
Solid-phase synthesis of coralmycin A/epi-coralmycin A and desmethoxycoralmycin A†
Paige M. E.
Hawkins
ab,
Dennis Y.
Liu
c,
Roger G.
Linington
 c and
Richard J.
Payne
*ab
c and
Richard J.
Payne
*ab
aSchool of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: richard.payne@sydney.edu.au
bAustralian Research Council Centre of Excellence for Innovations in Peptide and Protein Science, The University of Sydney, NSW 2006, Australia
cDepartment of Chemistry, Simon Fraser University, Burnaby, BC V5A 1S6, Canada
First published on 1st July 2021
Abstract
The total synthesis of the natural product coralmycin A/epi-coralmycin A, as well as a desmethoxy analogue is described. Synthesis was achieved via a divergent, bidirectional solid-phase strategy, including a key on-resin O-acylation, O to N acyl shift, and O-alkylation protocol to incorporate the unusual 4-amino-2-hydroxy-3-isopropoxybenzoic acid motifs. The synthetic natural product was generated as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of epimers at the central β-methoxyasparagine residue and exhibited potent antibacterial activity against a panel of ten Gram-negative and seven Gram-positive organisms. The desmethoxy analogue possessed significantly more potent antimicrobial activity against this panel with minimal inhibitory concentrations (MICs) as low as 50 nM.
:1 mixture of epimers at the central β-methoxyasparagine residue and exhibited potent antibacterial activity against a panel of ten Gram-negative and seven Gram-positive organisms. The desmethoxy analogue possessed significantly more potent antimicrobial activity against this panel with minimal inhibitory concentrations (MICs) as low as 50 nM.
Introduction
Antimicrobial resistance (AMR) is acknowledged as an emerging global health crisis. In 2017, the World Health Organization outlined the bacterial pathogens that most urgently need attention and included the Gram-negative bacteria Acinetobacter baumannii and Pseudomonas aeruginosa, as well as several members of the family Enterobacteriaceae.1 To catalyse the development of new therapeutic options in this area the WHO has recently partnered with the Drugs for Neglected Diseases initiative (DNDi) to develop the Global Antibiotic Research and Development Partnership (GARPD). This program is charged with developing new treatment options for drug-resistant infections, with a particular focus on serious infections, paediatric antibiotic development, and sexually transmitted infections. Development of international partnerships on this scale underscore the urgency of the issue surrounding AMR, and the continued need for the development of new first-in-class leads for advancement to the clinic.The development of antibiotics targeting Gram-negative bacteria has proved extremely challenging; this is owing to the presence of a unique cell wall that is notoriously impermeable to many classes of antibiotics.2 In addition to this physical barrier, many Gram-negative bacteria, e.g. P. aeruginosa, possess highly efficient efflux pumps that prevent the accumulation of drugs within the organism.3,4 As a result of these challenges, the last Gram-negative-targeting antibiotics approved for use (the quinolones and phosphonic antibiotics) were introduced more than 50 years ago.5–8 Given the emergence of resistance to the current arsenal of antibiotics that target Gram-negative organisms, it is crucial that new antibiotics with novel modes of action are developed.9
Coralmycin A (also known as cystobactamid 935-2) (1) is a natural product that was isolated from the soil myxobacterium Corallococcus coralloides M23 by Kim et al. following a screening campaign of Korean myxobacterial metabolites against Gram-negative bacteria (Fig. 1).10 The natural product was shown to exhibit potent antibacterial activity against a range of Gram-negative and Gram-positive bacteria (MICs of 0.02–4.0 μg mL−1) including pathogenic P. aeruginosa, A. baumannii, and Enterobacteriaceaes, E. coli and K. pneumoniae.10
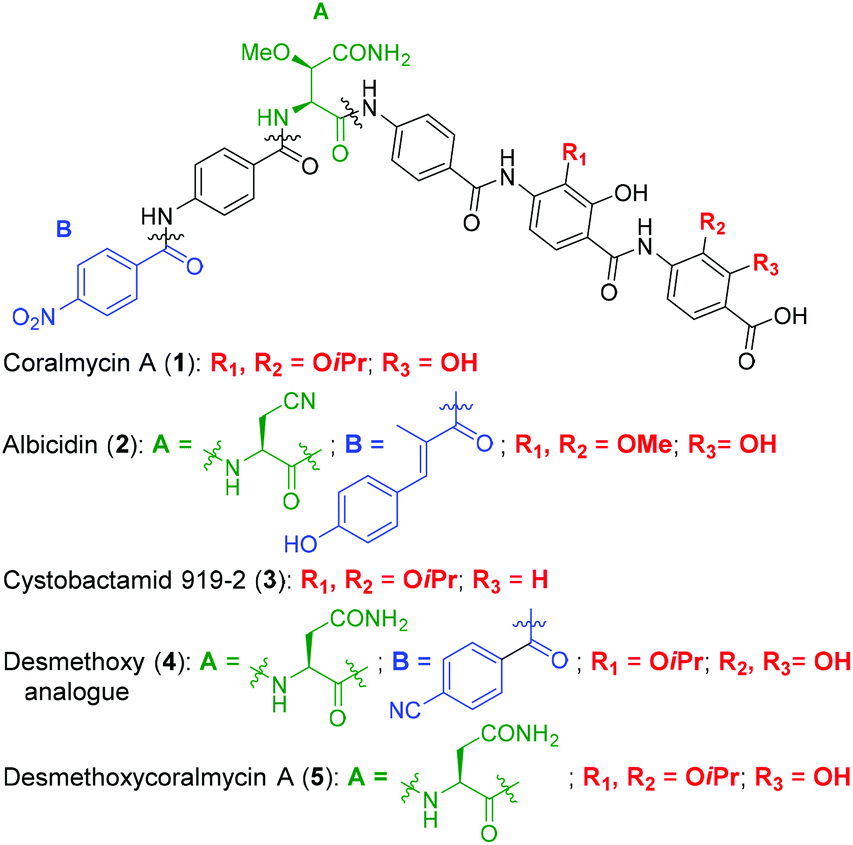 | ||
| Fig. 1 Structures of coralmycin A (1), albicidin (2), cystobactamid 919-2 (3), and desmethoxy analogue (4) reported by Brönstrup and co-workers.23 | ||
Structurally, coralmycin A is an entirely non-proteinogenic acyl pentapeptide, that predominantly comprises aromatic residues. Specifically, the natural product contains two p-aminobenzoic acid (PABA) moieties centred around an unusual erythro-β-methoxy-L-asparagine, an acyl p-nitrobenzoic acid at the pseudo-N-terminus, and two 4-amino-2-hydroxy-3-isopropoxybenzoic acids at the pseudo-C-terminus. Coralmycin A is structurally similar to a range of other acyl pentapeptides, namely, the cystobactamids and albicidin (2), which were isolated from the myxobacteria Cystobacter sp., Myxococcus sp., and the plant pathogen Xanthomonas albilineans, respectively.11–14
Albicidin (2), the cystobactamids, and coralmycin A have been independently shown to inhibit E. coli DNA gyrase and to a lesser extent, topoisomerase IV.11,15,16 Correct functioning of DNA gyrase is critical for transcription and replication, and is therefore essential for bacterial survival and is a well validated antibacterial target. Indeed, DNA gyrase is the target of the clinically approved fluoroquinolone family of antibiotics. Importantly, it has also been shown that the structurally related cystobactamid 919-2 (3) does not exhibit significant cytotoxicity against human liver hepatoblastoma cells (HepG2), fetal lung fibroblasts (MRC-9), and normal breast cells (MCF10A), with no activity against these lines observed at 50 μg mL−1.10
The potent antibacterial activity and promising selectivity profile of the cystobactamid/coralmycin/albicidin family of natural products has naturally led to significant attention on these molecules and the total synthesis of albicidin (2), and several members of the cystobactamid family have been reported.14,17–22 Very recently, this work was followed by an analogue study of the natural products, with one of the most promising desmethoxy analogues 4 (MIC 0.02 μg mL−1 against E. coli) synthesised on >100 mg scale and validated in an in vivo neutropenic thigh infection model in mice, with a four-log reduction in total CFU counts of E. coli at 20 mg kg−1 (administered as 4 × 6 hourly i.v. doses).23
Given the potent antibacterial activity of the coralmycin/cystobactamid family of natural products, we envisaged the development of a novel solid-phase synthetic route. It should be noted that all prior syntheses of these natural products employed solution-phase assembly.14,17–22 We rationalised that a solid-phase synthetic strategy would provide a robust means to access the natural products, but would also be amenable to rapid analogue generation on solid-support. In light of the impressive biological activity and lack of a current total synthesis, we chose to embark on the solid phase synthesis of coralmycin A (1), together with its desmethoxy analogue (5) as proof-of-principle for future analogue generation in this work (Fig. 1).
Results and discussion
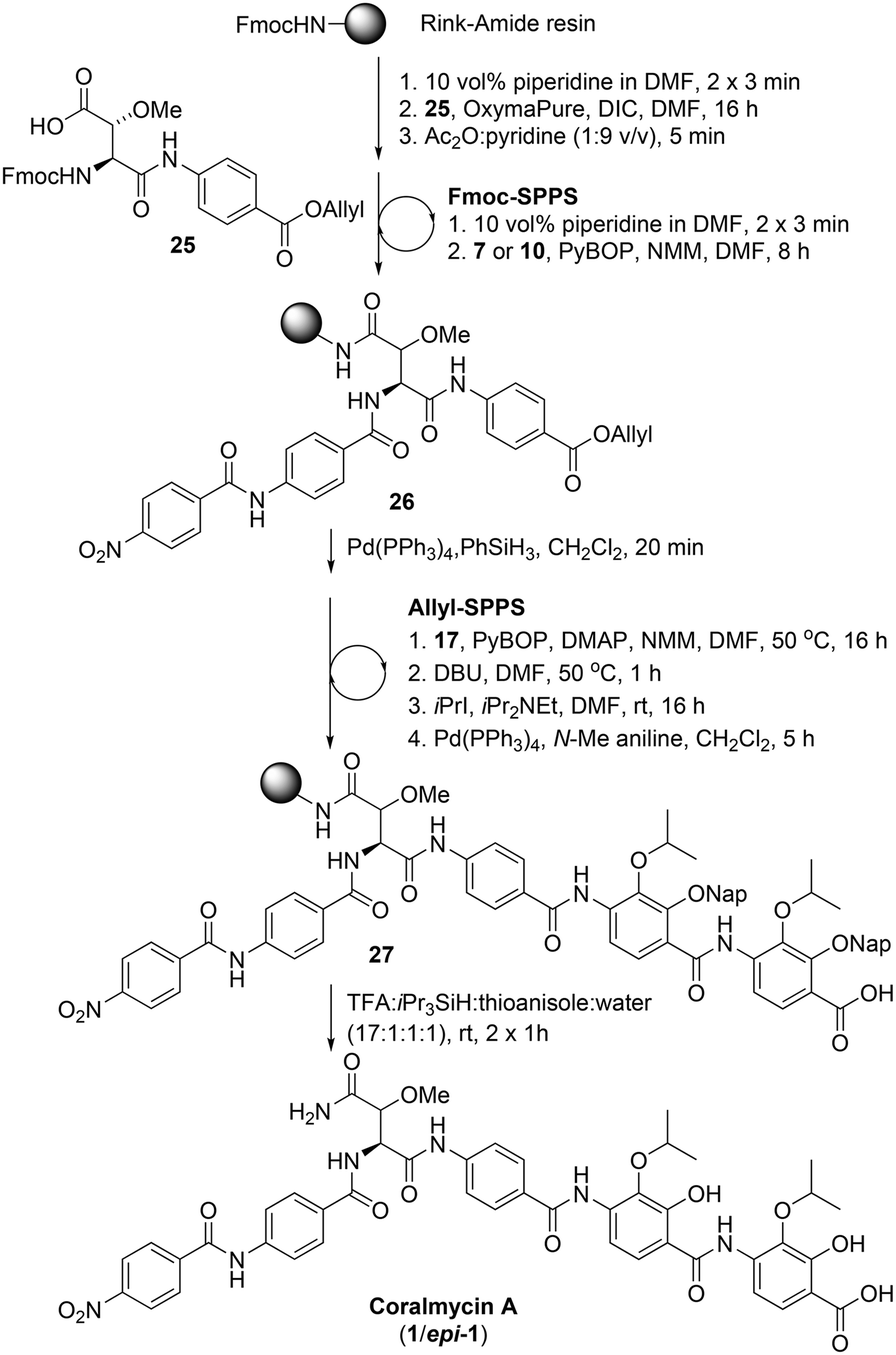
We proposed that coralmycin A (1) could be assembled on the solid-phase by immobilizing the central β-methoxy-asparagine (Asn) to resin and extending the linear peptide in both directions utilizing Fmoc-SPPS in the C to N direction and orthogonal ‘Allyl-SPPS’ in the N to C direction. It should be noted that ‘Allyl-SPPS’ as described refers to the proposed iterative Allyl-deprotection and subsequent amidation with the next amino acid. In order to investigate this strategy, we first attempted the synthesis of a simplified desmethoxy-model peptide, for which the requisite amino acid building blocks: Fmoc-L-Asp-PABA-OAll dipeptide (6), Fmoc-PABA-OH (7), and PABA-OAll (8) (a simplified derivative of 4-amino-2-hydroxy-3-isopropoxybenzoate) were all readily obtained in a few synthetic steps (see ESI† for full synthetic details).Synthesis of the model peptide commenced with the loading of the Fmoc-L-Asp-PABA-OAll dipeptide (6) onto deprotected Rink-amide polystyrene resin using N,N′-diisopropylcarbodiimide (DIC) and OxymaPure® (Scheme 1). It should be noted that we chose to load onto an acid-labile Rink-amide resin to provide the desired Asn functionality upon resin cleavage. Additionally, dipeptide 6 was loaded onto resin to prevent epimerization of the α-centre on-resin (as the amidation of anilines is known to be challenging and requires harsh coupling conditions).24 From here, the resin-bound peptide 9 could then be extended in either direction utilizing Fmoc-SPPS in the C to N direction to incorporate Fmoc-PABA-OH (7) and p-nitrobenzoic acid (10), and orthogonal ‘Allyl-SPPS’ in the N to C direction to incorporate two PABA-OAll (8) molecules in turn. While in principle either Fmoc-SPPS or ‘Allyl-SPPS’ could be performed first in the reaction sequence, we decided to begin by extending in the C to N direction via Fmoc-SPPS, as removal of the Fmoc-group would allow the resin-loading to be determined following the loading step. The peptide chain was extended in the C to N direction using standard Fmoc-SPPS conditions with benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate (PyBOP) as the coupling reagent and N-methylmorpholine (NMM) as the base to afford resin-bound peptide 11. At this stage, resin-bound peptide 11 was Allyl-deprotected via treatment with catalytic Pd(PPh3)4 and excess phenylsilane, after which coupling to PABA-OAll (8) in the N to C direction was investigated. However, attempts to forge this amide bond were unsuccessful (as judged by UPLC-MS analysis of a small amount of resin cleaved product); this was despite trialling an exhaustive range of coupling reagents and conditions including microwave irradiation and in situ acid fluoride generation (ESI Table S3†).25 This is likely due to a combination of steric and electronic effects, namely, the enhanced steric bulk around the reaction site, exacerbated by the pre-extension of the peptide in the C to N direction, and the poor nucleophilicity of the aniline coupled with the poorer electrophilicity of the resin-bound active ester (as compared to the corresponding active ester of p-nitrobenzoic acid that was coupled successfully in the C to N direction).
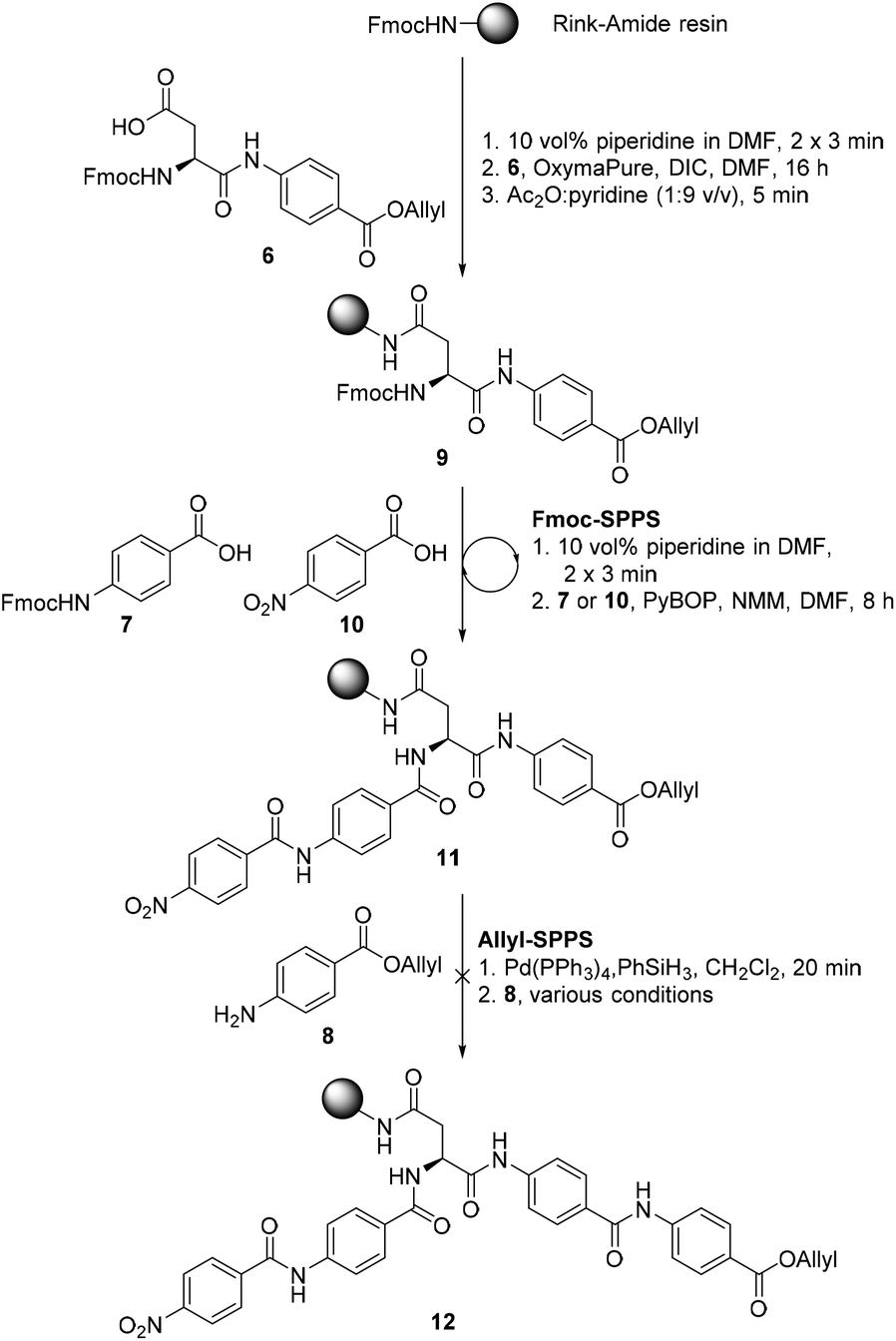 | ||
| Scheme 1 Attempted synthesis of the model peptide 12. | ||
To bypass the poor reactivity of the aniline nucleophile in the model system, we decided to make use of the superior reactivity of the ortho-phenol moiety to generate an O-acyl intermediate which could then be converted to the desired amide via an O to N acyl migration, as reported by Lee and Ahn in their solid-phase synthesis of tris-benzamides.26 This strategy is depicted in Scheme 2 and involves an on-resin O-acylation of a resin-bound benzoic acid (such as 13) to deliver the desired resin-bound O-acylated product 14, followed by a subsequent O to N shift sequence to deliver the requisite amide 15. The revealed phenol can then be alkylated on-resin to furnish the desired isopropyl ether (16) found in coralmycin A. We also envisaged that this synthetic approach would be amenable to the synthesis of natural product analogues as the on-resin O-alkylation would allow easy late-stage diversification.
 | ||
| Scheme 2 Proposed three-step O-acylation, O to N acyl shift, and O-alkylation to introduce 4-amino-2-hydroxy-3-isopropoxybenzoate to the resin-bound acid (PG = protecting group). | ||
To investigate this strategy, a suitably protected 4-amino-2,3-dihydroxybenzoate building block 17 was required (Scheme 3). Synthesis of 17 commenced from 2,3-dihydroxybenzaldehyde (18) with tert-butyldimethylsilyl (TBS)-protection at the 3-position proceeding cleanly with TBSCl and imidazole and subsequent Tosyl (Ts)-protection using TsCl and NEt3 providing the desired diprotected benzaldehyde 19 in 65% yield. Nitration using NaNO3, trifluoroacetic acid (TFA), and trifluoroacetic anhydride (TFAA) provided a single regioisomer (the regiochemistry of nitrobenzaldehyde 20 was confirmed via X-ray analysis of the crystalline compound, see ESI†).18 Benzaldehyde 20 was next protected with the photolabile nitroveratryl (Nv)-protecting group. Following Nv-protection, benzaldehyde 21 was treated with NaOMe to effect Ts deprotection and re-protected with a 2-naphthylmethyl (Nap) moiety, providing benzaldehyde 22 in quantitative yield. It should be noted that the Nap-protecting group was chosen due to its lability to the acid cleavage conditions used on-resin, allowing concomitant peptide cleavage and deprotection. At this point, benzaldehyde 22 was oxidized via Pinnick conditions, Allyl-protected, and Nv-deprotected under UV irradiation. Finally, treatment with Zn/NH4Cl facilitated the reduction of the aryl-nitro group providing the desired 4-amino-2,3-dihydroxybenzoate 17 in quantitative yield.
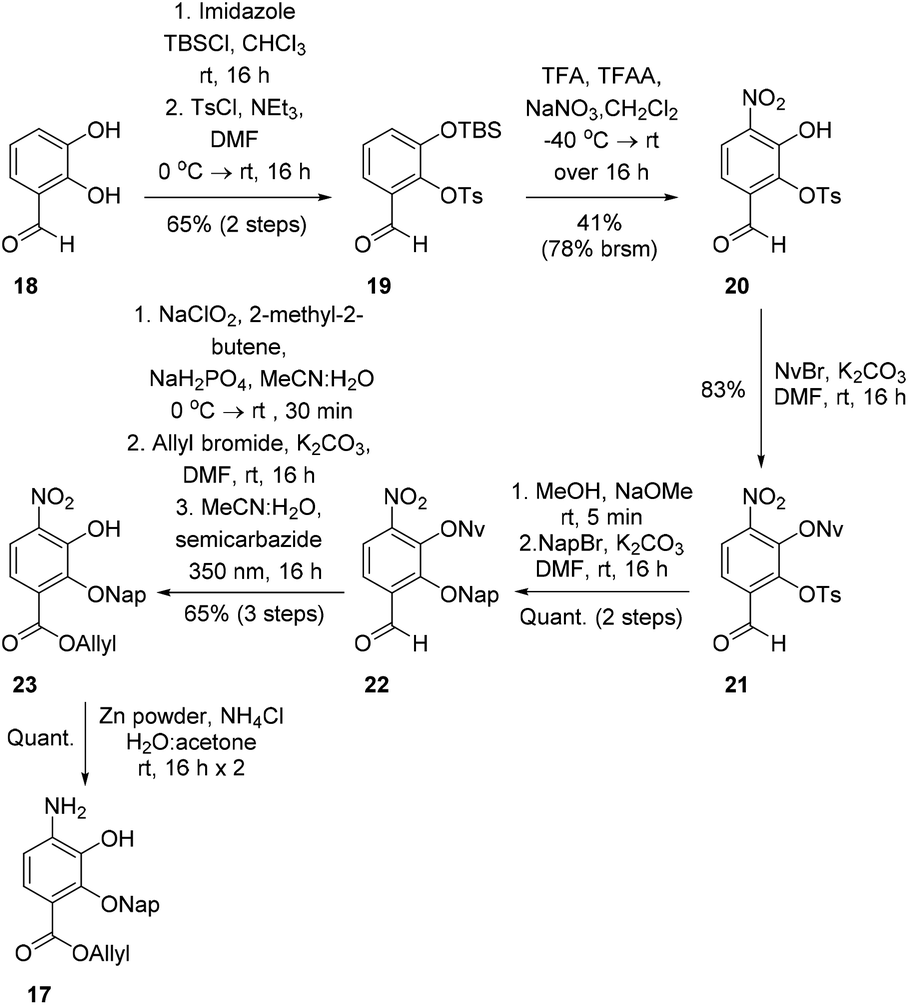 | ||
| Scheme 3 Synthesis of suitably protected 4-amino-2,3-dihydroxybenzoate 17. | ||
From here, the previously synthesised resin-bound 11 was extended to assess the new methodology for the synthesis of desmethoxycoralmycin A (5) (Scheme 4). To this end, 4-amino-2,3-dihydroxybenzoate 17 was coupled using PyBOP and 4-dimethylaminopyridine (DMAP) at 50 °C for 16 h to provide the O-acyl peptide. To effect the O to N shift, the resin was treated with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) for 1 h at 50 °C. To confirm O to N migration had occurred, several beads of resin from the starting material and product were separately treated with dilute hydrazine (2 vol% in H2O) for 30 min. As expected, the ester in the starting material was hydrazinolyzed to generate the acyl hydrazide with displacement of the amino acid. Meanwhile, the amide product was stable to hydrazine treatment. Next, alkylation was attempted, using iPrI and iPr2NEt, and gratifyingly this resulted in smooth conversion. Subsequent Allyl-deprotection using catalytic Pd(PPh3)4 and N-Me aniline as the scavenger (to prevent Nap-deprotection) was completed after 5 h. At this point, the previously described O-acylation, O to N migration, O-alkylation, and Allyl-deprotection were repeated to provide resin-bound peptide 24. Final cleavage from resin using TFA:iPr3SiH:H2O:thioanisole gave the desired peptide 5. It should be noted that the exclusion of thioanisole from the cleavage cocktail resulted in the formation of immutable Nap-adducts. Finally, reverse-phase HPLC purification, and lyophilization gave desmethoxycoralmycin A (5) in an 11% yield over 13 steps (average of 84% per step).
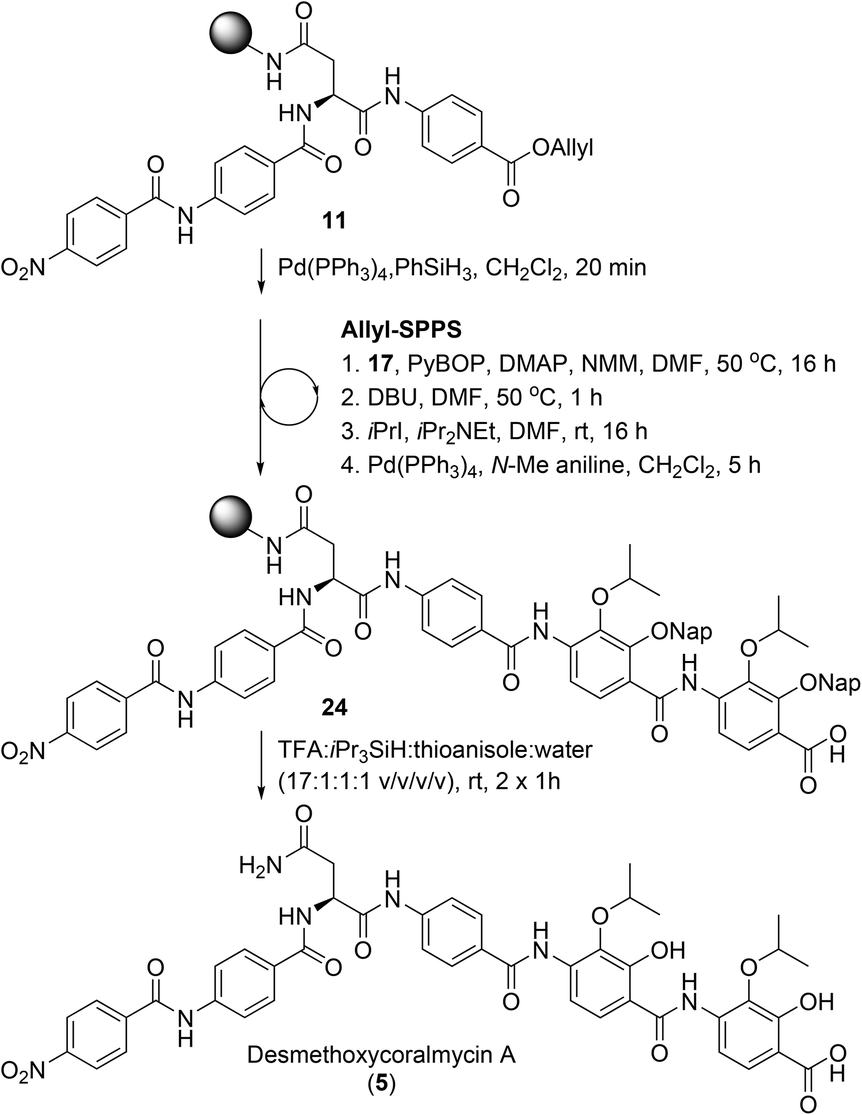 | ||
| Scheme 4 Solid-phase synthesis of desmethoxycoralmycin A (5). | ||
Following the successful synthesis of desmethoxycoralmycin A (5), the next goal was the synthesis of the natural product, coralmycin A (1). In order to achieve this, the Fmoc-erythro-β-methoxy-L-Asp-PABA-OAll dipeptide (25) was synthesised using a similar synthetic sequence to that previously reported by Wong and Taylor27 (see ESI† for full synthetic details). From here, using identical Fmoc-SPPS conditions as outlined previously and incorporating the dipeptide 25, the resin-bound peptide 26 was readily synthesized (Scheme 5). Likewise, Allyl-deprotection using catalytic Pd(PPh3)4 and phenylsilane and subsequent ‘Allyl-SPPS’ using the pre-optimized conditions (vide supra) instigated the stepwise O-acylation, O to N shift, O-alkylation, and Allyl-deprotection affording the resin-bound peptide 27. Finally, acidic resin-cleavage using TFA, thioanisole, iPr3SiH, and H2O; reversed-phase HPLC purification; and lyophilization gave the natural product coralmycin A in a 1.4% yield over 13 steps (average of 72% per step) as a 1:1 mixture of diastereomers (1/epi-1). It should be noted that the lower isolated yield of 1/epi-1 (as compared to 5) was due to poor recovery following purification by reverse-phase HPLC. Pleasingly, despite the appearance of an epimer, spectroscopic data for synthetic coralmycin A (1) was consistent with that reported for the isolated natural product.10 Comparison to the optical rotation and chiral HPLC data of the enantiopure desmethoxycoralmycin A (5) (see ESI†) suggests that the site of epimerization was at the β-centre of the β-methoxyAsn residue. The epimerization most likely occurred via an enolization pathway during extended treatment with DBU in the O to N shift (at 50 °C). While this was unexpected, this provided us with the opportunity to investigate whether this stereocentre was important for biological activity. As such, desmethoxycoralmycin A (5) and coralmycin A (1/epi-1), were assessed for their antibacterial activity against a panel of ten Gram-negative and seven Gram-positive bacteria (Table 1). These data showed that the epimeric mixture of coralmycin A exhibited potent antibacterial activity against the majority of the bacterial strains in the panel with MICs mostly in the low micromolar range. Pleasingly, the desmethoxycoralmycin A analogue (5) exhibited significantly more potent activity against all 17 of the organisms in the screen compared to 1/epi-1. This suggests that the removal of the functionality and stereochemistry at the β-centre of the β-methoxyAsn moiety is well tolerated. Importantly, desmethoxycoralmycin A displayed potent MICs against the WHO-listed critical Gram-negative pathogens A. baumannii, P. aeruginosa, and the Enterobacteriaceae: E. coli, K. aerogenes, K. pneumoniae, P. alcalifaciens, S. enterica, S. sonnei, and Y. pseudotuberculosis.1 Notably, 5 was more active against A. baumannii than the controls tested (5 = 1 μM; ampicillin = >128 μM; gentamicin = 8 μM; ciprofloxacin = 2 μM). This is especially promising given that A. baumannii has a high global prevalence of carbapenem-resistance and MDR strains and has an especially high mortality rate – it is the leading cause of ventilator-associated pneumonia and bloodstream/wound infections globally.1,9 Additionally, desmethoxycoralmycin A exhibited superior MICs against the WHO-listed high-priority Gram-positive bacteria MSSA, MRSA, and E. faecium (5 = 60 nM; penicillin G = 8 μM; vancomycin = 1 μM; and azithromycin = 10.7 μM against E. faecium).1
 | ||
| Scheme 5 Solid-phase synthesis of coralmycin A (1/epi-1). | ||
| MIC (μM) | ||
|---|---|---|
| Gram-negative bacteria | Coralmycin A (1/epi-1) | Desmethoxy coralmycin A (5) |
| E. coli | 2 | 0.83 |
| K. aerogenes | 21 | 1.7 |
| K. pneumoniae | >128 | 21 |
| P. alcalifaciens | 19 | 1 |
| S. enterica | 4 | 1 |
| S. sonnei | 1.5 | 0.15 |
| Y. pseudotuberculosis | 32 | 5.3 |
| A. baumanii | 8 | 1 |
| P. aeruginosa | 128 | 21 |
| V. cholerae | 1.3 | 0.46 |
| Gram-positive bacteria | ||
| B. subtilis | 2.2 | 0.5 |
| MSSA | 2.8 | 0.38 |
| MRSA | 3.3 | 0.8 |
| S. epidermidis | 2.7 | 0.5 |
| E. faecium | 0.33 | 0.06 |
| E. faecalis | 2 | 0.33 |
| S. pneumoniae | 0.13 | 0.05 |
Comparing the biological activity profiles of coralmycin A (1/epi-1) and desmethoxycoralmycin A (5) against commercial fluoroquinolone antibiotics that are known inhibitors of bacterial DNA gyrase and topoisomerase IV revealed unique biological attributes for the coralmycin class. Both the coralmycins and the fluoroquinolones exhibited similar activity profiles across the panel of bacterial pathogens, with potent broad-spectrum activities against most pathogens (ESI Table S5†). The fluoroquinolones displayed greater potency on average against Gram-negative organisms whereas coralmycins showed increased activity against Gram-positive bacteria. Strikingly however, the MIC values for (1/epi-1) and (5) did not differ appreciably between MSSA (1/epi-1 = 2.8 μM; 5 = 0.38 μM) and a clinical multidrug-resistant MRSA strain isolated from Portugal (1/epi-1 = 3.3 μM; 5 = 0.83 μM; Fig. 2). By contrast, the commercial fluoroquinolones demonstrated an average 32-fold increase in MICs between MSSA and the MDR-MRSA strain (Fig. 2). These results demonstrate that coralmycin A is a highly robust antibacterial scaffold with potent activity even against drug-resistant pathogens, and that the drug resistance profile for this scaffold is orthogonal to existing commercial antibiotics such as the fluoroquinolone class. For this reason, we believe desmethoxycoralmycin A to be an excellent candidate for further analogue generation against the critical Gram-negative and high priority Gram-positive pathogenic bacteria.
 | ||
| Fig. 2 Relative difference in MICs between coralmycin A (1/epi-1), desmethoxycoralmycin A (5), and commercial fluoroquinolones. MSSA = methicillin-sensitive S. aureus; MRSA = multidrug-resistant/methicillin-resistant S. aureus. Bars denote average MIC values across three biological replicates; error bars show standard error of the mean (SEM). Some compounds exhibit identical MICs across all replicates and SEMs of zero. | ||
Conclusion
Herein, we describe a practical solid-phase strategy towards coralmycin A (1/epi-1) and desmethoxycoralmycin A (5), employing a sidechain loaded Asp-dipeptide that could be subjected to bidirectional elongation by traditional Fmoc-SPPS, in tandem with an ‘Allyl-SPPS’ strategy. In addition, a three step: O-acylation, O to N shift, and O-alkylation strategy was developed that may serve as a useful means to perform late-stage modification of the phenolic sidechain for analogue generation in the future. Based on the potent antibacterial activity of desmethoxycoralmycin A, this work provides the starting point for the rapid generation of synthetic desmethoxy analogues which may stimulate the discovery of potential new antibacterial leads for the treatment of Gram-negative infections in the future.Experimental
Fmoc-SPPS general protocols
Fmoc-strategy solid-phase peptide synthesis (Fmoc-SPPS) procedures were employed on acid-labile Rink-amide polystyrene resin, within fritted syringes. All reagent equivalents are with respect to the amount of the initial amino acid loaded to resin.:100 with 10 vol% piperidine/DMF. The amount of compound loaded to resin was determined by measurement of the UV absorbance at λ = 301 nm of the diluted deprotection solution.
:thioanisole:H2O:iPr3SiH (17:1:1:1, v/v/v/v) 2 × 1 h. The cleavage solution was discharged, collected and concentrated under a stream of N2 and dried in vacuo.
2-((tert-Butyldimethylsilyl)oxy)-6-formylphenyl 4-methylbenzenesulfonate (19)
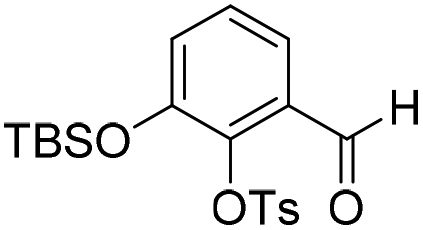
Ts-protection. The crude residue was dissolved in DMF (90 mL) and to this solution was added triethylamine (10 mL, 71.5 mmol) followed by TsCl (6.31 g, 33.1 mmol) at 0 °C. The resulting reaction mixture was warmed to room temperature and stirred for 16 h. The reaction solution was poured onto 2 M aqueous HCl (100 mL) and the organic layer was extracted with EtOAc (2 × 100 mL). The combined organic extracts were washed with water (100 mL), brine (100 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The crude residue was purified by flash chromatography (0–10% v/v EtOAc in n-hexane) to afford 19 (6.97 mg, 17.2 mmol, 65%) as a yellow gum. 1H NMR (CDCl3, 400 MHz) δ (ppm); 9.88 (s, 1H), 7.70 (m, 2H), 7.49 (m, 1H), 7.33 (m, 2H), 7.27 (m, 1H), 7.18 (m, 1H), 2.44 (s, 3H), 0.98 (s, 9H), 0.19 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ (ppm); 187.1, 149.3, 145.8, 142.3, 132.0, 131.4, 129.8, 128.3, 127.5, 126.8, 120.3, 25.4, 21.4, 18.1, −4.6; LR-MS: (+ESI) m/z 407 [M + H]+; HR-MS: (+ESI) calc. for C20H26O5SSi: 445.0912 [M + K]+, found: 445.0900 [M + K]+; IR (ATR):νmax = 2954, 2930, 2885, 2859, 1696, 1597, 1578, 1474, 1451, 1377, 1297, 1243 cm−1.
Aldehyde 19 (3.57 g, 8.79 mmol) and sodium nitrate (784 mg, 9.22 mmol) were dissolved in CH2Cl2 (8.6 mL) and to this solution was added trifluoroacetic anhydride (5.5 mL) dropwise at −40 °C, followed by trifluoroacetic acid (1.8 mL) dropwise at −40 °C. The resulting reaction mixture was stirred and warmed to room temperature over 16 h. The reaction solution was quenched by the slow addition of ice water. The reaction mixture was extracted with CH2Cl2 (2 × 100 mL). The combined organic extracts were washed with aqueous saturated NaHCO3 (100 mL), brine (100 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The crude residue was purified by flash chromatography (0–30% v/v EtOAc in n-hexane) to afford 20 (484 mg), 2-((tert-butyldimethylsilyl)oxy)-6-formyl-3-nitrophenyl 4-methylbenzenesulfonate intermediate (1.00 g) and recovered starting material (1.32 g). The 2-((tert-butyldimethylsilyl)oxy)-6-formyl-3-nitrophenyl 4-methylbenzenesulfonate intermediate (1.00 g) was treated with 2 M HCl
 :THF (1:1, v/v, 50 mL) and stirred at room temperature for 3 h, then extracted with EtOAc (2 × 50 mL). The combined organic extracts were washed with water (50 mL), brine (50 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo and combined with the title compound isolated previously to provide a total of (1.22 g, 3.63 mmol, 41%, 78% based on recovered starting material) as a bright yellow solid. 1H NMR (CDCl3, 500 MHz) δ (ppm); 10.53 (br s, 1H), 10.15 (s, 1H), 8.12 (d, J = 8.1 Hz, 1H), 7.87 (m, 2H), 7.50 (d, J = 8.1 Hz, 1H), 7.39 (m, 2H), 2.49 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ (ppm); 186.6, 150.0, 146.8, 141.6, 137.1, 136.0, 131.9, 130.2, 128.8, 123.4, 117.3, 21.9; LR-MS: (−ESI) m/z 336 [M − H]−; HR-MS: (−ESI) calc. for C14H11NO7S: 336.0184 [M − H]−, found: 336.0183 [M − H]−; IR (ATR):νmax = 3277, 1698, 1619, 1584, 1542, 1456, 1358, 1308, 1258 cm−1; mp: 137–143 °C. See crystallographic information file.
:THF (1:1, v/v, 50 mL) and stirred at room temperature for 3 h, then extracted with EtOAc (2 × 50 mL). The combined organic extracts were washed with water (50 mL), brine (50 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo and combined with the title compound isolated previously to provide a total of (1.22 g, 3.63 mmol, 41%, 78% based on recovered starting material) as a bright yellow solid. 1H NMR (CDCl3, 500 MHz) δ (ppm); 10.53 (br s, 1H), 10.15 (s, 1H), 8.12 (d, J = 8.1 Hz, 1H), 7.87 (m, 2H), 7.50 (d, J = 8.1 Hz, 1H), 7.39 (m, 2H), 2.49 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ (ppm); 186.6, 150.0, 146.8, 141.6, 137.1, 136.0, 131.9, 130.2, 128.8, 123.4, 117.3, 21.9; LR-MS: (−ESI) m/z 336 [M − H]−; HR-MS: (−ESI) calc. for C14H11NO7S: 336.0184 [M − H]−, found: 336.0183 [M − H]−; IR (ATR):νmax = 3277, 1698, 1619, 1584, 1542, 1456, 1358, 1308, 1258 cm−1; mp: 137–143 °C. See crystallographic information file.
6-Formyl-2-hydroxy-3-nitrophenyl 4-methylbenzenesulfonate (20) (868 mg, 2.58 mmol) was dissolved in DMF (5 mL) and to this solution was added 4,5-dimethoxy-2-nitrobenzylbromide (953 mg, 2.84 mmol) and K2CO3 (532 g, 3.87 mmol). The resulting reaction mixture was stirred at room temperature for 16 h. The reaction solution was acidified with 2 M aqueous HCl (100 mL) and the organic layer was extracted with EtOAc (2 × 100 mL). The combined organic extracts were washed with water (100 mL), brine (100 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The crude residue was purified by flash chromatography (0–30% v/v EtOAc in n-hexane) to afford the title compound (1.15 g, 2.15 mmol, 83%) as a bright yellow foam. 1H NMR (CDCl3, 500 MHz) δ (ppm); 9.97 (br s, 1H), 7.88 (d, J = 8.6 Hz, 1H), 7.83 (d, J = 8.6 Hz, 1H), 7.26 (s, 1H), 7.69 (m, 2H), 7.37 (s, 1H), 7.22 (m, 2H), 5.49 (s, 2H), 4.04 (s, 3H), 4.00 (3H), 2.39 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ (ppm); 185.7, 154.2, 148.2, 148.2, 147.1, 146.6, 146.2, 138.7, 134.5, 131.5, 130.4, 128.4, 127.7, 123.7, 123.6, 109.9, 107.8, 73.5, 56.8, 56.6, 21.9; LR-MS: (+ESI) m/z 555 [M + Na]+; HR-MS: (+ESI) calc. for C23H20N2O11S: 555.0680 [M + Na]+, found: 555.0680 [M + Na]+; IR (ATR):νmax = 3101, 2951, 1700, 1595, 1584, 1521 cm−1.
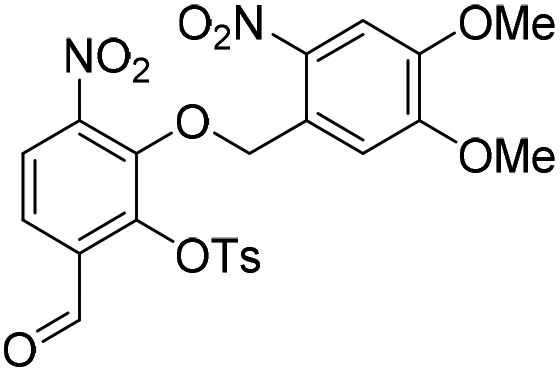

Ts-deprotection. To a stirred solution of aldehyde 21 (1.40 g, 2.62 mmol) was dissolved in MeOH (20 mL) was added 30% NaOMe in MeOH (2 mL). Upon addition the solution went from yellow to bright orange and after stirring at room temperature for a further 5 min the reaction solution was acidified with 2 M aqueous HCl (100 mL), before the organic layer was extracted with EtOAc (2 × 100 mL). The combined organic extracts were washed with water (100 mL), brine (100 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo.
Nap-protection. The crude residue was dissolved in DMF (7.5 mL) and to this solution was added 2-(bromomethyl)naphthalene (639 mg, 2.89 mmol) and K2CO3 (398 mg, 2.89 mmol). The resulting reaction mixture was stirred at room temperature for 16 h. The reaction solution was acidified with 2 M aqueous HCl (100 mL) and the organic layer was extracted with EtOAc (2 × 100 mL). The combined organic extracts were washed with water (100 mL), brine (100 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The crude residue was purified by flash chromatography (0–100% v/v CH2Cl2 in n-hexane) to afford naphthyl ether 22 (1.36 g, 2.62 mmol, quantitative over 2 steps) as a cream solid. 1H NMR (CDCl3, 400 MHz) δ (ppm); 10.23 (s, 1H), 7.80 (m, 1H), 7.77 (d, J = 8.5 Hz, 1H), 7.59–7.69 (m, 5H), 7.46–7.50 (m, 2H), 7.36 (m, 1H), 7.33 (s, 1H), 5.69 (s, 2H), 5.36 (s, 2H), 3.98 (s, 3H), 3.90 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ (ppm); 188.0, 156.2, 154.0, 148.7, 148.1, 146.2, 139.0, 133.8, 133.4, 133.1, 132.3, 128.9, 128.1, 127.9, 127.8, 127.7, 126.8, 126.7, 125.5, 123.6, 120.0, 109.6, 107.8, 78.3, 73.5, 56.7, 56.4; LR-MS: (+ESI) m/z 541 [M + Na]+; HR-MS: (+ESI) calc. for C27H22N2O9: 557.0957 [M + K]+, found: 557.0954 [M + K]+; IR (ATR):νmax = 2938, 1692, 1588, 1518 cm−1; mp: 143–146 °C.
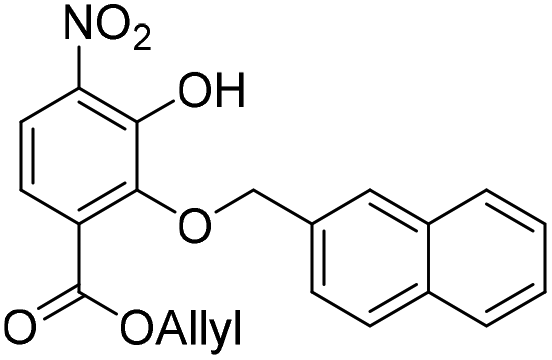
Oxidation. Aldehyde 22 (978 g, 1.90 mmol) was dissolved in MeCN
:H2O (15:1, v/v, 160 mL) before the addition of NaH2PO4 (1.35 g, 11.25 mmol), 2-methyl-2-butene (2 mL, 212 mmol) and sodium chlorite (288 mg, 3.18 mmol) at 0 °C. The resulting reaction mixture was warmed to room temperature and stirred for 30 min, then concentrated in vacuo. The crude residue was dissolved in EtOAc (100 mL) and washed with 2 M aqueous HCl (25 mL), water (25 mL) and brine (25 mL). The organic extract was dried over anhydrous Na2SO4, filtered and concentrated in vacuo.
Allyl-protection. The crude residue was dissolved in DMF (8 mL) and to this solution was added allyl bromide (242 μL, 2.8 mmol) and K2CO3 (389 mg, 2.81 mmol). The resulting reaction mixture was stirred at room temperature for 16 h. The reaction solution was acidified with 2 M aqueous HCl (100 mL) and the organic layer was extracted with EtOAc (2 × 100 mL). The combined organic extracts were washed with water (100 mL), brine (100 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo.
Nv-deprotection. The crude residue was dissolved in MeCN
:H2O (5:2, v/v, 185 mL) and to this solution was added semicarbazide hydrochloride (2.4 g, 21.5 mmol). The resulting reaction solution was irradiated for 16 h using 350 nm light bulbs. The crude residue was concentrated in vacuo and redissolved in water:EtOAc (1:1, v/v, 200 mL). The organic layer was separated, and the aqueous layer was further extracted with EtOAc (100 mL). The combined organic extracts were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The crude residue was purified by flash chromatography (0–10% v/v EtOAc in n-hexane) to afford the title compound 23 (469 mg, 1.24 mmol, 65% over 3 steps) as a bright yellow solid. 1H NMR (CDCl3, 400 MHz) δ (ppm); 10.85 (br s, 1H), 7.94 (m, 1H), 7.91 (d, J = 9.1 Hz, 1H), 7.84–7.90 (m, 3H), 7.67 (m, 1H), 7.50 (m, 2H), 7.28 (d, J = 9.1 Hz, 1H), 5.92 (ddt, J = 17.1, 11.6, 5.79, 1H), 5.37 (s, 2H), 5.29 (m, 2H), 4.76 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ (ppm); 164.6, 150.4, 148.6, 136.0, 134.1, 133.5, 133.4, 133.4, 131.5, 128.4, 128.2, 127.9, 127.7, 126.4, 126.4, 126.3, 119.8, 119.5, 119.4, 76.2, 66.7; LR-MS: (−ESI) m/z 378 [M − H]−; HR-MS: (−ESI) calc. for C21H17NO6: 378.0983 [M − H]−, found: 378.0983 [M − H]−; IR (ATR):νmax = 2917, 2849, 1716, 1535 cm−1; mp: 36–38 °C.
Compound 23 (109 g, 0.29 mmol) was dissolved in acetone
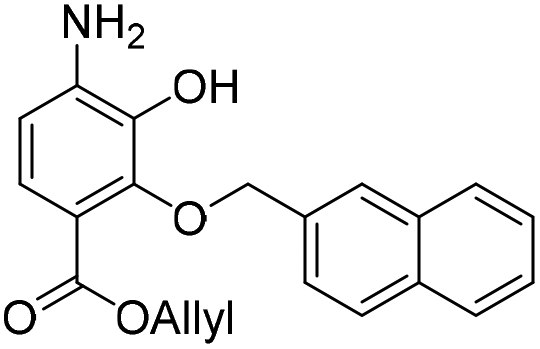 :H2O (5:1, v/v, 5.28 mL) and to this solution was added NH4Cl (236 mg, 4.41 mmol) and zinc solid (22 mg, 0.34 mmol). The resulting reaction mixture was stirred room temperature for 16 h. The reaction mixture was filtered and the precipitate washed with acetone (2 × 20 mL). The filtrate was concentrated in vacuo and resubjected to the above reaction conditions. After two treatments the reaction reached completion and was filtered and the precipitate washed with acetone (2 × 20 mL). The crude residue was dissolved in EtOAc (100 mL) and washed with water (50 mL) and brine (50 mL). The organic extract was dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The crude residue was purified by flash chromatography (0–20% v/v EtOAc in n-hexane) to afford the title compound 17 (100 mg, 0.29 mmol, quantitative) as a yellow oil. 1H NMR (CDCl3, 500 MHz) δ (ppm); 7.88–7.90 (m, 2H), 7.84–7.87 (m, 2H), 7.61 (m, 1H), 7.49–7.52 (m, 3H), 6.48 (d, J = 8.7 Hz, 1H), 6.05 (ddt, J = 17.3, 11.3, 5.6 Hz, 1H), 5.62 (br s, 1H), 5.33 (m, 2H), 5.20 (s, 2H), 4.81 (dt, J = 5.8, 1.3 Hz, 2H); 13C NMR (CDCl3, 125 MHz) δ (ppm); 165.1, 146.9, 140.0, 136.6, 134.7, 133.5, 132.7, 128.8, 128.2, 127.9, 127.7, 127.0 (HMBC), 126.5, 126.5, 126.2, 124.6, 118.3, 112.4, 110.1, 77.5, 65.3; LR-MS: (+ESI) m/z 350 [M + H]+; HR-MS: (+ESI) calc. for C21H19NO4: 372.1206 [M + Na]+, found: 372.1206 [M + Na]+; IR (ATR):νmax = 3485, 3377, 3057, 2980, 2932, 2882, 1699, 1618, 1510 cm−1.
:H2O (5:1, v/v, 5.28 mL) and to this solution was added NH4Cl (236 mg, 4.41 mmol) and zinc solid (22 mg, 0.34 mmol). The resulting reaction mixture was stirred room temperature for 16 h. The reaction mixture was filtered and the precipitate washed with acetone (2 × 20 mL). The filtrate was concentrated in vacuo and resubjected to the above reaction conditions. After two treatments the reaction reached completion and was filtered and the precipitate washed with acetone (2 × 20 mL). The crude residue was dissolved in EtOAc (100 mL) and washed with water (50 mL) and brine (50 mL). The organic extract was dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The crude residue was purified by flash chromatography (0–20% v/v EtOAc in n-hexane) to afford the title compound 17 (100 mg, 0.29 mmol, quantitative) as a yellow oil. 1H NMR (CDCl3, 500 MHz) δ (ppm); 7.88–7.90 (m, 2H), 7.84–7.87 (m, 2H), 7.61 (m, 1H), 7.49–7.52 (m, 3H), 6.48 (d, J = 8.7 Hz, 1H), 6.05 (ddt, J = 17.3, 11.3, 5.6 Hz, 1H), 5.62 (br s, 1H), 5.33 (m, 2H), 5.20 (s, 2H), 4.81 (dt, J = 5.8, 1.3 Hz, 2H); 13C NMR (CDCl3, 125 MHz) δ (ppm); 165.1, 146.9, 140.0, 136.6, 134.7, 133.5, 132.7, 128.8, 128.2, 127.9, 127.7, 127.0 (HMBC), 126.5, 126.5, 126.2, 124.6, 118.3, 112.4, 110.1, 77.5, 65.3; LR-MS: (+ESI) m/z 350 [M + H]+; HR-MS: (+ESI) calc. for C21H19NO4: 372.1206 [M + Na]+, found: 372.1206 [M + Na]+; IR (ATR):νmax = 3485, 3377, 3057, 2980, 2932, 2882, 1699, 1618, 1510 cm−1.
Compound 25 (157 μmol) was loaded to Rink-amide resin as per the general procedures. Resin loading was determined to be 37 μmol as per the general procedure.
The resin-bound compound (37 μmol) was elongated using the standard PyBOP coupling conditions and Fmoc-deprotection conditions, incorporating in turn: Fmoc-PABA-OH (7) and p-nitrobenzoic acid followed by allyl ester deprotection using the general procedure. Next, the resin-bound compound was elongated using the PyBOP/DMAP coupling conditions, incorporating the suitably protected 4-amino-2,3-dihydroxybenzoate 17. The O to N acyl shift was then performed, followed by OiPr alkylation and allyl ester deprotection according to the general procedures. The resin-bound compound was again elongated using the PyBOP/DMAP coupling conditions, incorporating the suitably protected 4-amino-2,3-dihydroxybenzoate 17. The O to N acyl shift, OiPr alkylation and allyl ester deprotection was performed once more via the general procedures. The resin-bound compound was then cleaved following the general procedure.
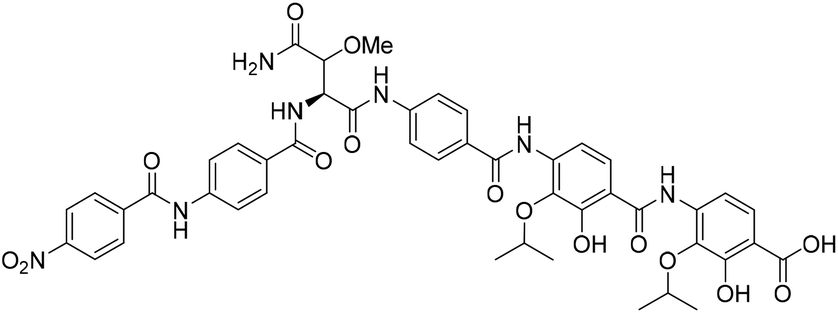
The crude compound was purified by preparative RP-HPLC using a Sunfire OBD 5 μm 19 × 150 mm (C18) column using a gradient of 48–60% MeCN in H2O (0.1% formic acid) over 36 min at a flow rate of 10 mL min−1 at 40 °C. The compound was lyophilized to give a 1:1 epimeric mixture of coralmycin A (1/epi-1) (0.49 mg, 1% over 13 steps from resin loading, average of 72% per step). 1H NMR for1andepi-1 (DMSO-d6, 500 MHz) δ (ppm); 10.92 (s, 1H), 10.60 (s, 0.5H), 10.51 (s, 0.5H), 9.16 (m, 0.5H), 8.55 (d, J = 6.3 Hz, 0.5H), 8.36 (d, J = 7.5 Hz, 2H), 8.22 (m, 2H), 7.42–7.92 (m, 12H), 5.02 (m, 0.5H, epi-1), 4.92 (m, 0.5H, 1), 4.69 (m, 1H), 4.65 (m, 1H), 4.27 (m, 0.5H, epi-1), 4.14 (d, J = 7.9 Hz, 0.5H, 1), 3.36 (s, 1.5H, epi-1), 3.30 (s, 1.5H, 1), 1.25 (d, J = 6.0 Hz, 6H), 1.23 (d, J = 6.1 Hz, 6H); 13C NMR for1andepi-1 (DMSO-d6, 125 MHz) δ (ppm); 165.3, 163.3, 149.6, 149.1, 140.1, 129.1, 128.1, 127.2, 125.6, 124.6, 124.1, 123.4, 119.5, 119.0, 108.5, 80.7 (epi-1), 80.0 (1), 72.7, 58.5 (epi-1), 57.8 (1), 56.2 (epi-1), 55.7 (1), 22.0; HR-MS: (−ESI) calc. for C46H45N7O15: 934.2901 [M − H]−, found: 934.2911 [M − H]−; IR (ATR):νmax = 3494, 3398, 2965, 1705, 1677, 1598, 1504 cm−1; [α]20D = +7.5° (c = 0.15 in THF:MeOH, 1:1, v/v); (see Table S1† for NMR characterization). This data is in agreement with that previously reported by Kim et al.10
Compound 6 (470 μmol) was loaded to Rink-amide resin as per the general procedures. Resin loading was determined to be 234 μmol as per the general procedure. The resin-bound compound (234 μmol) was elongated using the standard PyBOP coupling conditions and Fmoc-deprotection conditions, incorporating in turn: Fmoc-PABA-OH (7) and p-nitrobenzoic acid. The resin-bound compound was Allyl-deprotected via the general procedure. The resin was then divided and a small portion of resin (58 μmol) was carried onto the next step. Resin-bound compound (58 μmol) was elongated using the PyBOP/DMAP coupling conditions, incorporating the suitably protected 4-amino-2,3-dihydroxybenzoate 17. The O to N acyl shift was then performed, followed by OiPr alkylation and allyl ester deprotection via the general procedures. The resin-bound compound was again elongated using the PyBOP/DMAP coupling conditions, incorporating the suitably protected 4-amino-2,3-dihydroxybenzoate 17. O to N acyl shift, OiPr alkylation and allyl ester deprotection was then performed via the general procedures. The resin-bound compound was then cleaved following the general procedure.
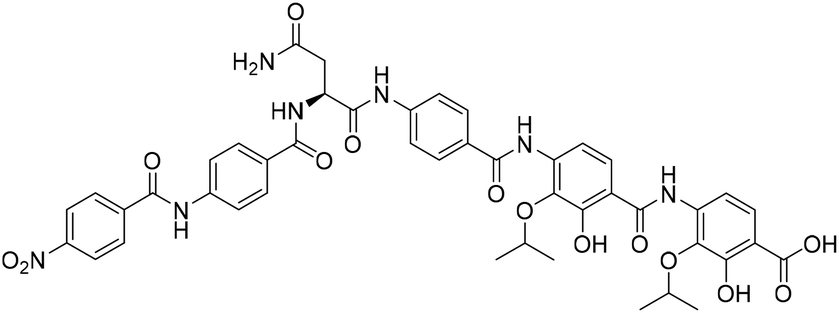
The crude compound was purified by preparative RP-HPLC using a Sunfire OBD 5 μm 19 × 150 mm (C18) column using a gradient of 48–60% MeCN in H2O (0.1% formic acid) over 36 min at a flow rate of 10 mL min−1 at 40 °C. The compound was lyophilized to give the natural product analogue 5 (5.80 mg, 11% over 13 steps from resin loading, average of 84% per step). 1H NMR (DMSO-d6, 500 MHz) δ (ppm); 11.28 (s, 1H), 10.81 (s, 1H), 9.6 (m, 1H), 8.69 (d, J = 7.3 Hz, 1H), 8.38 (d, J = 8.4 Hz, 2H), 8.22 (d, J = 8.4 Hz, 2H), 7.99 (m, 1H), 7.98 (m, 2H), 7.96 (m, 2H), 7.92 (m, 2H), 7.82 (m, 2H), 7.79 (m, 2H), 7.51 (d, J = 8.7 Hz, 1H), 7.42 (m, 1H), 7.42 (s, 1H), 6.98 (s, 1H), 4.92 (dd, J = 7.1, 7.1 Hz, 1H), 4.70 (m, 1H), 4.32 (m, 1H), 2.70 (d, J = 6.9 Hz, 2H), 1.25 (m, 12H); 13C NMR (DMSO-d6, 125 MHz) δ (ppm); 171.3, 170.6, 165.7, 164.3, 164.2, 163.6, 163.5, 151.7, 150.4, 149.3, 142.3, 141.5, 140.3, 138.2, 137.3, 136.2, 135.2, 129.3, 129.2, 128.4, 128.4, 128.3, 124.8, 123.7, 123.5, 119.6, 118.8, 117.3, 116.3, 115.0, 111.1, 71.5, 71.4, 51.7, 36.8, 21.9, 21.9; HR-MS: (−ESI) calc. for C45H43N7O14: 904.2795 [M − H]−, found: 904.2803 [M − H]−; IR (ATR): νmax = 3321, 2961, 2892, 1673, 1649, 1598, 1526, 1506 cm−1; [α]20D = −96° (c = 0.15 in THF:MeOH, 3:1, v/v); (see Table S2† for NMR characterization).
:1 dilution series and pinned into each assay plate (200 nL) using a high throughput pinning robot (Tecan Freedom EVO 100) to achieve final screening concentrations ranging from 128 μM to 3.91 nM per compound. In each 384-well plate; lane 1 was reserved for DMSO vehicle and culture medium; lane 2 reserved for DMSO vehicle, culture medium and target bacteria; lanes 23 and 24 reserved for antibiotic controls, DMSO vehicle, culture medium and target bacteria. After compound pinning, assay plates were read as T0 at OD600 using an automated plate reader (BioTek Synergy Neo2), sealed with lid and placed in a humidity-controlled incubator at 37 °C for 18–20 hours before optical density is obtained for T20. S. pneumoniae was incubated in a separate incubator (37 °C; 5% CO2). Resulting growth curves for each dilution series were used to determine the MIC values for all test compounds following standard procedures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Australian Research Council (through the Centre of Excellence for Innovations in Peptide and Protein Science, CE200100012; RJP) and the Natural Sciences and Engineering Research Council of Canada (NSERC Discovery; RGL) for funding. The authors acknowledge Dr Ian Luck (Sydney Analytical, The University of Sydney) for technical support for NMR spectroscopy, Dr Nick Proschogo (The University of Sydney) for technical support for mass spectrometry, and Dr Peter Turner (Sydney Analytical, The University of Sydney) for technical support for X-ray Crystallography. The authors also acknowledge the Australian Postgraduate Award and the John A. Lamberton Research Scholarship for PhD funding (PH).References
- WHO Prioritization of pathogens to guide discovery, research and development of new antibiotics for drug-resistant bacterial infections, including tuberculosis. (2017). World Health Organization < https://www.who.int/medicines/areas/rational_use/prioritization-of-pathogens/en/>.
- L. Brown, J. M. Wolf, R. Prados-Rosales and A. Casadevall, Nat. Rev. Microbiol., 2015, 13, 620–630 CrossRef CAS PubMed.
- L. L. Silver, Clin. Microbiol. Rev., 2011, 24, 71–109 CrossRef CAS PubMed.
- M. Bassetti, A. Vena, A. Croxatto, E. Righi and B. Guery, Drugs Context, 2018, 7, 212527–212544 Search PubMed.
- K. Lewis, Nat. Rev. Drug Discovery, 2013, 12, 371–387 CrossRef CAS PubMed.
- R. Raz, Clin. Microbiol. Infect., 2012, 18, 4–7 CrossRef CAS PubMed.
- Y. Imai, K. J. Meyer, A. Iinishi, Q. Favre-Godal, R. Green, S. Manuse, M. Caboni, M. Mori, S. Niles, M. Ghiglieri, C. Honrao, X. Ma, J. J. Guo, A. Makriyannis, L. Linares-Otoya, N. Böhringer, Z. G. Wuisan, H. Kaur, R. Wu, A. Mateus, A. Typas, M. M. Savitski, J. L. Espinoza, A. O'Rourke, K. E. Nelson, S. Hiller, N. Noinaj, T. F. Schäberle, A. D'Onofrio and K. Lewis, Nature, 2019, 576, 459–464 CrossRef CAS PubMed.
- 2020 Antibacterial agents in clinical and preclinical development: an overview and analysis. (2021). World Health Organization < https://www.who.int/medicines/areas/rational_use/prioritization-of-pathogens/en/>.
- H. W. Boucher, G. H. Talbot, J. S. Bradley, J. E. Edwards, D. Gilbert, L. B. Rice, M. Scheld, B. Spellberg and J. Bartlett, Clin. Infect. Dis., 2009, 48, 1–12 CrossRef PubMed.
- Y. K. Kim, H.-J. Kim, G.-W. Kim, K. Cho, S. Takahashi, H. Koshino and W.-G. Kim, J. Nat. Prod., 2016, 79, 2223–2228 CrossRef CAS PubMed.
- S. Baumann, J. Herrmann, R. Raju, H. Steinmetz, K. I. Mohr, S. Hüttel, K. Harmrolfs, M. Stadler and R. Müller, Angew. Chem., Int. Ed., 2014, 53, 14605–14609 CrossRef CAS PubMed.
- S. Cociancich, A. Pesic, S. Petras, S. Uhlmann, J. Kretz, V. Schubert, L. Vieweg, S. Duplan, M. Marguerettaz, J. Noëll, I. Pieretti, M. Hügelland, S. Kemper, A. Mainz, P. Rott, M. Royer and R. D. Süssmuth, Nat. Chem. Biol., 2015, 11, 195–197 CrossRef CAS PubMed.
- D. Petras, D. Kerwat, A. Pesic, B.-F. Hempel, L. von Eckardstein, S. Semsary, J. Arasté, M. Marguerettaz, M. Royer, S. Cociancich and R. Süssmuth, ACS Chem. Biol., 2016, 11, 1198–1204 CrossRef CAS PubMed.
- S. Hüttel, G. Testolin, J. Herrmann, T. Planke, F. Gille, M. Moreno, M. Stadler, M. Brönstrup, A. Kirschning and R. Müller, Angew. Chem., Int. Ed., 2017, 56, 12760–12764 CrossRef PubMed.
- S. M. Hashimi, M. K. Wall, A. B. Smith, A. Maxwell and R. G. Birch, Antimicrob. Agents Chemother., 2007, 51, 181–187 CrossRef CAS PubMed.
- B.-M. Kim, N. V. Minh, H.-Y. Choi and W.-G. Kim, Molecules, 2019, 24, 1390–1402 CrossRef CAS PubMed.
- J. Kretz, D. Kerwat, V. Schubert, S. Grätz, A. Pesic, S. Semsary, S. Cociancich, M. Royer and R. D. Süssmuth, Angew. Chem., Int. Ed., 2015, 54, 1969–1973 CrossRef CAS PubMed.
- B. Cheng, R. Müller and D. Trauner, Angew. Chem., Int. Ed., 2017, 56, 12755–12759 CrossRef CAS PubMed.
- M. Moeller, M. D. Norris, T. Planke, K. Cirnski, J. Herrmann, R. Müller and A. Kirschning, Org. Lett., 2019, 21, 8369–8372 CrossRef CAS PubMed.
- T. Planke, M. Moreno, S. Hüttel, J. Fohrer, F. Gille, M. D. Norris, M. Siebke, L. Wang, R. Müller and A. Kirschning, Org. Lett., 2019, 21, 1359–1363 CrossRef CAS PubMed.
- W. A. M. Elgaher, M. M. Hamed, S. Baumann, J. Herrmann, L. Siebenbürger, J. Krull, K. Cirnski, A. Kirschning, M. Brönstrup, R. Müller and R. W. Hartmann, Chem. – Eur. J., 2020, 26, 7219–7225 CrossRef CAS PubMed.
- L. von Eckardstein, D. Petras, T. Dang, S. Cociancich, S. Sabri, S. Grätz, D. Kerwat, M. Seidel, A. Pesic, P. C. Dorrestein, M. Royer, J. B. Weston and R. D. Süssmuth, Chem. – Eur. J., 2017, 23, 15316–15321 CrossRef CAS PubMed.
- G. Testolin, K. Cirnski, K. Rox, H. Prochnow, V. Fetz, C. Grandclaudon, T. Mollner, A. Baiyoumy, A. Ritter, C. Leitner, J. Krull, J. van den Heuvel, A. Vassort, S. Sordello, M. M. Hamed, W. A. M. Elgaher, J. Herrmann, R. W. Hartmann, R. Müller and M. Brönstrup, Chem. Sci., 2020, 11, 1316–1334 RSC.
- R. M. de Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed.
- H. Wenschuh, M. Beyermann, R. Winter, M. Bienert, D. Ionescu and L. A. Carpino, Tetrahedron Lett., 1996, 37, 5483–5486 CrossRef CAS.
- T.-K. Lee and J.-M. Ahn, ACS Comb. Sci., 2011, 13, 107–111 CrossRef CAS PubMed.
- D. Wong and C. M. Taylor, Tetrahedron Lett., 2009, 50, 1273–1275 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2087457. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ob01062j |
| This journal is © The Royal Society of Chemistry 2021 |