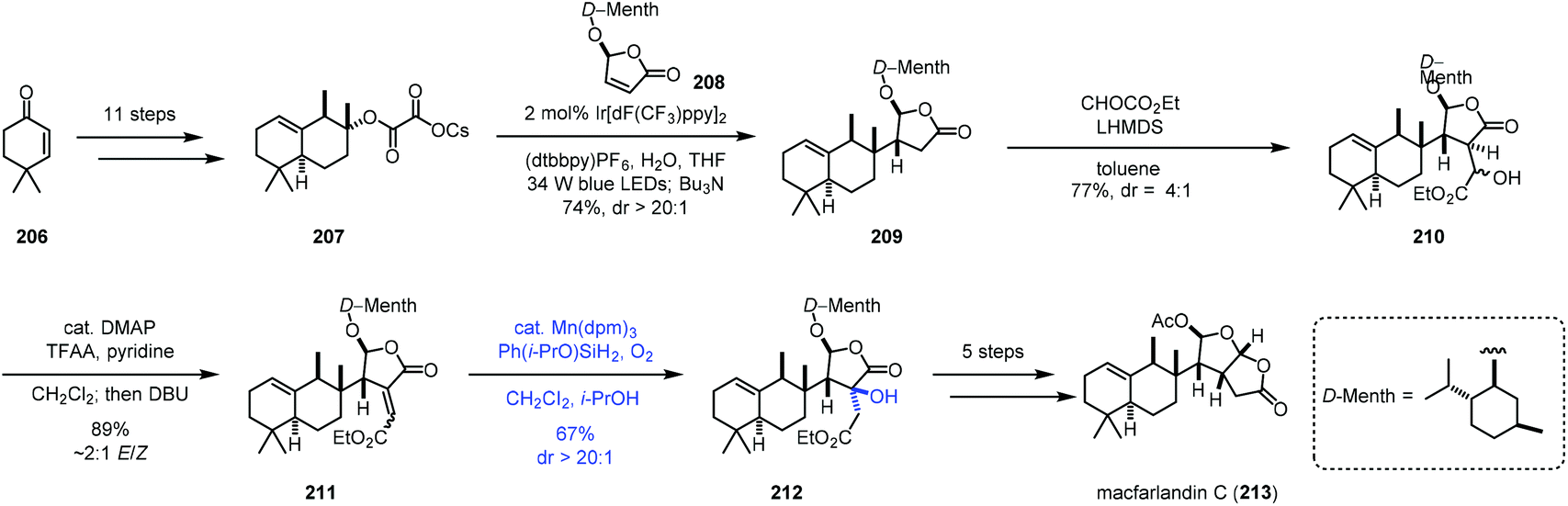 | ||
| Scheme 26 Overman's synthesis of macfarlandin C (213). | ||
The above-mentioned examples are related to the hydration of exocyclic double bonds. Next, the hydration of endocyclic double bonds will be summarized.
In 2017, Shenvi and co-workers reported a seven-step synthesis of (−)-11-O-debenzoyltashironin (222) from butenolide (Scheme 27).49 Butenolide heterodimerization of 215 to 214, followed by the addition of titanium tetraisopropoxide and lithium diisopropylamide, gave tetracyclic 216. To invert C4, the authors planned to eliminate the lactone to an alkene, and then use Mukaiyama hydration reaction to construct the tertiary hydroxyl group with the desired configuration. In accordance with the strategy, a two-step sequence afforded 217/218, which was converted to 219 featuring a dehydration step. Seven-membered lactone 220 was afforded in two more steps. Mukaiyama hydration of the endocyclic double bond of 220 with Co(acac)2 followed by the addition of p-toluenesulfonic acid produced 222 with a trans-hydrindane framework in 72% yield. trans-Hydrindane is thought to be thermodynamically disfavoured. The authors suspected that, in the current case, C10 alcohol might determine the stereochemistry of hydration as C10 alcohol shields the β-face. In addition, the authors mentioned that the hydration had to be arranged at the late stage since attempts with earlier intermediates resulted in a complex mixture or the undesired cis-hydrindane product.
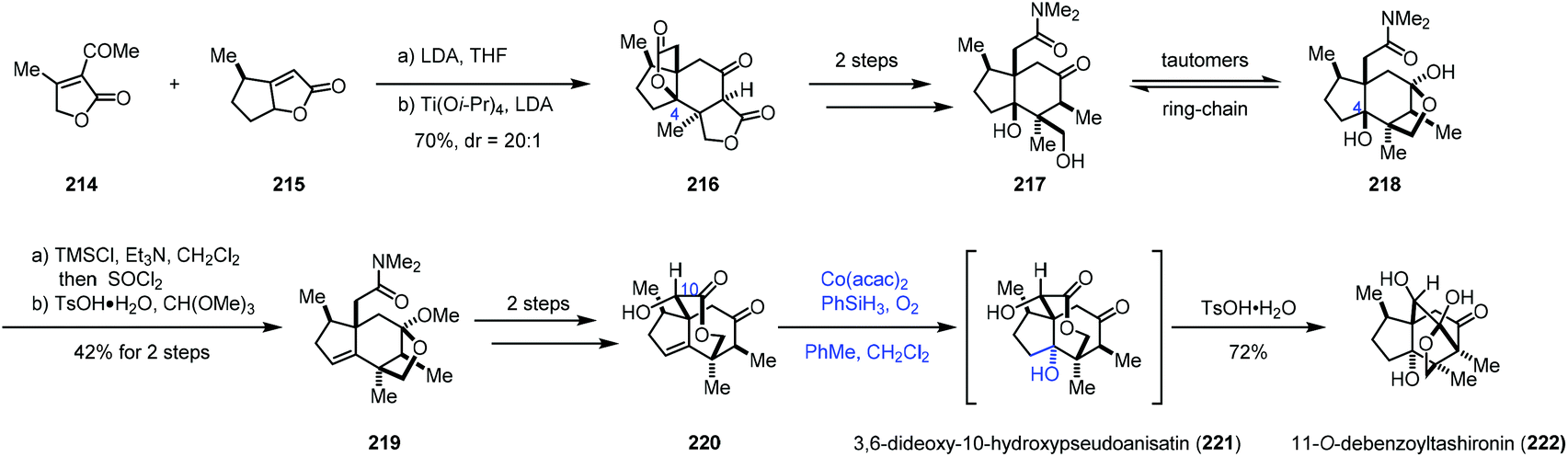 | ||
| Scheme 27 Shenvi's synthesis of (−)-11-O-debenzoyltashironin (222). | ||
In 2017, Fan and co-workers communicated their synthesis of lycodoline-type Lycopodium alkaloids (Scheme 28).50 In this case, subjecting the common intermediate anhydrolycodoline (229) to different hydration conditions could give different types of products. This synthesis commenced with the preparation of 224 from 4-methylcyclohexone 223. Treatment of 224 with KHMDS and TMSCl, followed by tandem oxidative dehydrogenation and oxa-Michael reaction, gave 225/226. Then tandem reductive amination and bridgehead aminolysis reaction were performed to give 227. Lastly, lycodoline (228) was obtained through a sequence of Oppenauer oxidation, aldol condensation and hydrogenation of the olefin. Anhydrolycodoline (229) was furnished via dehydration from lycodoline (228).
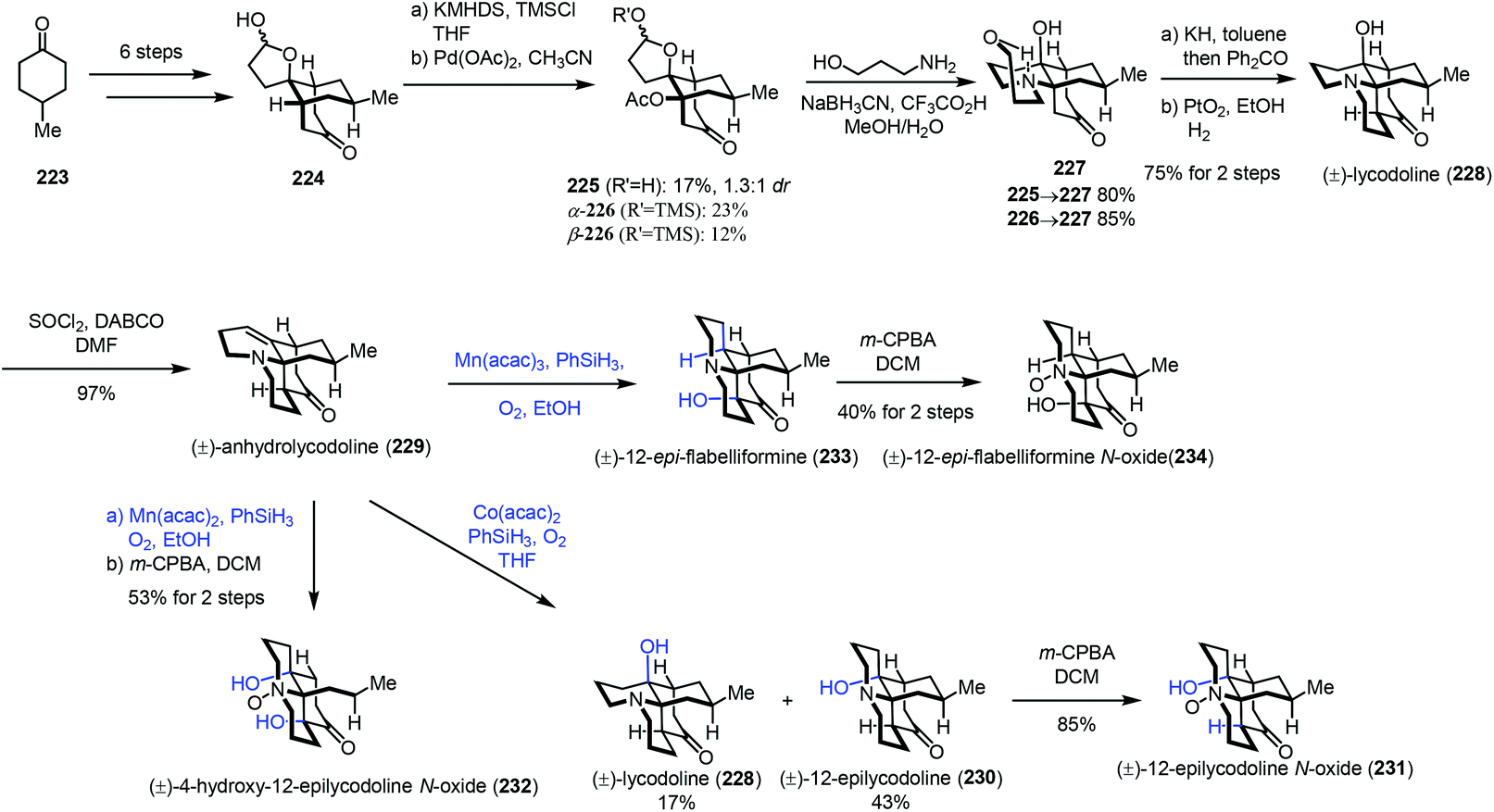 | ||
| Scheme 28 Fan's synthesis of lycodoline-type Lycopodium alkaloids. | ||
Under Mukaiyama hydration conditions with Co(acac)2 in THF, anhydrolycodoline (229) could be elaborated to 12-epilycodoline (230) and lycodoline (228). 230 could be further oxidized with m-CPBA to provide 12-epilycodoline N-oxide (231). In comparison, when anhydrolycodoline (229) was treated with Mn(acac)2 and phenylsilane in EtOH, followed by oxidation with m-CPBA, it turned out that not only the C–C double bond was hydrated, but also α-hydroxylation occurred, providing 4-hydroxy-12-epilycodoline N-oxide (232). It is interesting to note that hydration with Mn(acac)3 gave the α-hydroxylation product 12-epi-flabelliformine (233), which could be oxidized into 234.
In 2019, Shenvi and co-workers described a solvent-controlled Mukaiyama hydration in total synthesis of (−)-bilobalide (240) (Scheme 29).51a,b In the process of the synthesis of bilobalide, with respect to the hydration step of 236 to 237, Shenvi and co-workers observed that there is strong correlation between solvent polarity and diastereoselectivity. For example, under standard Mukaiyama conditions, Mn(dpm)3/PhSiH3/i-PrOH, 237 was delivered with no diastereoselectivity. Switching to Ph(i-PrO)SiH2 and ethereal solvent such as tert-butyl methyl ether led to undesired diastereomer as the major product (dr = 1:1.8), while methylcyclohexane as solvent favoured the desired product 237 in a 3:1 ratio. An additional 4 steps gave 238. Shenvi and co-workers then developed a new method for alkyne oxidation to form 239. Finally, (−)-bilobalide (240) was accessed through a skeletal rearrangement and oxidation.
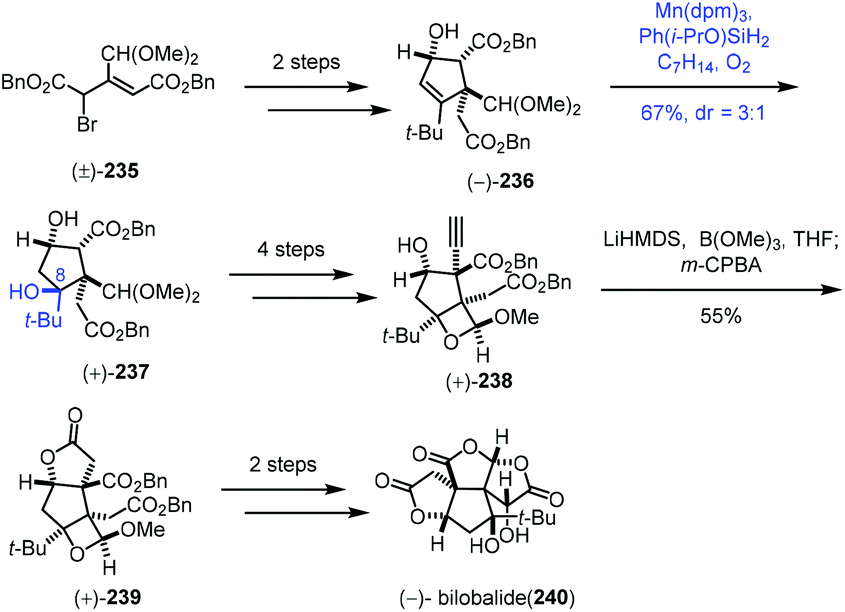 | ||
| Scheme 29 Shenvi's synthesis of (−)-bilobalide (240). | ||
In 2019, Maimone and co-workers published a semisynthetic strategy to the Illicium sesquiterpenes from cedrol by their site-selective C(sp3)–H bond functionalization reactions. They employed Mn or Co-catalyzed hydration to install the C4 hydroxyl group (Scheme 30).52 A hypoiodite photolysis of 241 with PhI(OAc)2 and I2 furnished a strained tetrahydrofuran intermediate by oxidation of C14 of cedrol, which underwent alkylation and concomitant elimination to provide 242. The C4 oxidation precursor 243 was furnished from 242 in four steps. Fe-Catalyzed selective C–H oxidation of C4 afforded 244, which was then transformed to alkene 245. Under Co-catalyzed conditions, 246 was afforded in 29% yield as the minor product through the hydration process due to the modest diastereocontrol. Meanwhile, cedrol (241) could be converted to 247 in thirteen steps. A hydration reaction was conducted with a Mn-catalyst to give 248 in 50% yield.
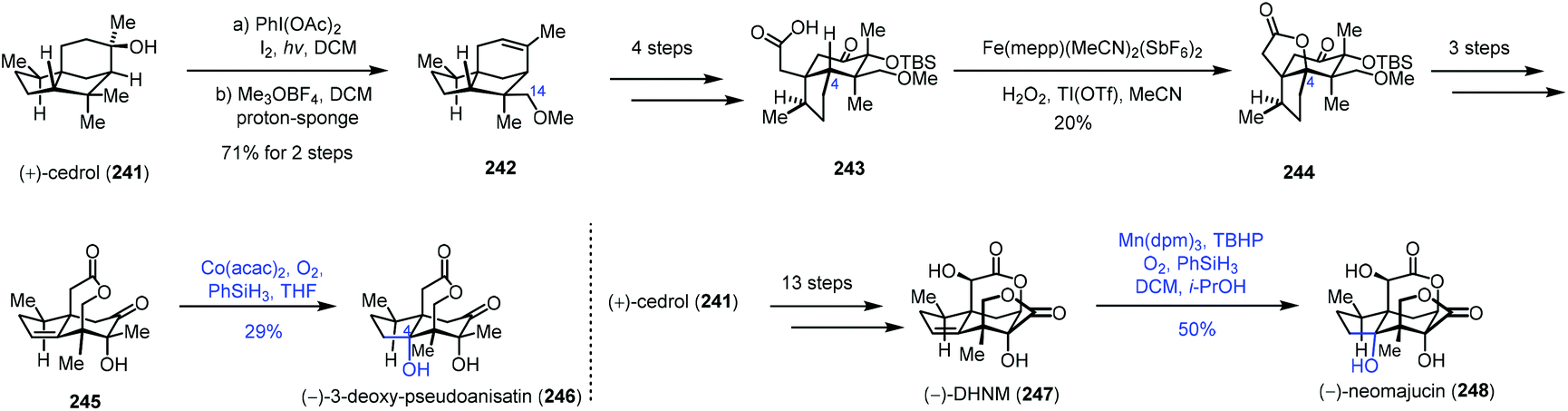 | ||
| Scheme 30 Maimone's synthesis of Illicium sesquiterpenes. | ||
In 2019, Ding and co-workers accomplished the synthesis of cembrane ditepenionds, (+)-sarcophytin (255) (Scheme 31).53 They employed Mukaiyama hydration to introduce the tertiary alcohol. The synthesis commenced with the construction of bicyclic 252, which was accessed through double-Mukaiyama Michael addition between 250 and the Rawal diene 251 and elimination. 253 was then furnished in three steps from 252. The authors speculated that the subsequent hydration would take place from the β-face since the proximate pseudoaxial hydroxy group blocks the α-face. As expected, subjecting 253 to Shenvi's optimized hydration conditions (Mn(dpm)3, Ph(i-PrO)SiH2, O2, and THF) formed 254 and its epimer in a 5.6:1 ratio with 80% yield, favouring desired 254. In comparison, using other hydride sources (EtSiH3, PhSiH3, and NaBH4) and metal catalysts (Co(acac)2, Co(tfa)2, Mn(acac)3, and Fe(acac)3) led to inferior yield (15–60%) and diastereoselectivity (1.2:1–4.5:1). Necessary functional group transformations eventually led to (+)-sarcophytin (255).
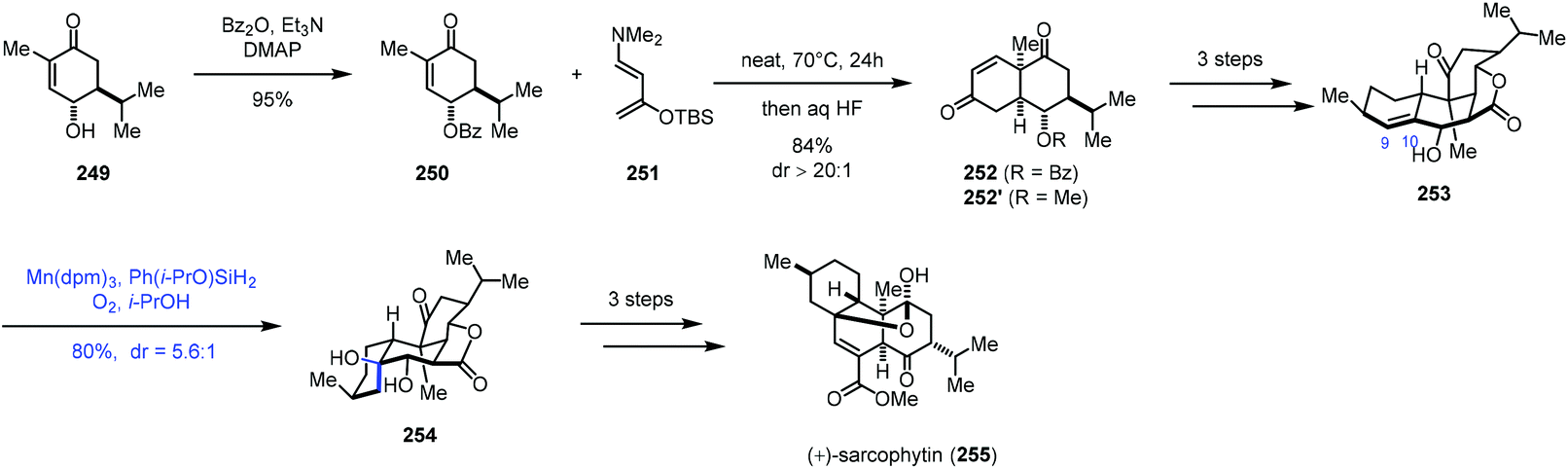 | ||
| Scheme 31 Ding's synthesis of (+)-sarcophytin (255). | ||
In 2019, Li and co-workers reported the synthesis of arcutinidine, arcutinine, and arcutine. Mukaiyama hydration was adopted (Scheme 32).54 Alcohol 257 was made from alcohol 256 through sequential intermolecular and anionic Diels–Alder reactions. A dehydration reaction of 257 with SOCl2 furnished a trisubstituted olefin which could further be converted to 258 through a Prins/Wagner–Meerwein cascade reaction. Mukaiyama hydration of 258 with Mn(acac)2 and PhSiH3 furnished tertiary alcohol 259 with the desired stereochemistry after site-selective C–H oxidation. In addition, the authors found that Mn(acac)2 as a precatalyst was superior to Co(acac)2 for this olefin hydration. Diketoaldehyde 260 was then prepared in five steps. Chemoselective condensation and 1,2-reduction provided the corresponding oxime, followed by cleavage of the N–O bond and reduction giving arcutinidine (261).
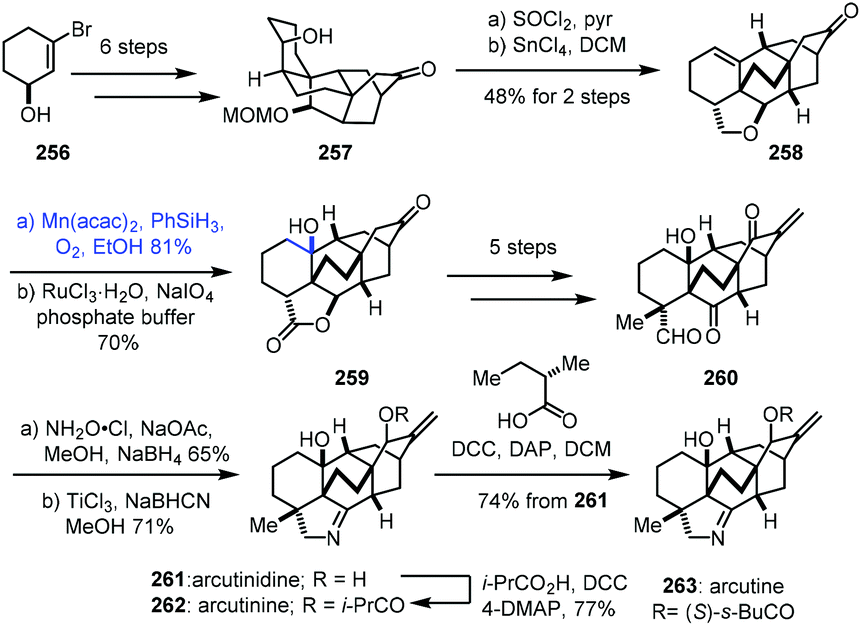 | ||
| Scheme 32 Li's synthesis of arcutinidine (261), arcutinine (262) and arcutine (263). | ||
Arcutinine (262) and arcutine (263) were afforded from arcutinidine (261) via acylation, respectively.
In 2019, Sarpong and co-workers reported total synthesis of xiamycins A (268), C (270), F (271), and H (272) and oridamycin A (269) (Scheme 33).55 In this case, they employed Mukaiyama hydration to achieve functionalization of endocyclic disubstituted olefins. Their synthesis commenced with the assembly of the trans-decalin framework. (R)-Carvone was transformed to 264 and 265 in four steps. Diol 266 was furnished from 264. Dehydration of 266, followed by oxidation and key photocyclization–desulfonylation, led to aldehyde 267. Xiamycin A (268) was obtained after two more steps. Subjecting 267 to Mukaiyama hydration conditions (Co(acac)2, PhSiH3, THF, and O2) followed by Pinnick oxidation provided xiamycin C (270) and its C-19 epimer and xiamycin F (271). Meanwhile, xiamycin H (272) was generated from 267 in steps.
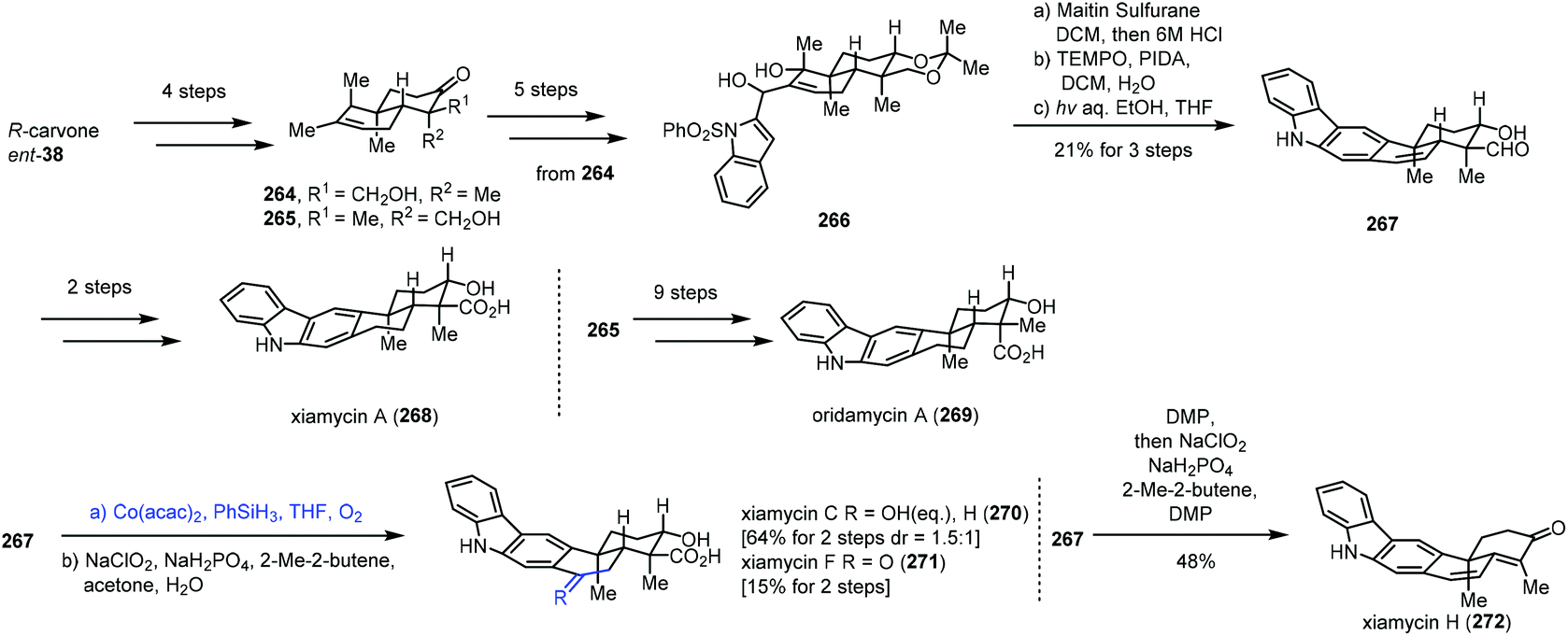 | ||
| Scheme 33 Sarpong's synthesis of xiamycins A (268), C (270), F (271), and H (272) and oridamycin A (269). | ||
A similar conversion was performed by Yang and co-workers in the synthesis of the core structure of euphorikanin A (Scheme 34).56 The 7/3-bicyclic 274 was synthesized from (+)-3-carene 273 in four steps. Three more steps of conversions gave the key dienyne precursor 275. A domino RCM reaction of 275 with Grubbs second-generation catalyst 279 proceeded well to deliver the tetracyclic skeleton 276 in excellent yield. To realize the transformation of the conjugated double bonds into the α,β-unsaturated ketone, they tested diene 276 with various Wacker reactions, such as PdCl2 and CuCI, which led to no reaction, probably due to the high rigidity of the ring system. Then they resorted to HAT-initiated oxidation. Screening of different Fe(II) catalysts, silane reagents, solvents, temperatures and reaction times revealed that FeCl2/PMHS/EtOH was optimal, and the desired enone 277 could be obtained successfully in 56% yield.
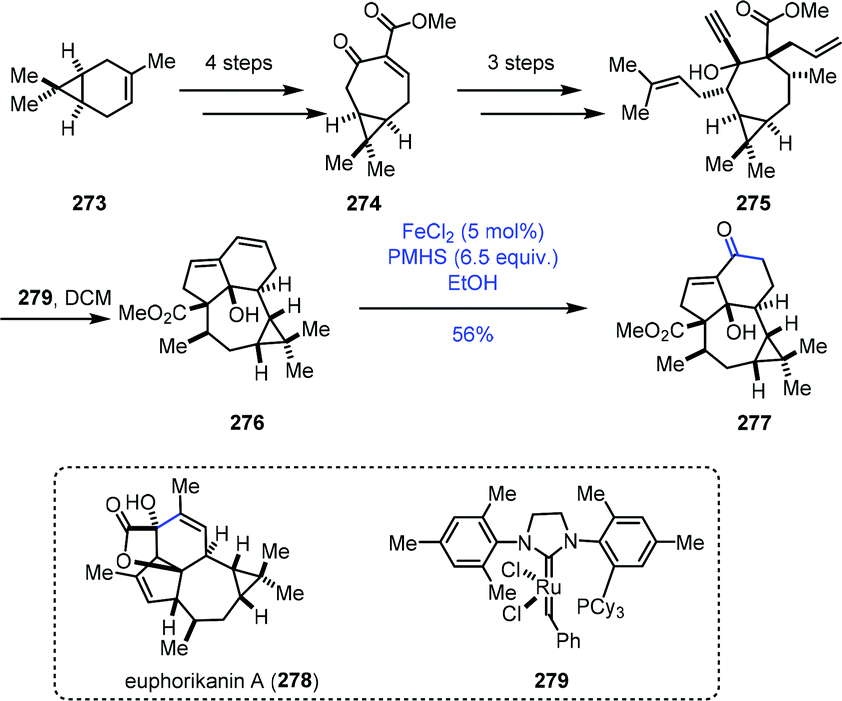 | ||
| Scheme 34 Yang's synthesis of the core structure of euphorikanin A. | ||
In 2021, Lu and co-workers described the synthesis of dysiherbol A (286) (Scheme 35).57 The Wieland–Miescher ketone derivative 280 could be advanced to ketone 281 in five steps. The Pd-catalyzed intramolecular Heck reaction was carried out to obtain tetracyclic 282, which was advanced to diene 283 in four steps. Mono-alkene 284 was created from diene 283via chemo- and diastereoselective reduction. The authors observed that the epoxidation of alkene 284 with H2O2, m-CPBA, TBHP or Vo(acac)2 and TBHP led to the skeleton rearrangement presumably due to the strain of the 6/5-fused ring system. Dihydroxylation of alkene 284 with OsO4, K2OsO4·H2O or AD-mix-α also failed.
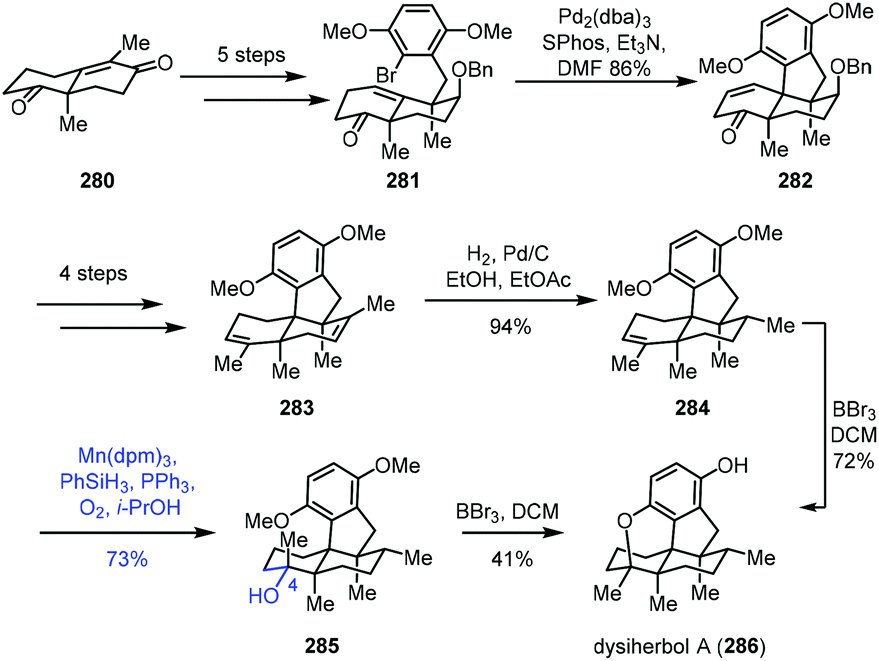 | ||
| Scheme 35 Lu's synthesis of dysiherbol A (286). | ||
Fortunately, Mukaiyama hydration of 284 worked, giving 285 under optimized conditions (Mn(dpm)3, PhSiH3, O2, and PPh3) in 73% yield. They further separated the natural product dysiherbol A (286) upon exposure of 285 to BBr3. Monitoring of the reaction revealed that the tertiary alcohol of 285 dehydrated in the presence of BBr3 to return to compound 284. Therefore, 284 was then transformed into dysiherbol A (286) directly with BBr3.
In 2021, Dethe and Nirpal disclosed the synthesis of japonicol C (292) (Scheme 36).58289 was obtained from 287 through Friedel–Crafts reaction, deprotection and acetylation. The authors then tested the hydration reaction using various metal catalysts. Treatment of 289 with Mn or Fe complexes in the presence of PhSiH3 or EtSiH3 in solvents like 1,2-dichloroethane, EtOH or i-PrOH led to no reaction. Under Mukaiyama hydration conditions, Co(acac)2 and phenylsilane in THF under O2, undesired tertiary alcohol 290 was isolated in 68% yield. The authors proposed that the stereochemical outcome of the hydration could be due to the less hindered β-face. Deacetylation of 290 gave 291, a regioisomer of the natural product japonicol C. Since 290 turned out to be an undesired hydration product, to overcome the regioselectivity issue, the authors attempted Fuch's C–H oxidation method and accessed the desired tertiary alcohol from 289, which was finally elaborated to japonicol C (292).
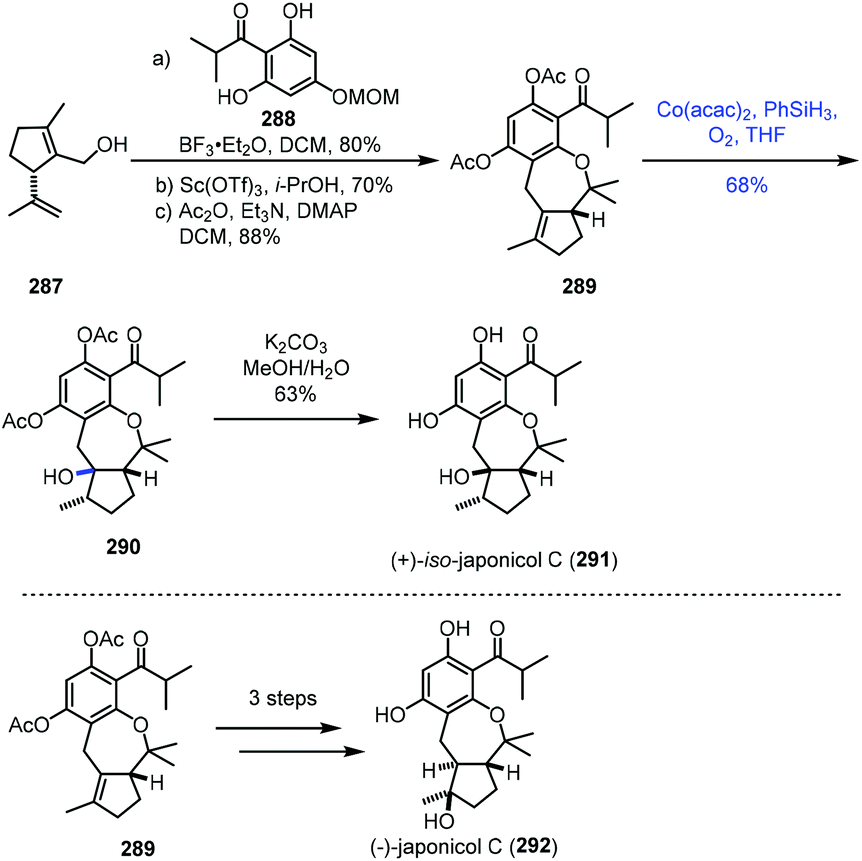 | ||
| Scheme 36 Dethe's synthesis of japonicol C (292). | ||
3.2 Hydroperoxidation
In this sub-section, we will introduce hydroperoxidation of olefins and its application in natural product synthesis. A number of important works have been summarized in other reviews,9,10 such as those by Maimone59 and Inoue.60 We will focus on the studies published in recent years.In 2018, Tang and co-workers used the Co-catalyzed HAT to oxygenation of vinylcyclobutane in the syntheses of (+)-gracilioether A (298) and (−)-gracilioether E (300) (Scheme 37).61 A Horner–Wadsworth–Emmons reaction of 293 with 294 furnished a diene, which cyclized into vinylcyclobutane 295 through [2 + 2] photocycloaddition, which was advanced to the key compound 297. They initially tried to construct a six-membered peroxy ring through a radical process as reported by Feldman;62 however, they failed to get the desired result. Inspired by Maimone's59 experience in the synthesis of (+)-cardamom peroxide using the Mn-catalyzed HAT to construct a peroxy group, the authors tested Maimone's MHAT conditions (Mn(dpm)3, PhSiH3, TBHP, O2, and i-PrOH/DCM), and only 5% of gracilioether A (298) and 7% of gracilioether E (300) were isolated. Further optimization revealed that the Mukaiyama/Isayama hydrosilyl peroxidation reaction system was superior (Co(acac)2, PhSiH3, TBHP, O2, and i-PrOH/DCE), and 297 could be converted to gracilioether A (298), 299 and gracilioether E (300) in acceptable yields. Mechanistically, the authors proposed that the radical is generated from the terminal alkene 297 by HAT from HCo(III), which undergoes cyclobutane ring-opening, dioxygen trapping of the resulting radical, 6-exo-trig cyclization, and second dioxygen insertion to give the corresponding peroxy radical. Subsequent hydrogen abstraction and peroxide reduction deliver 298 and 300.
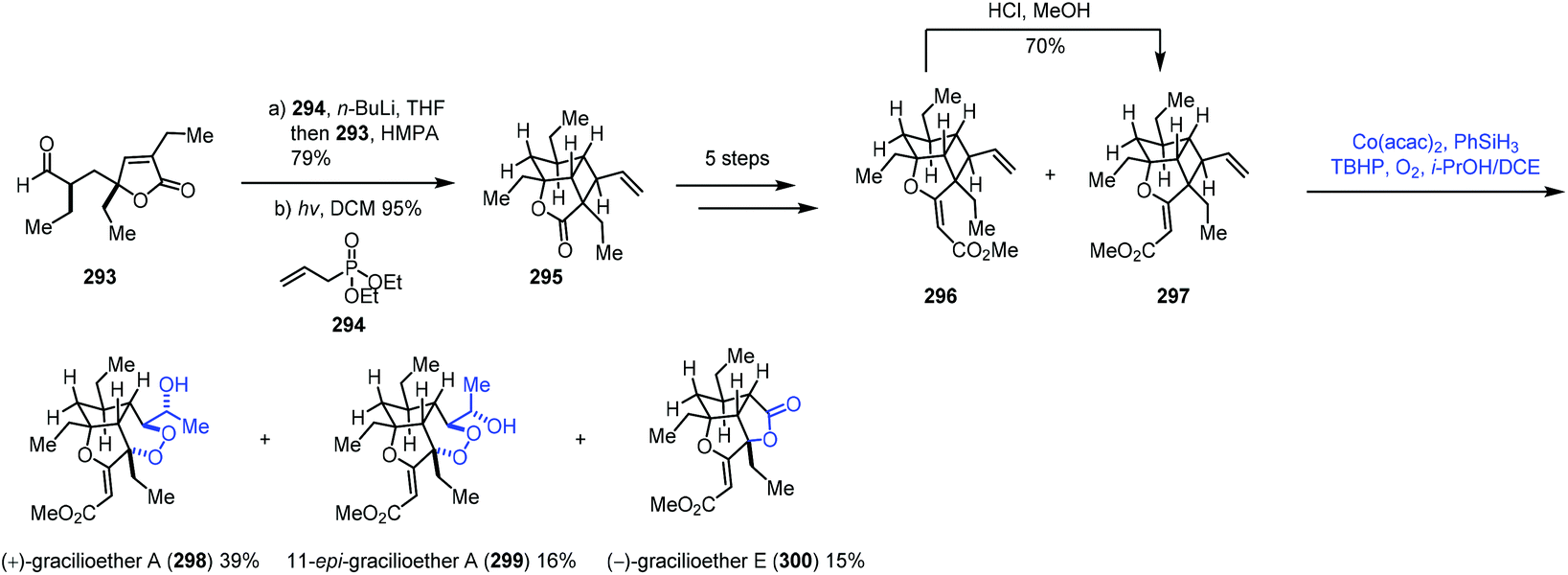 | ||
| Scheme 37 Tang's synthesis of (+)-gracilioether A (298) and (−)-gracilioether E (300). | ||
In 2019, Maimone and co-workers reported the synthesis of complex guaianolide sesquiterpenes (Scheme 38).63 In the synthesis of nortrilobolide (308), they creatively employed the radical tandem reaction under Co-catalyzed HAT conditions to introduce three hydroxyl groups through reduction of the peroxy bond generated.
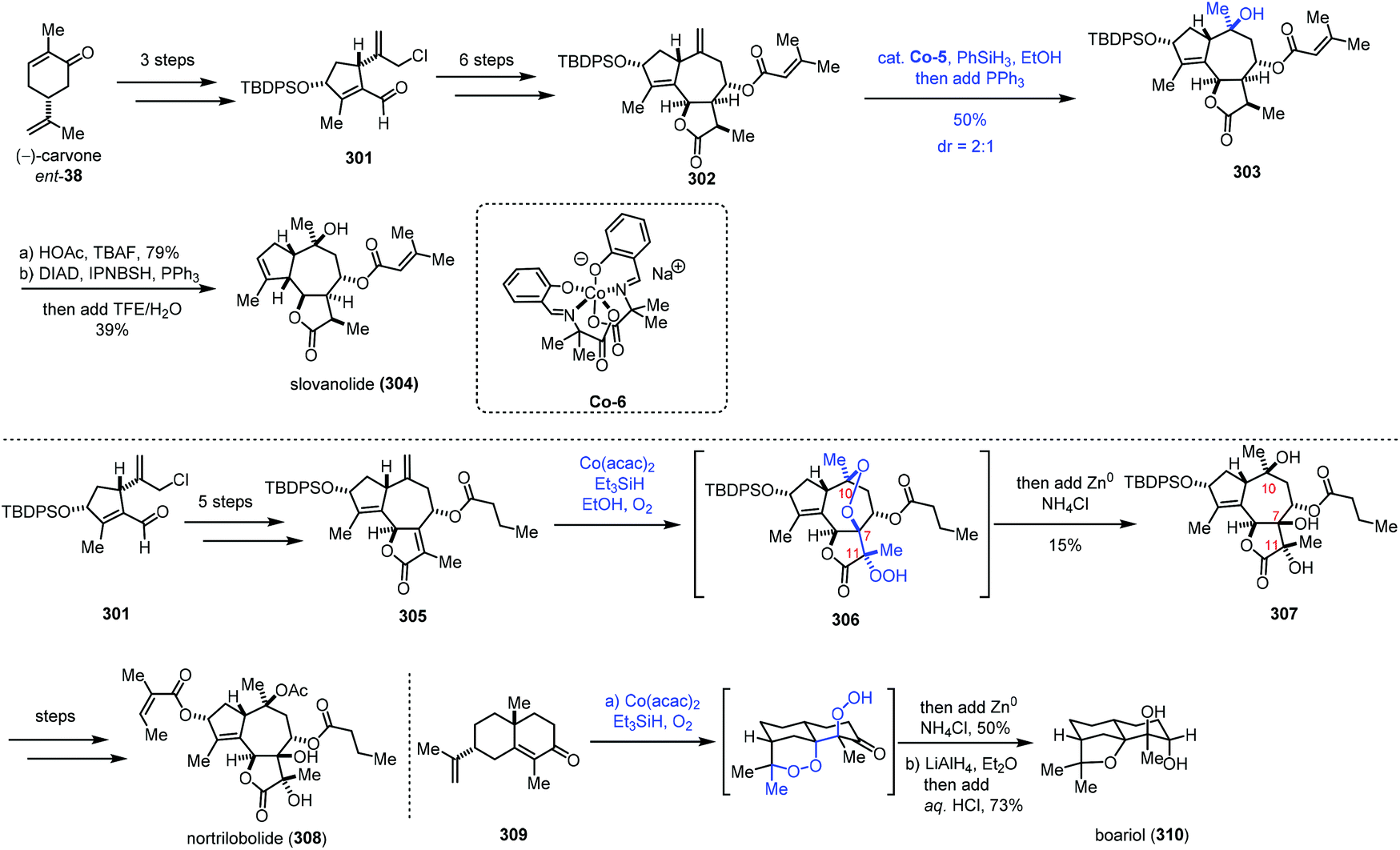 | ||
| Scheme 38 Maimone's synthesis of complex guaianolide sesquiterpenes. | ||
301 was prepared from carvone in three steps. Then 301 was converted to the 5,7,5-fused ring system 302 through a six-step sequence. Under MHAT conditions of a Co complex, PhSiH3 and PPh3 in EtOH, the exocyclic olefin was transformed to the corresponding tertiary alcohol, furnishing 303. Deprotection and reductive allylic transposition were conducted to give slovanolide (304). Regarding the synthesis of heavily oxidized nortrilobolide (308), they prepared 305 from 301 at first. Inspired by their synthesis of cardamom peroxide,59 the authors envisioned that a polyoxygenation cascade with molecular oxygen may be applicable to secure the requisite hydroxyl groups at C10, C7, and C11 of nortrilobolide. Subjecting 305 to Co-catalyzed Mukaiyama hydroperoxidation conditions, followed by reduction with metal Zn provided 307 in 15% yield. Although the yield of the step is low, the cascade process is rather impressive as three hydroxyl groups were installed with the desired configurations in a single operation. The authors noted that any variation of these conditions did not lead to desired 307. Based on the experimental results, the authors found that the major side reactions are premature reduction of the first peroxy radical intermediate and formation of an unstable peroxide. Lastly, 307 could be transformed to nortrilobolide (308). Maimone and co-workers also completed the synthesis of boariol via a similar radical process.
In 2021, Sarpong reported divergent syntheses of cephanolides A–D via the common intermediate 316 (Scheme 39).64 They employed Co-catalyzed radical hydroperoxidation to achieve olefin functionalization. The indanone derivative 312 was created from 7-hydroxy-4-methylindanone (311). Exposure of 312 to two equivalents of TMSOTf induced Diels–Alder cycloaddition conditions to provide the core skeleton 313. Functionalization of the bridging olefin group of 313 proved unfruitful. Due to the electron deficiency of olefins, conventional methods such as hydroboration and epoxidation all failed. Thus the authors turned their attention to the MHAT process. A modified Mukaiyama hydration process by Inoue was feasible (Co(thd)2, O2, Et3SiH, and TBHP), giving ketone 315 in 42% yield after treatment of the mixture with DBU. The authors proposed that the regioselectivity of the hydrocobaltation likely correlates with the proximal oxygen lone pair of the lactone. Next, olefination with a combination of Ti(i-PrO)2Cl2 and Nysted reagent gave 316, which could be elaborated to cephanolides A–D, respectively.
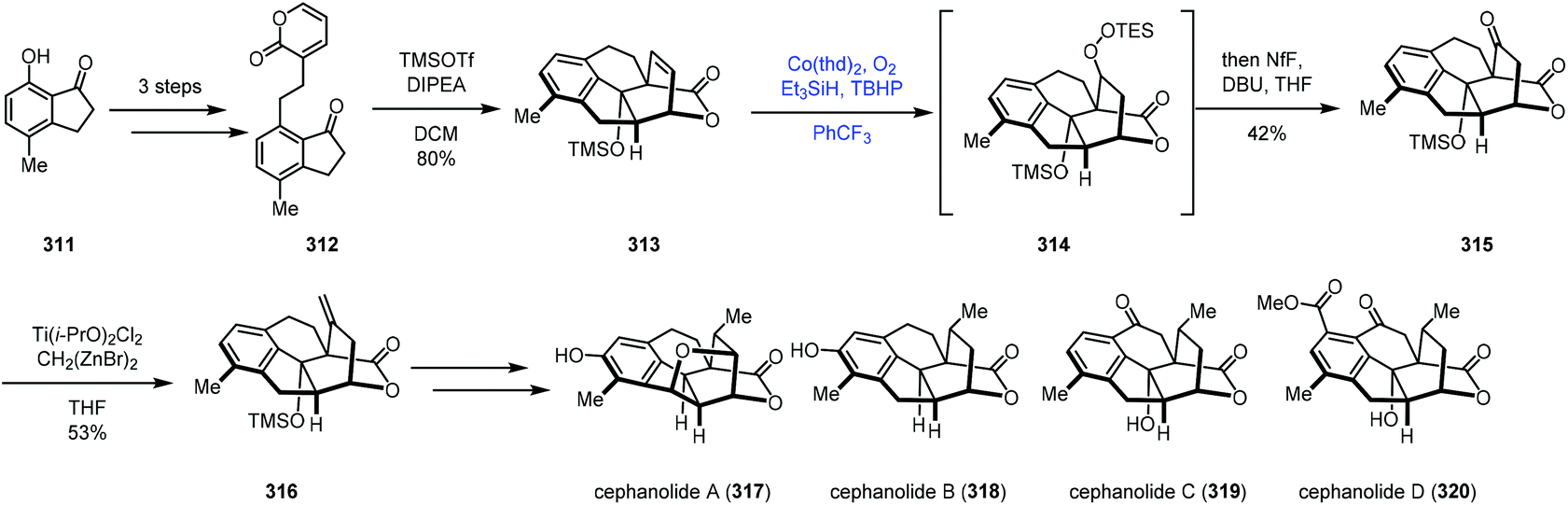 | ||
| Scheme 39 Sarpong's synthesis of cephanolides A–D. | ||
4. Hydrogenation
In the process of studying the hydration reaction of olefins with Co catalysts in secondary alcohols in 1989, Mukaiyama noted that the hydrogenation product was obtained as the minor product.2 In 2000, Magnus discovered that under the conditions of Mn(dpm)3/phenylsilane with the exclusion of air, α,β-unsaturated ketones could be transformed to saturated ketones.65 In 2014, Shenvi5a and Herzon6 independently reported using MHAT to achieve hydrogenation of unactivated olefins or alkenyl halides, greatly expanding the substrate scope. Since then, hydrogenation of olefins under MHAT conditions has been serving as an alternative to the conventional methods, and has been widely used in organic synthesis.In their synthesis of hippolachnin A (326) in 2015, Carreira tested MHAT-based hydrogenation of exocyclic double bonds (Scheme 40).66 Cyclopentenone 321 could be transformed to the bicyclic compound 322via a five-step sequence. An ene cyclization with BF3·Et2O furnished 323. When Mn(dpm)3 or Co(acac)2 or Fe(acac)3 was used as the catalyst in the presence of PhSiH3, they found that the undesired thermodynamic product 324 predominated. In contrast, the use of Pearlman's catalyst under a H2 atmosphere gave a mixture of the desired kinetic product 325 and 324 (dr = 3:1), favouring 325. Hippolachnin A (326) could be obtained from 325 in two steps.
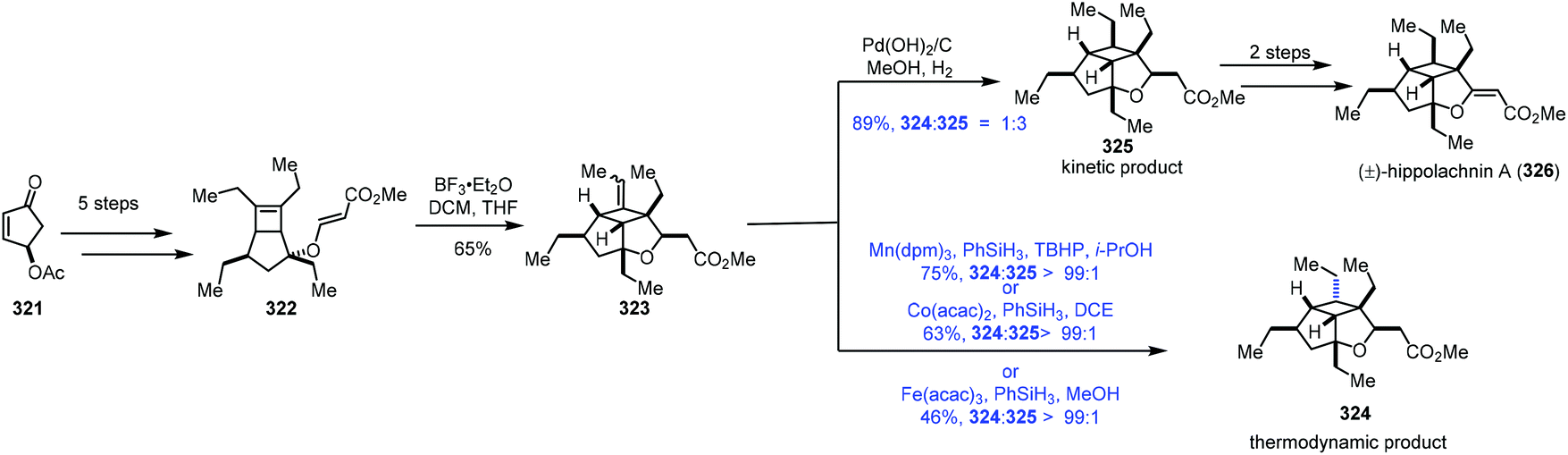 | ||
| Scheme 40 Carreira's synthesis of hippolachnin A (326). | ||
In 2016, Luo and co-workers finished the synthesis of various iboga alkaloids,67 in which iron-catalyzed HAT was performed to achieve hydrogenation and hydration of alkenes (Scheme 41). Starting from tryptamine, tertiary amine 327 was obtained in seven steps, which could be transformed to 330via gold-catalyzed oxidation, alkylation and stevens rearrangement. The Wittig reaction furnished alkene 331, and the double bond could be reduced to ibogamine (332) and epiibogamine (333) via Fe-catalyzed HAT-based hydrogenation, slightly preferring undesired 333. Manganese and cobalt-based catalysts were inferior in this case since 333 was afforded as the predominant product with these catalysts. Moreover, activated Pd/C and H2 conditions also gave 333 in high yield. In the synthesis of vinblastine (337), the advanced intermediate 334 was furnished from L-tryptophan in eight steps. Wittig reaction of 334 furnished the fragment 335. Using the conditions reported by Boger and co-workers,8 Fe(III)-promoted coupling of 335 with the commercially available vindoline 336 gave the coupled product, which was treated with an Fe(III)-NaBH4/O2 solution to selectively hydrate the resulting trisubstituted alkene and reduce the iminium ion, providing vinblastine (337) in 50% yield.
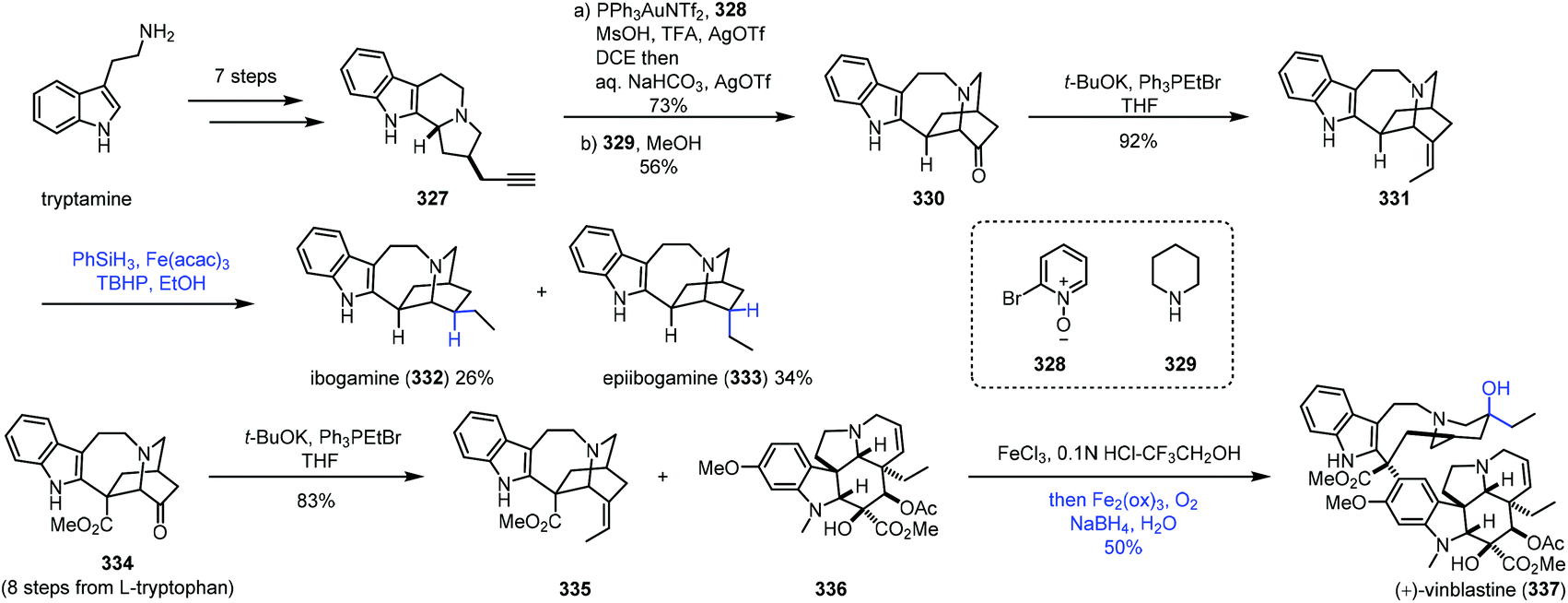 | ||
| Scheme 41 Luo's synthesis of various iboga alkaloids. | ||
In Reisman's synthesis of pleuromutilin,68 regarding the HAT-based hydrogenation step, an intramolecular abstraction of protons to rationalize the formation mechanism of the unexpected product 345 was proposed (Scheme 42). Bicycle 339 was synthesized from enone 338 in eight steps. Asymmetric allylboration with Z-340 gave desired 341, which could be transformed to 342 by a three-step sequence. An eight-membered ring was formed through a SmI2-promoted cyclization, delivering tricyclic 343. Enol ether 344 was then prepared. To finish the synthesis of pleuromutilin, chemoselective reduction of the C10–C17 exocyclic olefin was required. However, under the standard hydrogenation conditions with a cationic transition metal, hydrogenation of the more accessible C19–C20 vinyl group occurred. Thus the authors turned to MHAT reactions. HAT-type hydrogenation of 344 under the conditions Mn(dpm)3/PhSiH3/TBHP/i-PrOH led to unexpected 345 in 55% yield, in which the C10–C17 olefin was reduced and C14 alcohol was oxidized to ketone. Only trace amounts of products arising from C19–C20 vinyl reduction were noticed, which is in stark contrast to the standard hydrogenation conditions with a cationic transition metal. The authors thus proposed that a transannular [1,5]-HAT process occurs and cleavage of O–H bonds to form a ketone is a driving force of this conversion. The hypothesis is supported by deuterium-labeling experimental results. Besides, HAT-type hydrogenation of the protected C14 alcohol substrate gave the corresponding C10 alcohol as a mixture of diastereomers with only 6–10% conversion. Finally, pleuromutilin (346) was obtained after reduction, acylation and deprotection.
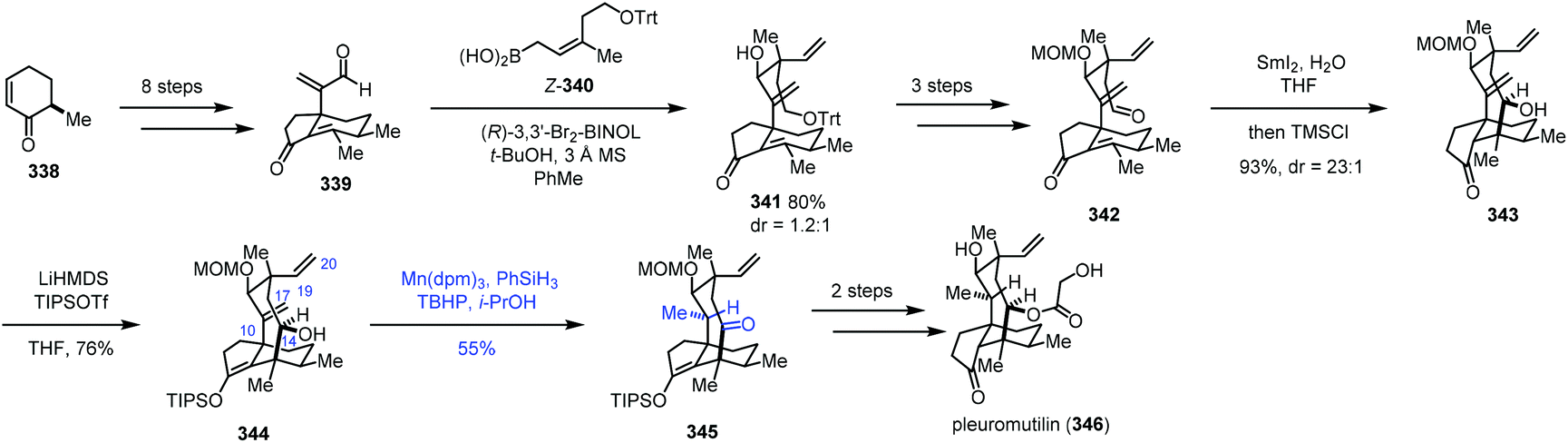 | ||
| Scheme 42 Reisman's synthesis of pleuromutilin (346). | ||
In 2019, Snyder and co-workers reported the synthesis of arborisidine (Scheme 43).69 They conducted a Mn-catalyzed hydrogenation process to realize the 1,4-reduction of dienoate. In their synthesis, the racemic 348 was obtained from tryptamine (353) via Pictet–Spengler reaction, while an enantioselective synthesis of 348 was achieved by a three-step sequence from D-tryptophan methyl ester. 348 was then advanced to tetracyclic dienoate 349. Conventional dienoate hydrogenation and conjugate reduction methods failed with 349. Under such circumstances, they tried radical hydrogenation conditions with Mn(dpm)3/PhSiH3, and the desired 1,4-reduction product 350 could be produced. The optimization results showed that the best conversion required using Mn(dpm)3 in a 50% loading, excess amounts of PhSiH3 and trace air as the activator of the catalyst. Arborisidine (352) was thus synthesized after lactamization and functional group conversions.
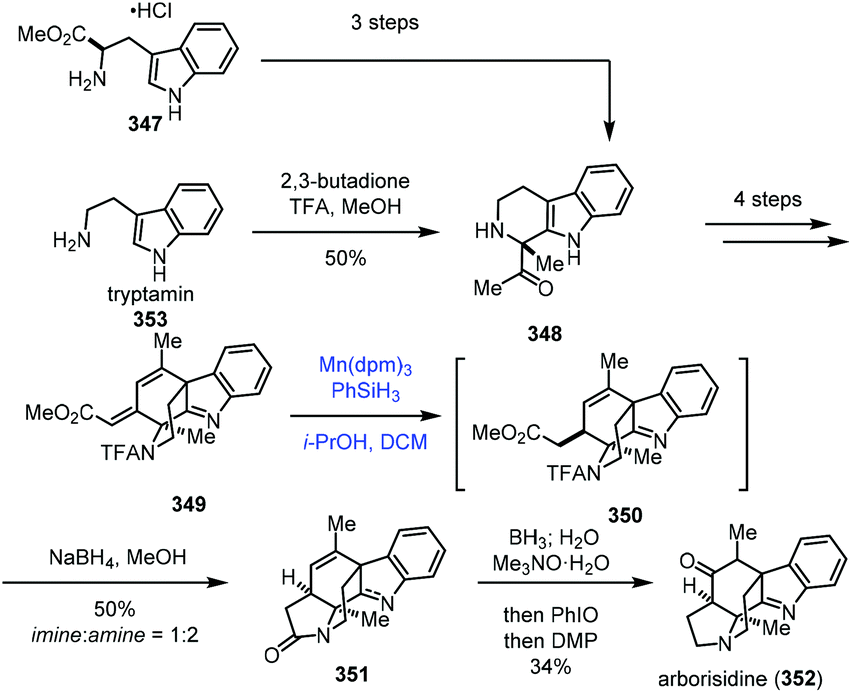 | ||
| Scheme 43 Snyder's synthesis of arborisidine (352). | ||
In Yang's synthesis of waihoensene (359) in 2020 (Scheme 44),70 it is interesting to note that HAT-type hydrogenation gave a higher yield of the desired product with the tetracyclic substrate 357. The tetracyclic substrate 357 was synthesized from enone 354 through Conia-ene type cyclization and Pauson–Khand reaction. Conventional hydrogenation with common metal catalysts led to an undesired diastereomer. HAT-type hydrogenation with Fe(acac)3 as the catalyst, in contrast, generated desired 358 in 75% yield. The authors envisioned that the resulting C9 radical from 357 would abstract a proton from the proximal C3 next to the C2 carbonyl group through an intramolecular HAT, and the proposed pathway might account for the high diastereoselective reduction. They also conducted a density functional theory (DFT) experiment to further rationalize the observed selectivity. Waihoensene (359) was then furnished via methylation and methylenation.
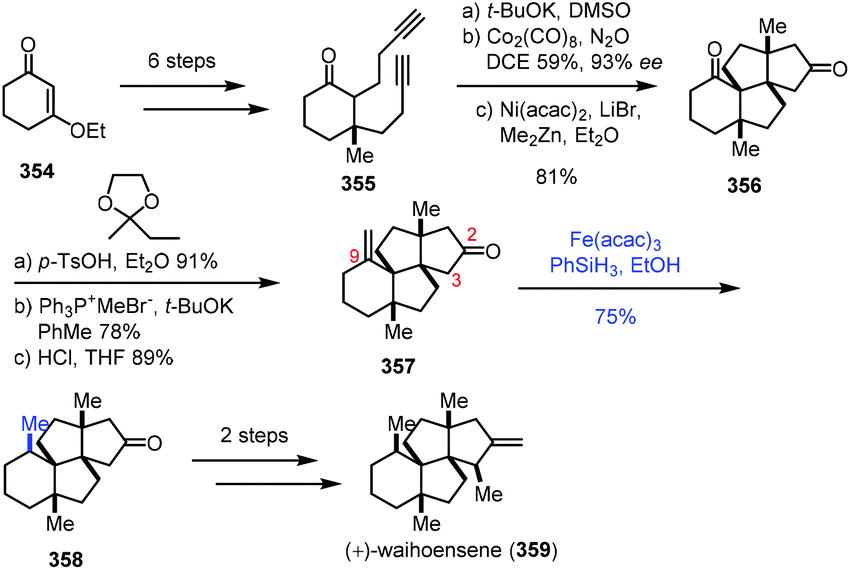 | ||
| Scheme 44 Yang's synthesis of waihoensene (359). | ||
In 2016, Shenvi and co-workers accomplished the synthesis of (+)-7,20-diisocyanoadociane (Scheme 45).71 They employed Mn-catalyzed radical hydrogenation of a conjugated tetrasubstituted olefin to access the thermodynamic product. Starting materials 360 and 361 were obtained from dioxenone and 4-bromo-2-butynone, respectively. An intermolecular Diels–Alder cycloaddition between 360 and 361 with Cu(OTf)2 furnished 362, which was transformed to tricyclic 363 by an intramolecular Diels–Alder cycloaddition at 180 °C and removal of the auxiliary at 200 °C. Methylation generated the corresponding alcohol. Upon exposure of the alcohol to a Pd, Pt, or Rh catalyst, undesired deconjugation occurred. They eventually found that the hydrogenation of the highly strained alkene could be realized under MHAT conditions (Mn(dpm)3, TBHP, PhSiH3, and i-PrOH), delivering the thermodynamic product 364 in 51% yield as the major isomer. Tricyclic 364 was advanced to (+)-7,20-diisocyanoadociane (366) in eight steps.
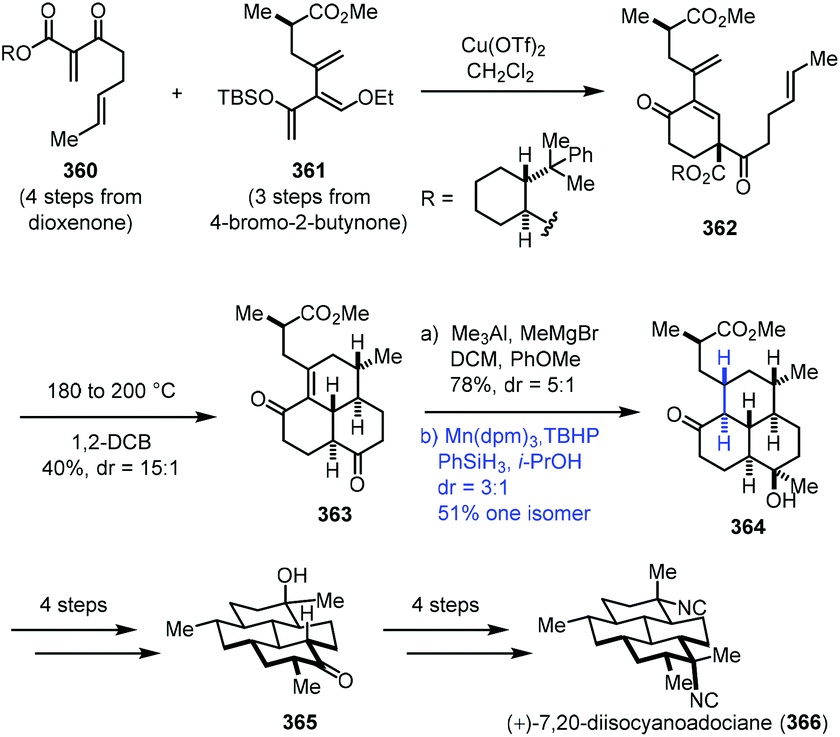 | ||
| Scheme 45 Shenvi's synthesis of (+)-7,20-diisocyanoadociane (366). | ||
In the synthesis of aplydactone in 2017,72 Zhang and co-workers employed a strategy to realize hydrogenation of vinyl bromide (Scheme 46). Coupling of 367 and 368 led to 369. Irradiation of 369 afforded tricyclic 370via [2 + 2] photocycloaddition, which was converted to 371 in seven steps. Transannular C–H insertion was performed to form the skeletal structure of aplydactone 373 as the minor product. Traditional hydrogenation conditions with Pd/C or PtO2 as catalysts under H2 turned out to be ineffective for the reduction of vinyl bromide of 373. To their delight, subjecting 373 to Shenvi's conditions5b afforded uneventfully aplydactone (374) and its C8-epimer 375 in 43 and 28% yield, respectively.
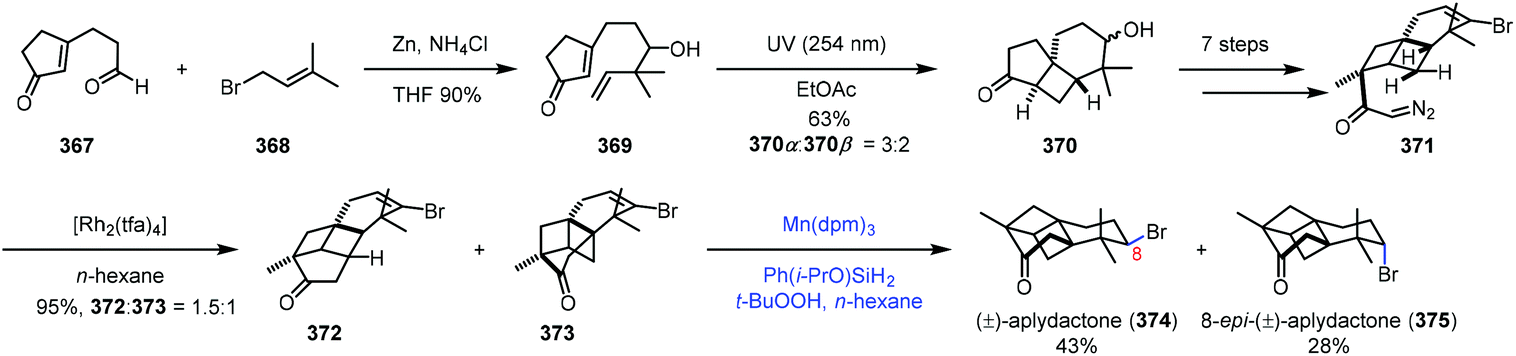 | ||
| Scheme 46 Zhang's synthesis of aplydactone (374). | ||
In 2018, Dethe completed the synthesis of (+)-taondiol (381) (Scheme 47).73 Enone 376 was prepared through ketal protection and Robinson-type annulation. Treatment of 376 with a four-step sequence gave 377. Under MHAT conditions with Mn(dpm)3 and phenylsilane, hydrogenation of the olefin was achieved to give 378. It should be noted that the usual hydrogenation conditions with Pd/C and H2 were able to reduce the olefin; however, undesired debenzylation occurred concomitantly. Moving forward, Friedel–Crafts reaction of 379 and 383 with BF3·Et2O was carried out to attain pentacyclic 380, which was then advanced to (+)-taondiol (381).
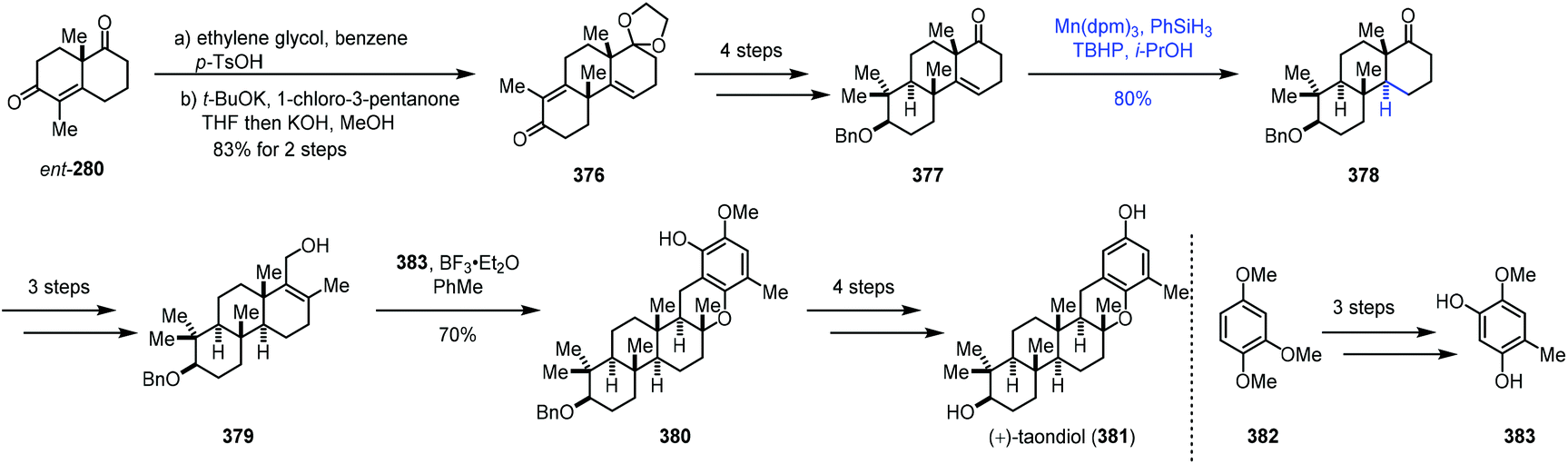 | ||
| Scheme 47 Dethe's synthesis of (+)-taondiol (381). | ||
Using the MHAT-based hydrogenation approach, Krische and co-workers completed the synthesis of andrographolide (Scheme 48).74 Diene 385 was attained from 5-hydroxy-2-pentanone (384) in five steps. Diels–Alder cycloaddition of diene 385 and dimethyl acetylene dicarboxylate followed by hydrolysis gave diol 387. Chemo- and stereoselective reduction of the alkene was achieved by the Mn-catalyzed hydrogenation reaction in the presence of phenylsilane to give trans-decalin 388 in 63% yield. In comparison, a cis-decalin product was exclusively formed upon subjecting acetonide 386 to identical reaction conditions. Andrographolide (389) was obtained from 388 in seven steps.
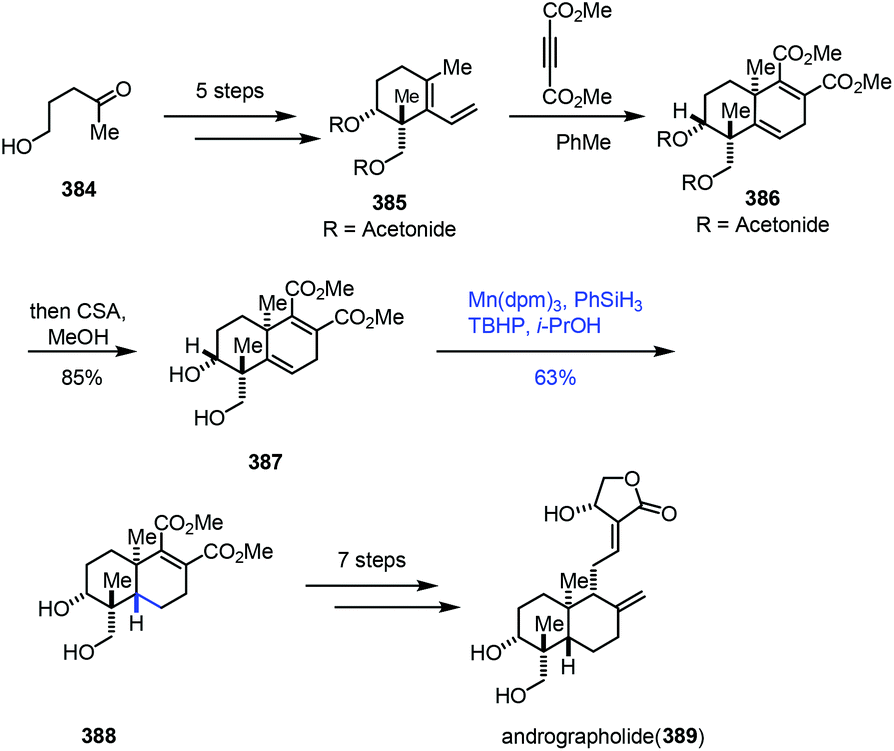 | ||
| Scheme 48 Krische's synthesis of andrographolide (389). | ||
In 2020, Renata and co-workers applied biocatalytic C–H oxidations and MHAT reactions as the main methods to achieve the synthesis of eight meroterpenoid natural products (Scheme 49).75 The key common intermediate 391 was obtained from sclareolide 390 by a five-step sequence, featuring biocatalytic C3-hydroxylation. A formal [3 + 3] union of 391 and 392 led to 394. Under the conventional hydrogenation conditions, with Rh, Pt and Pd as catalysts, the reductions occurred at the pyrone moiety. The authors reasoned that MHAT-based reactions would be feasible. The bulky metal catalyst might suppress the reduction of the tetrasubstituted alkene. Among two trisubstituted alkenes, reduction of the C9–C11 alkene would be preferred due to the greater stability of the resulting C9 tertiary radical. Besides, Shenvi discovered that MHAT-based hydrogenation could provide the thermodynamically stable trans-decalin product. In practice, the authors discovered that arisugacin F (395) could be accessed by Mn-catalyzed HAT-based hydrogenation (Mn(dpm)3, TBHP, and PhSiH3) in 87% yield. In addition, HAT-based intramolecular Giese coupling served as the key step in the synthesis of decaturin E (399), in which the trans-decalin product 398 was afforded from 397 with complete diastereoselectivity and excellent yield under the conditions of Fe(acac)3 and PhSiH3.
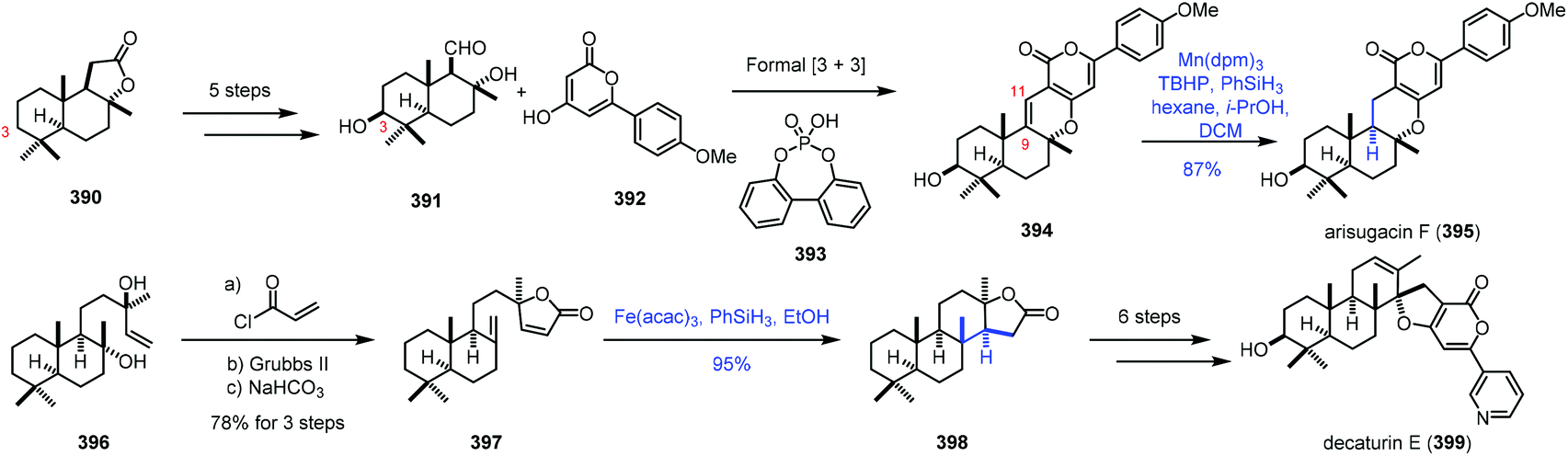 | ||
| Scheme 49 Renata's synthesis of oxidized meroterpenoids. | ||
In 2021, Reisman reported the synthesis of ritterazine B (Scheme 50).76 The advanced diene intermediate 401 was prepared in twelve steps from trans-dehydroandrosterone 400. MHAT-based reduction of the C5–C6 and C14–C15 alkenes of 401 proceeded well to give rise to 402 after oxidation. In this case, cis-fusion at the C/D ring-junction would be a thermodynamically stable configuration as supported by DFT studies. The authors found that it was difficult to reduce the C14–C15 double bond under conventional hydrogenation conditions. Ritterazine B (405) was accessed by heterodimerization of two advanced fragments 403 and 404 and deprotection.
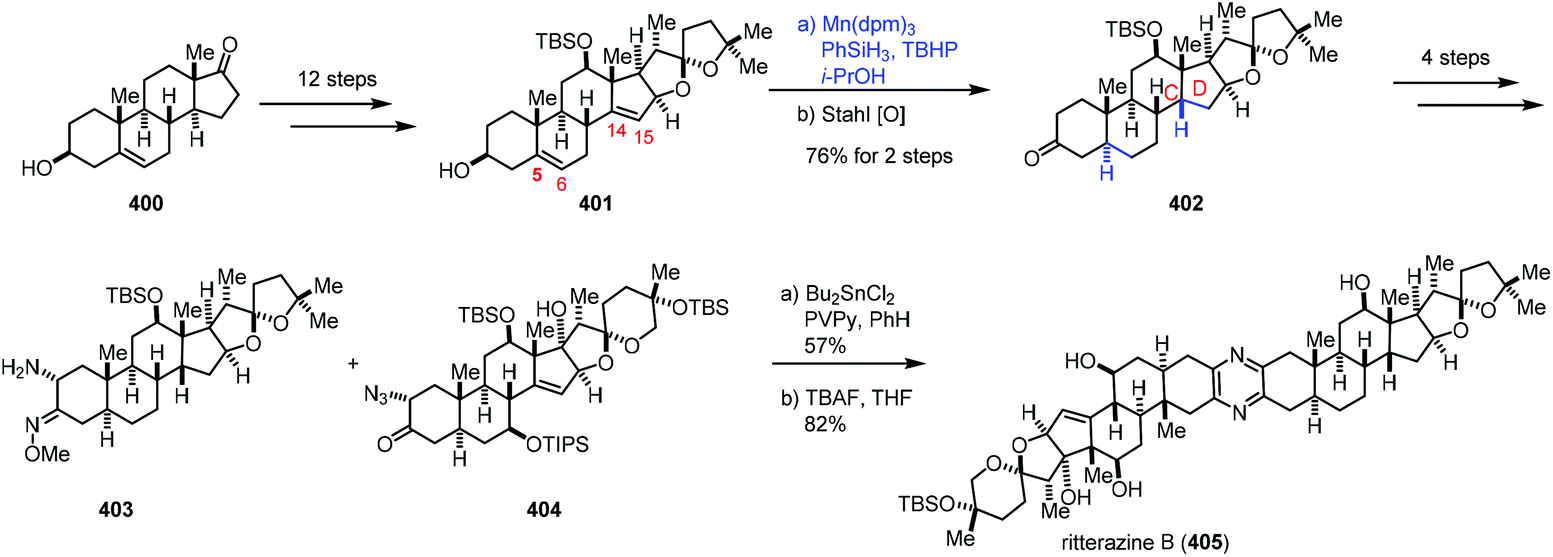 | ||
| Scheme 50 Reisman's synthesis of oxidized meroterpenoids. | ||
5. Isomerization
The isomerization of olefins is a redox neutral process, which could avoid surplus oxidation state manipulation. Mechanistically, the isomerization of olefins mainly proceeds with two different pathways: either a metal hydrogen mechanism or a π-allyl mechanism.77,78 However, the isomerization of olefins through MHAT follows a different pathway. In 2014, Holland reported Co-catalyzed isomerization of terminal alkenes to internal Z-alkenes via most likely the metal hydrogen mechanism.79 In 2017, Shenvi reported that Co-catalyzed HAT was used to isomerize a terminal alkene to an internal olefin by one position.5aMetz and co-workers subsequently employed Shenvi's method in their synthesis of 3β-hydroxy-7β-kemp-8(9)-en-6-one (416) in 2017 (Scheme 51).80 The synthesis commenced with Wieland–Miescher ketone 406, which was transformed to 407via chemoselective thioketalization, alkylation and epimerization. The carbonyl group underwent olefination to give compound 409, followed by acid hydrolysis to lead to 410. Prenylation provided 411. A ten-step sequence provided ketone 412. An olefin metathesis with Grubbs catalyst 413 gave tetracyclic dienol 414, which could be transformed to 415via a seven-step sequence. Upon treatment of 415 with aqueous hydrogen peroxide followed by heating the resulting mixture, 416 and the exocyclic olefin 417 were isolated. Under the conditions of the cobalt complex Co-4 and PhSiH3, the exocyclic double bond of 417 could be isomerized to an endocyclic double bond to provide 416 in 64% yield. It should be mentioned that 417 could be used to produce 415 through Co-catalyzed Markovnikov addition of thiophenol.
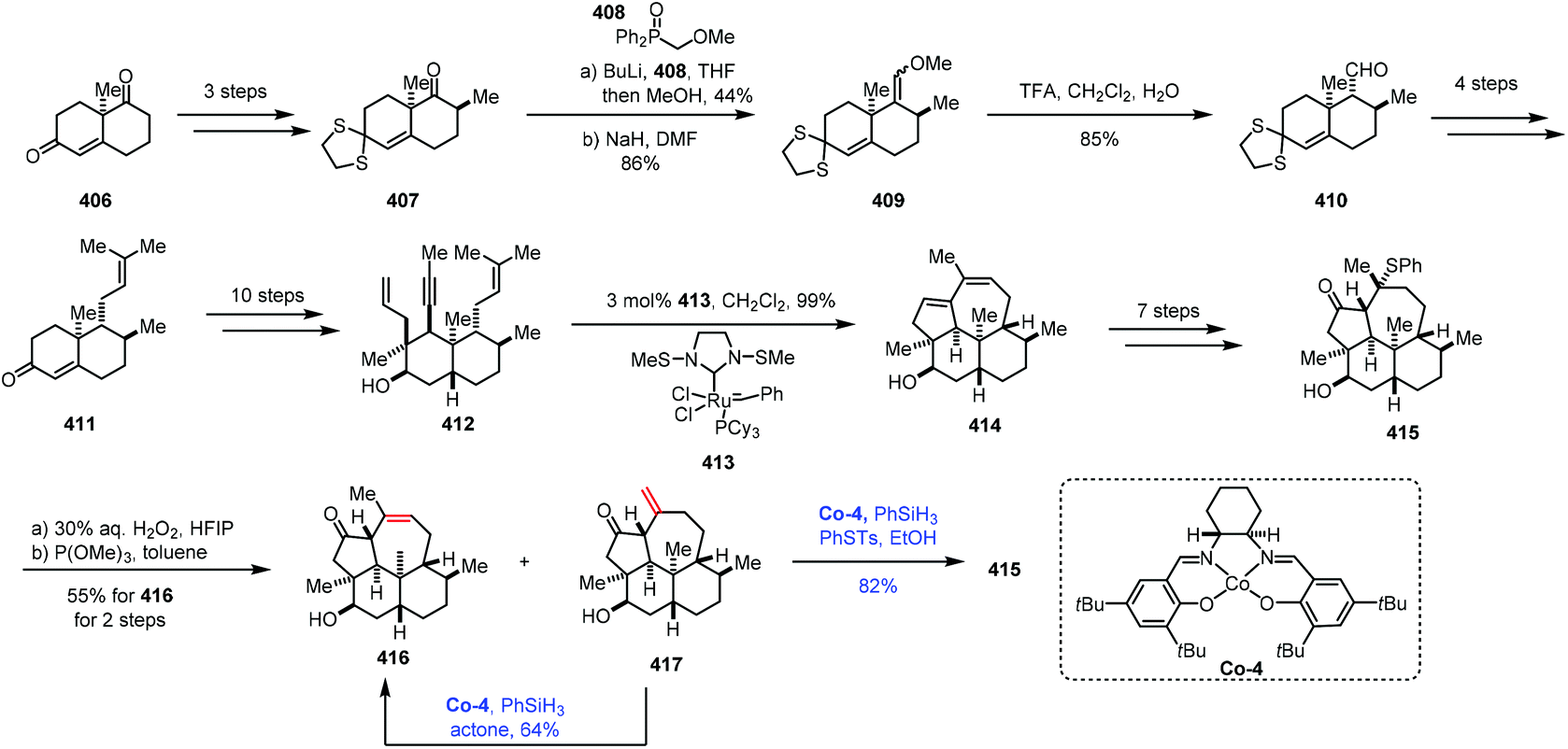 | ||
| Scheme 51 Metz's synthesis of 3β-hydroxy-7β-kemp-8(9)-en-6-one (416). | ||
6. Conclusion
The metal-hydride hydrogen atom transfer (MHAT) reaction is a powerful method to functionalize olefins. Here we summarized the applications of MHAT in natural product synthesis. As demonstrated by the aforementioned examples, in many cases, MHAT reactions occur under mild conditions, with high chemoselectivity and high tolerance of functional groups. Besides, Fe, Co, and Mn, the often used metal catalysts in MHAT reactions, are relatively cheap. Based on the aforementioned examples, presumably due to the different reactivities of the metal catalysts, reductive coupling reactions like the Giese reaction and reductive coupling with carbonyl or cyano groups often involve the use of Fe complexes. Hydrogenations use both Mn and Fe metals. With respect to the redox neutral process and hydration and hydroperoxidation reactions, Co and Mn complexes are usually adopted. In particular, Co complexes are the preferred catalysts in hydroarylation and isomerization reactions.Many reports have shown that C–N or C–X bonds could be formed by MHAT-induced reactions, such as Carreira's hydrohydrazination and Shigehisa's intramolecular hydroamination. However, regarding synthetic applications, there are just limited examples, indicating that in-depth application of these newly developed methods in complex molecule synthesis needs to be exploited in future. Furthermore, a few examples like Liu's synthesis of hispidanin A and Vanderwal's synthesis of plebedipene B have demonstrated the power of MHAT-based polyene cyclization. As such, more sophisticated designs of the MHAT-based cascade strategy in polycyclic or caged architectures, in which multiple bonds could be forged in a single operation, are highly expected.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (21871098, 22071064, and 21672073), the National Program on Key Research Project (2016YFA0602900), Guangdong Province Science Foundation (2017B090903003), the Science and Technology Program of Guangzhou (201707010073), and the Fundamental Research Funds for the Central Universities, SCUT for the financial support.References
- I. Tabushi and N. Koga, P-450 Type Oxygen Activation by Porphyrin-Manganese Complex, J. Am. Chem. Soc., 1979, 101, 6456 CrossRef CAS.
- (a) T. Mukaiyama, S. Isayama, S. Inoki, K. Kato, T. Yamada and T. Takai, Oxidation-Reduction Hydration of Olefins with Molecular Oxygen and 2-Propanol Catalyzed by Bis(acetylacetonato)cobalt(II), Chem. Lett., 1989, 18, 449–452 CrossRef; (b) S. Inoki, K. Kato, T. Takai, S. Isayama, T. Yamada and T. Mukaiyama, Bis(trifluoroacetylacetonato)cobalt(II) Catalyzed Oxidation-Reduction Hydration of Olefins Selective Formation of Alcohols from Olefins, Chem. Lett., 1989, 18, 515 CrossRef.
- S. Isayama and T. Mukaiyama, Hydration of Olefins with Molecular Oxygen and Triethylsilane Catalyzed by Bis(trifluoroacetylacetonato)cobalt(II), Chem. Lett., 1989, 18, 569 CrossRef.
- (a) J. Waser and E. M. Carreira, Convenient Synthesis of Alkylhydrazides by the Cobalt-Catalyzed Hydrohydrazination Reaction of Olefins and Azodicarboxylates, J. Am. Chem. Soc., 2004, 126, 5676 CrossRef CAS PubMed; (b) J. Waser and E. M. Carreira, Catalytic Hydrohydrazination of a Wide Range of Alkenes with a Simple Mn Complex, Angew. Chem., Int. Ed., 2004, 43, 4099 CrossRef CAS PubMed; (c) J. Waser, H. Nambu and E. M. Carreira, Cobalt-Catalyzed Hydroazidation of Olefins: Convenient Access to Alkyl Azides, J. Am. Chem. Soc., 2005, 127, 8294 CrossRef CAS PubMed; (d) J. Waser, B. Gaspar, H. Nambu and E. M. Carreira, Hydrazines and Azides via the Metal-Catalyzed Hydrohydrazination and Hydroazidation of Olefins, J. Am. Chem. Soc., 2006, 128, 11693 CrossRef CAS PubMed; (e) B. Gaspar and E. M. Carreira, Mild Cobalt-Catalyzed Hydrocyanation of Olefins with Tosyl Cyanide, Angew. Chem., Int. Ed., 2007, 46, 4519 CrossRef CAS PubMed; (f) B. Gaspar and E. M. Carreira, Catalytic Hydrochlorination of Unactivated Olefins with para-Toluenesulfonyl Chloride, Angew. Chem., Int. Ed., 2008, 47, 5758 CrossRef CAS PubMed; (g) B. Gaspar and E. M. Carreira, Cobalt Catalyzed Functionalization of Unactivated Alkenes: Regioselective Reductive C-C Bond Forming Reactions, J. Am. Chem. Soc., 2009, 131, 13214 CrossRef CAS PubMed.
- (a) S. W. M. Crossley, F. Barabé and R. A. Shenvi, Simple, Chemoselective, Catalytic Olefin Isomerization, J. Am. Chem. Soc., 2014, 136, 16788 CrossRef CAS PubMed; (b) K. Iwasaki, K. K. Wan, A. Oppedisano, S. W. M. Crossley and R. A. Shenvi, Simple, Chemoselective Hydrogenation with Thermodynamic Stereocontrol, J. Am. Chem. Soc., 2014, 136, 1300 CrossRef CAS PubMed.
- S. M. King, X.-S. Ma and S. B. Herzon, A Method for the Selective Hydrogenation of Alkenyl Halides to Alkyl Halides, J. Am. Chem. Soc., 2014, 136, 6884 CrossRef CAS PubMed.
- (a) J. C. Lo, J.-H. Gui, Y. Yabe, C. M. Pan and P. S. Baran, Functionalized olefin cross-coupling to construct carbon––carbon bonds, Nature, 2014, 516, 343 CrossRef CAS PubMed; (b) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, Radicals: Reactive Intermediates with Translational Potential, J. Am. Chem. Soc., 2016, 138, 12692 CrossRef CAS PubMed; (c) J. C. Lo, Y. Yabe and P. S. Baran, A Practical and Catalytic Reductive Olefin Coupling, J. Am. Chem. Soc., 2014, 136, 1304 CrossRef CAS PubMed.
- (a) H. Ishikawa, D. A. Colby, S. Seto, P. Va, A. Tam, H. Kakei, T. J. Rayl, I. Hwang and D. L. Boger, Total Synthesis of Vinblastine, Vincristine, Related Natural Products, and Key Structural Analogues, J. Am. Chem. Soc., 2009, 131, 4904 CrossRef CAS PubMed; (b) J. E. Sears and D. L. Boger, Total Synthesis of Vinblastine, Related Natural Products, and Key Analogues and Development of Inspired Methodology Suitable for the Systematic Study of Their Structure−Function Properties, Acc. Chem. Res., 2015, 48, 653 CrossRef CAS PubMed.
- (a) S. W. M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins, Chem. Rev., 2016, 116, 8912 CrossRef CAS PubMed; (b) S. A. Green, S. W. M. Crossley, J. L. M. Matos, S. Vasquez-Cespedes, S. L. Shevick and R. A. Shenvi, The High Chemofidelity of Metal-Catalyzed Hydrogen Atom Transfer, Acc. Chem. Res., 2018, 51, 2628 CrossRef CAS PubMed.
- (a) S. H. Gao and Y. Y. Qiu, Advances of Radical and Photo Reactions in Natural Products Synthesis, Sci. China: Chem., 2016, 59, 1093 CrossRef CAS; (b) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, Radicals: Reactive Intermediates with Translational Potential, J. Am. Chem. Soc., 2016, 138, 12692 CrossRef CAS PubMed; (c) J. E. Zweig, D. E. Kim and T. R. Newhouse, Methods Utilizing First-Row Transition Metals in Natural Product Total Synthesis, Chem. Rev., 2017, 117, 11680 CrossRef CAS PubMed; (d) S. L. Shevick, C. V. Wilson, S. Kotesova, D. Kim, P. L. Holland and R. A. Shenvi, Catalytic Hydrogen Atom Transfer to Alkenes: A Roadmap for Metal Hydrides and Radicals, Chem. Sci., 2020, 11, 12401 RSC; (e) A. Simonneau and M. Oestreich, Fascinating Hydrogen Atom Transfer Chemistry of Alkenes Inspired by Problems in Total Synthesis, Angew. Chem., Int. Ed., 2015, 54, 3556 CrossRef CAS PubMed.
- B. Giese and J. Meister, Die Addition von Kohlenwasserstoffen an Olefine Eine neue synthetische Methode, Chem. Ber., 1977, 110, 2588 CrossRef CAS.
- T. Taniguchi, N. Goto, A. Nishibata and H. Ishibashi, Iron-Catalyzed Redox Radical Cyclizations of 1,6-Dienes and Enynes, Org. Lett., 2010, 12, 112 CrossRef CAS PubMed.
- D. T. George, E. J. Kuenstner and S. V. Pronin, A Concise Approach to Paxilline Indole Diterpenes, J. Am. Chem. Soc., 2015, 137, 15410 CrossRef CAS PubMed.
- G. Xu, M. Elkin, D. J. Tantillo, T. R. Newhouse and T. J. Maimone, Traversing Biosynthetic Carbocation Landscapes in the Total Synthesis of Andrastin and Terretonin Meroterpenes, Angew. Chem., Int. Ed., 2017, 56, 12498 CrossRef CAS PubMed.
- C. P. Ting, G. Xu, X. Zeng and T. J. Maimone, Annulative Methods Enable a Total Synthesis of the Complex Meroterpene Berkeleyone A, J. Am. Chem. Soc., 2016, 138, 14868 CrossRef CAS PubMed.
- H. Shigehisa, T. Aoki, S. Yamaguchi, N. Shimizu and K. Hiroya, Hydroalkoxylation of Unactivated Olefins with Carbon Radicals and Carbocation Species as Key Intermediates, J. Am. Chem. Soc., 2013, 135, 10306 CrossRef CAS PubMed.
- T. Amatov, R. Pohl, I. Cisařová and U. Jahn, Sequential Oxidative and Reductive Radical Cyclization Approach toward Asperparaline C and Synthesis of Its 8-Oxo Analogue, Org. Lett., 2017, 19, 1152 CrossRef CAS PubMed.
- Z. H. Lu, X. Zhang, Z. C. Guo, Y. Chen, T. Mu and A. Li, Total Synthesis of Aplysiasecosterol A, J. Am. Chem. Soc., 2018, 140, 9211 CrossRef CAS PubMed.
- P. F. Hu, H. M. Chi, K. C. DeBacker, X. Gong, J. H. Keim, I. T. Hsu and S. A. Snyder, Quaternary-Centre-Guided Synthesis of Complex Polycyclic Terpenes, Nature, 2019, 569, 703 CrossRef CAS PubMed.
- G. Xu, J. Wu, L. Li, Y. Lu and C. Li, Total Synthesis of (−)-Daphnezomines A and B, J. Am. Chem. Soc., 2020, 142, 15240 CrossRef CAS PubMed.
- X. H. Zeng, V. Shukla and D. L. Boger, Divergent Total Syntheses of (−)-Pseudocopsinine and (−)-Minovincinine, J. Org. Chem., 2020, 85, 14817 CrossRef CAS PubMed.
- P. Q. Chen, C. Wang, R. Yang, H. J. Xu, J. H. Wu, H. F. Jiang, K. Chen and Z. Q. Ma, Asymmetric Total Synthesis of Dankasterones A and B and Periconiastone A through Radical Cyclization, Angew. Chem., Int. Ed., 2021, 60, 5512 CrossRef CAS PubMed.
- Y. Hu, M. Bai, Y. Yang, J. Y. Tian and Q. H. Zhou, Rapid Access to Tetracyclic Core of Wortmannin via an Intramolecular Reductive Olefin Coupling Strategy, Org. Lett., 2020, 22, 6308 CrossRef CAS PubMed.
- H. P. Deng, W. Cao, R. Liu, Y. H. Zhang and B. Liu, Asymmetric Total Synthesis of Hispidanin A, Angew. Chem., Int. Ed., 2017, 56, 5849 CrossRef CAS PubMed.
- W. Cao, H. P. Deng, Y. Sun, B. Liu and S. Qin, Asymmetric Synthesis of Hispidanin A and Related Diterpenoids, Chem. – Eur. J., 2018, 24, 9120 CrossRef CAS PubMed.
- H. M. Boehm, S. Handa, G. Pattenden, L. Roberts, A. J. Blake and W. S. Li, Cascade Radical Cyclisations Leading to Steroid Ring Constructions. Regio- and Stereo-chemical Studies Using Ester- and Fluoro-Alkene Substituted Polyene Acyl Radical Intermediates, J. Chem. Soc., Perkin Trans. 1, 2000, 3522 RSC.
- K. Yu, F. Yao, Q. Zeng, H. Xie and H. Ding, Asymmetric Total Syntheses of (+)-Davisinol and (+)-18-Benzoyldavisinol: A HAT-Initiated Transannular Redox Radical Approach, J. Am. Chem. Soc., 2021, 143, 10576 CrossRef CAS PubMed.
- W. P. Thomas, D. J. Schatz, D. T. George and S. V. Pronin, A Radical-Polar Crossover Annulation to Access Terpenoid Motifs, J. Am. Chem. Soc., 2019, 141, 12246 CrossRef CAS PubMed.
- J. R. Andreatta, B. A. McKeown and T. B. Gunnoe, Transition Metal Catalyzed Hydroarylation of Olefins Using Unactivated Substrates: Recent Developments and Challenges, J. Organomet. Chem., 2011, 696, 305 CrossRef CAS.
- Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Transition-Metal-Catalyzed C−H Alkylation Using Alkenes, Chem. Rev., 2017, 117, 9333 CrossRef CAS PubMed.
- Y. Ji, Z. Y. Xin, H.-B. He and S. H. Gao, Total Synthesis of Viridin and Viridiol, J. Am. Chem. Soc., 2019, 141, 16208 CrossRef CAS PubMed.
- Z. Y. Xin, H. Wang, H. B. He, X. L. Zhao and S. H. Gao, Asymmetric Total Synthesis of Norzoanthamine, Angew. Chem., Int. Ed., 2021, 60, 12807 CrossRef CAS PubMed.
- B. X. Zhang, W. F. Zheng, X. Q. Wang, D. Q. Sun and C. Z. Li, Total Synthesis of Notoamides F, I, and R and Sclerotiamide, Angew. Chem., Int. Ed., 2016, 55, 10435 CrossRef CAS PubMed.
- D. Vrubliauskas, B. M. Gross and C. D. Vanderwal, Stereocontrolled Radical Bicyclizations of Oxygenated Precursors Enable Short Syntheses of Oxidized Abietane Diterpenoids, J. Am. Chem. Soc., 2021, 143, 2944 CrossRef CAS PubMed.
- (a) H. Y. Jang and M. J. Krische, Catalytic C-C Bond Formation via Capture of Hydrogenation Intermediates, Acc. Chem. Res., 2004, 37, 653 CrossRef CAS PubMed; (b) S. Isayama and T. Mukaiyama, Cobalt(II) Catalyzed Coupling Reaction of α,β-Unsaturated Compounds with Aldehydes by the Use of Phenylsilane. New Method for Preparation of β-Hydroxy Nitriles, Amides, and Esters, Chem. Lett., 1989, 18, 2005 CrossRef.
- (a) M. Saladrigas, C. Bosch, G. V. Saborit, J. Bonjoch and B. Bradshaw, Radical Cyclization of Alkene-Tethered Ketones Initiated by Hydrogen-Atom Transfer, Angew. Chem., Int. Ed., 2018, 57, 182 CrossRef CAS PubMed; (b) O. J. Turner, J. A. Murphy, D. J. Hirst and E. Talbot, Hydrogen Atom Transfer-Mediated Cyclisations of Nitriles, Chem. – Eur. J., 2018, 24, 18658 CrossRef CAS PubMed.
- G. G. Liu, Z. J. Zhang, S. M. Fu and B. Liu, Asymmetric Total Synthesis of Rumphellclovane E, Org. Lett., 2021, 23, 290 CrossRef CAS PubMed.
- J. Liu and D. Ma, AUnified Approach for the Assembly of Atisine- and Hetidine-type Diterpenoid Alkaloids: Total Syntheses of Azitine and the Proposed Structure of Navirine C, Angew. Chem., Int. Ed., 2018, 57, 6676 CrossRef CAS PubMed.
- B. Xu, W. Xun, S.-B. Su and H. B. Zhai, Total Syntheses of (−)-Conidiogenone B, (−)-Conidiogenone, and (−)-Conidiogenol, Angew. Chem., Int. Ed., 2020, 59, 16475 CrossRef CAS PubMed.
- M. Tanaka, C. Mukaiyama, H. Mitsuhashi, M. Maruno and T. Wakamatsu, Synthesis of Optically Pure Gomisi Lignans: The Total Synthesis of (+)-Schizandrin, (+)-Gomisin A, and (+)-Isoschizandrin in Naturally Occurring Forms, J. Org. Chem., 1995, 60, 4339 CrossRef CAS.
- Y. Matsushita, K. Sugamoto, T. Nakama, T. Sakamoto, T. Matsui and M. Nakayama, γ-Selective Hydroxylation of α,β,γ,δ-Unsaturated Carbonyl Compounds and Its Application to Syntheses of (±)-6-Hydroxyshogaol and Related Furanoids, Tetrahedron Lett., 1995, 36, 1879 CrossRef CAS.
- H. C. Liu, X. W. Zhang, D. Shan, M. Pitchakuntla, Y. F. Ma and Y. X. Jia, Total Syntheses of Festuclavine, Pyroclavine, Costaclavine, epi-Costaclavine, Pibocin A, 9-Deacetoxyfumigaclavine C, Fumigaclavine G, and Dihydrosetoclavine, Org. Lett., 2017, 19, 3323 CrossRef CAS PubMed.
- S. A. Liu and D. Trauner, Asymmetric Synthesis of the Antiviral Diterpene Wickerol A, J. Am. Chem. Soc., 2017, 139, 9491 CrossRef CAS PubMed.
- S. Y. Pan, S. Chen and G. B. Dong, Divergent Total Syntheses of Enmein-Type Natural Products: (−)-Enmein, (−)-Isodocarpin, and (−)-Sculponin R, Angew. Chem., Int. Ed., 2018, 57, 6333 CrossRef CAS PubMed.
- M. J. Cheng, L. P. Zhong, C. C. Gu, X. J. Zhu, B. Chen, J. S. Liu, L. Wang, W. C. Ye and C. C. Li, Asymmetric Total Synthesis of Bufospirostenin A, J. Am. Chem. Soc., 2020, 142, 12602 CrossRef CAS PubMed.
- S. W. M. Crossley, G. H. Tong, M. J. Lambrecht, H. E. Burdge and R. A. Shenvi, Synthesis of (−)-Picrotoxinin by Late-Stage Strong Bond Activation, J. Am. Chem. Soc., 2020, 142, 11376 CrossRef CAS PubMed.
- T. K. Allred, A. P. Dieskau, P. Zhao, G. L. Lackner and L. E. Overman, Enantioselective Total Synthesis of Macfarlandin C, a Spongian Diterpenoid Harboring a Concave-Substituted cis-Dioxabicyclo-[3.3.0]octanone Fragment, Angew. Chem., Int. Ed., 2020, 59, 6268 CrossRef CAS PubMed.
- C. Obradors, R. M. Martinez and R. A. Shenvi, Ph(i-PrO)SiH2: An Exceptional Reductant for Metal-Catalyzed Hydrogen Atom Transfers, J. Am. Chem. Soc., 2016, 138, 4962 CrossRef CAS PubMed.
- M. Ohtawa, M. J. Krambis, R. Cerne, J. M. Schkeryantz, J. M. Witkin and R. A. Shenvi, Synthesis of (−)-11-O-Debenzoyltashironin: Neurotrophic Sesquiterpenes Cause Hyperexcitation, J. Am. Chem. Soc., 2017, 139, 9637 CrossRef CAS PubMed.
- X. H. Zhao, Q. Zhang, J. Y. Du, X. Y. Lu, Y. X. Cao, Y. H. Deng and C. A. Fan, Total Synthesis of (±)-Lycojaponicumin D and Lycodoline-Type Lycopodium Alkaloids, J. Am. Chem. Soc., 2017, 139, 7095 CrossRef CAS PubMed.
- (a) A. Baker, R. M. Demoret, M. Ohtawa and R. A. Shenvi, Concise Asymmetric Synthesis of (−)-Bilobalide, Nature, 2019, 575, 643 CrossRef PubMed; (b) R. M. Demoret, M. A. Baker, M. Ohtawa, S.-M. Chen, C. C. Lam, S. Khom, M. Roberto, S. Forli, K. N. Houk and R. A. Shenvi, Synthetic, Mechanistic, and Biological Interrogation of Ginkgobiloba Chemical Space En Route to (−)-Bilobalide, J. Am. Chem. Soc., 2020, 142, 18599 CrossRef CAS PubMed.
- K. Hung, M. L. Condakes, L. F. T. Novaes, S. J. Harwood, T. Morikawa, Z. Yang and T. J. Maimone, Development of a Terpene Feedstock-Based Oxidative Synthetic Approach to the Illicium Sesquiterpenes, J. Am. Chem. Soc., 2019, 141, 3083 CrossRef CAS PubMed.
- C. He, J. Xuan, P. R. Rao, P. P. Xie, X. Hong, X. F. Lin and H. F. Ding, Total Syntheses of (+)-Sarcophytin, (+)-Chatancin, (−)-3-Oxochatancin, and (−)-PavidolideB: A Divergent Approach, Angew. Chem., Int. Ed., 2019, 58, 5100 CrossRef CAS PubMed.
- S. P. Zhou, K. F. Xia, X. B. Leng and A. Li, Asymmetric Total Synthesis of Arcutinidine, Arcutinine, and Arcutine, J. Am. Chem. Soc., 2019, 141, 13718 CrossRef CAS PubMed.
- M. Pfaffenbach, I. Bakanas, N. R. O'Connor, J. L. Herrick and R. Sarpong, Total Syntheses of Xiamycins A, C, F, H and Oridamycin A and Preliminary Evaluation of their Anti-Fungal Properties, Angew. Chem., Int. Ed., 2019, 58, 15304 CrossRef CAS PubMed.
- L. L. Shi, Y. D. He, J. X. Gong and Z. Yang, Concise Gram-Scale Synthesis of Euphorikanin a Skeleton through A Domino Ring-Closing Metathesis Strategy, Chem. Commun., 2020, 56, 531 RSC.
- C. K. Chong, Q. L. Zhang, J. Ke, H. M. Zhang, X. D. Yang, B. J. Wang, W. Ding and Z. Y. Lu, Total Synthesis of Anti-Cancer Meroterpenoids Dysideanone B and Dysiherbol A and Structural Reassignment of Dysiherbol A, Angew. Chem., Int. Ed., 2021, 60, 13807 CrossRef CAS PubMed.
- D. H. Dethe and A. K. Nirpal, Enantiospecific Total Synthesis of (−)-Japonicol C, Org. Lett., 2021, 23, 2648 CrossRef PubMed.
- X. Hu and T. J. Maimone, Four-Step Synthesis of the Antimalarial Cardamom Peroxide via an Oxygen Stitching Strategy, J. Am. Chem. Soc., 2014, 136, 5287 CrossRef CAS PubMed.
- M. Nagatomo, M. Koshimizu, K. Masuda, T. Tabuchi, D. Urabe and M. Inoue, Total Synthesis of Ryanodol, J. Am. Chem. Soc., 2014, 136, 5916 CrossRef CAS PubMed.
- Q. K. Zhao, A. Peuronen, K. Rissanen, D. Enders and Y. F. Tang, Enantioselective Total Syntheses of (+)-Hippolachnin A, (+)-Gracilioether A, (−)-Gracilioether E, and (−)-Gracilioether F, J. Am. Chem. Soc., 2018, 140, 1937 CrossRef PubMed.
- (a) K. S. Feldman and M. Parvez, Synthesis of Polyoxygenated Hydrocarbons via Radical-Mediated Oxygenation of Vinylcyclopropanes, J. Am. Chem. Soc., 1986, 108, 1328 CrossRef CAS; (b) K. S. Feldman and R. E. Simpson, Oxygenation of Substituted Vinylcyclopropanes: Preparative and Mechanistic Studies, J. Am. Chem. Soc., 1989, 111, 4878 CrossRef CAS; (c) K. S. Feldman and C. M. Kraebel, Vinylcyclopropane Oxygenation. Anti Diastereoselectivity through an Unexpected Transition-State Geometry, J. Org. Chem., 1992, 57, 4574 CrossRef CAS.
- X. Hu, A. J. Musacchio, X. Shen, Y. Tao and T. J. Maimone, Allylative Approaches to the Synthesis of Complex Guaianolide Sesquiterpenes from Apiaceae and Asteraceae, J. Am. Chem. Soc., 2019, 141, 14904 CrossRef CAS PubMed.
- M. Haider, G. Sennari, A. Eggert and R. Sarpong, Total Synthesis of the Cephalotaxus Norditerpenoids (±)-Cephanolides A−D, J. Am. Chem. Soc., 2021, 143, 2710 CrossRef CAS PubMed.
- P. Magnus, A. H. Payne, M. J. Waring, D. A. Scott and V. Lynch, Conversion of α,β-Unsaturated Ketones into α-Hydroxy Ketones Using an MnIII Catalyst, Phenylsilane and Dioxygen: Acceleration of Conjugate Hydride Reduction by Dioxygen, Tetrahedron Lett., 2000, 41, 9725 CrossRef CAS.
- S. A. Ruider, T. Sandmeier and E. M. Carreira, Total Synthesis of (±)-Hippolachnin A, Angew. Chem., Int. Ed., 2015, 54, 2378 CrossRef CAS PubMed.
- Y. Zhang, Y. Xue, G. Li, H. Yuan and T. Luo, Enantioselective Synthesis of Iboga, Alkaloids and Vinblastine via Rearrangements of Quaternary Ammoniums, Chem. Sci., 2016, 7, 5530 RSC.
- E. P. Farney, S. S. Feng, F. Schafers and S. E. Reisman, Total Synthesis of (+)-Pleuromutilin, J. Am. Chem. Soc., 2018, 140, 1267 CrossRef CAS PubMed.
- Z. Y. Zhou, A. X. Gao and S. A. Snyder, Total Synthesis of (+)-Arborisidine, J. Am. Chem. Soc., 2019, 141, 7715 CrossRef CAS PubMed.
- Y. Z. Qu, Z. Y. Wang, Z. C. Zhang, W. D. Zhang, J. Huang and Z. Yang, Asymmetric Total Synthesis of (+)-Waihoensene, J. Am. Chem. Soc., 2020, 142, 6511 CrossRef CAS PubMed.
- H. H. Lu, S. V. Pronin, Y. Antonova-Koch, S. Meister, E. A. Winzeler and R. A. Shenvi, Synthesis of (+)-7,20-Diisocyanoadociane and Liver-Stage Antiplasmodial Activity of the Isocyanoterpene Class, J. Am. Chem. Soc., 2016, 138, 7268 CrossRef CAS PubMed.
- C. G. Liu, R. Z. Chen, Y. Shen, Z. H. Liang, Y. H. Hua and Y. D. Zhang, Total Synthesis of Aplydactone by a Conformationally Controlled C−H Functionalization, Angew. Chem., Int. Ed., 2017, 56, 8187 CrossRef CAS PubMed.
- D. H. Dethe, S. Mahapatra and S. K. Sau, Enantioselective Total Synthesis and Assignment of the Absolute Configuration of the Meroterpenoid (+)-Taondiol, Org. Lett., 2018, 20, 2766 CrossRef CAS PubMed.
- L. Yang, T. Wurm, B. S. Poudel and M. J. Krische, Enantioselective Total Synthesis of Andrographolide and 14-Hydroxy-Colladonin: Carbonyl Reductive Coupling and trans-Decalin Formation by Hydrogen Transfer, Angew. Chem., Int. Ed., 2020, 59, 23169 CrossRef CAS PubMed.
- J. Li, F. Z. Li, E. King-Smith and H. Renata, Merging Chemoenzymatic and Radical-Based Retrosynthetic Logic for Rapid and Modular Synthesis of Oxidized Meroterpenoids, Nat. Chem., 2020, 12, 173 CrossRef CAS PubMed.
- Y. Nakayama, M. R. Maser, T. Okita, A. V. Dubrovskiy, T. L. Campbell and S. E. Reisman, Total Synthesis of Ritterazine B, J. Am. Chem. Soc., 2021, 143, 4187 CrossRef CAS PubMed.
- B. M. Trost, Atom Economy-A Challenge for Organic Synthesis: Homogeneous Catalysis Leads the Way, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS.
- E. Larionov, H.-H. Lia and C. Mazet, Well-Defined Transition Metal Hydrides in Catalytic Isomerizations, Chem. Commun., 2014, 50, 9816 RSC.
- C. Chen, T. R. Dugan, W. W. Brennessel, D. J. Weix and P. L. Holland, Z-Selective Alkene Isomerization by High-Spin Cobalt(II) Complexes, J. Am. Chem. Soc., 2014, 136, 945 CrossRef CAS PubMed.
- Y. Wang, A. Jager, M. Gruner, T. Lubken and P. Metz, Enantioselective Total Synthesis of 3b-Hydroxy-7b-kemp-8(9)-en-6-one, a Diterpene Isolated from Higher Termites, Angew. Chem., Int. Ed., 2017, 56, 15861 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2021 |