
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring polymer dispersity by mixing ATRP initiators†
Kostas
Parkatzidis‡
 ,
Manon
Rolland‡
,
Nghia P.
Truong
and
Athina
Anastasaki
*
,
Manon
Rolland‡
,
Nghia P.
Truong
and
Athina
Anastasaki
*
Laboratory for Polymeric Materials, Department of Materials, ETH Zürich, Vladimir-Prelog-Weg 5, 8093 Zürich, Switzerland. E-mail: athina.anastasaki@mat.ethz.ch
First published on 21st September 2021
Abstract
Herein we present a simple batch method to control polymer dispersity using a mixture of two ATRP initiators with different reactivities. A wide dispersity spectrum (Đ ∼ 1.1–1.7) is achieved by altering the ratio between the two ATRP initiators while sustaining fairly monomodal molecular weight distributions. Depending on the targeted dispersity, a distinct kinetic profile was observed and, in contrast to previous ATRP approaches, near-quantitative conversions were obtained even for the highest dispersities. High end-group fidelity was also maintained in all cases as exemplified by chain extensions. Significantly, this method can be applied to various ATRP protocols as well as to various monomer classes.
One of the main variables of polymers is their molecular weight distribution (MWD), commonly measured as dispersity (Đ). It is defined as the ratio of the weight average molar mass (Mw) over the number average molar mass (Mn).1 Dispersity is always greater than one due to the heterogeneity of synthetic polymer chains. This is in contrast to biomacromolecules, such as proteins and DNA, which have a dispersity of 1 owing to their perfect uniformity.2 Controlled radical polymerization, also referred to as reversible deactivation radical polymerization, has traditionally focused on minimizing the Đ of polymers with low Đ becoming a trademark for high livingness and a successful polymerization.3,4 However, polymers of higher and intermediate Đs can also have advantageous physicochemical characteristics and properties.5–10 To this end, the first examples of methods to tailor polymer Đ while maintaining high end-group fidelity have recently emerged.2,5,11
To control the breadth of molecular weight distributions, two main approaches have been employed. First, the so-called “engineering” methods rely on using flow, feeding or mixing strategies based on theoretical calculations and modelling. For example, Fors and co-workers pioneered the field by introducing a clever feeding method to tune the shape and the breadth of molecular weight distributions. In particular, the initiating species were accurately fed in nitroxide-mediated,12 anionic,13 coordination14 and RAFT15 polymerizations at predetermined rates resulting in a continued initiation process and full control over the MWDs. The same group subsequently studied the effect of skewness and shape of MWDs in mechanical properties and bulk self-assembly.14,16,17 Other noticeable examples include blending approaches,9,18 data encryption via MWD fingerprints developed by the Junkers group,19–22 and the use of elegant flow chemistry methods employed by the groups of Frey, Boyer, Leibfarth, Guironnet, and Lu.23–29 The vast majority of these methodologies include the design of suitable equations and modelling systems to precisely tailor MWDs and provide polymers with a high range of Đs.
Alternatively, “chemistry” methodologies have also been developed in which the focus is to deviate from “ideal”/established polymerization conditions in order to controllably tailor the MWDs. For instance, Goto's group added a small amount of an acrylic co-monomer during the organocatalyzed polymerization of methacrylates and efficiently tuned polymer dispersity in both linear and branched block copolymers. Mechanistically, the presence of the co-monomer led to retardation of the growing polymer chains, without compromising the end-group fidelity.30 In ATRP, tailoring polymer dispersity is possible by altering the deactivator concentration, as independently demonstrated by Matyjaszewski's group and our group.31–34 In particular, the lower the deactivator concentration, the higher the dispersity achieved and the produced polymers displayed high livingness regardless of their dispersity. Other efficient examples to tailor polymer Đ include the use of reducing agents,35 polymerization retarders,36–39 photochromic initiators,40 and termination agents.41,42 In RAFT polymerization, our group developed a simple strategy whereby a mixture of two RAFT agents with vastly different reactivity were employed.43 The ratio between the high and low activity RAFT agent was precisely tailored to control polymer dispersity for a wide range of polymer classes and the concept could be successfully applied in both thermal and PET-RAFT polymerization.44
Inspired by our previous work in RAFT polymerization, we present here a method to tailor polymer dispersity through mixing two commercially available ATRP initiators with different reactivities (Fig. 1a). To realize our concept, two suitable initiators have to be identified: one to enable the synthesis of polymers with narrow molecular weight distributions (e.g. Đ ∼ 1.1) and one to generate polymers with much broader molecular weight distributions (e.g. Đ ∼ 1.7) under otherwise identical conditions. The selection of the high reactivity ATRP initiator is relatively simple as it has been a major focus of the literature.45 For instance, 2-bromopropionate (EBP) is a commonly used initiator leading to excellent control over the MWDs and low dispersity values. However, the choice of the lower activity ATRP initiator can be more challenging. We envisioned that ethyl 2-chloropropionate (ECP) could be an ideal candidate due to the chlorine end-group which would, in principle, lead to lower deactivation rate than the bromine analogue under the same catalyst loading.45–47 Importantly, apart from the terminal halogen (chlorine versus bromine) both initiators possess the same degree of substitution (i.e. secondary initiators) and an identical α-end group, without the latter being the main determining characteristic.
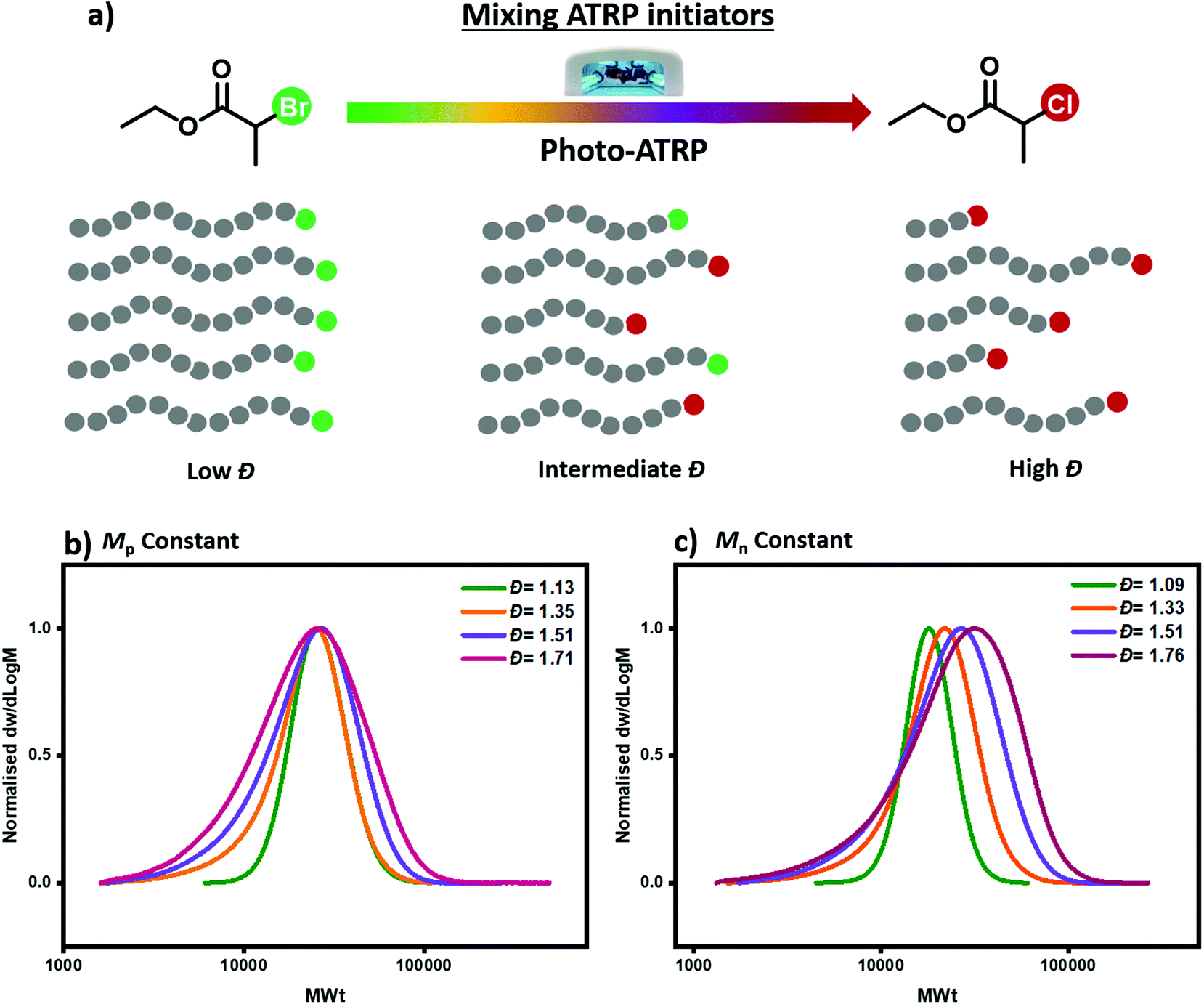 | ||
| Fig. 1 (a) Schematic representation of mixing ATRP initiators to tune polymer dispersity, (b) SEC analysis of PMA synthesized via photo-ATRP, illustrating the variation in dispersity as EBP and ECP are mixed in different ratios (aligned by Mp value) and (c) SEC analysis of PMA synthesized via photo-ATRP, illustrating the variation in dispersity as EBP and ECP are mixed in different ratios (aligned by Mn value). | ||
To demonstrate the concept, we first employed photo-ATRP as a model polymerization system. All reactions were performed under UV light irradiation (36 W, λmax = 360 nm) using methyl acrylate (MA) as a monomer, tris[2-(dimethylamino)ethyl]amine (Me6TREN) as a ligand, CuBr2 as the copper source and dimethylsulfoxide (DSMO) as a solvent. When EBP was employed as the alkyl halide initiator, a well-controlled polymerization took place yielding poly(methyl acrylate) (PMA) with narrow molecular weight distributions (Đ = 1.13, Fig. 1b and Table S1,† entry 4), as characterized by size exclusion chromatography (SEC). This result is in line with previous reports.48 On the other hand, when EBP was replaced with ECP, a much broader yet monomodal MWD was obtained, (Đ = 1.71, Fig. 1b and Table S1,† entry 1). This verifies our initial hypothesis that ECP would yield polymers with higher dispersity values due to its relatively low deactivation rate. It is noted that given the very low amounts of catalyst employed (0.01 equivalents with respect to the initiator), halogen exchange is not expected to be the key factor which controls polymer dispersity. As we have identified the upper and lower Đ limits of our system, we subsequently altered the ratio of these two initiators aiming to tailor polymer dispersity (Fig. 1b and c). As depicted in Fig. 1, a series of polymers with different Đ and constant Mn or Mp values could be successfully synthesized following our strategy. For example, a ratio of EBP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ECP 20:80 (mol %) resulted in PMA with Đ = 1.35 (Fig. 1b and Table S1,† entry 3), while when the EBP:ECP ratio was 5:95, the obtained PMA displayed higher dispersity Đ = 1.51 (Fig. 1b and Table S1,† entry 2). These initial experiments demonstrate that by simply mixing two commercially available alkyl halide initiators with different reactivities, polymers with a range of Đs can be synthesized through photo-ATRP.
:ECP 20:80 (mol %) resulted in PMA with Đ = 1.35 (Fig. 1b and Table S1,† entry 3), while when the EBP:ECP ratio was 5:95, the obtained PMA displayed higher dispersity Đ = 1.51 (Fig. 1b and Table S1,† entry 2). These initial experiments demonstrate that by simply mixing two commercially available alkyl halide initiators with different reactivities, polymers with a range of Đs can be synthesized through photo-ATRP.
To gain a deeper insight into the polymerization mechanism when different ATRP initiators were employed, we conducted comprehensive kinetic experiments. Through the exclusive use of EBP, a conventional linear first order kinetic profile was observed (Fig. 2a) accompanied by a gradual decrease of the Đ throughout the polymerization (from 1.23 to 1.08), as expected from a controlled/living polymerization. The molecular weight distributions shifted gradually to higher molecular weights (MW) and a relatively good agreement between theoretical and experimental molecular weights was achieved (when compared to PMMA calibrants, Table S3†). A similar kinetic profile was also observed when mixtures of the two ATRP initiators were employed (Tables S3–5†). For instance, under the ratio [EBP]:[ECP]=[40]:[60], a linear increase of Mn with conversion was also obtained as well as a linear dependence of ln([M]0/[M]) on time (Fig. 2b, Table S4†). However, a slightly worse agreement between theoretical and experimental MW was noticed, which was attributed to slow initiation of the chlorine initiator. Indeed, we found that when just ECP was employed, slower initiation was observed when compared to EBP: with ECP only 15% of the initiator was consumed at 17% of conversion while with EBP 83% of the initiator was consumed at 18% of monomer conversion (Fig. S1†). In addition, when ECP was solely employed, a distinct kinetic profile was noticed. Although the conversion and ln([M]0/[M]) still increased over time, the Mn reached its’ highest value at a very early stage of the polymerization without a significant further increase throughout the reaction. The Đ was maintained at >1.6 regardless of the monomer conversions. We attribute these findings to the low kdeact of ECP. Slow deactivation enforces polymer chains to achieve their final MW at an early stage of polymerization, without undergoing multiple activation-deactivation steps. Importantly, regardless of the final Đ (i.e. low, intermediate or high), near-quantitative conversions could be reached in all cases (>95%). This is an important advantage over previous approaches in which only moderate conversions (15–60%) could be achieved for either intermediate or high Đ polymers due to the cessation of the polymerization as a result of the low catalyst loading.32,34 Instead, by employing either a lower ATRP initiator or mixtures of high/low ATRP initiators, no reaction plateau is observed, thus allowing for the polymerizations to reach completion. The linear kinetic profile suggests high end-group fidelity regardless of the initial Đ. In order to further examine the livingness of the synthesized polymers, we also conducted chain extension experiments. In particular, macro-initiators with Đ = 1.49 and 1.70 were initially synthesized and subsequently chain extended by adding a second aliquot of MA. SEC traces shifted to higher molecular weights thus indicating successful chain extensions in both cases. (Fig. 3, Table S6,† entries 1–4).
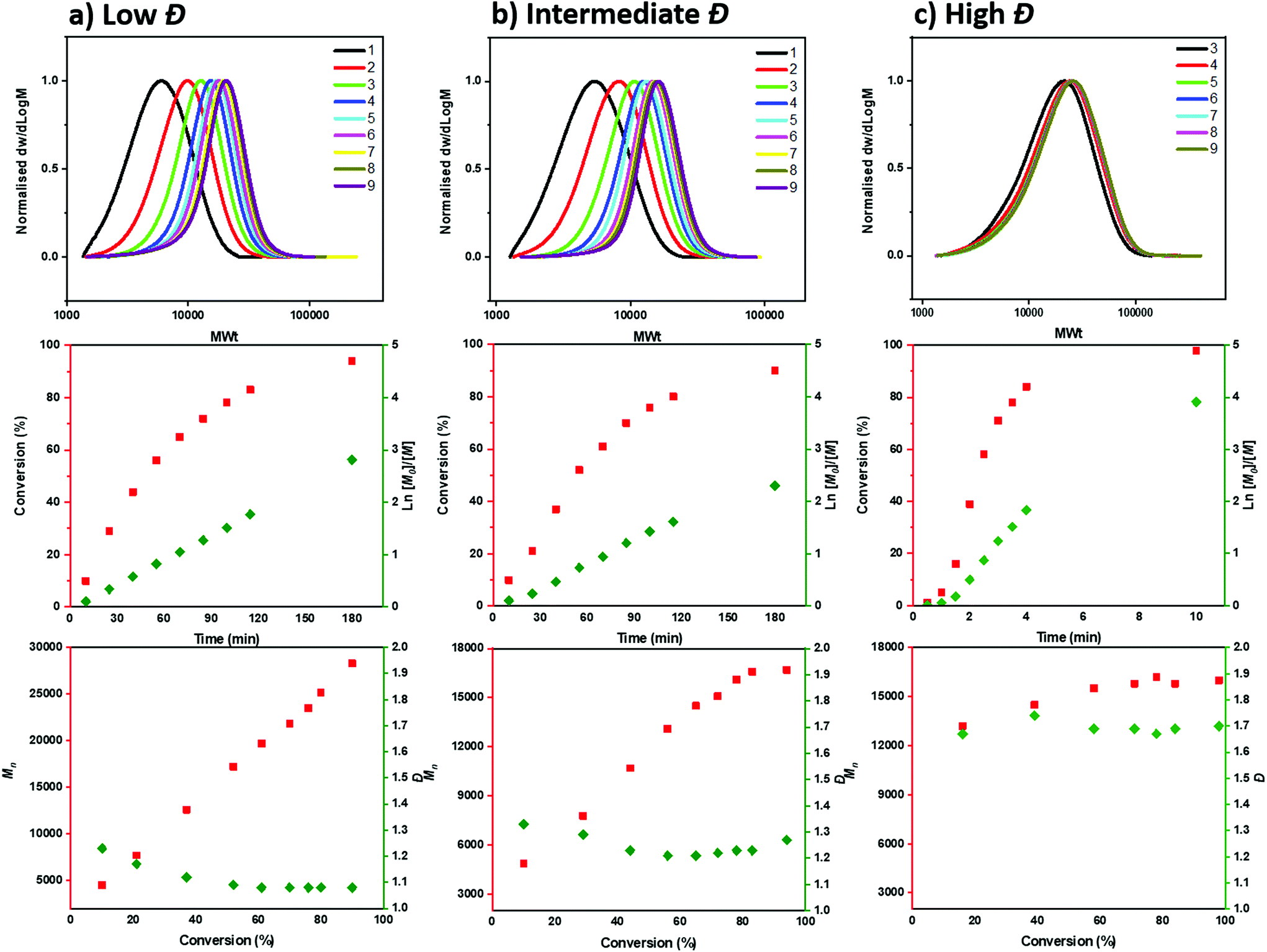 | ||
| Fig. 2 SEC and kinetic analysis for the synthesis of PMA with different initiators: (a) only EBP, (b)a mixture of EBP:ECP in a ratio 40:60 and (c) only ECP. | ||
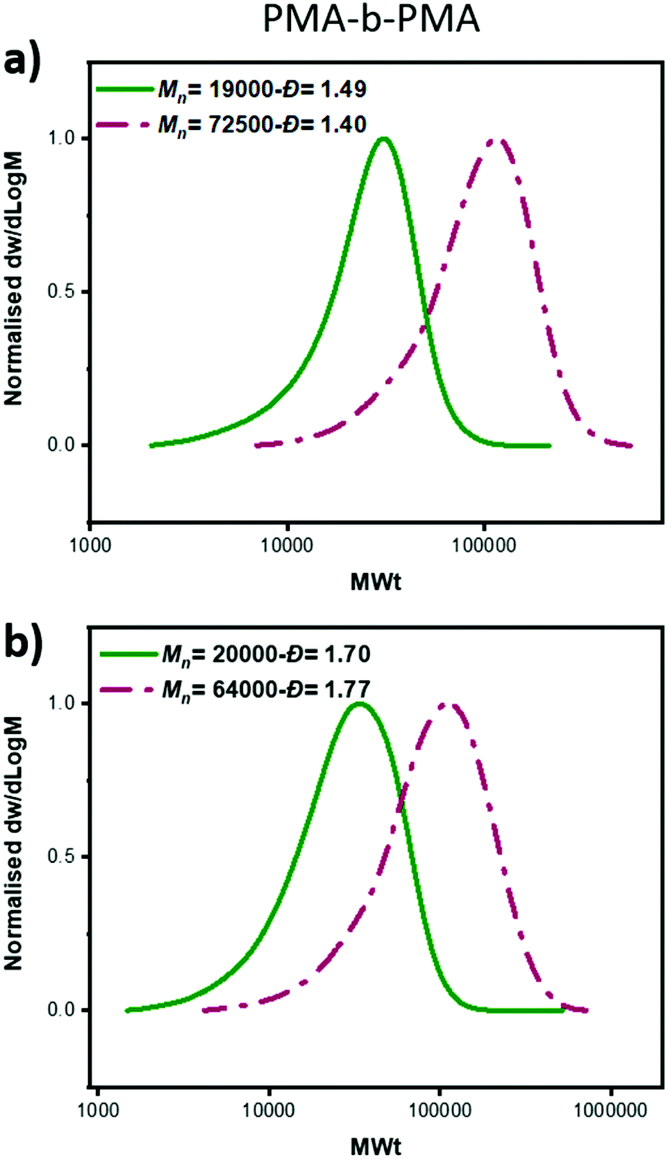 | ||
| Fig. 3 SEC analysis of the block copolymers formed from chain extension of a PMA macro-initiators prepared with (a) EBP:ECP 60:40 (b) EBP:ECP 5:95. | ||
One of the advantages of our developed approach is that it should, in principle, be compatible with any ATRP method as it relies on the selection of two initiators with different reactivity. Instead, previous ATRP approaches are limited to systems that start with only the deactivator (e.g. CuBr2) and tune the Đ by altering the catalyst concentration. Thus, previous approaches would be incompatible with, for example, Cu(0)-RDRP.49–53 To test the robustness of our methodology, EBP, ECP and mixtures thereof were utilized as initiators in Cu(0)-RDRP using copper wire and otherwise identical conditions with the photo-ATRP experiments. Pleasingly, by changing the ratio between the two initiators the Đ could be efficiently controlled and chain extension experiments were also successful, thus suggesting high end-group fidelity (Fig. 4, Tables S7–8†). These experiments also expand the scope of our method as we are no longer limited to external stimuli, such as light, in order to control polymer Đ which could enable additional opportunities and directions.32,33,54 To further probe the potential of our method we also conducted preliminary experiments to control the Đ of other monomer classes such as methyl methacrylate (MMA). By using mixtures of methyl α-bromophenylacetate (MBPA) with EBP, polymers with different Đ could be obtained (Đ ∼ 1.3–1.67, Fig. S9 and Table S2†), albeit with a more pronounced tailing when compared to the acrylate polymerizations. The observed tailing was attributed to the polymerization of MMA and in line with previous reports.48 Thus, upon selection of an appropriate system, the concept of mixing ATRP initiators can apply in different copper polymerization strategies.
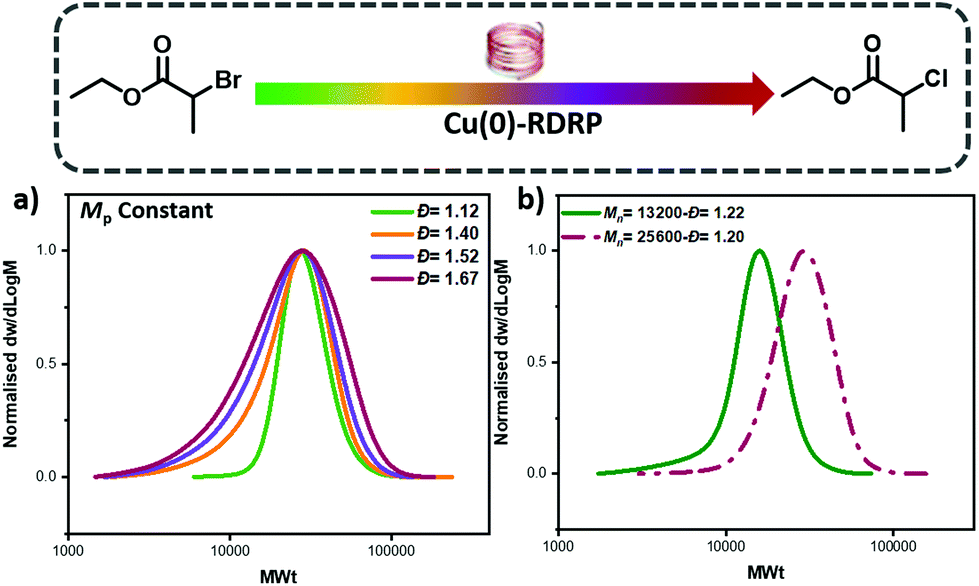 | ||
| Fig. 4 SEC analysis of the polymerization of MA utilizing Cu(0)-RDRP, illustrating (a) the variation in dispersity as EBP and ECP are mixed in different ratios (aligned by Mp value) and (b) a chain extension of a PMA macroCTA prepared with EBP:ECP 60:40. | ||
Conclusions
In summary, we have developed a simple batch method based on mixing two alkyl halide initiators with different activities, which allows to tune the Đ of polymers. Dispersities between ∼1.1–1.7 can be obtained while maintaining monomodal molecular weight distributions and high livingness, as verified through kinetic experiments and successful chain extensions. Importantly, our developed approach can reach very high monomer conversions regardless of the targeted dispersity value, and can be applied to different ATRP approaches and monomer families.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A.A gratefully acknowledges ETH Zurich for financial support. N.P.T. acknowledges the award of a DECRA Fellowship and DP from the ARC (DE180100076 and DP200100231). KP thanks Onasis Foundation as this scientific paper was partially supported by the Onassis Foundation – Scholarship ID: FZQ051-1/2020-2021.References
- R. F. Stepto, Pure Appl. Chem., 2009, 81, 351–353 Search PubMed.
- D. T. Gentekos, R. J. Sifri and B. P. Fors, Nat. Rev. Mater., 2019, 4, 761–774 CrossRef.
- K. Parkatzidis, H. S. Wang, N. P. Truong and A. Anastasaki, Chem, 2020, 6, 1575–1588 CAS.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- R. Whitfield, N. P. Truong, D. Messmer, K. Parkatzidis, M. Rolland and A. Anastasaki, Chem. Sci., 2019, 10, 8724–8734 RSC.
- N. A. Lynd, A. J. Meuler and M. A. Hillmyer, Prog. Polym. Sci., 2008, 33, 875–893 CrossRef CAS.
- S. George, R. Champagne-Hartley, G. Deeter, D. Campbell, B. Reck, D. Urban and M. Cunningham, Macromolecules, 2015, 48, 8913–8920 CrossRef CAS.
- V. Yadav, Y. A. Jaimes-Lizcano, N. K. Dewangan, N. Park, T.-H. Li, M. L. Robertson and J. C. Conrad, ACS Appl. Mater. Interfaces, 2017, 9, 44900–44910 CrossRef CAS PubMed.
- S. Xu, F. J. Trujillo, J. Xu, C. Boyer and N. Corrigan, Macromol. Rapid Commun., 2021, 2100212 CrossRef CAS.
- T.-H. Li, V. Yadav, J. C. Conrad and M. L. Robertson, ACS Macro Lett., 2021, 10, 518–524 CrossRef CAS.
- T. Junkers, Macromol. Chem. Phys., 2020, 221, 2000234 CrossRef CAS.
- D. T. Gentekos, L. N. Dupuis and B. P. Fors, J. Am. Chem. Soc., 2016, 138, 1848–1851 CrossRef CAS PubMed.
- V. Kottisch, D. T. Gentekos and B. P. Fors, ACS Macro Lett., 2016, 5, 796–800 CrossRef CAS.
- R. J. Sifri, O. Padilla-Vélez, G. W. Coates and B. P. Fors, J. Am. Chem. Soc., 2019, 142, 1443–1448 CrossRef PubMed.
- S. I. Rosenbloom, R. J. Sifri and B. P. Fors, Polym. Chem., 2021, 12, 4910–4915 RSC.
- D. T. Gentekos, J. Jia, E. S. Tirado, K. P. Barteau, D.-M. Smilgies, R. A. DiStasio Jr. and B. P. Fors, J. Am. Chem. Soc., 2018, 140, 4639–4648 CrossRef CAS.
- S. I. Rosenbloom and B. P. Fors, Macromolecules, 2020, 53, 7479–7486 CrossRef CAS.
- R. Whitfield, N. Truong and A. Anastasaki, Angew. Chem., Int. Ed., 2021, 60, 19383–19388 CrossRef CAS.
- T. Junkers and J. H. Vrijsen, Eur. Polym. J., 2020, 134, 109834 CrossRef CAS.
- J. H. Vrijsen, M. Rubens and T. Junkers, Polym. Chem., 2020, 11, 6463–6470 RSC.
- M. Rubens and T. Junkers, Polym. Chem., 2019, 10, 5721–5725 RSC.
- M. Rubens and T. Junkers, Polym. Chem., 2019, 10, 6315–6323 RSC.
- J. Morsbach, A. H. Müller, E. Berger-Nicoletti and H. Frey, Macromolecules, 2016, 49, 5043–5050 CrossRef CAS.
- N. Corrigan, A. Almasri, W. Taillades, J. Xu and C. Boyer, Macromolecules, 2017, 50, 8438–8448 CrossRef CAS.
- N. Corrigan, R. Manahan, Z. T. Lew, J. Yeow, J. Xu and C. Boyer, Macromolecules, 2018, 51, 4553–4563 CrossRef CAS.
- K. Liu, N. Corrigan, A. Postma, G. Moad and C. Boyer, Macromolecules, 2020, 53, 8867–8882 CrossRef CAS.
- M. H. Reis, T. P. Varner and F. A. Leibfarth, Macromolecules, 2019, 52, 3551–3557 CrossRef CAS.
- D. J. Walsh, D. A. Schinski, R. A. Schneider and D. Guironnet, Nat. Commun., 2020, 11, 3094 CrossRef CAS.
- H. Liu, Y.-H. Xue, Y.-L. Zhu, F.-L. Gu and Z.-Y. Lu, Macromolecules, 2020, 53, 6409–6419 CrossRef CAS.
- X. Liu, C. G. Wang and A. Goto, Angew. Chem., Int. Ed., 2019, 58, 5598–5603 CrossRef CAS PubMed.
- Z. Wang, J. Yan, T. Liu, Q. Wei, S. Li, M. Olszewski, J. Wu, J. Sobieski, M. Fantin and M. R. Bockstaller, ACS Macro Lett., 2019, 8, 859–864 CrossRef CAS.
- R. Whitfield, K. Parkatzidis, M. Rolland, N. P. Truong and A. Anastasaki, Angew. Chem., Int. Ed., 2019, 58, 13323–13328 CrossRef CAS.
- M. Rolland, N. P. Truong, R. Whitfield and A. Anastasaki, ACS Macro Lett., 2020, 9, 459–463 CrossRef CAS.
- M. Rolland, V. Lohmann, R. Whitfield, N. P. Truong and A. Anastasaki, J. Polym. Sci., 2021 DOI:10.1002/pol.20210319.
- A. Plichta, M. Zhong, W. Li, A. M. Elsen and K. Matyjaszewski, Macromol. Chem. Phys., 2012, 213, 2659–2668 CrossRef CAS.
- R. Jia, Y. Tu, M. Glauber, Z. Huang, S. Xuan, W. Zhang, N. Zhou, X. Li, Z. Zhang and X. Zhu, Polym. Chem., 2021, 12, 349–355 RSC.
- M. Chen, J. Li, K. Ma, G. Jin, X. Pan, Z. Zhang and J. Zhu, Angew. Chem., 2021, 133, 1–6 CrossRef.
- M. Zhang, J. Li, M. Chen, X. Pan, Z. Zhang and J. Zhu, Macromolecules, 2021, 54(13), 6502–6510 CrossRef CAS.
- M. A. Wallace and L. R. Sita, Angew. Chem., Int. Ed., 2021, 60, 19671–19678 CrossRef CAS PubMed.
- D. Liu, A. D. Sponza, D. Yang and M. Chiu, Angew. Chem., 2019, 131, 16356–16362 CrossRef.
- C.-G. Wang, A. M. L. Chong and A. Goto, ACS Macro Lett., 2021, 10, 584–590 CrossRef CAS.
- V. Yadav, N. Hashmi, W. Ding, T.-H. Li, M. K. Mahanthappa, J. C. Conrad and M. L. Robertson, Polym. Chem., 2018, 9, 4332–4342 RSC.
- R. Whitfield, K. Parkatzidis, N. P. Truong, T. Junkers and A. Anastasaki, Chem, 2020, 6, 1–13 Search PubMed.
- K. Parkatzidis, N. P. Truong, M. N. Antonopoulou, R. Whitfield, D. Konkolewicz and A. Anastasaki, Polym. Chem., 2020, 11, 4968–4972 RSC.
- W. Tang and K. Matyjaszewski, Macromolecules, 2007, 40, 1858–1863 CrossRef CAS.
- C. Fang, M. Fantin, X. Pan, K. de Fiebre, M. L. Coote, K. Matyjaszewski and P. Liu, J. Am. Chem. Soc., 2019, 141, 7486–7497 CrossRef CAS PubMed.
- W. Tang, Y. Kwak, W. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 CrossRef CAS PubMed.
- G. R. Jones, R. Whitfield, A. Anastasaki, N. Risangud, A. Simula, D. J. Keddie and D. M. Haddleton, Polym. Chem., 2018, 9, 2382–2388 RSC.
- D. S. Maurya, A. Malik, X. Feng, N. Bensabeh, G. Lligadas and V. Percec, Biomacromolecules, 2020, 21, 1902–1919 CrossRef CAS PubMed.
- X. Feng, D. S. Maurya, N. Bensabeh, A. Moreno, T. Oh, Y. Luo, J. n. Lejnieks, M. Galià, Y. Miura and M. J. Monteiro, et al. , Biomacromolecules, 2019, 21, 250–261 CrossRef PubMed.
- N. P. Truong, W. Gu, I. Prasadam, Z. Jia, R. Crawford, Y. Xiao and M. J. Monteiro, Nat. Commun., 2013, 4, 1902 CrossRef PubMed.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5109–5119 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2016, 116, 835–877 CrossRef CAS PubMed.
- M. Rolland, R. Whitfield, D. Messmer, K. Parkatzidis, N. P. Truong and A. Anastasaki, ACS Macro Lett., 2019, 8, 1546–1551 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py01044a |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2021 |